An Off-the-Shelf Approach for the Production of Fc Fusion Proteins by Protein Trans-Splicing towards Generating a Lectibody In Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
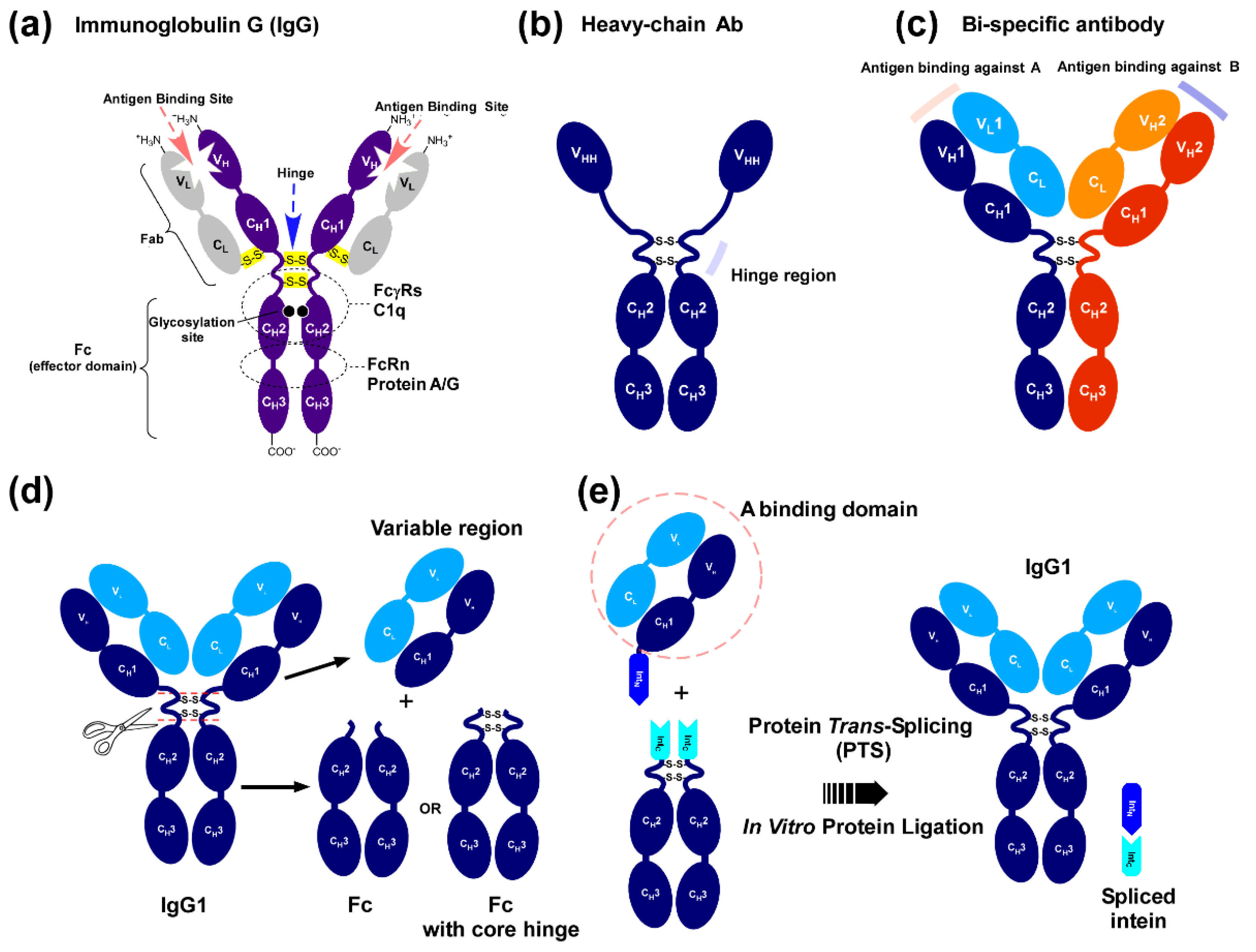
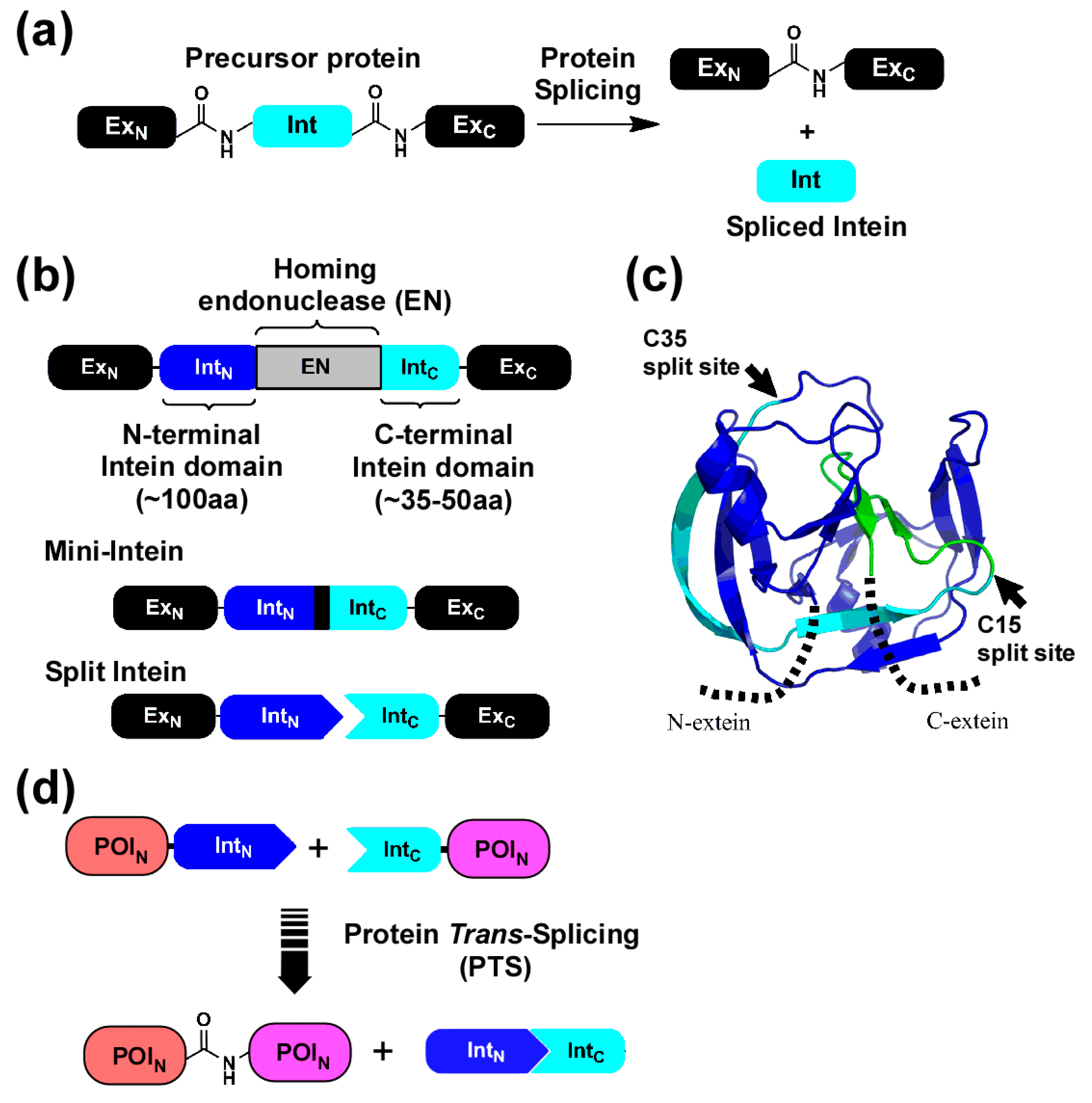
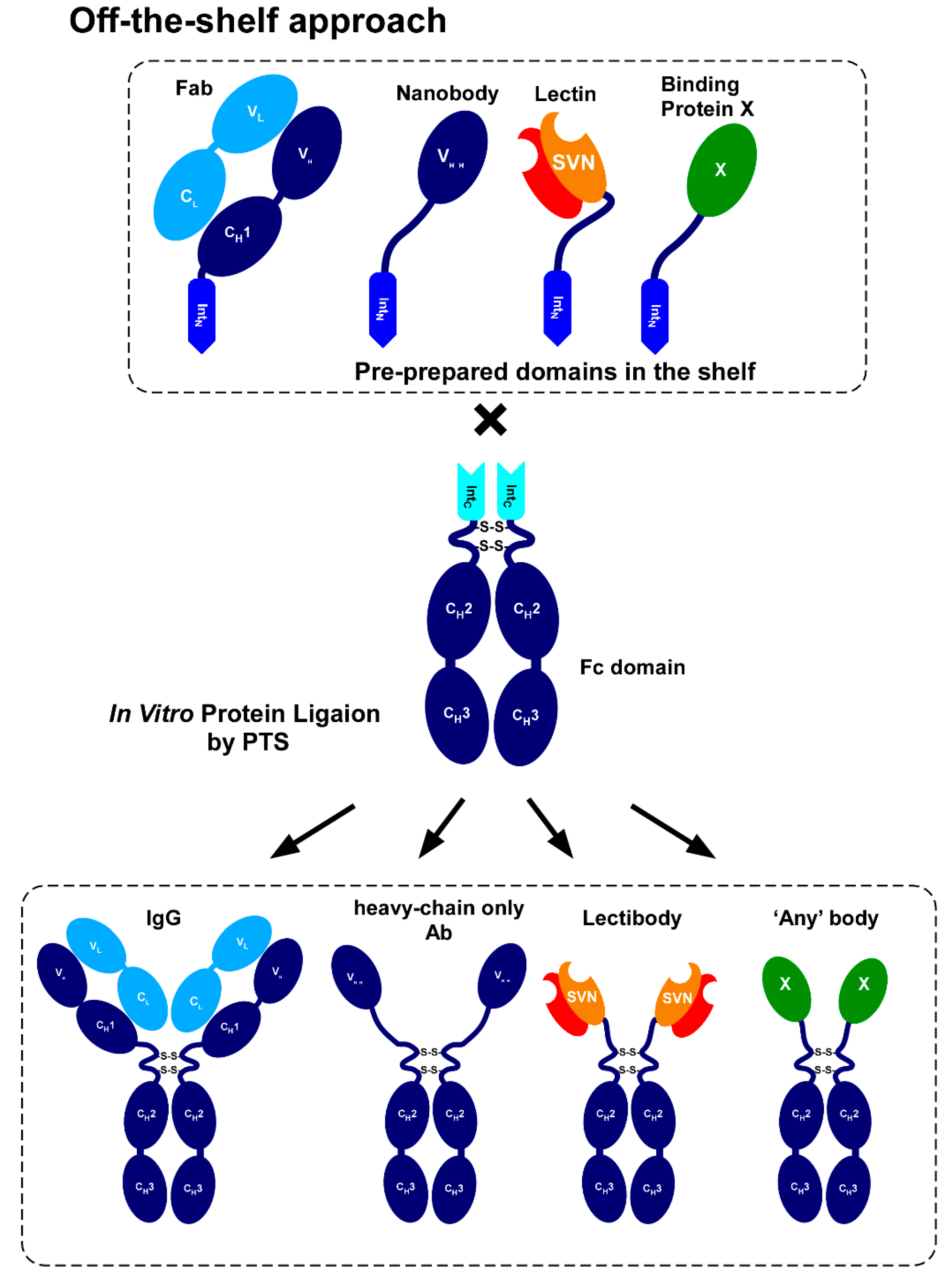
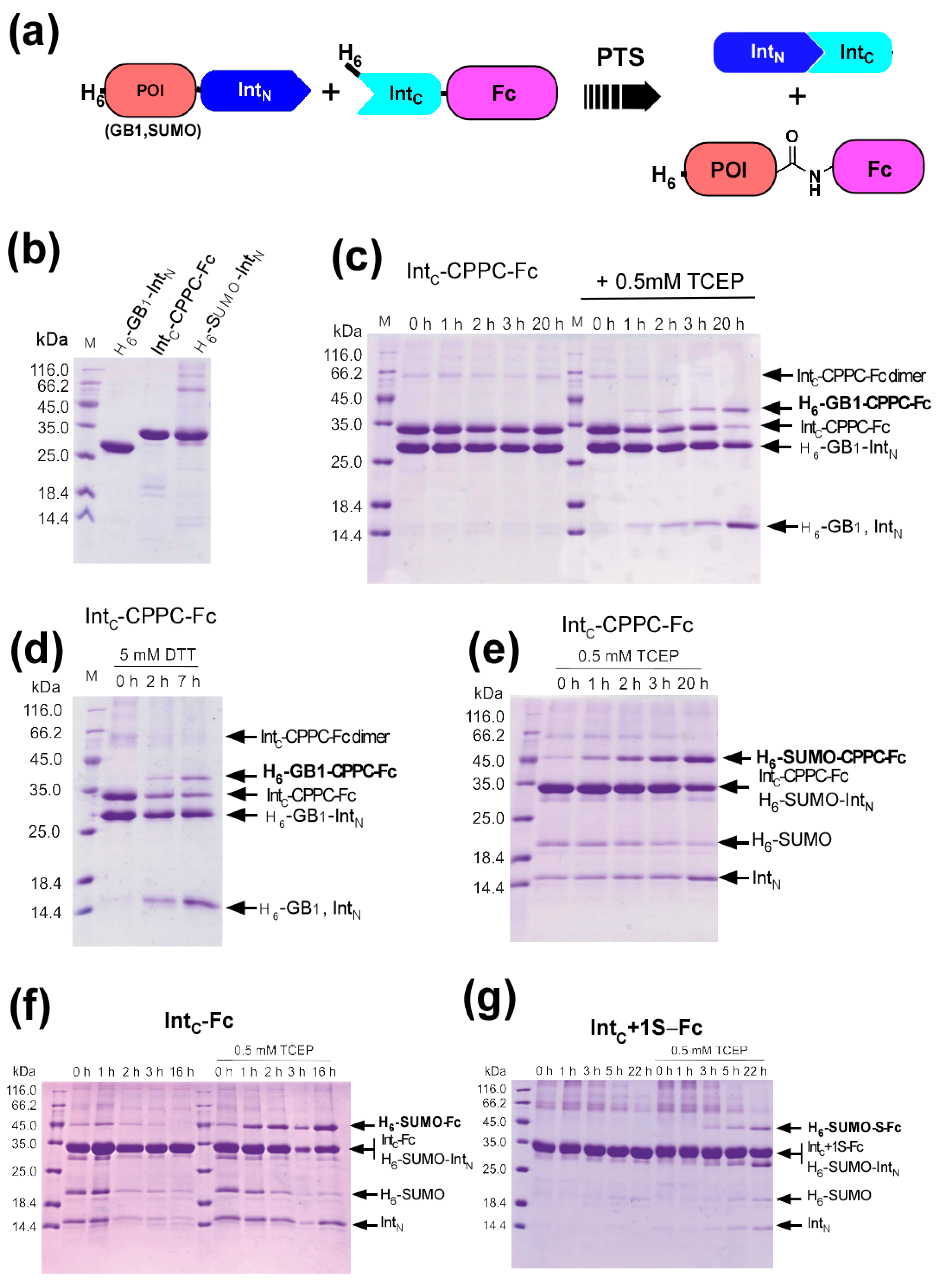
2.1. Strategy to Produce Fc Fusions In Vitro
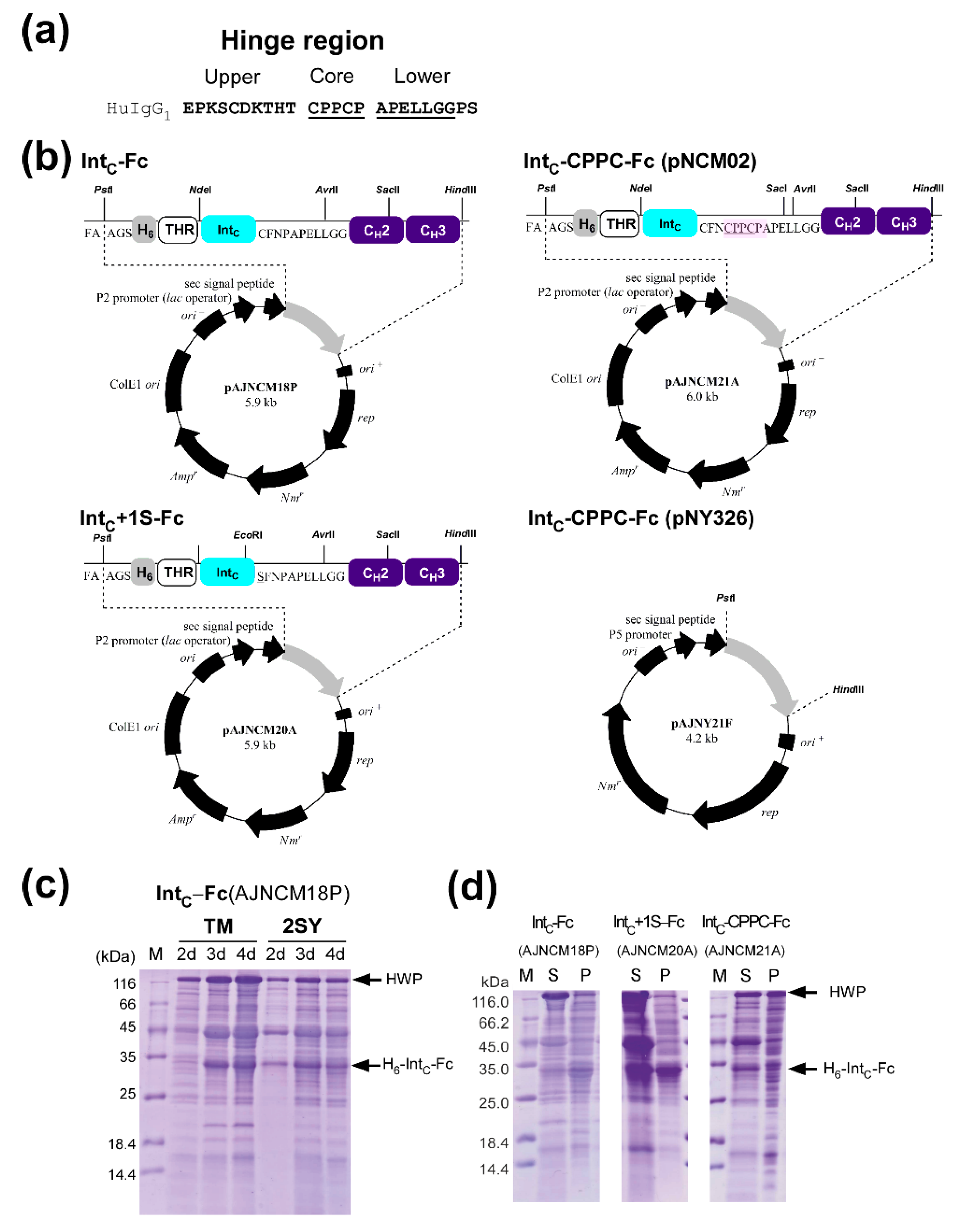
2.2. Cloning and Protein Expression of IntC-Fc in Brevibacillus choshinensis
2.3. Production of IntC-Fc for Protein Trans-Splicing
2.4. Purification of IntC-Fc Fusions
2.5. Model Proteins for Protein Ligation with IntC-Fc
2.6. Protein Ligation of IntC-CPPC-Fc by Protein Trans-Splicing
2.7. The Influence of the Core Hinge on Protein Trans-Splicing
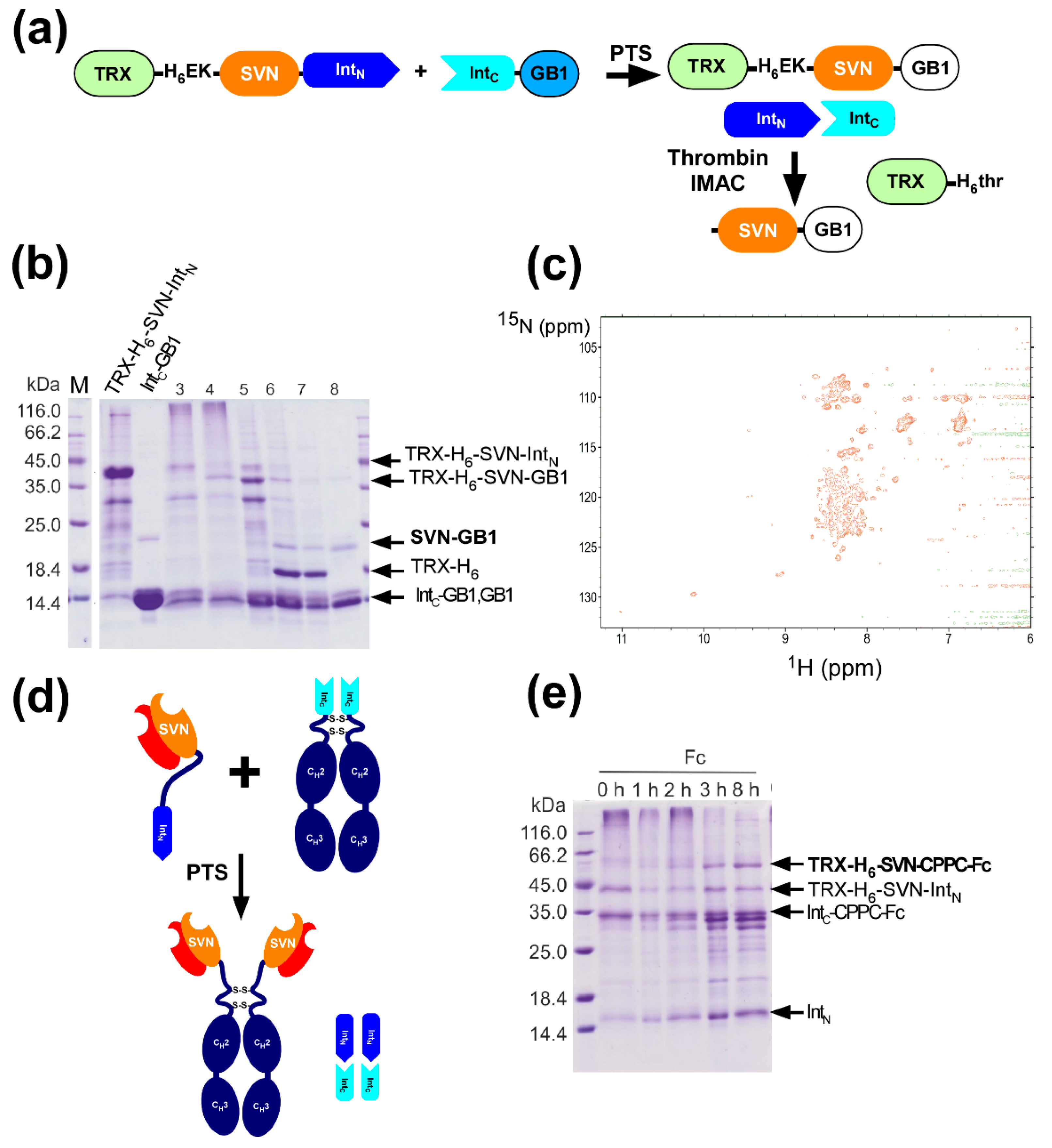
2.8. In Vitro Production of the Lectin Fusion Protein by Protein Trans-Splicing
2.9. In vitro Production of a Lectibody by Protein Trans-Splicing
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Construction of Plasmids
4.3. Expression and Purification of Fc Fusions from B. choshinensis
4.4. Expression and Purification of the N-Terminal Model Domains
4.5. Protein Trans-Splicing Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| VH | variable heavy chain domain |
| VL | variable light chain domain |
| VHH | the variable domain of heavy chain-only Ab |
| mAb | monoclonal antibody |
| BsAb | bispecific antibody |
| scFv | single-chain variable fragment |
| BiTE | bispecific T-cell engager |
| TRX | thioredoxin |
| NMR | nuclear magnetic resonance |
| HSQC | heteronuclear single quantum coherence |
| SVN | scytovirin |
| Fc | the Fc domain of Immunoglobulin G |
| GB1 | the B1 domain of IgG binding protein G |
| PTS | Protein trans-splicing |
References
- Schroeder, H.W., Jr.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [Green Version]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Kolkman, J.A.; Law, D.A. Nanobodies—From llamas to therapeutic proteins. Drug Discov. Today Technol. 2010, 7, e139–e146. [Google Scholar] [CrossRef] [PubMed]
- Beghein, E.; Gettemans, J. Nanobody technology: A versatile toolkit for microscopic imaging, Protein–Protein interaction analysis, and protein function exploration. Front. Immunol. 2017, 8, 771. [Google Scholar] [CrossRef]
- Jaakkonen, A. Production of Antibody Fc Fusion Proteins by Intein-Mediated Splicing. Master’s Thesis, Aalto University, Espoo, Finland, 2012. INSSI record number: 44596. Available online: http://web.lib.aalto.fi/en/oa/db/INSSI/ (accessed on 8 April 2020).
- Strohl, W.R. Current progress in innovative engineered antibodies. Protein Cell 2018, 9, 86–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Vijayasankaran, N.; Shen, A.Y.; Kiss, R.; Amanullah, A. Cell culture processes for monoclonal antibody production. MAbs 2010, 2, 466–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemoto, N.; Kumachi, S.; Arai, H. In vitro selection of single-domain antibody (VHH) using cDNA Display. In Methods in Molecular Biology; Nevoltris, D., Chames, P., Eds.; Antibody Engineering; Humana Press: New York, NY, USA, 2018; p. 1827. [Google Scholar]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Devel Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Hirata, R.; Ohsumk, Y.; Nakano, A.; Kawasaki, H.; Suzuki, K.; Anraku, Y. Molecular structure of a gene, VMA1, encoding the catalytic subunit of H(+)-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 6726–6733. [Google Scholar] [PubMed]
- Gogarten, J.P.; Senejani, A.G.; Zhaxybayeva, O.; Olendzenski, L.; Hilario, E. Inteins: Structure, function, and evolution. Annu. Rev. Microbiol. 2002, 56, 263–287. [Google Scholar] [CrossRef] [Green Version]
- Perler, F.B.; Davis, E.O.; Dean, G.E.; Gimble, F.S.; Jack, W.E.; Neff, N.; Noren, C.J.; Thorner, J.; Belfort, M. Protein splicing elements: Inteins and exteins--a definition of terms and recommended nomenclature. Nucleic Acids Res. 1994, 22, 1125–1127. [Google Scholar] [CrossRef] [PubMed]
- Aranko, A.S.; Wlodawer, A.; Iwaï, H. Nature’s recipe for splitting inteins. Protein Eng. Des. Sel. 2014, 27, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.W.; Camarero, J.A. Intein applications: From protein purification and labeling to metabolic control methods. J. Biol. Chem. 2014, 289, 14512–14519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciragan, A.; Aranko, A.S.; Tascon, I.; Iwaï, H. Salt-inducible protein splicing in cis and trans by inteins from extremely halophilic archaea as a novel protein-engineering tool. J. Mol. Biol. 2016, 428, 4573–4588. [Google Scholar] [CrossRef]
- Aranko, A.S.; Oeemig, J.S.; Zhou, D.; Kajander, T.; Wlodawer, A.; Iwaï, H. Structure-based engineering and comparison of novel split inteins for protein ligation. Mol. Biosyst. 2014, 10, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Chen, J.; Ding, K.; Zong, H.; Xie, Y.; Jiang, H.; Zhang, B.; Lu, H.; Yin, W.; Gilly, J.; et al. Efficient generation of bispecific IgG antibodies by split intein mediated protein trans-splicing system. Sci. Rep. 2017, 7, 8360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haga, N.; Asano, R.; Nakazawa, H.; Hattori, T.; Takeda, D.; Sugiyama, A.; Kurotani, R.; Kumagai, I.; Umetsu, M.; Makabe, K. Generation of camelid VHH bispecific constructs via in-cell intein-mediated protein trans-splicing. Protein Eng. Des. Sel. 2017, 30, 15–21. [Google Scholar] [CrossRef]
- Oeemig, J.S.; Aranko, A.S.; Djupsjöbacka, J.; Heinämäki, K.; Iwaï, H. Solution structure of DnaE intein from Nostoc punctiforme: Structural basis for the design of a new split intein suitable for site-specific chemical modification. FEBS Lett. 2009, 583, 1451–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, H.; Züger, S.; Jin, J.; Tam, P.H. Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. Febs Lett. 2006, 580, 1853–1858. [Google Scholar] [CrossRef] [Green Version]
- Aranko, A.S.; Züger, S.; Buchinger, E.; Iwaï, H. In vivo and in vitro protein ligation by naturally occurring and engineered split DnaE inteins. PLoS ONE 2009, 4, e5185. [Google Scholar] [CrossRef] [Green Version]
- Udaka, S.; Yamagata, H. High-level secretion of heterologous proteins by Bacillus brevis. Methods Enzym. 1993, 217, 23–33. [Google Scholar]
- Takagi, H.; Kadowaki, K.; Udaka, S. Screening and characterization of protein-hyperproducing bacteria without detectable exoprotease activity. Agric. Biol. Chem. 1989, 53, 691–699. [Google Scholar]
- Bies, C.; Lehr, C.M.; Woodley, J.F. Lectin-mediated drug targeting: History and applications. Adv. Drug Deliv. Rev. 2004, 56, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Akkouh, O.; Ng, T.B.; Singh, S.S.; Yin, C.; Dan, X.; Chan, Y.S.; Pan, W.; Cheung, R.C. Lectins with anti-HIV activity: A review. Molecules 2015, 20, 648–668. [Google Scholar] [CrossRef]
- McFeeters, R.L.; Xiong, C.; O’Keefe, B.R.; Bokesch, H.R.; McMahon, J.B.; Ratner, D.M.; Castelli, R.; Seeberger, P.H.; Byrd, R.A. The novel fold of scytovirin reveals a new twist for antiviral entry inhibitors. J. Mol. Biol. 2007, 369, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Xiong, C.; O’Keefe, B.R.; Botos, I.; Wlodawer, A.; McMahon, J.B. Overexpression and purification of scytovirin, a potent, novel anti-HIV protein from the cultured cyanobacterium Scytonema varium. Protein. Expr. Purif. 2006, 46, 233–239. [Google Scholar] [CrossRef]
- Hamorsky, K.T.; Kouokam, J.C.; Dent, M.W.; Grooms, T.N.; Husk, A.S.; Hume, S.D.; Rogers, K.A.; Villinger, F.; Morris, M.K.; Hanson, C.V.; et al. Engineering of a lectibody targeting high-mannose-type glycans of the HIV envelope. Mol. Ther. 2019, 2, 2038–2052. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Matsunaga, C.; Yoshino, A.; Kato, K.; Arata, Y. Dynamical structure of the hinge region of immunoglobulin G as studied by 13C nuclear magnetic resonance spectroscopy. J. Mol. Biol. 1994, 236, 300–309. [Google Scholar] [CrossRef]
- Verma, R.; Boleti, E.; George, A.J. Antibody engineering: Comparison of bacterial, yeast, insect and mammalian expression systems. J. Immunol. Methods 1998, 216, 165–181. [Google Scholar] [CrossRef]
- Gong, R.; Vu, B.K.; Feng, Y.; Prieto, D.A.; Dyba, M.A.; Walsh, J.D.; Prabakaran, P.; Veenstra, T.D.; Tarasov, S.G.; Ishima, R.; et al. Engineered human antibody constant domains with increased stability. J. Biol. Chem. 2009, 284, 14203–14210. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Cocco, M.J.; Rosenfied, R.; Lewis, J.K.; Ren, D.; Li, L.; Remmele, R.L., Jr.; Brems, D.N. Assignment of backbone 1H, 13C and 15N resonances of human IgG1 Fc (51.4 kDa). Biomol. Nmr. Assign. 2007, 1, 233–235. [Google Scholar] [CrossRef]
- Yashiro, K.; Lowenthal, J.W.; O’Neil, T.E.; Ebisu, S.; Takagi, H. High-level production of recombinant chicken interferon-g by Brevibacillus choshinensis. Protein. Expr. Purif. 2001, 23, 113–120. [Google Scholar] [CrossRef]
- Takara Brevibacillus Expression System; Product Manual; Takara Bio. Inc.: Shiga, Japan, 2015.
- Züger, S.; Iwai, H. Intein-based biosynthetic incorporation of unlabeld protein tags into isotopically labeled proteins for NMR studies. Nat. Biotech. 2005, 23, 736–740. [Google Scholar] [CrossRef]
- Muona, M.; Aranko, A.S.; Raulinaitis, V.; Iwaï, H. Segmental isotopic labelling of multi-domain and fusion proteins by protein trans-splicing in vivo and in vitro. Nat. Prot. 2010, 5, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.A.; Ramessar, K.; O’Keefe, B.R. Antiviral lectins: Selective inhibitors of viral entry. Antivir. Res. 2017, 142, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Hust, M.; Schirrmann, T. Expression of recombinant antibodies. Front. Immunol. 2013, 4, 217. [Google Scholar] [CrossRef] [Green Version]
- Kunert, R.; Reinhart, D. Advances in recombinant antibody manufacturing. Appl. Microbiol. Biotechnol. 2016, 100, 3451–3461. [Google Scholar] [CrossRef] [Green Version]
- Mohlmann, S.; Bringmann, P.; Greven, S.; Harrenga, A. Site-specific modification of ED-B-targeting antibody using intein-fusion technology. BMC Biotechnol. 2011, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, T.; Schmidt, J.; Ciesielski, E.; Becker, S.; Rysiok, T.; Schütte, M.; Toleikis, L.; Kolmar, H.; Doerner, A. Intein mediated high throughput screening for bispecific antibodies. MAbs 2020, 12, e1731938. [Google Scholar] [CrossRef] [Green Version]
- Busche, A.E.L.; Aranko, A.S.; Talebzadeh-Farooji, M.; Bernhard, F.; Dötsch, V.; Iwaï, H. Segmental isotopic labelling of a central domain in a multi-domain protein by protein trans-splicing using only one robust DnaE intein. Angew. Chem. Int. Ed. 2009, 48, 6128–6131. [Google Scholar] [CrossRef]
- Garrison, A.R.; Giomarelli, B.G.; Lear-Rooney, C.M.; Saucedo, C.J.; Yellayi, S.; Krumpe, L.R.; Rose, M.; Paragas, J.; Bray, M.; Olinger, G.G.; et al. The cyanobacterial lectin scytovirin displays potent in vitro and in vivo activity against Zaire Ebola virus. Antivir. Res. 2014, 112, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulaei, T.; Alexandre, K.B.; Shenoy, S.R.; Meyerson, J.R.; Krumpe, L.R.; Constantine, B.; Wilson, J.; Buckheit, R.W., Jr.; McMahon, J.B.; Subramaniam, S.; et al. Griffithsin tandemers: Flexible and potent lectin inhibitors of the human immunodeficiency virus. Retrovirology 2015, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Perler, F.B. InBase: The Intein Database. Nucleic Acids Res. 2002, 30, 383–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, A.; Kosugi, A.; Koizumi, Y.; Yanagida, F.; Udaka, S. High-efficiency transformation of Bacillus brevis by electroporation. Biosci. Biotechnol. Biochem. 1997, 61, 202–203. [Google Scholar] [CrossRef]
- Guerrero, F.; Ciragan, A.; Iwaï, H. Tandem SUMO fusion vectors for improving soluble protein expression and purification. Protein Expr. Purif. 2015, 116, 42–49. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaakkonen, A.; Volkmann, G.; Iwaï, H. An Off-the-Shelf Approach for the Production of Fc Fusion Proteins by Protein Trans-Splicing towards Generating a Lectibody In Vitro. Int. J. Mol. Sci. 2020, 21, 4011. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114011
Jaakkonen A, Volkmann G, Iwaï H. An Off-the-Shelf Approach for the Production of Fc Fusion Proteins by Protein Trans-Splicing towards Generating a Lectibody In Vitro. International Journal of Molecular Sciences. 2020; 21(11):4011. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114011
Chicago/Turabian StyleJaakkonen, Anniina, Gerrit Volkmann, and Hideo Iwaï. 2020. "An Off-the-Shelf Approach for the Production of Fc Fusion Proteins by Protein Trans-Splicing towards Generating a Lectibody In Vitro" International Journal of Molecular Sciences 21, no. 11: 4011. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114011