2.3.1. Docking into Homology Model
Ligand docking is a commonly used approach to identify ligand–protein interactions. However, in the case of P-gp, this could be challenging due to the high degree of flexibility and the large binding cavity consisting of multiple binding sites [
19,
32]. In addition to this, P-gp can bind more than one ligand simultaneously [
33,
34] and until 2019, when the cryoEM structure of human ABCB1 was resolved (PDB ID: 6QEX) [
12], there was a lack of a high resolution crystal structure of
hP-gp which rendered necessary the use of protein homology models, adding additional layers of uncertainty to the process. Nonetheless, a recent study has reported the use of homology models in virtual screening applications with a superior performance in comparison to crystal structures [
35]. This fact is explained by the conformational flexibility provided by homology models which allows a better accommodation of diverse ligands and therefore a better screening performance.
Two docking runs were performed utilizing two different algorithms, CDOCKER [
36] and GOLD [
37,
38]. In order to analyse the binding pocket of the
hP-gp, the simulations started with the docking of thirteen compounds, among them eight well-known molecules which interact with P-gp as substrates, inhibitors or both: cyclosporin A (CsA), amiodarone (AM), doxorubicin (DOX), digoxine (DIG), loperamide (LPM), rifampin (RMP), verapamil (VER), carvedilol (CAR), and five non-interacting compounds with P-gp: valproic acid (VPA), busulfan (BU), gentamicin (GEN), pamidronate (APD), and paraquat (PQ). The large binding pocket, observed in the
mP-gp crystal structure (PDB ID: 4M1M), binds the ligands at different sites with partially overlapping residues; some of them identical to those involved in rhodamine and verapamil binding [
39,
40,
41,
42]. Therefore, when defining the binding site for performing the docking simulations, the entire transmembrane (TM) region was considered. The binding region was delineated by those atoms within a radius of 15 Å and 24.7 Å, for CDOCKER and GOLD calculations respectively, utilizing the experimental coordinates of the co-crystallized
mP-gp ligand (PDB ID: 4XWK). The selection of the binding site and the settings for the docking simulations were validated via re-docking (ligand reproduction) procedure, obtaining heavy-atoms RMDS values of 1.5697 Å, 1.6021 Å, 1.6427 Å for CDOCKER calculations and 0.5527 Å, 0.7988 Å, 0.8498 Å for GOLD calculations (
Table S2, and Figure S4 in Supplementary Materials). The RMSD results are in agreement with the accepted threshold of 2 Å.
The resulting docking poses were subsequently rescored with fourteen additional scoring functions implemented in Discovery Studio 4.1 Client. The main fitness function used during the docking runs and the rescoring functions calculated for the resulting poses are listed in
Table 9.
Based on the sum of ranking differences (SRD) results, the best ranking poses were selected using the consensus ranking scheme, fusing the six best performing scoring fitness functions and using the geometric mean for computing the fused rank (
Table 10).
According to the SRD results, the best performing scoring functions for the CDOCKER run presented a very low probability that their performance was of random character, with values greater than 2.91E-15% and smaller or equal to 0.92% (see
Table 11); they performed better than random ranking, as they do not overlap with the cumulative relative frequency curve of a random ranking shown in
Figure 7a. For the GOLD results, the best performing scoring functions presented a probability of being of random character greater than 2.64%–10% and smaller or equal to 1.01%, as you can observe in
Table 12. Additionally, in this case, they do not overlap with the cumulative relative frequency curve of random ranking shown in
Figure 7b.
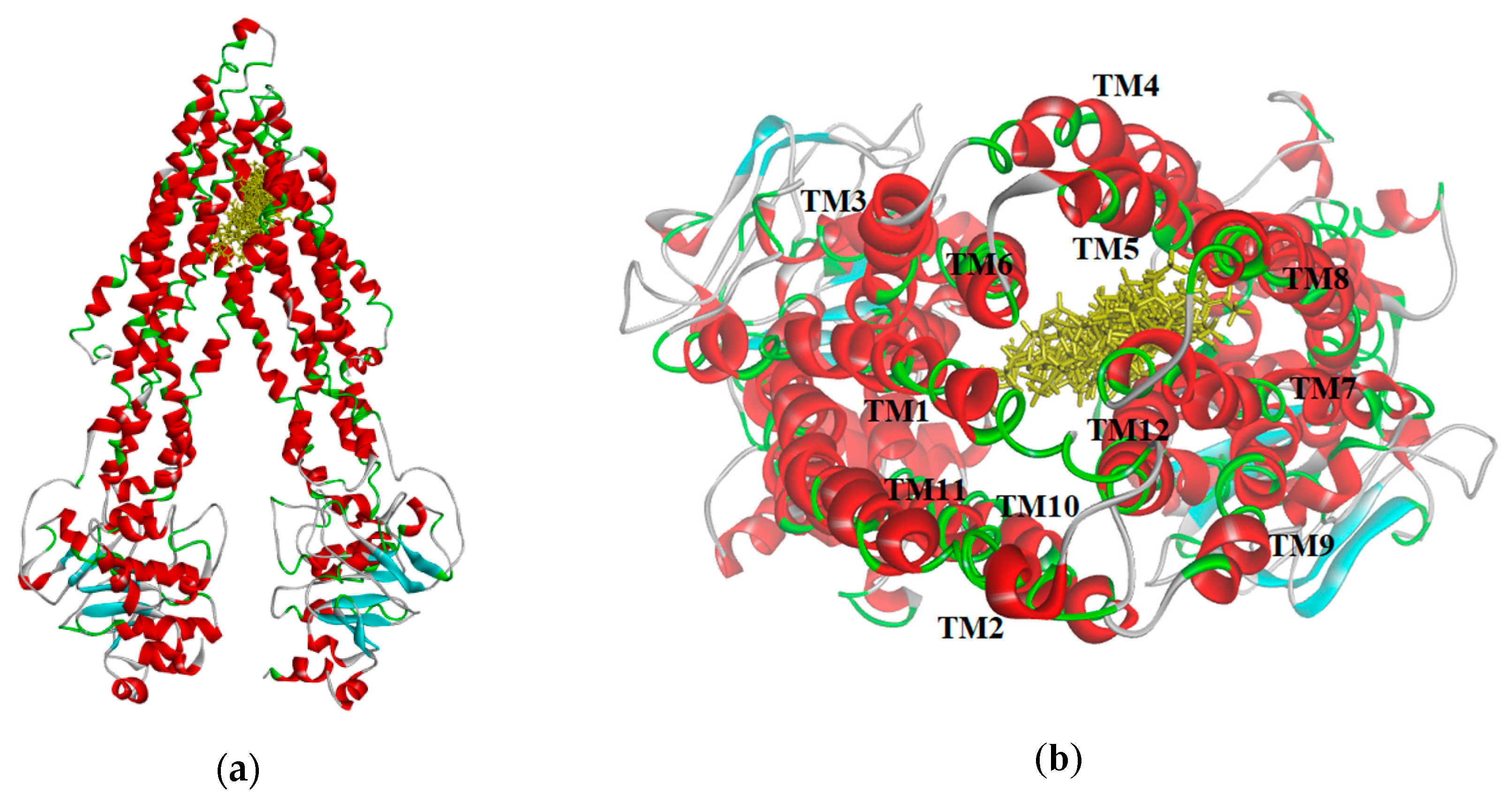
The resulting poses were distributed within the TM regions of P-gp (
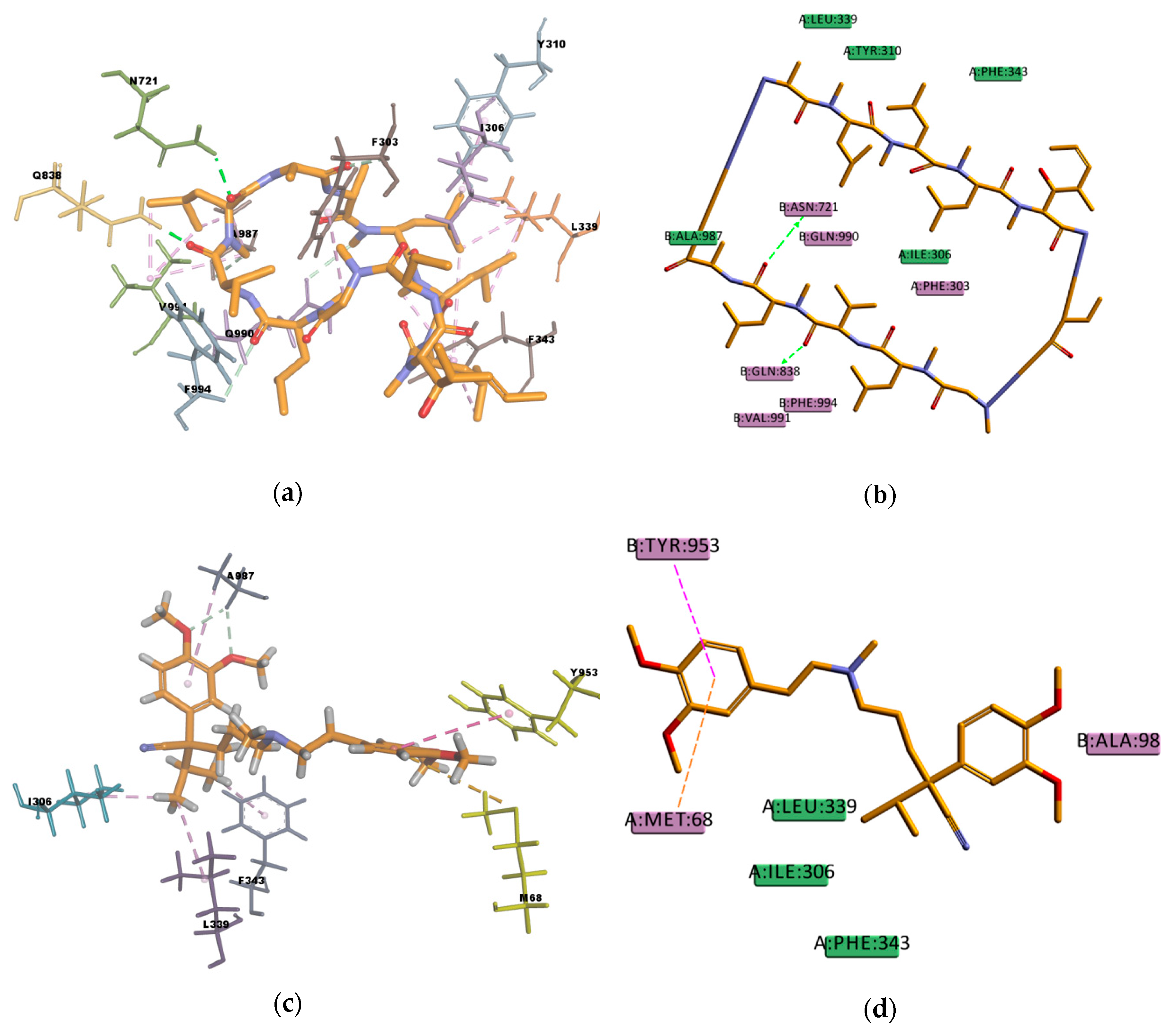
Figure 8), showing interactions with protein residues of multiple TM helices located throughout the binding region. Interacting amino acids were identified with the tool “ligand interactions” in Discovery Studio 4.1 Client. For the CDOCKER results, residues primarily located on TM helices 5, 6, 7, 8, 10, and 12 were involved in binding, while for the GOLD results additionally residues in TM 1, 9, and 11 were involved in the binding of VER and CsA. The pose obtained for CsA is showing one conventional hydrogen bond with Q838 (TM9) and the pose obtained for VER is showing one π–sulphur interaction with M68 (TM1) and one π–π interaction with Y953 (TM11) (
Figure 9).
The docking results obtained with the CDOCKER algorithm were comparable with those obtained with the GOLD algorithm in terms of calculated binding energies, as well as type of interactions between the docked compounds and
hP-gp. The estimated binding energies for the docking set, calculated using the Calculate Binding Energies protocol in Discovery Studio 4.1 and shown in
Table 13, are in close agreement in the two methodologies employed. The type of interactions was essentially the same in both cases (
Table 14,
Table S1); mainly hydrophobic π–sigma, π–alkyl, and π–π type of interaction with the presence of some hydrogen bond interactions.
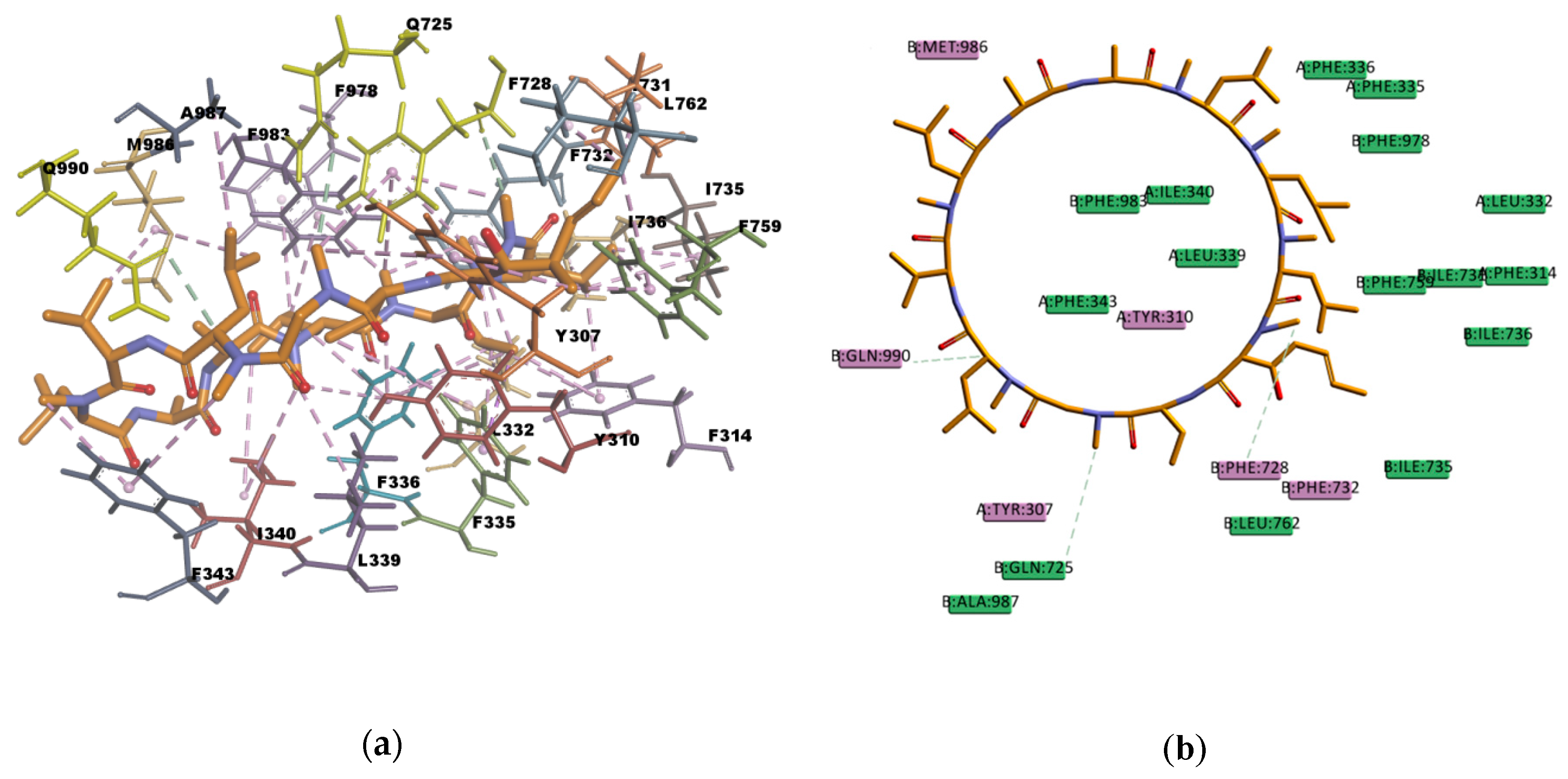
Based on the visual inspection of the selected docking poses, the interactions between the ligands and residues in the binding pocket are mainly of hydrophobic character. In the case of CsA, there are at least 20 residues involved in hydrophobic interactions (Y307, Y310, F314, L332, F335, F336, L339, I340, I343, F728, I731, F732, I735, I736, F759, L762, F978, F983, M986, and A987) which are reported in
Table 13 and can be seen in
Figure 10. CsA, though a big molecule, was found to have the lowest binding energy (‒268.516 kcal/mol) in the set of docked molecules. The stability of CsA in the binding site could be attributed to the large number of π interactions present, such as π–alkyl interactions, and to the simultaneous presence of hydrogen bonds in the binding pose. Even if the three hydrogen bonds present in the docked pose are carbon hydrogen bonds type C–H…O, i.e., weak interactions with a greater dispersive component, they may also play a role in stabilizing the ligand–protein complex. CsA is known to be a high affinity substrate of P-gp [
43,
44], hence the docking results obtained are in agreement with the available literature and with the experimental transport assay results reported in
Figure 11, where CsA leads to the lower excretion rate of rhodamine 123 (R123) out of Caco-2 cells. Nevertheless, it should be noted that Caco-2 cells also express other ABC transporters such as ABCG2 and ABCC1 which participate in the active efflux of R123 out of the cells and could be inhibited by CsA as these transporters share many similarities with P-gp.
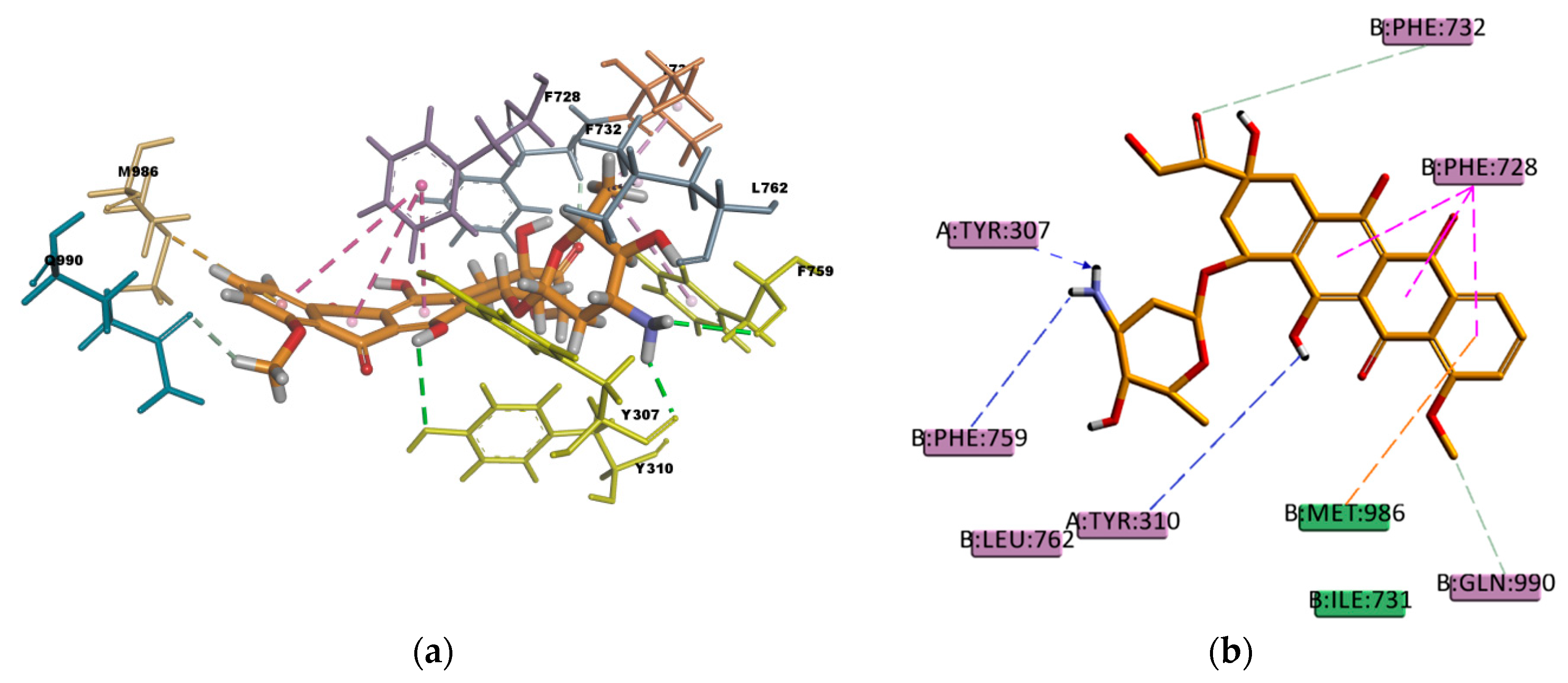
Similar as CsA, RMP is a big molecule as well, with a very favorable estimated binding energy of -214.872 kcal/mol. The docking pose reported in
Figure 12 shows hydrophobic interactions with 11 residues in the binding pocket (I340, M986, Y310, F732, Y307, F335, F336, F343, F759, F983, and F728. RMP presents a high number of π interactions in its binding mode, among them π–sigma, π–alkyl, and π–π interactions. Hydrogen bond interactions of weak character are also present in the binding pose, such as weak carbon hydrogen bonds, and one π–donor hydrogen bond between the hydroxyl group (donor) in RMP and the π electron cloud over the aromatic ring in F759 (acceptor). The sum of these interactions undoubtedly creates a strong cohesive environment, thereby stabilizing the complex formed. The docking results obtained are in agreement with experimental transport assay results reported in reference [
45] and with the literature available regarding RMP and P-gp interactions. RMP is known to be a substrate [
46] and inducer [
47] of P-gp.
In
Figure 13 the binding pose of DIG can be observed. DIG binding mode involves hydrophobic interactions with eight residues in the binding pocket (A987, F336, F343, F728, F732, F983, Y307, and Y310) and four hydrogen bond interactions. Three of them are conventional, electrostatic N–H…O or O–H…O type, of strong character, and one is π–donor hydrogen bond interaction of weaker character. The sum of these interactions contributes to the stability of the complex which is reflected in the value of the estimated binding energy of -211.755 kcal/mol. This results are in agreement with the literature available that reports DIG as a high affinity P-gp substrate [
48].
AM docked pose, shown in
Figure 14, presents many hydrophobic interactions involving ten residues in the binding pocket (Y307, Y310, F314, F728, I731, F732, I735, I736, F759, and F983), among them many π–alkyl type of interactions but also π–sigma and π–π type of interactions. The estimated binding energy is favorable but of smaller magnitude compared with CsA or RMP values. The difference in the binding energies could be explained by the lower number of hydrogen bonds in the binding mode; there is only one weak carbon hydrogen bond type of interaction C–H…O with the residue I731. This is also the case of LPM, which shows hydrophobic interactions with nine residues in the binding pocket (L762, F732, Y307, Y310, F728, F759, L339, I340, and F314) but just one weak carbon hydrogen bond type of interaction with the residue F728 (
Figure 15). The calculated binding energy of LPM is also favorable but smaller than CsA or RMP energy values, indicating less binding affinity and stability in comparison with them. The results obtained are in agreement with the available literature and with the experimental transport assay results reported in
Figure 11 and reference [
44] for AM and in reference [
45] for LPM. AM and LPM are known substrates of P-gp [
44,
49,
50].
On the other hand, DOX that forms hydrophobic interactions with just five residues in the binding pocket (I731, l762, F759, F728, and M986) has a very favorable binding energy of -196.862 kcal/mol. The stability in the binding pocket could be attributed to the presence of hydrogen bonds interactions involving five different residues in the docked pose (
Figure 16), three of them are strong conventional type of hydrogen bond, and two are weak carbon type of hydrogen bond. Additionally, DOX may be stabilized by the π–sulphur type of interaction present between the π electron cloud of one of the aromatic rings in the structure and the lone pair of electrons cloud of the sulphur atom in M986. DOX is known to be a substrate of P-gp [
51,
52] so the docking results obtained are in agreement with the literature available and with the experimental transport assay results reported in
Figure 11.
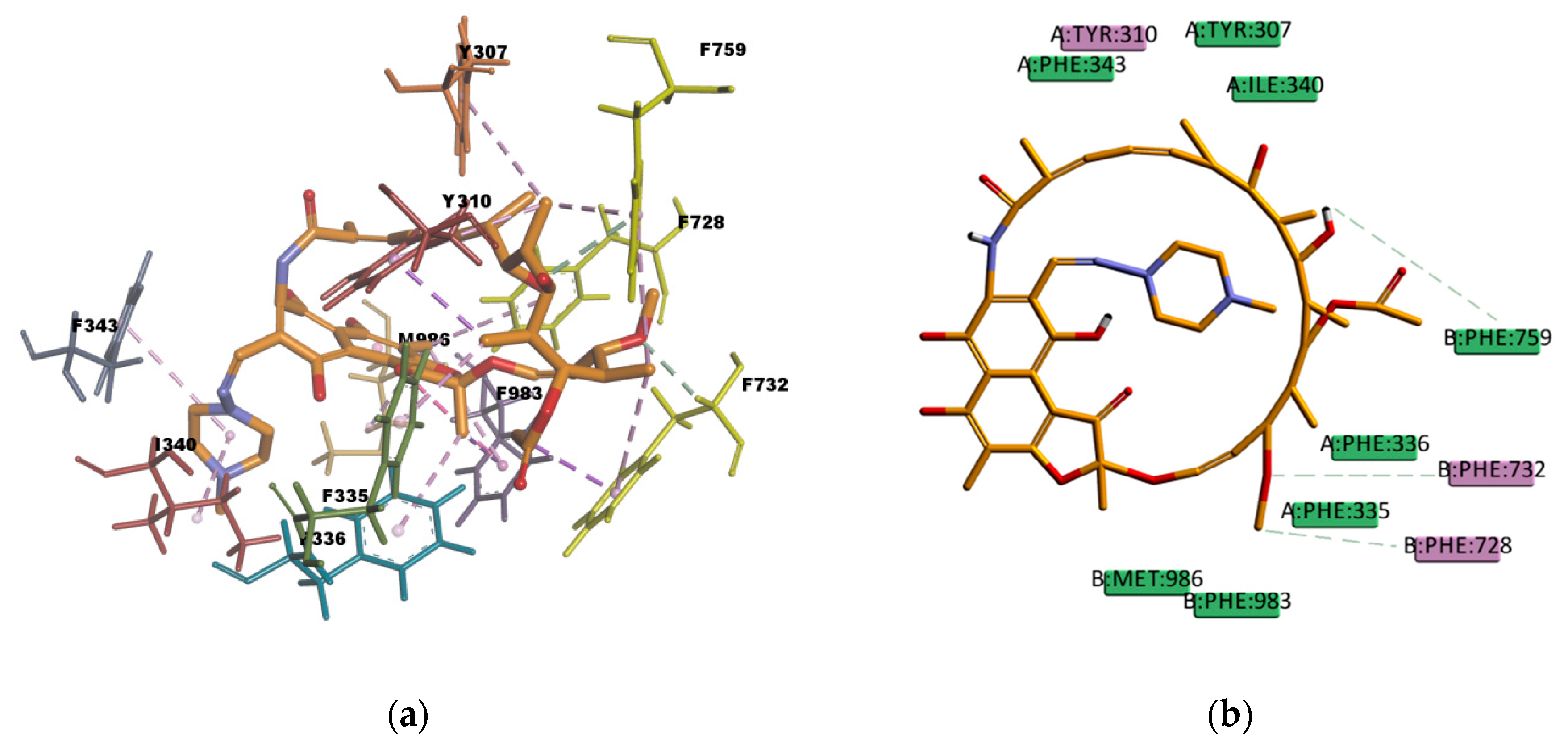
CAR, another well-known substrate of P-gp [
50], forms hydrophobic interactions with three residues (I306, F314, and F759) in the binding pocket; these interactions are mainly of π character, two of them are π–π type of interaction and one is π–alkyl type of interaction. The docking pose is also forming one π–sulphur type of interaction (
Figure 17) between the π electron cloud of one of the aromatic rings in the carbazol structure and the lone pair of electrons cloud of the sulphur atom in M986; π–sulphur interactions have been well-recognized to play an important role in chemical and biological recognition, as well as in drug development [
53,
54], thus they may have a big contribution in stabilizing the molecule into the receptor binding site. The binding mode of the VER pose (
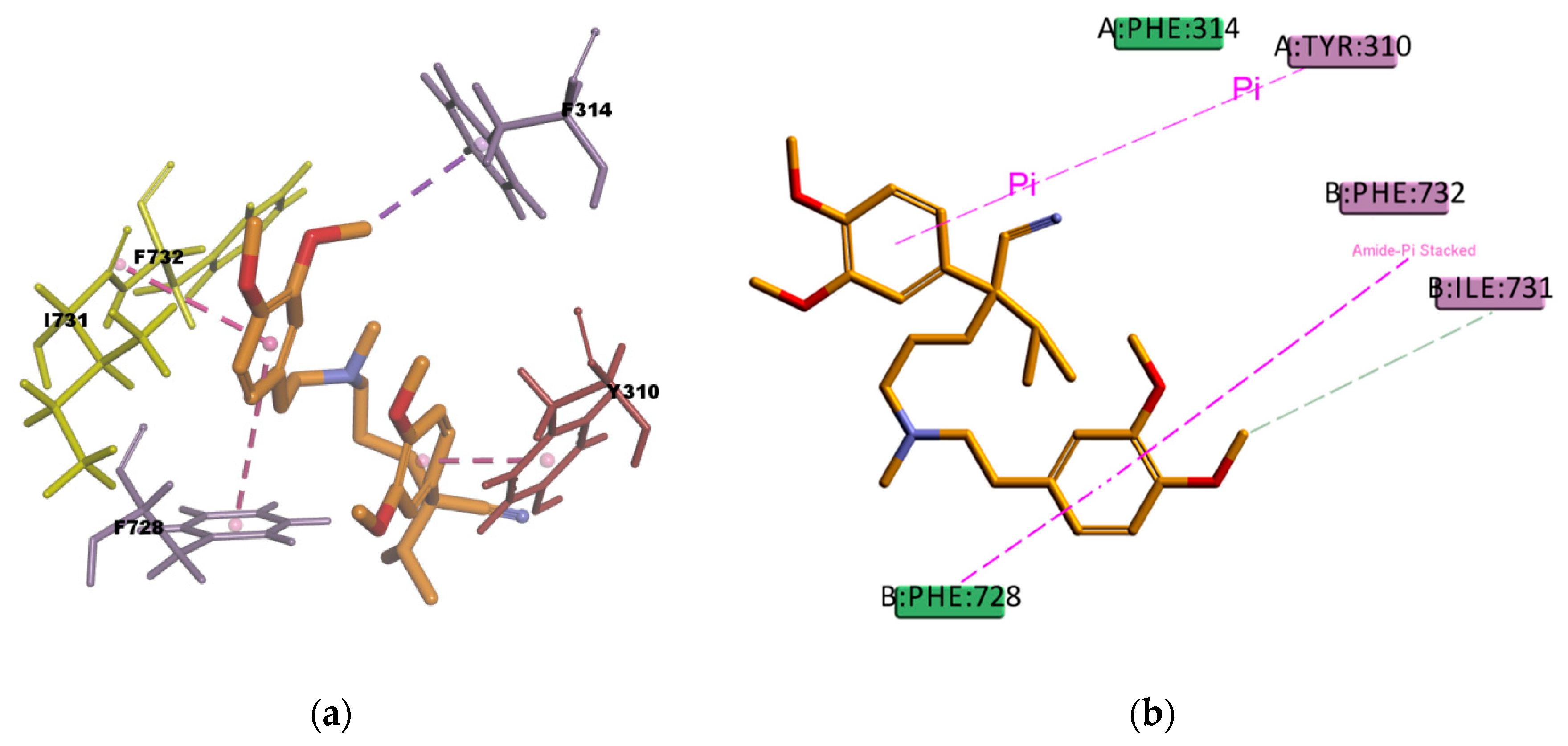
Figure 18) shows hydrophobic interactions with four residues (Y310, F314, F732, and F728) in the binding pocket, all of them π type of interactions, one π–sigma type of interaction, two π–π type of interaction, and one amide···π stacking interaction, in which the π-surface of the amide bond between residues I731 and F732 stacks against the π-surface of the one aromatic ring in VER. Amide···π stacking interactions are common and significant in protein structures [
55] and sometimes they play an important role in ligand binding [
56,
57]. VER binding mode also involves a weak carbon hydrogen bond interaction with residue I731, which contributes in stabilizing the ligand–protein complex. The estimated binding energies of CAR and VER, shown in
Table 14, indicate that the complexes are stable and with a good binding affinity. The docking results are in agreement with the literature available as CAR and VER are well-known substrates of P-gp [
44,
50].
Regarding PQ and GEN compounds, even though the experimental transport assay results in
Figure 11 show that these compounds at 50 µM do not interfere with the P-gp mediated efflux of R123 out of Caco-2 cells, the estimated binding energies have very favorable values. The literature available reveals different results regarding PQ and P-gp interaction (e.g., some authors stated that PQ is transported by P-gp [
58] while others stated that it is not a P-gp substrate [
59]). In the resulting binding pose (
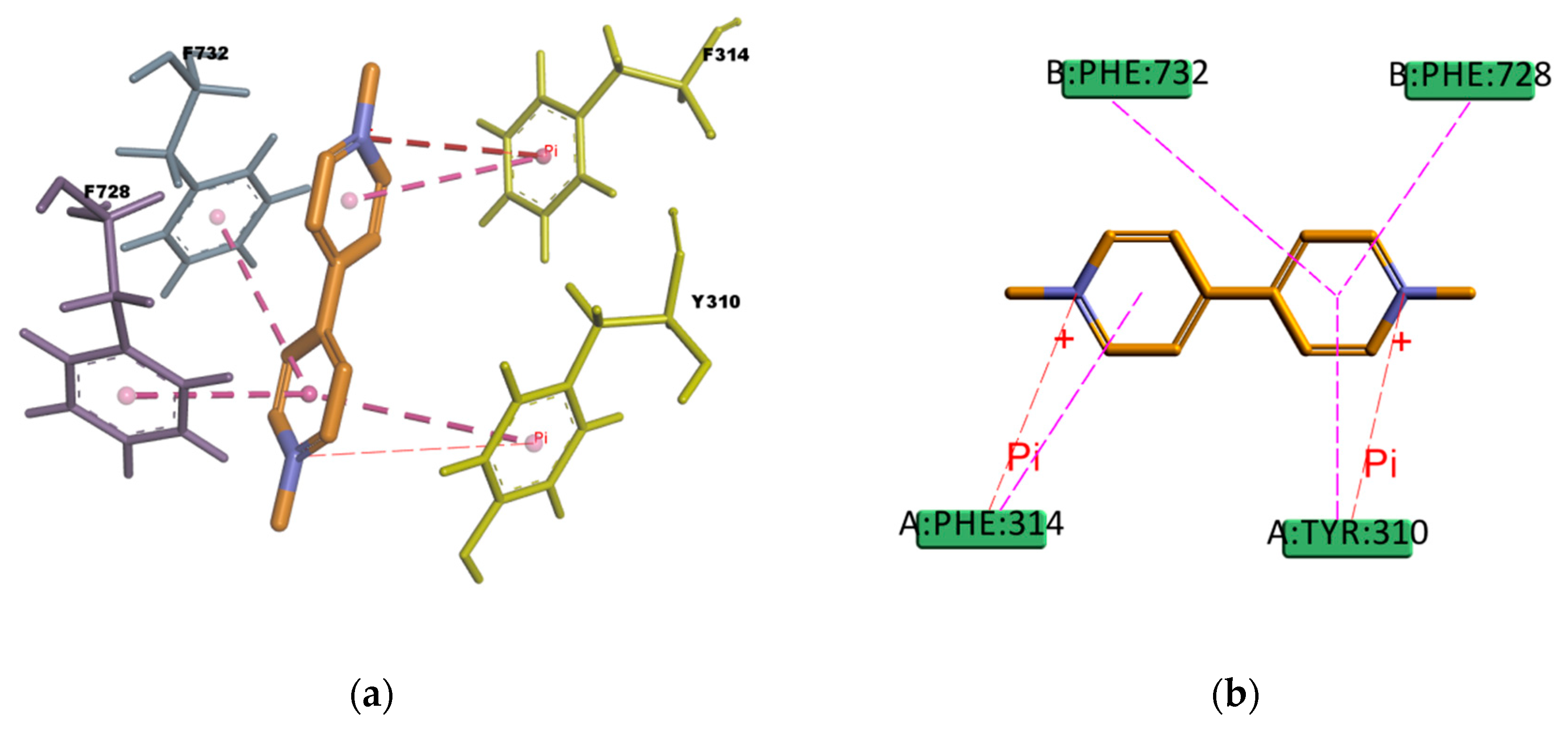
Figure 19), PQ forms hydrophobic interactions involving four residues in the binding pocket (Y310, F314, F728, and 732), all of them π–π type of interaction. It also presents a cation–π interaction between the positively charged nitrogen of PQ and the polarizable π electron cloud of the aromatic ring in F314 and Y310. These are essentially electrostatic interactions due to the negatively charged electron cloud of π systems, and are involved in many drug–receptor interactions, demonstrating that they play an important role in ligand–binding affinity [
60]. On the other hand, GEN binding pose (
Figure 20) forms hydrophobic interactions with three residues in the binding pocket (I736, F314, and F732), two π–Alkyl type of interactions and one Alkyl type of interaction, besides, it forms one conventional strong type of hydrogen bond with Y310 and one weak carbon type of hydrogen bond with I731, interactions which may explain the stability in the binding pocket reflected by the favorable binding energy. Despite of the binding energies which reflect certain stability in the binding site, both PQ and GEN are hydrophilic compounds, property which may interfere with the capability of both compounds in reaching the binding pocket due to its highly hydrophobic environment.
VPA and BU compounds resulted with the highest estimated binding energies in the set of docked compounds, as well as not transported by P-gp in the experimental transport assay results (
Figure 11). However, looking at the selected docking pose, VPA is forming π–Alkyl type of interactions with four residues in the binding pocket (Y310, F336, F728, and F759) (
Figure 21), i.e., interactions which may explain why in some literature articles VPA is reported as a P-gp inducer [
61] or a weak affinity inhibitor [
62]. On the other hand, the resulting docked pose of BU (
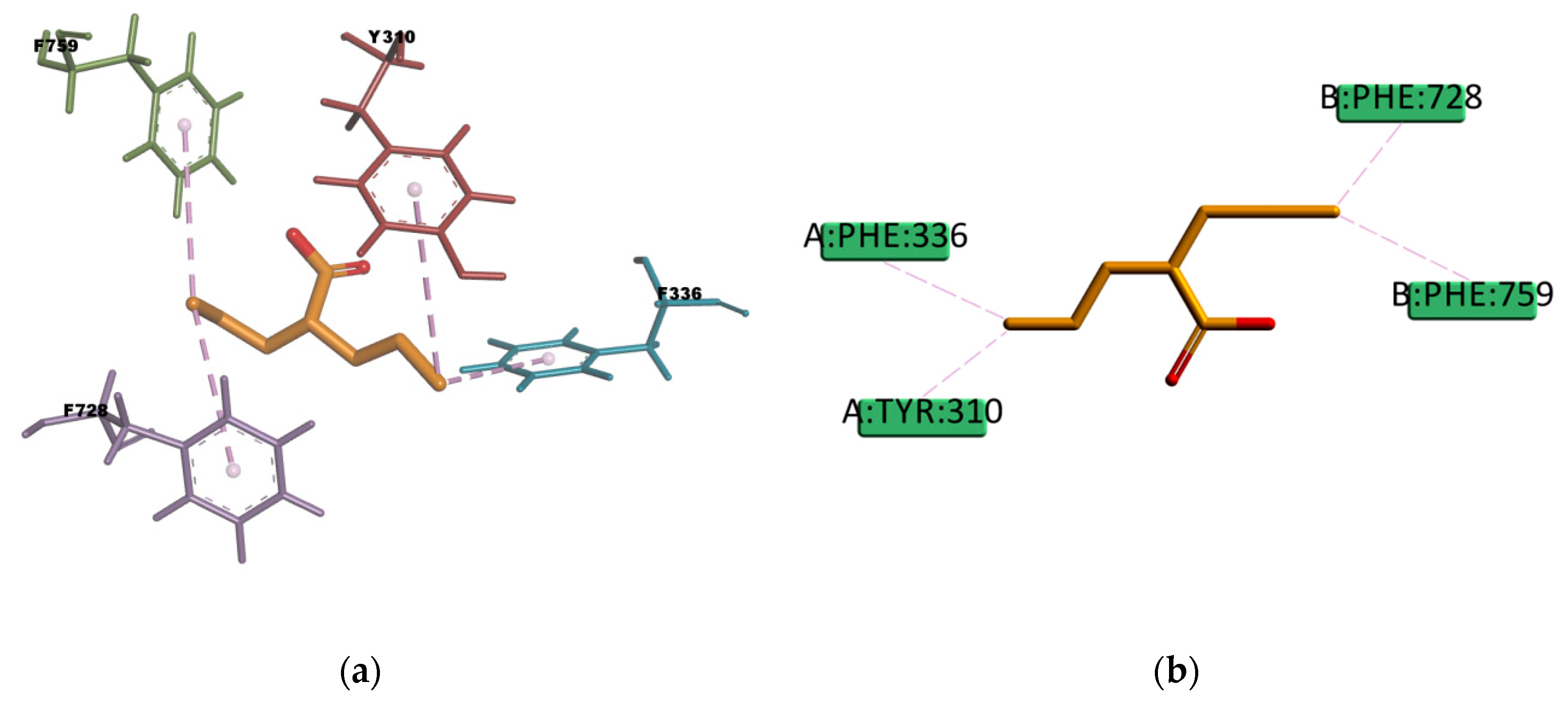
Figure 22) involves four π–sulphur type of interactions with residues F336, F728, F314, and F759, and one weak carbon hydrogen bond type of interaction with F732 in the binding mode. In experimental conditions, due to the slightly hydrophilic nature, BU may experience difficulties reaching the binding site, which, as previously mentioned, is located in a highly hydrophobic environment. The APD compound instead, presents just one π–donor hydrogen bond type of interaction between the π electron cloud of the aromatic ring in Y310 and the hydrogen atom of the amine group in APD (
Figure 23). This only interaction seems to confer certain stability to the complex according to the calculated binding energy (
Table 14), despite that, the hydrophilic nature of APD may certainly interfere in reaching the binding place as the results of the experimental transport assay did not show any interaction between APD and P-gp.
Interestingly, six of the eight known active compounds of P-gp in the docking set (CsA, AM, DIG, DOX, LPM and RMP) involved simultaneous interactions with residues Y307, Y310, F728, and F732 in the binding mode, indicating that these residues may play a crucial role in ligand recognition and binding. These four residues were also found to interact with the inhibitor PBDE (polybrominated diphenyl ether)-100 in the co-crystallized structure of mP-gp (PDB ID: 4XWK) demonstrating their relevance in ligand binding and the consistency between the amino acids predicted by molecular docking and the experimental data available.
Some compounds, such as PQ and GEN might not interact with the P-gp as shown in our study, despite the calculated binding energies that reflect a certain stability in the binding site. Although this could be seen as evidence of the lack of predictability of this approach to identify compounds that interact with P-gp, it should also be noted that these results highlight the importance of the physicochemical properties of the compounds (particularly their lipophilicity) that can prevent them from reaching the binding pocket of P-gp. Moreover, it should be noted that the predictive value of the model applied is still very good for compounds that have been found not to bind to P-gp, since for these compounds the ability to reach the binding pocket of P-gp would not make a difference.

 ,
, 
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}