Switching Homes: How Cancer Moves to Bone



Department of Biotechnological and Applied Clinical Sciences, University of L’Aquila, 67100 L’Aquila, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(11), 4124; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114124
Submission received: 30 April 2020
/
Revised: 4 June 2020
/
Accepted: 5 June 2020
/
Published: 9 June 2020
(This article belongs to the Special Issue Cellular and Molecular Mechanisms of Bone Metastasis 2.0)
{kind=link}
Abstract
:Bone metastases (BM) are a very common complication of the most prevalent human cancers. BM are extremely painful and may be life-threatening when associated with hypercalcaemia. BM can lead to kidney failure and cardiac arrhythmias and arrest, but why and how do cancer cells decide to “switch homes” and move to bone? In this review, we will present what answers science has provided so far, with focus on the molecular mechanisms and cellular aspects of well-established findings, such as the concept of “vicious cycle” and “osteolytic” vs. “osteosclerotic” bone metastases; as well as on novel concepts, such as cellular dormancy and extracellular vesicles. At the molecular level, we will focus on hypoxia-associated factors and angiogenesis, the Wnt pathway, parathyroid hormone-related peptide (PTHrP) and chemokines. At the supramolecular/cellular level, we will discuss tumour dormancy, id est the mechanisms through which a small contingent of tumour cells coming from the primary site may be kept dormant in the endosteal niche for many years. Finally, we will present a potential role for the multimolecular mediators known as extracellular vesicles in determining bone-tropism and establishing a premetastatic niche by influencing the bone microenvironment.
1. Introduction
Metastatic bone disease is currently considered incurable [1]. Bone metastases (BM) are extremely common, and patients report unbearable pain in late stages, which makes palliative therapy with analgesics of the highest grade of the World Health Organisation analgesic ladder, i.e., strong opioids, a necessity [2]. BM incidence has been reported to be 65–90% in prostate cancer, 65–75% in breast cancer, 17–64% in lung cancer and 10% in colorectal cancer [3,4]. In addition to pain, common complications include pathological fractures, spinal cord and nerve compression syndromes and hypercalcaemia, which can lead to kidney dysfunction, cardiac arrhythmias and death [5]. However, it is important to notice that at least in breast cancer patients, BM generally present with better prognosis compared to visceral metastases. Therefore, improving the quality of life and reducing skeletal-related events (SREs) of BM patients would allow them to live close-to-normal lives, both in span and quality.
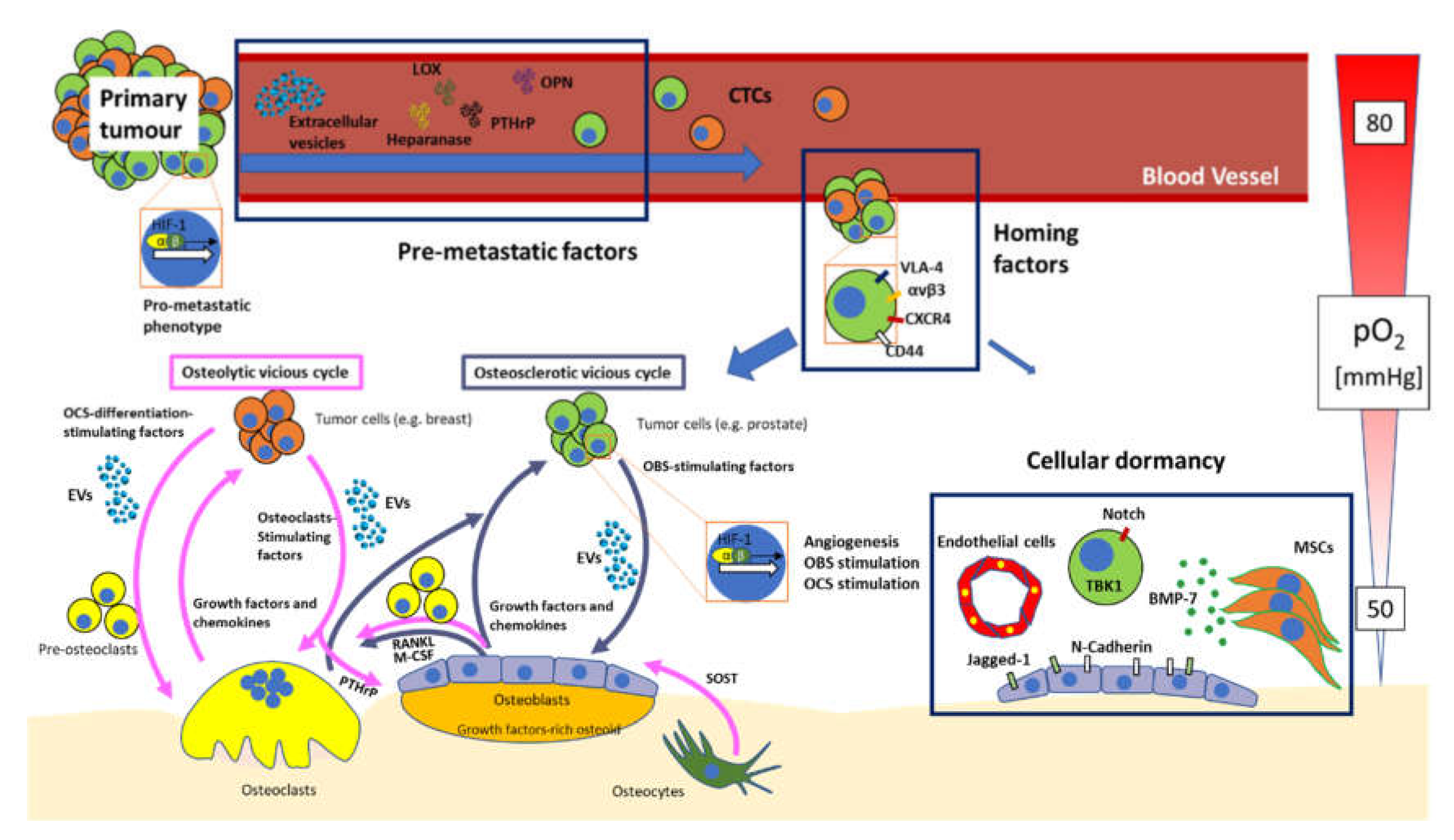
There are three main ways through which cancer cells manifest their presence in the bone microenvironment: osteosclerotic (a.k.a. osteoblastic) lesions, osteolytic lesions and mixed lesions [6]. Osteosclerotic lesions are typically related to prostate cancer BM and are identified as radio-dense spots on x-ray, while osteolytic lesions are present in BM from breast and most other types of cancers and are visually apparent as voids in areas where bone should be present [5,6]. Nowadays, the general idea is that even osteosclerotic lesions have an osteolytic component, that is either local, which gives rise to mixed lesions or can even be located far from the osteosclerosis. In fact, the consensus is that a primary osteolytic lesion is required to give rise to the osteosclerotic one, which explains why antiresorptive drugs such as bisphosphonates work also against this type of metastasis [7,8]. Intriguingly, breast cancer lesions can also present with mixed features [7,8].
The purpose of this review is to give an overview of the field, presenting well-established concepts, as well as new research that might be important for its future.
2. Cellular Players in the Bone/Bone Marrow Tumour Microenvironment
Osteoblasts are the cells that produce and mineralise bone matrix [9]. They arise from the differentiation of mesenchymal-lineage progenitors after activation of key osteoblast differentiation genes, with the master gene being Runt-related transcription factor 2 (Runx2) [2,10,11]. Runx2 can activate transcription of osteoblast-characterising genes, such as bone-specific alkaline phosphatase (BALP), osteocalcin (OCN), Collagen I and bone sialoprotein (BSP). Commitment towards the osteogenic program is driven by members of the Wingless-type MMTV integration site (Wnt) family, which activates the β-catenin pathway in osteo/chondroprogenitors in a paracrine fashion, leading to the aforementioned activation of Runx2 [12,13,14]. Osteoblasts are the major paracrine regulators of osteoclasts formation and function by producing a plethora of growth factors, cytokines and chemokines, which however can be exploited by cancer cells for their growth and engraftment, as we will discuss in detail below.
After osteoblasts finish their life cycle, they have three possibilities: (i) undergoing apoptosis, (ii) becoming a quiescent bone-lining cell or (iii) differentiating into osteocytes [9,15]. When the program to follow the latter option is activated, osteoblasts depose bone matrix around themselves, until the point where they are encased in it, becoming bona fide osteocytes [15,16,17]. This cell type vastly outnumbers the other bone-resident ones, and its main function is sensing mechanical variations in the microenvironment through a complex network of interconnected canaliculi, able to perceive shear stress by means of the primary cilium, which requires a protein complex composed by the cilium and the cilia-associated proteins Polycystin 1 and 2 [18,19,20]. Thanks to their sensing features, osteocytes are able to coordinate (and in specific circumstances, perform) bone resorption or deposition in response to both physiological environmental cues such as mechanical loading or lactation, as well as pathological ones, including micro- and macroscopic fractures [18,21,22,23,24]. Molecularly, osteocytes do this mainly through sclerostin (SOST), an antagonist of the canonical Wnt pathway that reduces bone formation when it is not needed [15] and RANKL, which allows them to control osteoclastogenesis as well [25].
Osteoclasts are multinucleated, giant, bone-resorbing cells [26]. At variance with osteoblasts, they derive from haematopoietic precursors of the monocyte/macrophage lineage, and their differentiation is a multistep process involving the activation of a series of transcription factors, including Microphthalmia Transcription Factor (MITF) and PU.1 [27,28,29]. In the last stages, osteoclast precursors express the macrophage-colony stimulating factor (M-CSF), which binds to its receptor c-fms eventually promoting their survival and growth. Costimulation by receptor activator of nuclear factor kappa B (NfĸB) ligand (RANKL) leads to activation of NfĸB, which in turns stimulates nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent (NFATc1), leading to fusion of precursors and generation of mature, resorbing osteoclasts [30,31,32,33]. Moreover, a plethora of inflammatory cytokines regulates osteoclastogenesis [34]. This feature is often exploited by cancer cells to induce osteoclastic activity and bone resorption [35].
A resident population of bone and bone marrow macrophages (BMMs) is present in both these tissues [34]. The formers are also known as osteal macrophages or osteomacs. Both cell types are F4/80-positive and TRAcP-negative in mice and are able to form functional osteoclasts in vitro [36]. Intriguingly, osteomacs have also been shown to produce BMPs, thus controlling osteoblast activity in vitro and in vivo [37,38]. They represent almost 20% of the bone marrow cells, and together with BMMs can become tumour-associated macrophages, eventually supporting tumour growth and immune escape, which is particularly important in the bone marrow, as it has been suggested in prostate cancer [39]. However, the role of these cells in breast cancer bone metastasis and dormancy is still to be elucidated.
Endothelial cells in the bone marrow have peculiar features, being organised in fenestrated sinusoids that allow for relatively easy intra- and extravasation of bone marrow and circulating cells, respectively [40]. The cross-regulation between bone cells and endothelial cells is known, as well as the concept that angiogenesis and osteogenesis are coupled during development and bone regeneration, where bone cells produce angiotropic factors, and endothelial cells produce angiocrine factors that stimulate bone modelling [41,42]. Importantly, bone marrow endothelial cells have been demonstrated to be involved in keeping cancer cells and HSCs dormant [40,43]. Recently, bone marrow endothelial cells have been suggested to promote prostate cancer progression through the CCL5/CCR5/autophagy axis, which is even able to reduce androgen receptor expression [44]. Furthermore, a recent emerging concept proposed by Lin and colleagues [45], is that endothelial cells can transdifferentiate into osteoblast-like cells in the metastatic prostate cancer context, thus inducing or contributing to osteosclerotic metastases [45].
Even though the commonly used mouse models have almost exclusively red marrow in their long bones, in adult humans this is mostly replaced by adipose yellow marrow. It is, therefore, conceivable that being the most abundant cell type in adult bone marrow, bone marrow adipocytes (BMAs) could have a crucial role in metastasis. In normal bone physiology, BMAs and osteoblasts both come from a common progenitor, i.e., the bone marrow mesenchymal stromal/stem cell [46], and a differentiation shift from osteoblasts to adipocytes is thought to be one of the determining factors in the bone mass accrual reduction during ageing [47]. Moreover, BMA-derived fatty acids are able to increase osteoclast differentiation and survival [48,49]. BMAs also support haematopoiesis, driving differentiation of CD34+ precursors towards the myeloid and lymphoid lineages [50]. In cancer, numerous reports suggest that BMA-derived fatty acids and factors such as fatty acids binding protein 4 (FABP4) and leptin, might drive tumour progression in prostate and breast cancer [51,52,53,54]. Intriguingly, adipose tissue-derived leptin has been positively associated with PI3K/Akt pathway activation and radio/chemotherapy resistance in cancer cells, further confirming the importance of BMAs in the bone marrow metastatic microenvironment [55,56]. In addition to this, as stated, BMAs can increase osteoclast activity and decrease osteoblast function, while providing nutrients for cancer cells in the form of fatty acids. This has been shown to be happening in metastatic cancer, through at least the PTH1R/PTHrP signalling pathway [47,53]. Although BMAs are also thought to be part of the endosteal niche, limited or no evidence is present to date about their role in cancer or HSC dormancy/reactivation [57].
3. Bone Metastasis: Established and Novel Concepts
Stephen Paget, more than 130 years ago, gathered data from over 700 autopsies of patients who died of cancer, and noticed a discrepancy between the metastatisation probability between similarly blood-perfused organs, such as spleen vs. liver. For breast cancer patients he stated that “In cancer of the breast the bones suffer in a special way, which cannot be explained by any theory of embolism alone” [58]. He stated that this was probably because the “seed” (i.e., the cancer cell) found a more favourable “soil” (i.e., the microenvironment) in certain organs, and, therefore, metastasis was more frequent in those districts [58]. This was a strong paradigm shift, since although Paget himself stated that the “seed” is more important, studying the “soil” could give valuable insights. Today, we know bone is a particularly attractive milieu for cancer cells: the bone matrix contains an abundance of growth factors of the transforming growth factor (TGF)-β superfamily, insulin-like growth factors (IGFs), bone morphogenetic proteins (BMPs), platelet-derived growth factor (PDGF), fibroblast growth factors (FGFs) and calcium (Ca2+) [59]. Importantly, these factors are not only stored in the bone matrix, but are also actively produced by osteoblasts [2,60].
3.1. “Virtuous” vs. “Vicious” Cycle of the Bone
Bone tissue undergoes continuous cycles of matrix renewal, which is carried out by bone-resident cells in a process termed “bone remodelling” [61]. To accomplish this process properly, bone cells cross-communicate in a tightly controlled manner, which guarantees the well-keeping of the delicate balance between bone deposition and resorption [2,15,62,63]. The cycle starts with a stimulus (mechanical or molecular) that primes periosteal or endosteal bone-lining cells, which move away from each other, exposing the bone surface to be remodelled, to the action of osteoclasts. This stimulates osteoclast precursors, which differentiate into osteoclasts in situ, attach and seal the exposed bone portion, and degrade it [26,64]. The consequence is the release of growth factors and chemoattractants, that summon reverse cells, macrophage-like cells that clean out the resorbed lacuna of debris and promote osteoblast differentiation. The latter phase is the longest one and culminates in the newly formed bone being covered in bone-lining cells again, closing the so-called “virtuous cycle” of bone [26,61]. Being such a well-structured process, which involves a large number of molecular players, cancer cells can hijack it in different phases to promote their own growth. Whatever the type of modification to this cycle, when a cancer cell corrupts it to its own benefit, the “virtuous cycle” becomes “vicious”, thus marking the establishment of bone metastasis [5,6].
In the “osteoblastic vicious cycle”, prostate cancer cells migrate to bone, and persuade osteoblasts to produce large amounts of growth factors-rich osteoid, which is then mineralised into primary bone, that never becomes lamellar [65]. Prostate cancer cells do this through several secreted and membrane factors, including Insulin-like growth factor (IGF), WNT1 and 3A, tumour necrosis factor (TNF)-α, BMPs, Endothelin (ET)-1 and parathyroid hormone-related peptide (PTHrP) [3,65,66,67,68,69]. In turn, osteoblasts produce factors that favour tumour growth, such as IGFs, chemokines, interleukin (IL)-6 and TGF-β [70]. Moreover, osteoblasts also promote osteoclast differentiation and activity through production of RANKL and M-CSF, which further stimulates the release of IGF-1, BMPs, PDGF, TGF-β from the bone matrix because of osteoclastic bone resorption, hence promoting tumour growth, restarting the vicious cycle [2,4,6,60,65,66]. Additionally, Ca2+ ions released from the bone matrix by osteoclasts have a role in the vicious cycle, since, in addition to causing potentially fatal hypercalcaemia, they can stimulate calcium-sensing receptor (CaSr)-positive cancer cells, which promotes their growth, apoptosis escape ability, and PTHrP production [71,72].
In the “osteolytic vicious cycle”, tumour cells produce factors (i.e., IL-6, TNF-α, IL-1β) that stimulate differentiation of osteoclast precursors into mature osteoclasts directly, or indirectly, such as PTHrP, that acts by increasing osteoblastic RANKL production, eventually stimulating osteoclast formation [73]. the final outcome is an exacerbation in bone resorption, which is not balanced by bone deposition [7,60,65], causing the release of the aforementioned growth factors encased in the bone matrix, which stimulate tumour growth, closing the vicious cycle.
3.2. From Primary Site to Bone: A Bumpy Ride
To establish the vicious cycle, cancer cells first have to migrate from the primary site to the bone, which is classically simplified in the following steps: (i) local invasion, (ii) transfer to bloodstream or lymphatic system which requires the ability to intravasate and survive in it, (iii) arrest and extravasation in distant site, (iv) immune resistance and establishment of a supportive niche in the secondary site (settlement), (v) activation of growth and overt metastasis, which unfortunately happens in a short period of time in the majority of cases, but can also have a latency of years [74]. All these steps make metastatisation a difficult journey, thus explaining the very low efficiency of the process, which is estimated to be less than 1%, with authors reporting values as low as 0.01% metastasis efficiency [74,75,76]. However, it is important to notice that cancer dissemination, classically thought as a late process in cancer development, starts very early. In fact, over time, even small tumours can shed millions of circulating tumour cells (CTCs), which are now being exploited as a diagnostic tool (liquid biopsies). CTCs may then engraft in distant sites, including the bone marrow, becoming disseminated tumour cells (DTCs) [74,77]. Every step of metastatisation requires a specific set of molecules, some of which may be common among different solid cancers, such as matrix metalloproteinase (MMP)-9 in tumour intravasation [78,79,80,81,82,83], and some others being specific for different cancers or type of metastasis, such as PTHrP for skeletal metastases [73,84].
3.3. Premetastatisation
An important step in bone metastatisation is the establishment of a “premetastatic niche”, i.e., tumour cells preparing the bone microenvironment to host them, before “switching homes” and moving to bone [77]. Much research has been devoted to this topic in lung and liver metastasis, but research on the bone premetastatic niche (PMN) seems to be lagging behind, with only scarce reports present. A factor that seems to be important in the establishment of the bone PMN is lysyl oxidase (LOX), an enzyme that deals with crosslinking collagen and elastin in the extracellular matrix to increase tensile strength [85]. LOX has been shown to increase matrix stiffness, modulate the microenvironment and promote the establishment of a metastasis-permissive network of crosslinked fibres in bone, both in colon [86] and, importantly, in breast [87] cancer. On the other hand, before the concept of PMN was even coined, Kelly and colleagues found that primary tumour-derived heparanase was able to influence the bone microenvironment and increase bone resorption [88], which could prime the bone microenvironment for metastasis starting the vicious cycle from a distance before metastasis are established. Similarly, PTHrP, which normally acts in a paracrine fashion, can act from the primary tumour to cause a net increase in bone resorption, priming the bone microenvironment to metastasis, while causing tumour-induced hypercalcaemia [73,89]. TGF-β has also been shown to have a similar effect [90].
Bone marrow-derived cells seem to have a crucial role in establishing premetastatic niches in other organs [91,92,93,94]. Whether this is true for the bone marrow is still not demonstrated, although other niches located there are important for tumour dormancy and homing. Osteopontin (OPN), a bone matrix protein produced by osteoblasts, has also been studied in the field of bone metastasis for years [95]. In fact, OPN can also be expressed by cancer cells (because of osteomimicry [7]) and is a known driver of bone metastases, whose inhibition has even been hypothesised as a BM-inhibiting therapy [96]. It partakes in different stages of BM, including the establishment of the premetastatic niche, by increasing the expression of growth and chemotactic factors in the BM, such as b-FGF, hepatocyte growth factor (HGF) and TGF-β [95,96].
3.4. Homing
This process expresses the ability of cancer cells to set roots in a distant organ’s microenvironment, surviving in it, and either becoming dormant or proliferating. This process is obviously very complex, but many molecular mediators have been identified and characterised. A key player is αvβ3, an integrin that is well known for mediating the adhesion of osteoclasts to the bone matrix [26], which has been shown to favour bone homing and metastasis when expressed in breast cancer cells [97,98,99,100,101]. Similar actions have been proposed for very late antigen (VLA)-4, which interacts with vascular cell adhesion molecule (VCAM)-1, constitutively expressed in bone marrow endothelial and stromal cells [102]. Another possible player in cancer homing to bone is connective tissue growth factor (CTGF), which has been implicated in BM development in a seminal large-scale study more than 15 years ago [103]. This study exploited differently osteotropic cancer cell lines to identify factors that would be important for bone metastatisation by analysis of differentially expressed genes, and CTGF was one of the top hits. CTGF has been studied by different groups over the years with mixed results [104,105], and is now making a comeback in the context of prostate cancer, where it seems to drive a RUNX2-RANKL axis to promote BM and tumour progression [106].
An important topic, which still has momentum despite many years of research have already been put in it, is the commonality between the homing mechanism of haematopoietic stem cells (HSCs) and cancer cells [107]. A key molecular player seems to be C-X-C chemokine receptor type (CXCR)-4, the ligand for stromal-derived factor (SDF)-1. CXCR4 is crucial for haematopoietic stem cells (HSC) homing [108,109], but also cancer cells can exploit this pathway in order to exit the bloodstream and interact with the bone marrow stromal components [77,110,111].
CD44 is the major cell ligand for hyaluronan, and has been proposed as a stemness marker in breast cancer [112,113,114,115]. Intriguingly, both CD44 and αvβ3 are receptors for OPN, a major component of the bone matrix [115]. Therefore, CD44 may act in concert with αvβ3 integrin to home cancer cells to bone. Furthermore, CD44 can interact with MMP-9, providing the cancer cell with enhanced invasion potential [116]. Another function of this protein, which may be important for driving cancer cells in the bone marrow niche, is that CD44 can also bind CXCR4, which is crucial in retaining HSC in the niche, and might be important for cancer cells as well [117]. Annexin (ANXA) II can also bind SDF-1 [118] and is another crucial factor that helps localise HSC in the niche [119]. Unsurprisingly, this protein can be also exploited by cancer cells to “steal” the HSCs’ spot in the bone microenvironment [120,121]. Other factors, which have been linked to HSC and tumour dormancy will be presented in a separate section.
The involvement of the Notch pathway in bone metastasis has been poorly investigated, and only in recent years have specific reports emerged on the topic [122,123,124]. Jagged-1, a ligand for Notch family members, is expressed by metastatic cancer cells, and can bind Notch on osteoblasts and preosteoclasts. This, in turn, triggers the cleavage of the Notch intracellular domain (NICD) and its nuclear translocation, which activates downstream effectors of the pathway, eventually leading to IL-6 production by osteoblasts, and preosteoclast differentiation into mature osteoclasts, both directly and through IL-6 [123,124]. Despite a few years of relative quietness on the topic, Jagged-1 has proven itself to be a viable target in bone metastasis by Zheng and colleagues [122] especially in combination with chemotherapy.
Dickkopf Wnt signalling pathway inhibitor (DKK)1 is another novel factor in bone metastasis, which shows an interesting dichotomy in lung vs. bone metastasis. In fact, a recent report by Zhuang and colleagues [125] shows that DKK1-high tumours preferentially metastatise to bone, while DKK1-low tumours tend to migrate to the lungs. The dichotomy is maintained in the pathways hindered by DKK1: in lung this protein impairs noncanonical Wnt pathway, which would lead to immune cells chemotaxis and thus slower growth, while in bone it acts on osteoblasts to reduce canonical Wnt pathway activation [125], which tilts the osteoblast-osteoclast balance towards the osteoclasts, leading to increased bone resorption [125].
A relationship between PTHrP and CCL-2 (a.k.a. Monocyte Chemoattractant Protein-1, MCP-1) has emerged in the last decade and could be important in BM [126,127]. In fact, this protein is a potent osteoclastogenesis inducer, that can even overcome the antiosteoclastogenesis effect given by granulocyte-monocyte (GM)-CSF [128]. CCL-2 appears to be a “textbook” example of a vicious cycle protein. In fact, it is induced by PTHrP, induces osteoclastogenesis, and at the same time acts autocrinally to increase tumour production of vascular endothelial growth factor (VEGF)-A, eventually promoting angiogenesis and tumour growth [127,128]. However, inhibition of CCL-2 gave inconclusive results in reducing skeletal metastases, which shows just how complex and diverse the tumoural bone microenvironment is in patients [129,130].
3.5. Hypoxia, Angiogenesis and Blood Vessels in Bone Metastasis
Seminal works by Albert Lasker Basic Medical Research awardees and Nobel laureates Gregg L. Semenza, William G. Kaelin Jr and Sir Peter J. Ratcliffe [131,132] showed how important oxygen levels adaptation is in cells, which is mainly performed through the hypoxia-inducible factors (HIF) family [133]. This is true in physiological settings as well as pathological ones, such as cancer [134]. If we consider that arterial blood hold 130–80 mmHg and organs like kidney have 72 mmHg, we could define the endosteum as a hypoxic microenvironment, with pO2 dropping steeply to 50mmHg in close proximity to the endosteal niche [2,135]. Hypoxia seems to favour bone cells activity both directly, and through guaranteeing the coupling between angiogenesis and osteogenesis [136,137,138]. Unsurprisingly, the HIF family of factors and their many downstream targets are also important in breast cancer and BM. Recent evidence shows that HIF-1α signalling in osteoblasts increases breast cancer metastasis towards all districts, including bone [139]. The expression of the premetastatic factor LOX, discussed above, is strongly induced by HIF-1α [140,141], although it has been also reported to promote BM independently of HIF-1α in colon cancer [86]. In non-small-cell lung cancer (NSCLC), a correlation between HIF-1α and BM has been proposed as well in a retrospective study by Pezzuto et al. [142]. The intriguing aspect of this work is that HIF-1α positivity has been observed in the primary site rather than in the metastatic foci, characterising this factor not only as a local metastasis determinant but also as a player in the metastatic organotropism from the tumour cells’ side. Consistently, another previous study came to similar conclusions in breast cancer, showing that primary tumours that induce bone micrometastases have specific molecular signatures, which include the rat sarcoma (RAS) family and HIF-1α [143]. Moreover, experimental BM studies from almost 15 years ago reported that overexpressing a dominant-negative version of HIF-1α decreases BM development, whereas a constitutively active HIF-1α increases it, and targeting hypoxic cells with a caspase-3 activating protein selectively reduces BM [144,145].
In line with the key role of oxygen homeostasis in this context, we found that one of the most upregulated genes in bone metastatic samples from patients carrying also metastases to visceral organs vs. bone metastasis only patients was haemoglobin beta (HBB) [146]. Of course, this haem-protein is well known for its oxygen-binding and transport function in red blood cells, but its role in cancer was unknown. Further investigations revealed that HBB was able to increase cancer aggressiveness through HIF-1α, that it was increased in primary tumours of higher grade, and that blocking its oxygen-binding ability could abrogate some of the phenotypical effects observed [147]. We hypothesised this was due to what we called “oxygen-positive hypoxia”, a phenomenon through which oxygen is sequestered and rendered unavailable for the cell sensors, leading to the activation of HIF-1α target genes, such as VEGF [147]. Another excellent report by an independent group came to similar conclusions, although HIF-1α was not identified as the determinant of the phenotype. Nevertheless, the group found that oxidative stress was reduced in HBB-expressing cells, which is consistent with lower free intracellular pO2, and also agrees with our findings [146,148]. Arguably, the most evident consequence of bone metastasis is pain, and hypoxia has a role to play in this as well. In fact, HIF-1α activates the Warburg effect in cancer cells, which induces the release of lactate and protons in the bone microenvironment, which, in turn, signal to nociceptive terminals in the bone, causing pain [2,149]. Of course, another major source of protons is the osteoclast, which is activated by the vicious cycle, and targeting the V-ATPase reduces cancer-induced bone pain [149,150,151], also considering that tumour cells exploit V-ATPase to release protons in the bone microenvironment [152].
Another important aspect related to hypoxia and cancer is sustained angiogenesis. This is one of the hallmarks of cancer, and the concept that hypoxia increases angiogenesis has been known for more than 15 years [153]. In fact, HIF-1α can activate the expression of VEGF and its receptors, along with Angiopoietins, bFGF, PDGF, stem cell factor (SCF), osteopontin (OPN), MMPs, nitric oxide synthase (NOS)-2, cyclooxygenase (COX)-2 [154] and ET-1 (which is also important for osteoblastic bone metastases [67,69]) [155,156]. However, apart from the obvious advantage of having a vasculature transporting nutrients and oxygen, the bone metastatic tumour has other less obvious advantages that derive from activating the angiogenic program in the bone marrow: the establishment of the vascular niche [157], which will be discussed in the dormancy section and the ability of many angiogenic factors to fuel the vicious cycle. Key examples are VEGF, which on one hand promotes osteoblast differentiation and activity and can, therefore, fuel the osteoblastic vicious cycle [158,159,160,161] while, on the other hand, it activates osteoclastogenesis, potentially fuelling the osteolytic vicious cycle as well [162]. Similarly, bFGF can trigger osteoclast differentiation but also osteoblast activity, and, therefore, may contribute to both vicious cycles in BM [163,164,165,166].
3.6. Tumour Cellular Dormancy
Tumour dormancy is a behaviour that some cancer cells adopt, by which they remain dormant as single cells or small clones. This is achieved by either stopping to proliferate, or by equalising the rate of cell death with that of proliferation [167]. This status is akin to what fungi or bacteria do when they become spores, since dormant cancer cells are highly resistant to many external insults: they develop multidrug and radiotherapy resistance, are able to elude classical chemotherapy because they are not proliferating as well as the immune system, and they can be reactivated when a suitable stimulus/i (which are only partially known) is applied [167,168]. Perhaps the most incredible aspect of this process is that the cell can stay dormant for many years, usually between 5 and 20, as disseminated tumour cell (DTC) [169] before cancer tragically relapses and is often extremely hard to treat, because it arises from such a highly resistant clone [167,168,170,171].
The bone marrow is likely the most common site where cellular dormancy occurs, since many of the mechanisms that are thought to keep cancer cells dormant, are based on molecular pathways that are usually used by haematopoietic stem cells for maintaining their own quiescence [167,168,170,172]. Undoubtedly, cellular dormancy is an extremely complex phenomenon, that reflects the complexity of the bone marrow microenvironment and the many niches that reside there. These are often just a few microns away from one another, so one cell can participate in more than one niche at the same time. There are at least two niches in the bone marrow that allow cancer cells to be kept quiescent, and that rely on cell-cell contact as well as soluble factors: the endosteal niche and the vascular niche. Since the two are likely to coexist and cooperate in the bone marrow microenvironment, they will be discussed as one for the purpose of this review. This niche is formed by tumour cells, HSCs, MSC-osteoblast lineage cells, osteoclasts, endothelial cells and other players such as osteocytes and bone marrow adipocytes, which can all influence tumour cells dormancy [57].
In the early years, much work was done on prostate cancer cell dormancy. One of the first factors that was identified in cellular dormancy was BMP-7, which induces a reversible proliferation arrest in bone metastatic prostate cancer cells, through p21, p38 and n-myc downstream-regulated gene (NRDG)-1 [173]. This growth arrest could be reverted by BMP-7 removal, thus resulting in overt metastasis. Intriguingly, several years later an independent report showed that osteonectin activation in MSC-lineage cells promotes BMP-7, which could then lead to prostate cancer dormancy [174].
Another factor that proved to be important in tumour dormancy is tank-binding kinase 1 (TBK1). This is a noncanonical IKK family member that mediates part of the toll-like receptor-mediated innate immunity, leading to the transcription of target genes such as RANTES and interferon-β [175]. Intriguingly, Kim and colleagues found that prostate cancer cells contacted osteoblasts in the bone marrow, they activated TBK-1, eventually leading to an inhibition of the target of rapamycin (mTOR) pathway. This, in turn, gives rise to an arrest of protein synthesis and growth, drug resistance, and increased “stemness”, which fits the definition of cellular dormancy to the letter [175]. In breast cancer, other factors have been identified as important in determining niche-mediated cellular dormancy, such as N-cadherin. It is expressed in cancer cells after epithelial to mesenchymal transition, but also by a subset of osteoblasts called spindle-shaped N-Cadherin-positive osteoblasts (SNOs) [176,177,178]. Intriguingly, SNO cells have been implicated in the maintenance of HSC quiescence [179], albeit the role of N-cadherin (CDH2) has been ruled out as a determining factor [180] per se, while ablating all CDH2-positive cells using diphtheria toxin receptor models, caused a tremendous mobilisation of HSC, confirming that SNO cells are indeed important in the maintenance of the HSC bone marrow niche [179]. We found that SNO cells were able to reduce the proliferation of breast cancer cells, especially when selected for Notch2, consistent with the fact that SNO cells expressed high levels of the Notch ligand Jagged1 [176]. However, Abravanel and colleagues obtained different results when investigating the Notch family [181], likely because fine-tuning such an important pathway might cause dramatic differences, as is shown in other reports in the literature [176]. Another pathway that is still under investigation in this regard is the already cited SDF-1/CXCR4 pathway, which might partake in this process as well, although there is currently no consensus on the matter, and results seem to be pointing at different directions in different cancers [182,183,184,185].
Besides the knowledge of the molecular mechanisms allowing tumour dormancy, a crucial and very important issue is to identify which stimuli can revoke it, awakening the cancer cell into an overt metastasis. A general consideration is that the extracellular signal-regulated kinase (ERK) pathway has a crucial role in determining whether a cancer cell will remain dormant (inactive ERK pathway) or proliferate (activated ERK pathway). ERK inactivation is also linked to endoplasmic reticulum (ER)-stress, which also makes dormant cancer cells chemotherapy-resistant [186,187,188]. A number of studies suggest that integrin signalling might be crucial for the switch from a dormancy state to a reactivated metastasis. In particular, loss of integrin β1 signalling has been reported to be a key factor not only in the proliferation of primary and disseminated tumours [189,190,191] but also in the reactivation of dormant cancer cells, acting also through the Focal adhesion kinase (FAK)/SRC/ERK pathway [192]. Therefore, β1-integrin could be a potential therapeutic target, which can be exploited to keep tumour cells dormant, using neutralising molecules such as ATN-161, volociximab and JSM6427 [193]. Discoidin domain receptor (DDR)-1 can also trigger reactivation of cancer at multiple secondary sites including lung, brain and bone. This happens through tetraspanin transmembrane 4 L six family member 1 (TM4SF1), acting through the PKCα/JAK/STAT pathway to promote cell proliferation [194]. Intriguingly, DDR-1 noncanonical signalling may also be activated by binding to collagen, which could be another possible means of cancer cell awakening in bone. In fact, bone is highly enriched in collagen, which is usually buried in the mineral matrix, but can be exposed following osteoclastic bone resorption or acidification of the microenvironment [194]. Wnt signalling promotes G1-S transition through Cyclin E1 and D1, and c-myc [195] and Dikkopf-1 (DKK1), a Wnt signalling inhibitor, has been reported to promote tumour dormancy through an autocrine [196], but probably also paracrine signalling network. Factors that can have an opposite effect on the Wnt pathway, such as tenascin C and periostin (albeit indirectly) have opposite effects on tumour dormancy and can promote metastatic reactivation [197]. Finally, also inflammation can trigger reactivation of dormant cancer cells in distant sites, consistent with the fact that high circulating levels of acute-phase proteins correlate with distant-site recurrence in breast cancer [198].
3.7. Extracellular Vesicles in Bone Metastasis
Extracellular vesicles (EVs) are particles ranging from 20 to 1000 nm of diameter, which are delimited by a phospholipid bilayer that is compositionally and structurally reminiscent of a plasma membrane [199]. EVs contain multimolecular cargoes that can mirror the composition of the origin cell, or be enriched of specific molecules, such as miRNAs and other noncoding RNA, mRNAs, DNA, and specific membrane proteins, such as CD63, the most highly used marker of EVs. They have been implicated in a variety of physiopathological events and can be used by virtually every cell type as means of paracrine as well as long-distance communication [200,201]. Unsurprisingly, cancer cells can use EVs to influence their microenvironment, promoting tumour progression, metastasis, and organotropism [202]. A seminal report by Peinado et al. [203] showed that exosomes from melanoma cells could reprogram bone marrow progenitor cells, which would then migrate out and into the metastatic site, establishing a premetastatic niche in the target organ.
A large body of evidence is accumulating that exosomes can influence metastatic behaviour of many types of cancer, in different secondary sites [202]. We recently showed that EVs are indeed able to influence bone biology, and osteoblast-EV-shuttled RANKL is a novel way through which osteoblasts promote osteoclast survival without direct cell contact [204]. We also found that osteotropic breast cancer cells (MDA-MB-231) can reprogram osteoblasts so that they produce a higher amount of RANKL-EVs, which contributes to the vicious cycle by inducing osteoclastogenesis. Furthermore, we showed that MDA-EVs are able to also increase osteoclastogenesis directly, and that they strongly inhibit osteoblast activity, while activating angiogenesis in the bone marrow microenvironment [205]. This means that through a single delivery system, osteotropic cancer cells may be able to radically reprogram the bone microenvironment to their advantage, thereby increasing metastatic growth. Intriguingly, we show that systemic administration of MDA-EVs in mice results in the targeting of bone cells (osteoblasts and osteoclasts) with high specificity vs. the bone marrow, which only shows a small percentage of positive cells [205].
Another strong indication of the importance of EVs in metastatic tropism comes from Hoshino and colleagues, who showed in a very important paper for the field, that integrins present on the membrane of EVs are able to tell the organotropism of the origin tumour when it metastasises, because according to the integrin set, EVs will reprogram a specific microenvironment turning it into a permissive premetastatic niche [206]. In fact, the authors show that preconditioning mice with EVs presenting integrins associated with lung metastasis, resulted primarily in lung metastasis upon intracardiac injection of MDA cells, even if these were of a highly selected bone-targeting clone [206]. Nevertheless, the authors could not correlate any specific EV-integrin to BM. Other indications come from recent reports showing that lung cancer-derived EV-mediated transfer of Amphiregulin [207] or miR-21 [208] promote osteoclastogenesis, while another report identified an EV-miR signature enriched in metastatic lung cancer [209]. In prostate cancer, some advances in this field were also achieved, with Karlsson and colleagues finding that TRAMP-C1-EVs impaired osteoclastogenesis [210] and Inder et al. showing that Cavin-1/PTRF can alter the EV content of PC, which modifies its osteoblast- and osteoclast-activity-influencing behaviour [211]. Intriguingly, MSC-EVs have been shown to promote a dormancy-like proliferation reduction in metastatic breast cancer cells via miR-23b in vitro, which speaks volumes about the versatility of these multimolecular messengers, that could have a crucial role in bone metastasis.
3.8. Role of Immune Cells in Bone Biology and Metastases
Immune cells are among the most important regulators of bone physiology and pathology, as testified by the immense body of literature in the field of “osteoimmunology”, a term referred to the cross-regulation of bone and immune cells [212]. T-lymphocytes, especially Th17 cells, are strong inducers of osteoclastogenesis, both directly and indirectly through osteoblast induction of RANKL and M-CSF [213,214], while the immunosuppressive T-reg cells are antiosteoclastogenic [25]. B-cells produce RANKL, which acts as both an autocrine factor and an osteoclastogenesis inducer, and in fact, B-cell deficient mice are protected against ovariectomy-induced bone loss. Additionally, neutrophils can influence bone physiology, mainly by cytokine secretion and by attracting Th17 lymphocytes in situ [215]. Unsurprisingly, all these immune cells present in the bone marrow, influence the fate of bone metastatic cells in addition to influencing bone biology.
Circulating osteoclast precursors increase in number in bone metastatic patients, and coculturing them with T-cells results in osteoclastogenesis with no further stimulation by RANKL or M-CSF [216]. Furthermore, IL-7, a key cytokine produced by immune cells in the tumour context, is able to increase RANKL and other inflammatory cytokines such as TNFα in osteoblasts and bone marrow stromal cells, thus increasing osteoclastogenesis and fuelling bone degradation in the metastatic context [217,218,219]. Importantly, the RANKL-RANK contact between T-cells and tumour cells promotes tumour growth, invasion and metastasis [220]. To consolidate the link between T-cells and tumour burden in bone, increasing T-cells response by knocking out Lyn tyrosine kinase, causes a reduction in bone metastatic growth, despite increased osteoclastic bone resorption [221,222]. Another important cell type in metastatic bone growth is the myeloid-derived suppressor cell (MDSCs). This is a myeloid cell type characterised by coexpression of Gr-1 (od CD33 in humans) and CD11b, which can suppress both innate and adaptive immunity, thus increasing tumour growth in multiple cancers, including lung, breast and multiple myeloma [223,224]. It is, therefore, unsurprising that cells are a negative prognostic factor in these cancers, also considering they are precursors for osteoclasts and can differentiate into OCS after stimulation [225]. MDSCs are also able to activate tumour angiogenesis by secretion of VEGF and bFGF, which can be directly incorporated in the tumoural endothelium [226,227]. Immune cells are also involved in the establishment of two different types of tumour dormancy. On the one hand, potential dormant tumour cells have been reported to compete with haematopoietic stem cells (HSCs) for the endosteal niche [176,228], where they become dormant (see Section 3.6). On the other hand, immune cells are also able to entertain a fight with metastatic (or primary) cancer cells, balancing the rate of tumour proliferation with that of immune-mediated killing, thus inducing an apparently latent state in the tumour mass. This process is termed immune-mediated dormancy [229,230]. This equilibrium status seems to be mainly promoted by adaptive immune cells and factors, such as CD4+/CD8+ T-cells, perforin-mediated toxicity by NK cells, IL-12 and IFN-γ, while cancer cells respond by increasing Treg and MDSC production of TGF-β, IL-10, ROS and NO and upregulating their own programmed death-ligand 1 (PD-L1) expression [229,231]. Moreover, neutrophils and macrophages have been shown to be alternatively activated by cancer cells into an M2 or N2 phenotype, respectively, thus becoming tumour-associated neutrophils (TANs) or macrophages (TAMs), eventually leading to increased angiogenesis and metastatic dissemination in secondary sites including bone [232,233] Although interesting, this is surely not the full extent of cross-regulation between cancer cells and immune cells in the bone microenvironment. After all, if a tumour becomes able to grow in the immune “lion’s den” of the organism, its action on immunity cannot be anything short of incredible.
4. Discussion and Conclusions
Here, we presented an overview of key topics related to bone metastasis (summarised in Figure 1), focusing our attention on classical concepts related to bone marrow metastasis, as well as aspects that have emerged in the very last few years, such as the cellular dormancy and extracellular vesicles. Factors of key importance regulating the establishment of the bone marrow premetastatic niche have been presented. An additional phenomenon that we discussed in this review, is cancer cells homing and establishment of the vicious cycle in the bone marrow. This is one of the most investigated areas in bone metastasis, with many seminal reports showing the importance of factors such as αvβ3 and VLA-4 integrins, OPN, CD44, CXCR4 and more.
Hypoxia is another crucial factor in bone metastasis development. The bone marrow is a fundamentally hypoxic environment, and HIF-1α is able to promote bone metastatisation even when expressed in the primary site. HIF modulators such as HBB may also take part in bone and visceral metastatisation. The HIF family may also activate proton production in the bone microenvironment, thus signalling to nociceptive terminals and causing severe pain, which is one of the most terrible consequences of bone metastasis. Cancer cells also exploit HIF signalling to activate angiogenesis, which results in the production of factors like VEGF and bFGF, which can not only activate angiogenesis, but also osteoblast and osteoclast activity, which can fuel the osteoblastic and osteolytic vicious cycles.
With regards to cancer cellular dormancy and extracellular vesicles, these topics are very complex, and despite considerable efforts by the scientific community, there is still much work to do before we reach a reasonable comprehension of these two aspects, which are clearly important in the cancer field. In particular, understanding how cellular dormancy works may allow us to create drugs that keep the cells dormant, or awaken them so that they become sensitive to chemotherapy. As for extracellular vesicles, they have potential both as biomarkers and therapeutic drug delivery systems. The fact that EVs mirror the content of the cell of origin, has been exploited in cancer theranostics to detect the onset of target therapy resistance or efficacy by circulating cell-free DNA analysis [234,235,236,237] in order to adjust therapy in itinere and in an individualised manner. Cancer cells produce very high amounts of EVs compared to their normal counterparts [199] and are enriched in aggressiveness-related features in patients. This also holds true for high invasive MDA-MB-231 vs. low invasive MCF7 cell lines [238,239]. Although in general terms, inter-patient variability seems to be in line with that of other biomarkers [240], a biomarker-based pilot study to assess normal levels of EV-shuttled factors is crucial for most biomarkers (e.g., miRNAs), since the field is new, and normality ranges, biological variability and technical variability are still not established [241,242]. However, the fact that these are communication devices for cells, opens many possibilities in the drug delivery field. In fact, loading extracellular vesicles with drugs, could allow us to minimise side effects, while increasing the drug concentration in target cells. This would be particularly important in bone metastasis, since the endosteal niche is hypoxic and relatively less perfused, and targeting would help increase therapeutic efficacy of cytotoxic drugs as well as targeted therapy.
Much work has been carried out on the topic of bone metastases, with the identification of useful therapeutic options, such as bisphosphonates, and Denosumab (anti-RANKL mAb), both targeting the osteoclastic side of the vicious cycles, and being able to reduce skeletal-related events both in metastatic prostate [243] and breast [244] cancers. Bone metastases are, therefore, treatable, but still incurable. Nevertheless, ongoing research holds much promise for the future, and will try to provide novel actionable pathways and drugs, in hopes of overcoming this very common complication of the most prevalent human cancers.
Funding
This review was supported by the grant IG 2015 Id.16826 of the “Associazione Italiana per la Ricerca sul Cancro” to NR, and by the “Associazione Italiana per la Ricerca sul Cancro” Fellowship for Italy 2019 Id.24019 to MP for salary.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ANXA | Annexin |
| BALP | Bone-specific alkaline phosphatase |
| BM | Bone metastasis |
| BMA | Bone marrow adipocyte |
| BMM | Bone Marrow Macrophages |
| BMP | Bone morphogenetic proteins |
| BSP | Bone sialoprotein |
| CaSr | Calcium-sensing receptor |
| CCL | CC chemokine ligands |
| CCR | CC chemokine receptor |
| CDH2 | N-cadherin |
| COX | Cyclooxygenase |
| CTCs | Circulating tumour cells |
| CTGF | Connective tissue growth factor |
| CXCR | C-X-C chemokine receptor type |
| DDR | Discoidin domain receptor |
| DKK1 | Dikkopf-1 |
| DTCs | Disseminated tumour cells |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ET | Endothelin |
| EVs | Extracellular vesicles |
| FABP4 | Fatty acid-binding protein 4 |
| FAK | Focal adhesion kinase |
| FGF | Fibroblast growth factors |
| HBB | Haemoglobin beta |
| HGF | Hepatocyte growth factor |
| HIF | Hypoxia-inducible factors |
| HSC | Haematopoietic stem cells |
| IGF | Insulin-like growth factor |
| IL | Interleukin |
| LOX | Lysyl oxidase |
| MCP | Monocyte Chemoattractant Protein |
| M-CSF | Macrophage-colony stimulating factor |
| MDSC | Myeloid-derived suppressor cells |
| MITF | Microphthalmia Transcription Factor |
| MMP | Matrix metalloprotease |
| MSCs | Mesenchymal stromal cells |
| NFATc1 | Nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent |
| NfĸB | Nuclear factor kappa B |
| NICD | Notch intracellular domain |
| NO | Nitric Oxide |
| NOS | Nitric oxide synthase |
| NRDG | N-myc downstream-regulated gene |
| NSCLC | Nonsmall-cell lung cancer |
| OCN | Osteocalcin |
| OPN | Osteopontin |
| OPN | Osteopontin |
| PDGF | Platelet-derived growth factor |
| PD-L1 | Programmed death-ligand 1 |
| PMN | Premetastatic niche |
| PTHrP | Parathyroid hormone-related peptide |
| RANKL | Receptor activator of nuclear factor kappa B ligand |
| RAS | Rat sarcoma |
| Runx2 | Runt-related transcription factor 2 |
| SCF | Stem cell factor |
| SDF | Stromal-derived factor |
| SNOs | Spindle-shaped N-Cadherin-positive osteoblasts |
| SOST | Sclerostin |
| SREs | Skeletal-related events |
| TAM | Tumour-associated macrophages |
| TAN | Tumour-associated neutrophils |
| TBK1 | Tank-binding kinase 1 |
| TGF-β | Transforming growth factor-beta |
| TM4SF1 | Tetraspanin transmembrane 4 L six family member 1 |
| TNF-α | Tumour necrosis factor-alpha |
| TOR | Target of rapamycin |
| VCAM | Vascular cell adhesion molecule |
| VEGF | Vascular endothelial growth factor |
| VLA | Very late antigen |
| Wnt | Wingless-type MMTV integration |
References
- Buijs, T.J.; Kuijpers, C.H.J.C.; van der Pluijm, G. Targeted therapy options for treatment of bone metastases; beyond bisphosphonates. Curr. Pharm. Des. 2010, 16, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Aielli, F.; Ponzetti, M.; Rucci, N. Bone metastasis pain, from the bench to the bedside. Int. J. Mol. Sci. 2019, 20, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Oronzo, S.; Coleman, R.; Brown, J.; Silvestris, F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J. Bone Oncol. 2019, 15, 10025. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11, 43–49. [Google Scholar] [CrossRef]
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef] [Green Version]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Rucci, N.; Teti, A. Osteomimicry: How the seed grows in the soil. Calcif. Tissue Int. 2018, 102, 131–140. [Google Scholar] [CrossRef]
- Woodward, J.E.; Coleman, E.R. Prevention and treatment of bone metastases. Curr. Pharm. Des. 2010, 16, 2998–3006. [Google Scholar] [CrossRef]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Thirunavukkarasu, K.; Zhou, L.; Pastore, L.; Baldini, A.; Hecht, J.; Geoffroy, V.; Ducy, P.; Karsenty, G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat. Genet. 1997, 16, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Galán-Díez, M.; Isa, A.; Ponzetti, M.; Nielsen, M.F.; Kassem, M.; Kousteni, S. Normal hematopoiesis and lack of β-catenin activation in osteoblasts of patients and mice harboring Lrp5 gain-of-function mutations. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Cappariello, A.; Ponzetti, M.; Rucci, N. The “soft” side of the bone: Unveiling its endocrine functions. Horm. Mol. Biol. Clin. Investig. 2016, 28, 5–20. [Google Scholar] [CrossRef]
- Clarke, B. Normal bone anatomy and physiology. Clin. J. Am. Soc. Nephrol. 2008, 3 (Suppl. 3), S131–S139. [Google Scholar] [CrossRef] [Green Version]
- Bonewald, L.F. Osteocytes. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism; Rosen, C.J., Ed.; Wiley: Washington, DC, USA, 2008; pp. 22–27. [Google Scholar]
- Temiyasathit, S.; Jacobs, C.R. The osteocyte primary cilium and its role in bone mechanotransduction. Ann. N. Y. Acad. Sci. 2010, 1192, 422–428. [Google Scholar] [CrossRef]
- Vaughan, T.J.; Mullen, C.A.; Verbruggen, S.W.; McNamara, L.M. Bone cell mechanosensation of fluid flow stimulation: A fluid-structure interaction model characterising the role integrin attachments and primary cilia. Biomech. Model. Mechanobiol. 2015, 14, 703–718. [Google Scholar] [CrossRef]
- Nguyen, A.M.; Jacobs, C.R. Emerging role of primary cilia as mechanosensors in osteocytes. Bone 2013, 54, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Noble, B.S. The osteocyte lineage. Arch. Biochem. Biophys. 2008, 473, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Jähn, K.; Kelkar, S.; Zhao, H.; Xie, Y.; Tiede-Lewis, L.A.M.; Dusevich, V.; Dallas, S.L.; Bonewald, L.F. Osteocytes acidify their microenvironment in response to PTHrP in vitro and in lactating mice in vivo. J. Bone Miner. Res. 2017, 32, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Mori, G.; D’Amelio, P.; Faccio, R.; Brunetti, G. The interplay between the bone and the immune system. Clin. Dev. Immunol. 2013, 2013, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappariello, A.; Maurizi, A.; Veeriah, V.; Teti, A. The Great Beauty of the osteoclast. Arch. Biochem. Biophys. 2014, 558, 70–78. [Google Scholar] [CrossRef]
- Kawaguchi, N.; Noda, M. Mitf is expressed in osteoclast progenitors in vitro. Exp. Cell Res. 2000, 260, 284–291. [Google Scholar] [CrossRef]
- Tondravi, M.M.; McKercher, S.R.; Anderson, K.; Erdmann, J.M.; Quiroz, M.; Maki, R.; Teitelbaum, S.L. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature 1997, 386, 81–84. [Google Scholar] [CrossRef]
- Xu, F.; Teitelbaum, S.L. Osteoclasts: New insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [Green Version]
- Burgess, T.L.; Qian, Y.X.; Kaufman, S.; Ring, B.D.; Van, G.; Capparelli, C.; Kelley, M.; Hsu, H.; Boyle, W.J.; Dunstan, C.R.; et al. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J. Cell Biol. 1999, 145, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 1997, 11, 3482–3496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponzetti, M.; Rucci, N. Updates on osteoimmunology: What’s new on the cross-talk between bone and immune system. Front. Endocrinol. 2019, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef]
- Mohamad, S.F.; Xu, L.; Ghosh, J.; Childress, P.J.; Abeysekera, I.; Himes, E.R.; Wu, H.; Alvarez, M.B.; Davis, K.M.; Aguilar-Perez, A.; et al. Osteomacs interact with megakaryocytes and osteoblasts to regulate murine hematopoietic stem cell function. Blood Adv. 2017, 1, 2520–2528. [Google Scholar] [CrossRef] [Green Version]
- Vi, L.; Baht, G.S.; Whetstone, H.; Ng, A.; Wei, Q.; Poon, R.; Mylvaganam, S.; Grynpas, M.; Alman, B.A. Macrophages promote osteoblastic differentiation in-vivo: Implications in fracture repair and bone homeostasis. J. Bone Miner. Res. 2015, 30, 1090–1102. [Google Scholar] [CrossRef]
- Sousa, S.; Määttä, J. The role of tumour-associated macrophages in bone metastasis. J. Bone Oncol. 2016, 5, 135–138. [Google Scholar] [CrossRef] [Green Version]
- Soki, F.N.; Cho, S.W.; Kim, Y.W.; Jones, J.D.; Park, S.I.; Koh, A.J.; Entezami, P.; Daignault-Newton, S.; Pienta, K.J.; Roca, H.; et al. Bone marrow macrophages support prostate cancer growth in bone. Oncotarget 2015, 6, 35782–35796. [Google Scholar] [CrossRef] [Green Version]
- Kusumbe, A.P. Vascular niches for disseminated tumour cells in bone. J. Bone Oncol. 2016, 5, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, S.K.; Kusumbe, A.P.; Schiller, M.; Zeuschner, D.; Bixel, M.G.; Milia, C.; Gamrekelashvili, J.; Limbourg, A.; Medvinsky, A.; Santoro, M.M.; et al. Blood flow controls bone vascular function and osteogenesis. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Grosso, A.; Burger, M.G.; Lunger, A.; Schaefer, D.J.; Banfi, A.; Di Maggio, N. It takes two to tango: Coupling of angiogenesis and osteogenesis for bone regeneration. Front. Bioeng. Biotechnol. 2017, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Bei, X.; Yang, B.; Wang, X.; Jiang, C.; Shi, F.; Wang, X.; Zhu, Y.; Jing, Y.; Han, B.; et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.C.; Lee, Y.C.; Yu, G.; Cheng, C.J.; Zhou, X.; Chu, K.; Murshed, M.; Le, N.T.; Baseler, L.; Abe, J.; et al. Endothelial-to-osteoblast conversion generates osteoblastic metastasis of prostate cancer. Dev. Cell 2017, 41, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, P.; Robey, P.G. Marrow stromal stem cells. J. Clin. Investig. 2000, 105, 1663–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardaway, A.L.; Herroon, M.K.; Rajagurubandara, E.; Podgorski, I. Bone marrow fat: Linking adipocyte-induced inflammation with skeletal metastases. Cancer Metastasis Rev. 2014, 33, 527–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, M.; de Paula, F.J.A.; Rosen, C.J. New insights into osteoporosis: The bone-fat connection. J. Intern. Med. 2012, 272, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.R.; Sul, O.J.; Kim, Y.Y.; Kim, H.J.; Yu, R.; Suh, J.H.; Choi, H.S. Saturated fatty acids enhance osteoclast survival. J. Lipid Res. 2010, 51, 892–899. [Google Scholar] [CrossRef] [Green Version]
- Corre, J.; Planat-Benard, V.; Corberand, J.X.; Pénicaud, L.; Casteilla, L.; Laharrague, P. Human bone marrow adipocytes support complete myeloid and lymphoid differentiation from human CD34+ cells. Br. J. Haematol. 2004, 127, 344–347. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; He, Y.; Yu, X. Bone marrow adipocyte: An intimate partner with tumor cells in bone metastasis. Front. Endocrinol. 2018, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Hardaway, A.L.; Powell, K.; Turchick, A.; Feldmann, D.; Podgorski, I. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget 2013, 4, 2108–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, M.; Chen, J.; Ye, Y.; Tseng, H.-Y.; Lai, F.; Tay, K.H.; Jin, L.; Guo, S.T.; Jiang, C.C.; Zhang, X.D. Adipocytes contribute to resistance of human melanoma cells to chemotherapy and targeted therapy. Curr. Med. Chem. 2014, 21, 1255–1267. [Google Scholar] [CrossRef]
- Yu, W.; Cao, D.D.; Li, Q.B.; Mei, H.L.; Hu, Y.; Guo, T. Adipocytes secreted leptin is a pro-tumor factor for survival of multiple myeloma under chemotherapy. Oncotarget 2016, 7, 86075–86086. [Google Scholar] [CrossRef]
- Haider, M.T.; Smit, D.J.; Taipaleenmäki, H. The endosteal niche in breast cancer bone metastasis. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Fattore, A.D.; Teti, A.; Rucci, N. Bone cells and the mechanisms of bone remodelling. Front. Biosci. 2012, 4, 2302–2321. [Google Scholar] [CrossRef]
- Maurizi, A.; Rucci, N. The osteoclast in bone metastasis: Player and target. Cancers 2018, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Rucci, N.; Teti, A. The “love-hate” relationship between osteoclasts and bone matrix. Matrix Biol. 2016, 52–54, 176–190. [Google Scholar] [CrossRef]
- Rucci, N. Molecular biology of bone remodelling. Clin. Cases Miner. Bone Metab. 2008, 5, 49–56. [Google Scholar] [PubMed]
- Raisz, L.G. Physiology and pathophysiology of bone remodeling. Clin. Chem. 1999, 45, 1353–1358. [Google Scholar] [PubMed]
- Harada, S.I.; Rodan, G.A. Control of osteoblast function and regulation of bone mass. Nature 2003, 423, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, T.; Flamini, E.; Mercatali, L.; Sacanna, E.; Serra, P.; Amadori, D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 2010, 116, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Mohammad, K.S.; Käkönen, S.M.; Harris, S.; Wu-Wong, J.R.; Wessale, J.L.; Padley, R.J.; Garrett, I.R.; Chirgwin, J.M.; Guise, T.A. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 10954–10959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.L.; Kang, S.; MacDougald, O.A.; Keller, E.T. Role of Wnts in prostate cancer bone metastases. J. Cell. Biochem. 2006, 97, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Clines, G.A.; Mohammad, K.S.; Bao, Y.; Stephens, O.W.; Suva, L.J.; Shaughnessy, J.D.; Fox, J.W.; Chirgwin, J.M.; Guise, T.A. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol. Endocrinol. 2007, 21, 486–498. [Google Scholar] [CrossRef] [Green Version]
- Ottewell, P.D. The role of osteoblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Takyar, F.M.; Swan, K.; Jeong, J.; Vanhouten, J.; Sullivan, C.; Dann, P.; Yu, H.; Fiaschi-Taesch, N.; Chang, W.; et al. Calcium-sensing receptor promotes breast cancer by stimulating intracrine actions of parathyroid hormone-related protein. Cancer Res. 2016, 76, 5348–5360. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Wysolmerski, J.J. Calcium-sensing receptor in breast physiology and cancer. Front. Physiol. 2016, 7, 440. [Google Scholar] [CrossRef] [Green Version]
- Guise, T.A.; Yin, J.J.; Taylor, S.D.; Kumagai, Y.; Dallas, M.; Boyce, B.F.; Yoneda, T.; Mundy, G.R. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J. Clin. Investig. 1996, 98, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Waning, D.L.; Guise, T.A. Molecular mechanisms of bone metastasis and associated muscle weakness. Clin. Cancer Res. 2014, 20, 3071–3077. [Google Scholar] [CrossRef] [Green Version]
- Van Kempen, L.C.L.; Coussens, L.M. MMP9 potentiates pulmonary metastasis formation. Cancer Cell 2002, 2, 251–252. [Google Scholar] [CrossRef] [Green Version]
- Mendes, O.; Kim, H.T.; Stoica, G. Expression of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat model. Clin. Exp. Metastasis 2005, 22, 237–246. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Nakamura, K.; Iwai, S.; Murakami, M.; Itoh, T.; Kijima, H.; Shipley, J.M.; Senior, R.M.; Shibuya, M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2002, 2, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Owyong, M.; Chou, J.; van den Bijgaart, R.J.E.; Kong, N.; Efe, G.; Maynard, C.; Talmi-Frank, D.; Solomonov, I.; Koopman, C.; Hadler-Olsen, E.; et al. MMP9 modulates the metastatic cascade and immune landscape for breast cancer anti-metastatic therapy. Life Sci. Alliance 2019, 2, e201800226. [Google Scholar] [CrossRef] [Green Version]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Li, C.J.; Yiang, G.T.; Cheng, Y.L.; Tsai, A.P.Y.; Hou, Y.T.; Ho, Y.C.; Hou, M.F.; Chu, P.Y. Molecular Regulation of bone metastasis pathogenesis. Cell. Physiol. Biochem. 2018, 46, 1423–1438. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef]
- Reynaud, C.; Ferreras, L.; Di Mauro, P.; Kan, C.; Croset, M.; Bonnelye, E.; Pez, F.; Thomas, C.; Aimond, G.; Karnoub, A.E.; et al. Lysyl oxidase is a strong determinant of tumor cell colonization in bone. Cancer Res. 2017, 77, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.R.; Rumney, R.M.H.; Schoof, E.M.; Perryman, L.; Høye, A.M.; Agrawal, A.; Bird, D.; Latif, N.A.; Forrest, H.; Evans, H.R.; et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 2015, 522, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Kelly, T.; Suva, L.J.; Huang, Y.; MacLeod, V.; Miao, H.Q.; Walker, R.C.; Sanderson, R.D. Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Res. 2005, 65, 5778–5784. [Google Scholar] [CrossRef] [Green Version]
- Martin, T.J.; Johnson, R.W. Multiple actions of parathyroid hormone-related protein in breast cancer bone metastasis. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massagué, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Psaila, B.; Lyden, D. Bone marrow cells in the “pre-metastatic niche”: Within bone and beyond. Cancer Metastasis Rev. 2006, 25, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y. The lung metastatic niche. J. Mol. Med. 2015, 93, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Doglioni, G.; Parik, S.; Fendt, S.M. Interactions in the (pre)metastatic niche support metastasis formation. Front. Oncol. 2019, 9, 219–291. [Google Scholar] [CrossRef]
- Pang, X.; Gong, K.; Zhang, X.; Wu, S.; Cui, Y.; Qian, B.Z. Osteopontin as a multifaceted driver of bone metastasis and drug resistance. Pharmacol. Res. 2019, 144, 235–244. [Google Scholar] [CrossRef]
- Shevde, L.; Das, S.; Clark, D.; Samant, R. Osteopontin: An effector and an effect of tumor metastasis. Curr. Mol. Med. 2010, 10, 71–81. [Google Scholar] [CrossRef]
- Wang, L.; Song, L.; Li, J.; Wang, Y.; Yang, C.; Kou, X.; Xiao, B.; Zhang, W.; Li, L.; Liu, S.; et al. Bone sialoprotein-αvβ3 integrin axis promotes breast cancer metastasis to the bone. Cancer Sci. 2019, 110, 3157–3172. [Google Scholar] [CrossRef] [PubMed]
- Kwakwa, K.A.; Sterling, J.A. Integrin αvβ3 signaling in tumor-induced bone disease. Cancers 2017, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Sloan, E.K.; Pouliot, N.; Stanley, K.L.; Chia, J.; Moseley, J.M.; Hards, D.K.; Anderson, R.L. Tumor-specific expression of αvβ3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006, 8. [Google Scholar] [CrossRef] [Green Version]
- Karadag, A.; Ogbureke, K.U.E.; Fedarko, N.S.; Fisher, L.W. Bone sialoprotein, matrix metalloproteinase 2, and αvβ3 integrin in osteotropic cancer cell invasion. J. Natl. Cancer Inst. 2004, 96, 956–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchaud, O.; Baron, R.; Clément-Lacroix, P.; Clézardin, P. Tumor αvβ3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007, 67, 5821–5830. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.G.; Amend, S.R.; Weilbaecher, K.N. Integrins and bone metastasis: Integrating tumor cell and stromal cell interactions. Bone 2011, 48, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Sun, X.; Wang, K.; Feng, H.; Liu, Y.; Fei, C.; Wan, S.; Wang, W.; Luo, J.; Shi, Q.; et al. BMP9 inhibits the bone metastasis of breast cancer cells by downregulating CCN2 (connective tissue growth factor, CTGF) expression. Mol. Biol. Rep. 2014, 41, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Kubota, S.; Yoshioka, N.; Ibaragi, S.; Isowa, S.; Eguchi, T.; Sasaki, A.; Takigawa, M. Pathogenic role of connective tissue growth factor (CTGF/CCN2) in osteolytic metastasis of breast cancer. J. Bone Miner. Res. 2006, 21, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, H.; Jung, S.; Moon, A.; Noh, D.Y.; Lee, Z.H.; Kim, H.J.; Kim, H.H. A CTGF-RUNX2-RANKL axis in breast and prostate cancer cells promotes tumor progression in bone. J. Bone Miner. Res. 2020, 35, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Allocca, G.; Hughes, R.; Wang, N.; Brown, H.K.; Ottewell, P.D.; Brown, N.J.; Holen, I. The bone metastasis niche in breast cancer-potential overlap with the haematopoietic stem cell niche in vivo. J. Bone Oncol. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Sharma, M.; Afrin, F.; Satija, N.; Tripathi, R.P.; Gangenahalli, G.U. Stromal-derived factor-1/CXCR4 signaling: Indispensable role in homing and engraftment of hematopoietic stem cells in bone marrow. Stem Cells Dev. 2011, 20, 933–946. [Google Scholar] [CrossRef]
- Kucia, M.; Jankowski, K.; Reca, R.; Wysoczynski, M.; Bandura, L.; Allendorf, D.J.; Zhang, J.; Ratajczak, J.; Ratajczak, M.Z. CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J. Mol. Histol. 2004, 35, 233–245. [Google Scholar] [CrossRef]
- Zhou, Y.; Cao, H.B.; Li, W.J.; Zhao, L. The CXCL12 (SDF-1)/CXCR4 chemokine axis: Oncogenic properties, molecular targeting, and synthetic and natural product CXCR4 inhibitors for cancer therapy. Chin. J. Nat. Med. 2018, 16, 801–810. [Google Scholar] [CrossRef]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2014; Volume 124, pp. 31–82. [Google Scholar]
- Cho, Y.; Lee, H.W.; Kang, H.G.; Kim, H.Y.; Kim, S.J.; Chun, K.H. Cleaved CD44 intracellular domain supports activation of stemness factors and promotes tumorigenesis of breast cancer. Oncotarget 2015, 6, 8709–8721. [Google Scholar] [CrossRef] [Green Version]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as cancer stem cell markers: An enduring ambiguity. Clin. Dev. Immunol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stivarou, T.; Patsavoudi, E. Extracellular molecules involved in cancer cell invasion. Cancers 2015, 7, 238–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front. Cell Dev. Biol. 2017, 5, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Stamenkovic, I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999, 13, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Zöller, M. CD44, hyaluronan, the hematopoietic stem cell, and leukemia-initiating cells. Front. Immunol. 2015, 6, 235. [Google Scholar]
- Jung, Y.; Shiozawa, Y.; Wang, J.; Patel, L.R.; Havens, A.M.; Song, J.; Krebsbach, P.H.; Roodman, G.D.; Taichman, R.S. Annexin-2 is a regulator of stromal cell-derived factor-1/CXCL12 function in the hematopoietic stem cell endosteal niche. Exp. Hematol. 2011, 39. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Wang, J.; Song, J.; Shiozawa, Y.; Wang, J.; Havens, A.; Wang, Z.; Sun, Y.X.; Emerson, S.G.; Krebsbach, P.H.; et al. Annexin II expressed by osteoblasts and endothelial cells regulates stem cell adhesion, homing, and engraftment following transplantation. Blood 2007, 110, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Havens, A.M.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, J.; Lu, G.; Roodman, G.D.; Loberg, R.D.; et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell. Biochem. 2008, 105, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Wang, J.; Lee, E.; McGee, S.; Berry, J.E.; Yumoto, K.; Dai, J.; Keller, E.T.; Shiozawa, Y.; Taichman, R.S. Annexin 2-CXCL12 interactions regulate metastatic cell targeting and growth in the bone marrow. Mol. Cancer Res. 2015, 13, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Bae, Y.; Kasimir-Bauer, S.; Tang, R.; Chen, J.; Ren, G.; Yuan, M.; Esposito, M.; Li, W.; Wei, Y.; et al. Therapeutic antibody targeting tumor- and osteoblastic niche-derived jagged1 sensitizes bone metastasis to chemotherapy. Cancer Cell 2017, 32, 731–747. [Google Scholar] [CrossRef] [Green Version]
- Sethi, N.; Kang, Y. Notch signalling in cancer progression and bone metastasis. Br. J. Cancer 2011, 105, 1805–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, N.; Dai, X.; Winter, C.G.; Kang, Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 2011, 19, 192–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X.; Zhang, H.; Li, X.; Li, X.; Cong, M.; Peng, F.; Yu, J.; Zhang, X.; Yang, Q.; Hu, G. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat. Cell Biol. 2017, 19, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Ricarte, F.R.; Le Henaff, C.; Kolupaeva, V.G.; Gardella, T.J.; Partridge, N.C. Parathyroid hormone(1–34) and its analogs differentially modulate osteoblastic Rankl expression via PKA/SIK2/SIK3 and PP1/PP2A-CRTC3 signaling. J. Biol. Chem. 2018, 293, 20200–20213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Loberg, R.; Liao, J.; Ying, C.; Snyder, L.A.; Pienta, K.J.; McCauley, L.K. A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res. 2009, 69, 1685–1692. [Google Scholar] [CrossRef] [Green Version]
- Mulholland, B.S.; Forwood, M.R.; Morrison, N.A. Monocyte Chemoattractant Protein-1 (MCP-1/CCL2) drives activation of bone remodelling and skeletal metastasis. Curr. Osteoporos. Rep. 2019, 17, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianconi, V.; Sahebkar, A.; Atkin, S.L.; Pirro, M. The regulation and importance of monocyte chemoattractant protein-1. Curr. Opin. Hematol. 2018, 25, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G.; Ratcliffe, P.J.; Semenza, G.L. Pathways for oxygen regulation and homeostasis: The 2016 albert lasker basic medical research award. JAMA J. Am. Med. Assoc. 2016, 316, 1252–1253. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Dutta, B.; Laha, A.; Bandopadhyay, R. 2019 Nobel Prize in Physiology and Medicine: A long road from bench to bedside of cell in oxygen sensing and adaptive responses. Natl. Acad. Sci. Lett. 2020. [Google Scholar] [CrossRef]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51. [Google Scholar] [CrossRef]
- Pezzuto, A.; Carico, E. Role of HIF-1 in cancer progression: Novel insights. A review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, E.S.; Sund, S.; Homsi, E.N.; Challenger, L.F.; Rameshwar, P. A theoretical simulation of hematopoietic stem cells during oxygen fluctuations: Prediction of bone marrow responses during hemorrhagic shock. Shock 2004, 22, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulley, P.A.; Bishop, T.; Vernet, A.; Schneider, J.E.; Edwards, J.R.; Athanasou, N.A.; Knowles, H.J. Hypoxia-inducible factor 1-alpha does not regulate osteoclastogenesis but enhances bone resorption activity via prolyl-4-hydroxylase 2. J. Pathol. 2017, 242, 322–333. [Google Scholar] [CrossRef]
- Bentovim, L.; Amarilio, R.; Zelzer, E. HIF1α is a central regulator of collagen hydroxylation and secretion under hypoxia during bone development. Development 2012, 139, 4473–4483. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wan, C.; Deng, L.; Liu, X.; Cao, X.; Gilbert, S.R.; Bouxsein, M.L.; Faugere, M.C.; Guldberg, R.E.; Gerstenfeld, L.C.; et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Investig. 2007, 117, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Devignes, C.S.; Aslan, Y.; Brenot, A.; Devillers, A.; Schepers, K.; Fabre, S.; Chou, J.; Casbon, A.J.; Werb, Z.; Provot, S. HIF signaling in osteoblast-lineage cells promotes systemic breast cancer growth and metastasis in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E992–E1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Pez, F.; Dayan, F.; Durivault, J.; Kaniewski, B.; Aimond, G.; Le Provost, G.S.; Deux, B.; Clézardin, P.; Sommer, P.; Pouysségur, J.; et al. The HIF-1-inducible lysyl oxidase activates HIF-1 via the akt pathway in a positive regulation loop and synergizes with HIF-1 in promoting tumor cell growth. Cancer Res. 2011, 71, 1647–1657. [Google Scholar] [CrossRef] [Green Version]
- Pezzuto, A.; Perrone, G.; Orlando, N.; Citarella, F.; Ciccozzi, M.; Scarlata, S.; Tonini, G. A close relationship between HIF-1α expression and bone metastases in advanced NSCLC, a retrospective analysis. Oncotarget 2019, 10, 7071–7079. [Google Scholar] [CrossRef] [Green Version]
- Woelfle, U.; Cloos, J.; Sauter, G.; Riethdorf, L.; Jänicke, F.; Van Diest, P.; Brakenhoff, R.; Pantel, K. Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res. 2003, 63, 5679–5684. [Google Scholar]
- Hiraga, T.; Kizaka-Kondoh, S.; Hirota, K.; Hiraoka, M.; Yoneda, T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007, 67, 4157–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, H.; Hiraoka, M.; Kizaka-Kondoh, S. Antitumor effect of TAT-oxygen-dependent degradation-caspase-3 fusion protein specifically stabilized and activated in hypoxic tumor cells. Cancer Res. 2002, 62, 2013–2018. [Google Scholar] [PubMed]
- Capulli, M.; Angelucci, A.; Driouch, K.; Garcia, T.; Clement-Lacroix, P.; Martella, F.; Ventura, L.; Bologna, M.; Flamini, S.; Moreschini, O.; et al. Increased expression of a set of genes enriched in oxygen binding function discloses a predisposition of breast cancer bone metastases to generate metastasis spread in multiple organs. J. Bone Miner. Res. 2012, 27, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Ponzetti, M.; Capulli, M.; Angelucci, A.; Ventura, L.; Monache, S.D.; Mercurio, C.; Calgani, A.; Sanità, P.; Teti, A.; Rucci, N. Non-conventional role of haemoglobin beta in breast malignancy. Br. J. Cancer 2017, 117, 994–1006. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Miyamoto, D.T.; Wittner, B.S.; Sullivan, J.P.; Aceto, N.; Jordan, N.V.; Yu, M.; Karabacak, N.M.; Comaills, V.; Morris, R.; et al. Expression of β-globin by cancer cells promotes cell survival during blood-borne dissemination. Nat. Commun. 2017, 8, 14344. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Hiasa, M.; Nagata, Y.; Okui, T.; White, F.A. Acidic microenvironment and bone pain in cancer-colonized bone. Bonekey Rep. 2015, 4. [Google Scholar] [CrossRef]
- Falk, S.; Dickenson, A.H. Pain and nociception: Mechanisms of cancer-induced bone pain. J. Clin. Oncol. 2014, 32, 1647–1654. [Google Scholar] [CrossRef] [Green Version]
- Di Pompo, G.; Lemma, S.; Canti, L.; Rucci, N.; Ponzetti, M.; Errani, C.; Donati, D.M.; Russell, S.; Gillies, R.; Chano, T.; et al. Intratumoral acidosis fosters cancer-induced bone pain through the activation of the mesenchymal tumor-associated stroma in bone metastasis from breast carcinoma. Oncotarget 2017, 8, 54478–54496. [Google Scholar] [CrossRef]
- Yoneda, T.; Hiasa, M.; Nagata, Y.; Okui, T.; White, F. Contribution of acidic extracellular microenvironment of cancer-colonized bone to bone pain. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2677–2684. [Google Scholar] [CrossRef] [Green Version]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002, 16, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Maroni, P.; Matteucci, E.; Luzzati, A.; Perrucchini, G.; Bendinelli, P.; Desiderio, M.A. Nuclear co-localization and functional interaction of COX-2 and HIF-1α characterize bone metastasis of human breast carcinoma. Breast Cancer Res. Treat. 2011, 129, 433–450. [Google Scholar] [CrossRef] [Green Version]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [Green Version]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Luzzati, A.; Perrucchini, G.; Desiderio, M.A. Microenvironmental stimuli affect Endothelin-1 signaling responsible for invasiveness and osteomimicry of bone metastasis from breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Raymaekers, K.; Stegen, S.; van Gastel, N.; Carmeliet, G. The vasculature: A vessel for bone metastasis. Bonekey Rep. 2015. [Google Scholar] [CrossRef] [Green Version]
- Cook, L.M.; Shay, G.; Aruajo, A.; Lynch, C.C. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metastasis Rev. 2014, 33, 511–525. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Olsen, B.R. Osteoblast-derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J. Clin. Investig. 2016, 126, 509–526. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Kitagawa, Y.; Zhang, J.; Yao, Z.; Mizokami, A.; Cheng, S.; Nör, J.; McCauley, L.K.; Taichman, R.S.; Keller, E.T. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res. 2004, 64, 994–999. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, Y.; Dai, J.; Zhang, J.; Keller, J.M.; Nor, J.; Yao, Z.; Keller, E.T. Vascular endothelial growth factor contributes to prostate cancer-mediated osteoblastic activity. Cancer Res. 2005, 65, 10921–10929. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; McHugh, K.P.; Patntirapong, S.; Gu, X.; Wunderlich, L.; Hauschka, P.V. VEGF enhancement of osteoclast survival and bone resorption involves VEGF receptor-2 signaling and β3-integrin. Matrix Biol. 2008, 27, 589–599. [Google Scholar] [CrossRef]
- Hurley, M.M.; Lee, S.K.; Raisz, L.G.; Bernecker, P.; Lorenzo, J. Basic fibroblast growth factor induces osteoclast formation in murine bone marrow cultures. Bone 1998, 22, 309–316. [Google Scholar] [CrossRef]
- Collin-Osdoby, P.; Rothe, L.; Bekker, S.; Anderson, F.; Huang, Y.; Osdoby, P. Basic fibroblast growth factor stimulates osteoclast recruitment, development, and bone pit resorption in association with angiogenesis in vivo on the chick chorioallantoic membrane and activates isolated avian osteoclast resorption in vitro. J. Bone Miner. Res. 2002, 17, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Vignani, F.; Bertaglia, V.; Buttigliero, C.; Tucci, M.; Scagliotti, G.V.; Di Maio, M. Skeletal metastases and impact of anticancer and bone-targeted agents in patients with castration-resistant prostate cancer. Cancer Treat. Rev. 2016, 44, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Felber, K.; Elks, P.M.; Lecca, M.; Roehl, H.H. Expression of osterix is regulated by FGF and Wnt/β-catenin signalling during osteoblast differentiation. PLoS ONE 2015, 10, e0144982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, H.; Inoue, M. Dormancy in cancer. Cancer Sci. 2019, 110, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Jahanban-Esfahlan, R.; Seidi, K.; Manjili, M.H.; Jahanban-Esfahlan, A.; Javaheri, T.; Zare, P. Tumor cell dormancy: Threat or opportunity in the fight against cancer. Cancers 2019, 11, 1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef] [Green Version]
- Neophytou, C.M.; Kyriakou, T.C.; Papageorgis, P. Mechanisms of metastatic tumor dormancy and implications for cancer therapy. Int. J. Mol. Sci. 2019, 20, 6158. [Google Scholar] [CrossRef] [Green Version]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef]
- Byrne, N.M.; Summers, M.A.; McDonald, M.M. Tumor cell dormancy and reactivation in bone: Skeletal biology and therapeutic opportunities. JBMR Plus 2019, 3, e10125. [Google Scholar] [CrossRef]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Xing, F.; Liu, Y.; Wu, K.; Said, N.; Pochampally, R.; Shiozawa, Y.; Lin, H.K.; Balaji, K.C.; Watabe, K. Secreted protein acidic and rich in cysteine (sparc) mediates metastatic dormancy of prostate cancer in bone. J. Biol. Chem. 2016, 291, 19351–19363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.K.; Jung, Y.; Wang, J.; Joseph, J.; Mishra, A.; Hill, E.E.; Krebsbach, P.H.; Pienta, K.J.; Shiozawa, Y.; Taichman, R.S. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia 2013, 15, 1064–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capulli, M.; Hristova, D.; Valbret, Z.; Carys, K.; Arjan, R.; Maurizi, A.; Masedu, F.; Cappariello, A.; Rucci, N.; Teti, A. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br. J. Cancer 2019, 121, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, N.; Katayama, Y. Regulation of hematopoiesis in endosteal microenvironments. Int. J. Hematol. 2014, 99, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Zhao, M.; Tao, F.; Venkatraman, A.; Li, Z.; Smith, S.E.; Unruh, J.; Chen, S.; Ward, C.; Qian, P.; Perry, J.M.; et al. N-cadherin-expressing bone and marrow stromal progenitor cells maintain reserve hematopoietic stem cells. Cell Rep. 2019, 26, 652–669. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, A.M.; Revollo, L.D.; Woloszynek, J.R.; Civitelli, R.; Link, D.C. N-cadherin in osteolineage cells is not required for maintenance of hematopoietic stem cells. Blood 2012, 120, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Abravanel, D.L.; Belka, G.K.; Pan, T.C.; Pant, D.K.; Collins, M.A.; Sterner, C.J.; Chodosh, L.A. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J. Clin. Investig. 2015, 125, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Nobutani, K.; Shimono, Y.; Mizutani, K.; Ueda, Y.; Suzuki, T.; Kitayama, M.; Minami, A.; Momose, K.; Miyawaki, K.; Akashi, K.; et al. Downregulation of CXCR4 in metastasized breast cancer cells and implication in their dormancy. PLoS ONE 2015, 10, e0130032. [Google Scholar] [CrossRef]
- Gao, X.L.; Zheng, M.; Wang, H.F.; Dai, L.L.; Yu, X.H.; Yang, X.; Pang, X.; Li, L.; Zhang, M.; Wang, S.S.; et al. NR2F1 contributes to cancer cell dormancy, invasion and metastasis of salivary adenoid cystic carcinoma by activating CXCL12/CXCR4 pathway. BMC Cancer 2019, 19, 743. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, D.; Maier, P.; Laufs, S.; Wenz, F.; Zeller, W.J.; Fruehauf, S.; Allgayer, H. CXCR4 expression and treatment with SDF-1α or plerixafor modulate proliferation and chemosensitivity of colon cancer cells. Transl. Oncol. 2013, 6, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyvandi, S.; Lan, Q.; Lorusso, G.; Rüegg, C. Chemotherapy-induced immunological breast cancer dormancy: A new function for old drugs? J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38α/β signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef] [Green Version]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- White, D.E.; Kurpios, N.A.; Zuo, D.; Hassell, J.A.; Blaess, S.; Mueller, U.; Muller, W.J. Targeted disruption of β1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 2004, 6, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the malignant phenotype of human breast cells in three- dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 1997, 137, 231–245. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Nam, J.S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020. [CrossRef] [Green Version]
- Barkan, D.; Chambers, A.F. β1-integrin: A potential therapeutic target in the battle against cancer recurrence. Clin. Cancer Res. 2011, 17, 7219–7223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Chakraborty, G.; Zhang, Z.; Akalay, I.; Gadiya, M.; Gao, Y.; Sinha, S.; Hu, J.; Jiang, C.; Akram, M.; et al. Multi-organ Site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell 2016, 166, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaldis, P.; Pagano, M. Wnt signaling in mitosis. Dev. Cell 2009, 17, 749–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malladi, S.; MacAlinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; De Stanchina, E.; Massagué, J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.L.; Vallée, A. Multiple targets of the canonical WNT/β-catenin signaling in cancers. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.W. Chronic inflammation and breast cancer recurrence. J. Clin. Oncol. 2009, 27, 3418–3419. [Google Scholar] [CrossRef]
- Cappariello, A.; Rucci, N. Tumour-derived extracellular vesicles (EVs): A dangerous “message in a bottle” for bone. Int. J. Mol. Sci. 2019, 20, 4805. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Dudás, J.; Riechelmann, H.; Skvortsova, I.I. The role of exosomes in cancer metastasis. Semin. Cancer Biol. 2017, 44, 170–181. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappariello, A.; Loftus, A.; Muraca, M.; Maurizi, A.; Rucci, N.; Teti, A. Osteoblast-derived extracellular vesicles are biological tools for the delivery of active molecules to bone. J. Bone Miner. Res. 2018, 33, 517–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loftus, A.; Cappariello, A.; George, C.; Ucci, A.; Shefferd, K.; Green, A.; Paone, R.; Ponzetti, M.; Delle Monache, S.; Muraca, M.; et al. Extracellular vesicles from osteotropic breast cancer cells affect bone resident cells. J. Bone Miner. Res. 2020, 35, 396–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Taverna, S.; Pucci, M.; Giallombardo, M.; Di Bella, M.A.; Santarpia, M.; Reclusa, P.; Gil-Bazo, I.; Rolfo, C.; Alessandro, R. Amphiregulin contained in NSCLC-exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Liu, X.; Wang, H.; Li, J.; Dai, L.; Li, J.; Dong, C. Lung adenocarcinoma cell-derived exosomal miR-21 facilitates osteoclastogenesis. Gene 2018, 666, 116–122. [Google Scholar] [CrossRef]
- Valencia, K.; Luis-Ravelo, D.; Bovy, N.; Antón, I.; Martínez-Canarias, S.; Zandueta, C.; Ormazábal, C.; Struman, I.; Tabruyn, S.; Rebmann, V.; et al. MiRNA cargo within exosome-like vesicle transfer influences metastatic bone colonization. Mol. Oncol. 2014, 8, 689–703. [Google Scholar] [CrossRef]
- Karlsson, T.; Lundholm, M.; Widmark, A.; Persson, E. Tumor cell-derived exosomes from the prostate cancer cell line TRAMP-C1 impair osteoclast formation and differentiation. PLoS ONE 2016, 11, e0166284. [Google Scholar] [CrossRef]
- Inder, K.L.; Ruelcke, J.E.; Petelin, L.; Moon, H.; Choi, E.; Rae, J.; Blumenthal, A.; Hutmacher, D.; Saunders, N.A.; Stow, J.L.; et al. Cavin-1/PTRF alters prostate cancer cell-derived extracellular vesicle content and internalization to attenuate extracellular vesicle-mediated osteoclastogenesis and osteoblast proliferation. J. Extracell. Vesicles 2014, 3, 23784. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of bone resorption in periodontitis. J. Immunol. Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajishengallis, G.; Moutsopoulos, N.M.; Hajishengallis, E.; Chavakis, T. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin. Immunol. 2016, 28, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roato, I.; Grano, M.; Brunetti, G.; Colucci, S.; Mussa, A.; Bertetto, O.; Ferracini, R. Mechanisms of spontaneous osteoclastogenesis in cancer with bone involvement. FASEB J. 2005, 19. [Google Scholar] [CrossRef]
- Roato, I.; Brunetti, G.; Gorassini, E.; Grano, M.; Colucci, S.; Bonello, L.; Buffoni, L.; Manfredi, R.; Ruffini, E.; Ottaviani, D.; et al. IL-7 up-regulates TNF-α-dependent osteoclastogenesis in patients affected by solid tumor. PLoS ONE 2006, 1, e124. [Google Scholar] [CrossRef]
- Roato, I.; Gorassini, E.; Brunetti, G.; Grano, M.; Ciuffreda, L.; Mussa, A.; Ferracini, R. IL-7 modulates osteoclastogenesis in patients affected by solid tumors. Ann. New York Acad. Sci. 2007, 1117, 377–384. [Google Scholar] [CrossRef]
- Roato, I.; Caldo, D.; Godio, L.; D’Amico, L.; Giannoni, P.; Morello, E.; Quarto, R.; Molfetta, L.; Buracco, P.; Mussa, A.; et al. Bone invading NSCLC cells produce IL-7: Mice model and human histologic data. BMC Cancer 2010, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancermetastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Kim, S.; Cremasco, V.; Hirbe, A.C.; Novack, D.V.; Weilbaecher, K.; Faccio, R. CD8+ T cells regulate bone tumor burden independent of osteoclast resorption. Cancer Res. 2011, 71, 4799–4808. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.H.; Lee, Y.; Kim, H.J.; Lee, Z.H.; Hyung, S.W.; Lee, S.W.; Kim, H.H. Lyn inhibits osteoclast differentiation by interfering with PLCγ1-mediated Ca2+ signaling. FEBS Lett. 2009, 583, 1164–1170. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzoni, A.; Bronte, V.; Visintin, A.; Spitzer, J.H.; Apolloni, E.; Serafini, P.; Zanovello, P.; Segal, D.M. Myeloid suppressor lines inhibit T cell responses by an no-dependent mechanism. J. Immunol. 2002, 168, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Deshane, J.; Jules, J.; Lee, C.M.; Harris, B.A.; Feng, X.; Ponnazhagan, S. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res. 2013, 73, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawant, A.; Ponnazhagan, S. Myeloid-derived suppressor cells as osteoclast progenitors: A novel target for controlling osteolytic bone metastasis. Cancer Res. 2013, 73, 4606–4610. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef]
- Wang, H.; Wang, S.; Huang, M.; Liang, X.; Tang, Y.-J.; Tang, Y. Targeting Immune-Mediated Dormancy: A Promising Treatment of Cancer. Front. Oncol. 2019, 9, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef]
- Shaul, M.E.; Levy, L.; Sun, J.; Mishalian, I.; Singhal, S.; Kapoor, V.; Horng, W.; Fridlender, G.; Albelda, S.M.; Fridlender, Z.G. Tumor-associated neutrophils display a distinct N1 profile following TGFβ modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology 2016, 5, e1232221. [Google Scholar] [CrossRef] [Green Version]
- Capietto, A.H.; Faccio, R. Immune regulation of bone metastasis. Bonekey Rep. 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardelli, A.; Pantel, K. Liquid biopsies, what we do not know (Yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siravegna, G.; Bardelli, A. Blood circulating tumor DNA for non-invasive genotyping of colon cancer patients. Mol. Oncol. 2016, 10, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Bardelli, A. Genotyping cell-free tumor DNA in the blood to detect residual disease and drug resistance. Genome Biol. 2014, 15, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Stevic, I.; Buescher, G.; Ricklefs, F.L. Monitoring therapy efficiency in cancer through extracellular vesicles. Cells 2020, 9, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Rosenow, M.; Xiao, N.; Spetzler, D. Profiling plasma extracellular vesicle by pluronic block-copolymer based enrichment method unveils features associated with breast cancer aggression, metastasis and invasion. J. Extracell. Vesicles 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K. ISEV2018 abstract book. J. Extracell. Vesicles 2018, 7, 1461450. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Duval, M.X.; Kaimal, V.; Cuff, C.; Clarke, S.H. Assessment of methods for serum extracellular vesicle small RNA sequencing to support biomarker development. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef]
- Oeyen, E.; Willems, H.; ’T Kindt, R.; Sandra, K.; Boonen, K.; Hoekx, L.; De Wachter, S.; Ameye, F.; Mertens, I. Determination of variability due to biological and technical variation in urinary extracellular vesicles as a crucial step in biomarker discovery studies. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.I.; Amorim, M.G.; Gadelha, C.; Milic, I.; Welsh, J.A.; Freitas, V.M.; Nawaz, M.; Akbar, N.; Couch, Y.; Makin, L.; et al. Technical challenges of working with extracellular vesicles. Nanoscale 2018, 10, 881–906. [Google Scholar] [CrossRef] [Green Version]
- Fizazi, K.; Carducci, M.; Smith, M.; Damião, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; De Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Overview of the bone metastatisation process. The primary tumour gets access to the circulation, also thanks to the activation of the HIF pathway, which activates a pro-metastatic program in a subset of cancer cells. Before metastases take place, a number of premetastatic factors, including extracellular vesicles, lysyl oxidase (LOX), heparanase, parathyroid hormone-related peptide (PTHrP), and osteopontin (OPN) educate the bone microenvironment making it suitable for metastatic engraftment. Circulating tumour cells (CTCs) can then extravasate and engraft in the bone thanks to homing factors such as very late antigen (VLA)-4 and αvβ3 integrins, C-X-C chemokine receptor type (CXCR)-4 and CD44. Once in the bone microenvironment, most cells entertain a cross-communication with bone cells, changing the physiologic “virtuous cycle” between osteoblasts and osteoclasts into a “vicious cycle” that favours tumour growth. Two types of vicious cycles are known. The osteosclerotic vicious cycle, normally established by prostate cancer cells, exploits osteoblasts (OBS)-stimulating factors (including extracellular vesicles) to increase OBS production of growth factors, as well as receptor activator of nuclear factor kappa B ligand (RANKL) and macrophage-colony-stimulating factor (M-CSF), that, in turn, stimulate osteoclast differentiation, further allowing the release of growth factors from the bone matrix. The growth factors produced by OBS and released by OCS then signal back to cancer cells, increasing their growth, thus closing the vicious cycle. The osteolytic vicious cycle is established by many types of tumours, including breast cancer. In this cycle, tumour cells use osteoclast-stimulating factors (including extracellular vesicles) and osteoclast differentiation-stimulating factors (including extracellular vesicles) to increase bone resorption, leading to the release of growth factors from the bone matrix, thus furthering tumour growth and restarting the vicious cycle. PTHrP is also exploited by breast cancer cells to induce osteoclastogenesis indirectly, through osteoblastic RANKL and M-CSF. Osteocytes may also take a part in the osteolytic vicious cycle, by suppressing OBS activity through secretion of sclerostin (SOST). A crucial factor in the vicious cycle is HIF, that is activated partially thanks to the low pO2 of the bone marrow microenvironment, and causing increased angiogenesis, OBS stimulation and OCS stimulation. A very small contingent of cells may undertake an alternative path and progress through cellular dormancy. This process requires the stimulation of many membrane and soluble factors, such as bone morphogenic protein (BMP)-7, that induces prostate cancer dormancy, or Jagged-1 which proposedly promotes dormancy through Notch-2 in breast cancer cells. Mesenchymal stromal cells (MSCs), endothelial cells and spindle-shaped N-cadherin-positive osteoblasts (SNOs) seem to be crucial in this process, but also other cell types may be taking part in this process too.
Figure 1.
Overview of the bone metastatisation process. The primary tumour gets access to the circulation, also thanks to the activation of the HIF pathway, which activates a pro-metastatic program in a subset of cancer cells. Before metastases take place, a number of premetastatic factors, including extracellular vesicles, lysyl oxidase (LOX), heparanase, parathyroid hormone-related peptide (PTHrP), and osteopontin (OPN) educate the bone microenvironment making it suitable for metastatic engraftment. Circulating tumour cells (CTCs) can then extravasate and engraft in the bone thanks to homing factors such as very late antigen (VLA)-4 and αvβ3 integrins, C-X-C chemokine receptor type (CXCR)-4 and CD44. Once in the bone microenvironment, most cells entertain a cross-communication with bone cells, changing the physiologic “virtuous cycle” between osteoblasts and osteoclasts into a “vicious cycle” that favours tumour growth. Two types of vicious cycles are known. The osteosclerotic vicious cycle, normally established by prostate cancer cells, exploits osteoblasts (OBS)-stimulating factors (including extracellular vesicles) to increase OBS production of growth factors, as well as receptor activator of nuclear factor kappa B ligand (RANKL) and macrophage-colony-stimulating factor (M-CSF), that, in turn, stimulate osteoclast differentiation, further allowing the release of growth factors from the bone matrix. The growth factors produced by OBS and released by OCS then signal back to cancer cells, increasing their growth, thus closing the vicious cycle. The osteolytic vicious cycle is established by many types of tumours, including breast cancer. In this cycle, tumour cells use osteoclast-stimulating factors (including extracellular vesicles) and osteoclast differentiation-stimulating factors (including extracellular vesicles) to increase bone resorption, leading to the release of growth factors from the bone matrix, thus furthering tumour growth and restarting the vicious cycle. PTHrP is also exploited by breast cancer cells to induce osteoclastogenesis indirectly, through osteoblastic RANKL and M-CSF. Osteocytes may also take a part in the osteolytic vicious cycle, by suppressing OBS activity through secretion of sclerostin (SOST). A crucial factor in the vicious cycle is HIF, that is activated partially thanks to the low pO2 of the bone marrow microenvironment, and causing increased angiogenesis, OBS stimulation and OCS stimulation. A very small contingent of cells may undertake an alternative path and progress through cellular dormancy. This process requires the stimulation of many membrane and soluble factors, such as bone morphogenic protein (BMP)-7, that induces prostate cancer dormancy, or Jagged-1 which proposedly promotes dormancy through Notch-2 in breast cancer cells. Mesenchymal stromal cells (MSCs), endothelial cells and spindle-shaped N-cadherin-positive osteoblasts (SNOs) seem to be crucial in this process, but also other cell types may be taking part in this process too.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ponzetti, M.; Rucci, N. Switching Homes: How Cancer Moves to Bone. Int. J. Mol. Sci. 2020, 21, 4124. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114124
AMA Style
Ponzetti M, Rucci N. Switching Homes: How Cancer Moves to Bone. International Journal of Molecular Sciences. 2020; 21(11):4124. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114124
Chicago/Turabian StylePonzetti, Marco, and Nadia Rucci. 2020. "Switching Homes: How Cancer Moves to Bone" International Journal of Molecular Sciences 21, no. 11: 4124. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21114124
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.