1. Introduction
Abuse and overuse of antibiotics in human health care and animal breeding has greatly contributed to a worldwide resistance to antibiotics. Moreover, the fact that hardly any new classes of antibiotics have been introduced to the market for decades makes the situation even more alarming [
1,
2]. Multidrug-resistant bacteria, the so-called ‘superbugs’, threaten our ability to tackle even common infectious diseases, resulting in prolonged illnesses and death of tens of thousands of people only in Europe and the United States [
3,
4]. Therefore, a lot of effort is being made to develop new antimicrobial agents, including antimicrobial peptides (AMPs).
AMPs, also known as cationic host defense peptides, are molecules that are widespread in all branches of the tree of life [
5]. In multicellular organisms, they participate in the first line of defense against bacteria, fungi, protozoans and viruses, and can even target cancer cells [
6,
7]. In addition to their microbicidal, antiviral and antitumor activities, they have immunomodulatory properties and may have a role in autoimmune disorders, wound healing and angiogenesis [
8,
9,
10]. In microorganisms, i.e., unicellular eukaryotes, bacteria and archaeans, they are used for self-protection and microbial competition [
11,
12,
13].
AMPs are short, generally fewer than 50 amino acids, rich in positive and hydrophobic residues, which gives them an amphipathic character, and do not display any similarities in sequence composition [
14,
15,
16]. Due to their structural characteristics, AMPs preferentially interact with negatively charged components of bacterial membranes, but do not adversely affect other eukaryoticmembranes. The latter are rich in neutral phospholipids and stabilizing cholesterol. AMPs act on the lipid bilayer in a detergent-like manner, solubilizing it into micelles, and/or penetrating it by forming pores. Both interactions lead to transient membrane permeabilization and cytoplasmic leakage that, depending on the AMP concentration, may trigger cell death [
14,
16,
17,
18]. Another AMP mechanism for efficient microbial killing is to act intracellularly, thus inhibiting, e.g., proteases, cell division and biosynthesis of proteins, nucleic acids and components of the cell wall [
19]. The interaction of AMPs with so many components of the bacterial cell gives them an advantage over traditional antibiotics, i.e., makes them less prone to select for resistance [
20,
21,
22]. Taking into account the therapeutic potential of AMPs [
16,
23] and the fact that superbugs are on the rise [
1,
2], it is of vital importance to find new AMPs. Unfortunately, the experimental procedures to identify novel AMPs are time-consuming, expensive and, most importantly, ineffective given the staggering numbers of genomes being sequenced. Consequently, there is a demand for high throughput computational tools to screen proteomes for AMPs with high accuracy.
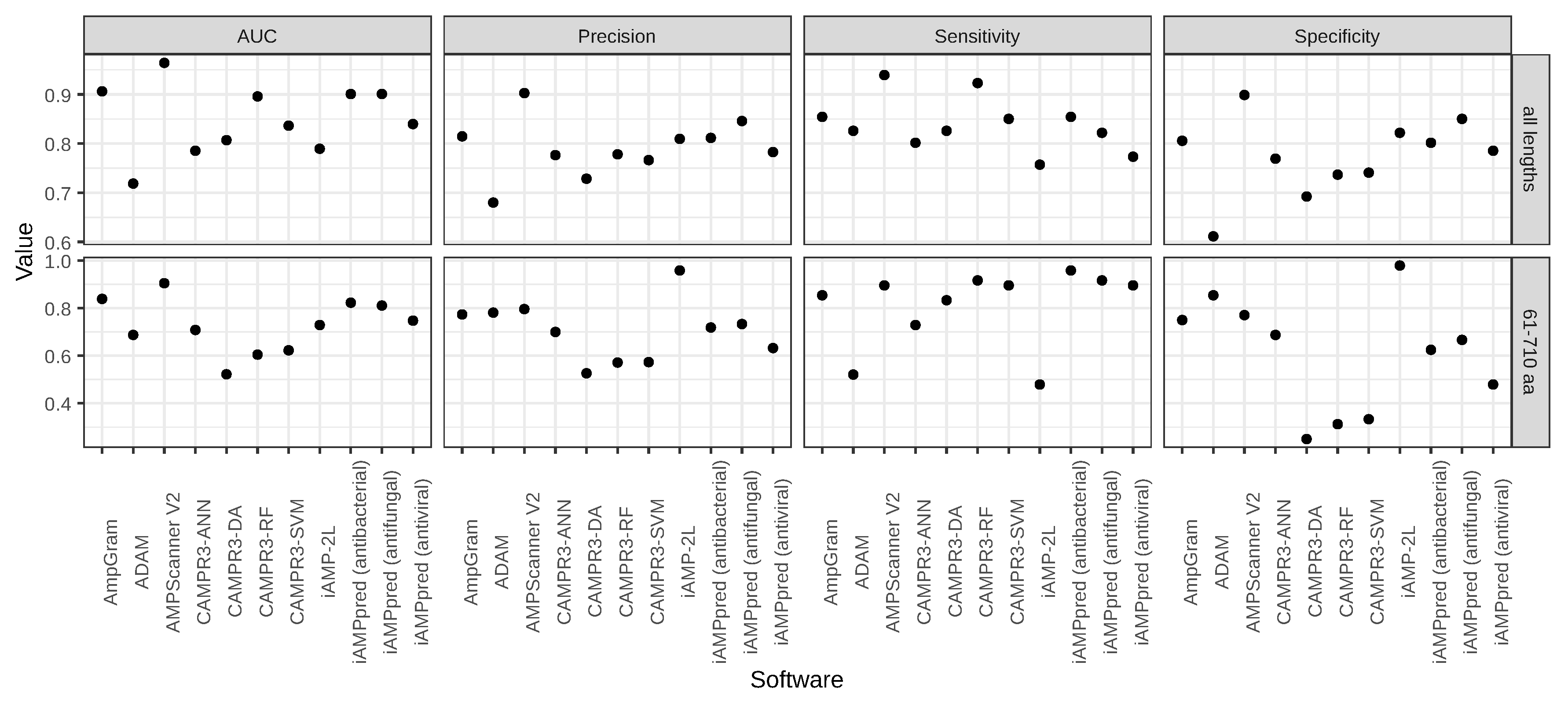
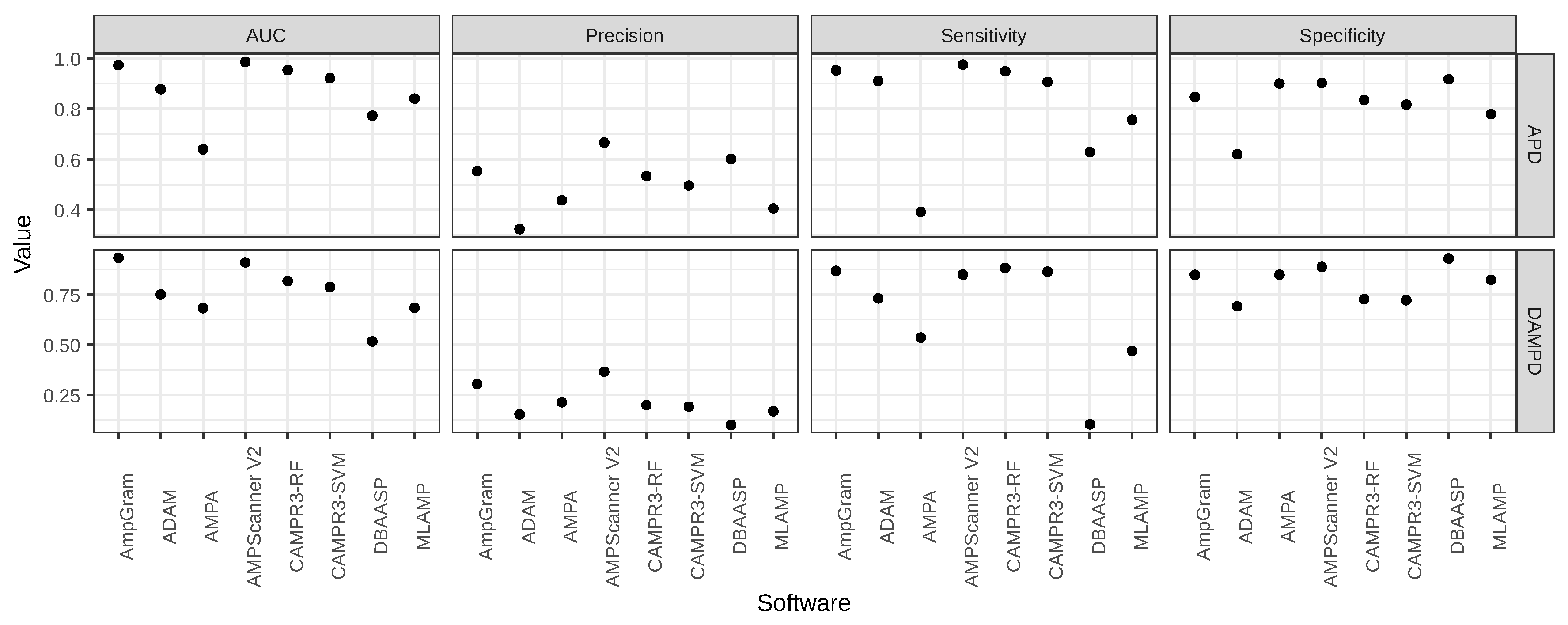
A variety of computational approaches have been adopted for AMP prediction, and over the years, the performance of new computational tools have gradually improved with machine learning algorithms in the lead. Many of the AMP prediction methods have also been made accessible as web servers and the top-ranking ones already accept multiple query sequences, e.g., AMPScanner [
24], ADAM [
25], iAMP-2L [
26], CAMPR3 [
27] and iAMPpred [
28]. However, there are still challenges to overcome, such as the prediction of longer AMPs. In their comparison of AMP prediction tools, Gabere and Noble [
29] indicated that many investigated classifiers, including accessible at that time ADAM [
25] and CAMPR3 [
27], exhibited strong length dependence, i.e., assigned very high scores for sequences over 100 amino acids regardless of whether they were AMPs or not.
In practice, there are plenty of longer peptides that do exhibit antimicrobial properties (
Table 1), e.g., milk proteins, such as
-lactoglobulin,
-lactoglobulin or lactoferrin [
30,
31,
32,
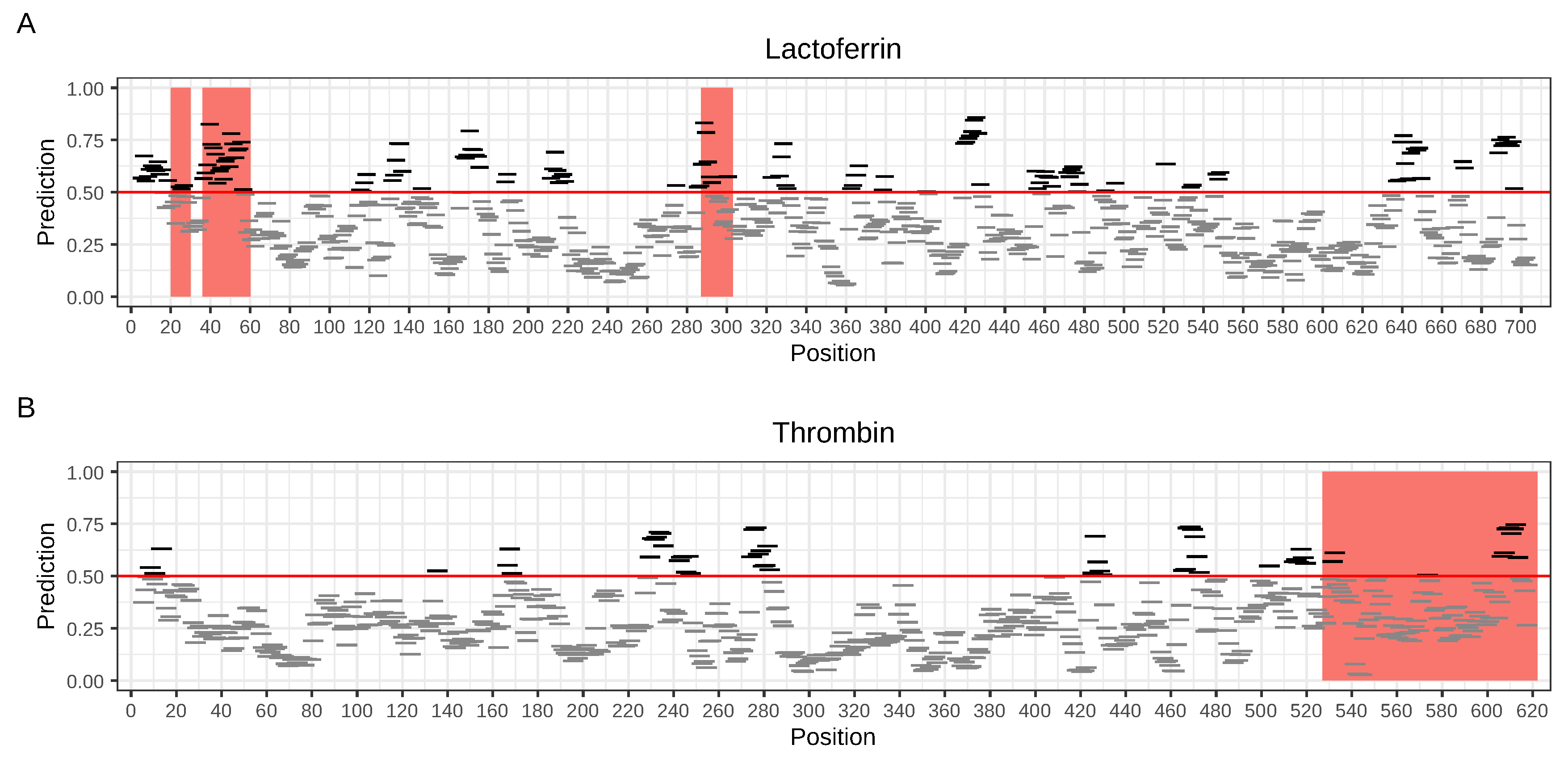
33]. The latter is especially of great importance, as literally hundreds of papers have been dedicated to its antimicrobial and antitumor activities. It is an 80 kDa iron-binding glycoprotein that, upon proteolitic processing, produces functional AMPs, such as 11 amino acid N-terminal lactoferrin fragment, lactoferricin (lactoferrin fragment 17–41) and lactoferrampin (lactoferrin fragment 268–284) [
32]. An important source of antimicrobial peptides and proteins are also animal venomes [
34,
35]. An example of such a protein is L-amino oxidase from pit vipers that upon proteolytic cleavage, similarily to lactoferrin, generates three functional AMPs [
36]. The most interesting are, however, proteins that do not exhibit any antimicrobial activities, such as human thrombin, but the products of their proteolysis do; they can be called ‘cryptic’ AMPs. In the case of human thrombin, its C-terminal peptides (527–622, 597–622, 604–622) constitute a novel class of AMPs produced during wounding and with therapeutic potential against infection and septic shock [
37].
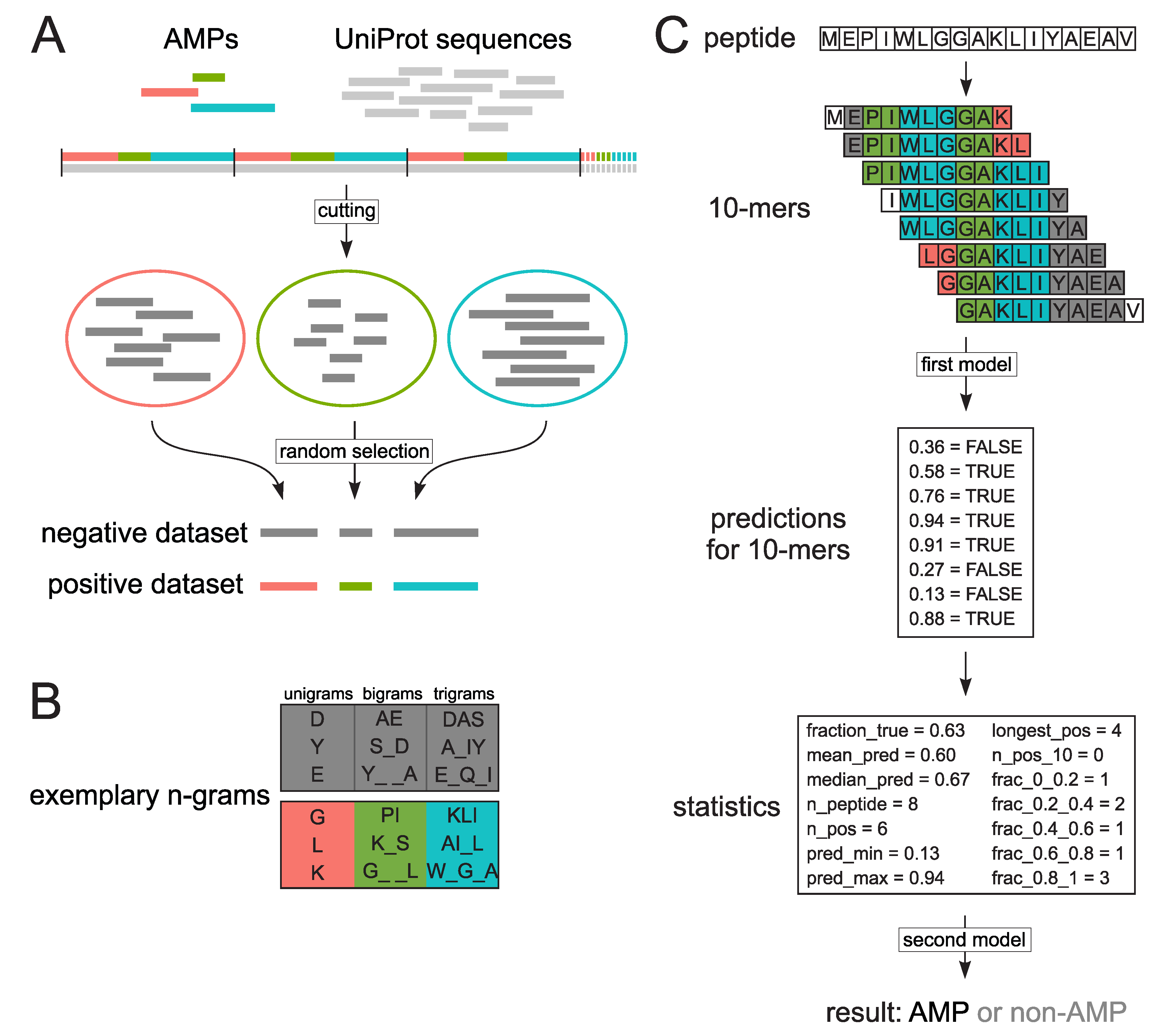
Our goal was to launch a high-throughput computational classifier, AmpGram, that could efficiently scan proteomes not only for typical AMPs but also longer proteins with AMP properties, including cryptic AMPs, and to indicate with high accuracy regions responsible for the AMP activity. AmpGram uses n-grams (amino-acid motifs) and random forests (a machine learning method) as an AMP classification algorithm. This methodology has already been used with success in our previous projects to create software for prediction of amyloid proteins [
40], signal peptides, [
41] and to assess optimal growth conditions for methanogens [
42].
A new approach that identifies protein AMP potential regions is needed, not only because of the alarming situation with the growing bacterial resistance but, because small peptides are easier and cheaper to synthesize and present fewer side effects as indicated, e.g., by pardaxin [
43]. Moreover, their activity can be easily improved by sequence modification that increases hydrophobicity and/or positive charge. Application of n-grams also allowed us to overcome the problem of high score–length dependency [
29]. The overprediction for longer AMPs could not have been solved by simply their inclusion in the positive training dataset because their amino acid composition is hardly distinguishable from other proteins in contrast to typical AMPs (
Supplementary Figure S1). The similarity in amino acid composition between longer AMPs and the negative dataset results from the fact that only short regions of proteins are responsible for their AMP properties.
4. Conclusions
AmpGram is a novel AMP predictor that uses n-grams to represent information hidden in amino acid sequences and random forests as the classification algorithm. In comparison to other top-ranking AMP predictors, including AMPScanner, CAMPR3R and iAMPpred, AmpGram performs better at detecting AMPs. To the best of our knowledge, AmpGram is the first AMP classifier created for the prediction of longer AMPs and high-throughput proteomic screening. The application of n-grams made it possible to overcome the problem of high score–length dependency that was first indicated by Gabere and Noble [
29] and also confirmed in our research. AmpGram not only allows to predict AMPs with high accuracy, but also precisely indicates peptide/protein fragments and regions that do have AMP potential. In order to test how AmpGram predictions relate to actual biological activity, we performed analyses for lactoferrin and thrombin; the former is a well-knownantimicrobial protein and the latter represents a cryptic AMP. Cryptic AMPs do not exhibit any AMP properties as mature proteins but their proteolytic products do. As expected, AmpGram identified both lactoferrin and thrombin as AMPs and indicated their potential AMP fragments and regions, including the sequences previously verified experimentally as AMPs [
32,
37]. The examples of lactoferrin and thrombin prove that antimicrobial fragments and regions predicted by AmpGram are good candidates for further investigation in terms of bactericidal activity, stability, toxicology, pharmacokinetics and the rational design of new AMPs; their antimicrobial activity can be further improved by amino acid modification to balance the peptide hydrophobicity and positive charge vital for disrupting bacterial membranes [
58]. Moreover, the small size of AmpGram predicted fragments makes them easy to synthesize and exhibit potentially fewer side effects compared to longer AMPs [
43].
AmpGram is available as a web server for multiple query sequences; however, for high-throughput proteomic screening, the users are encouraged to use its stand-alone version (see
Appendix A). Therefore, we have also implemented AmpGram as an easy-to-use R package.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}