Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition

 , , and
, , and 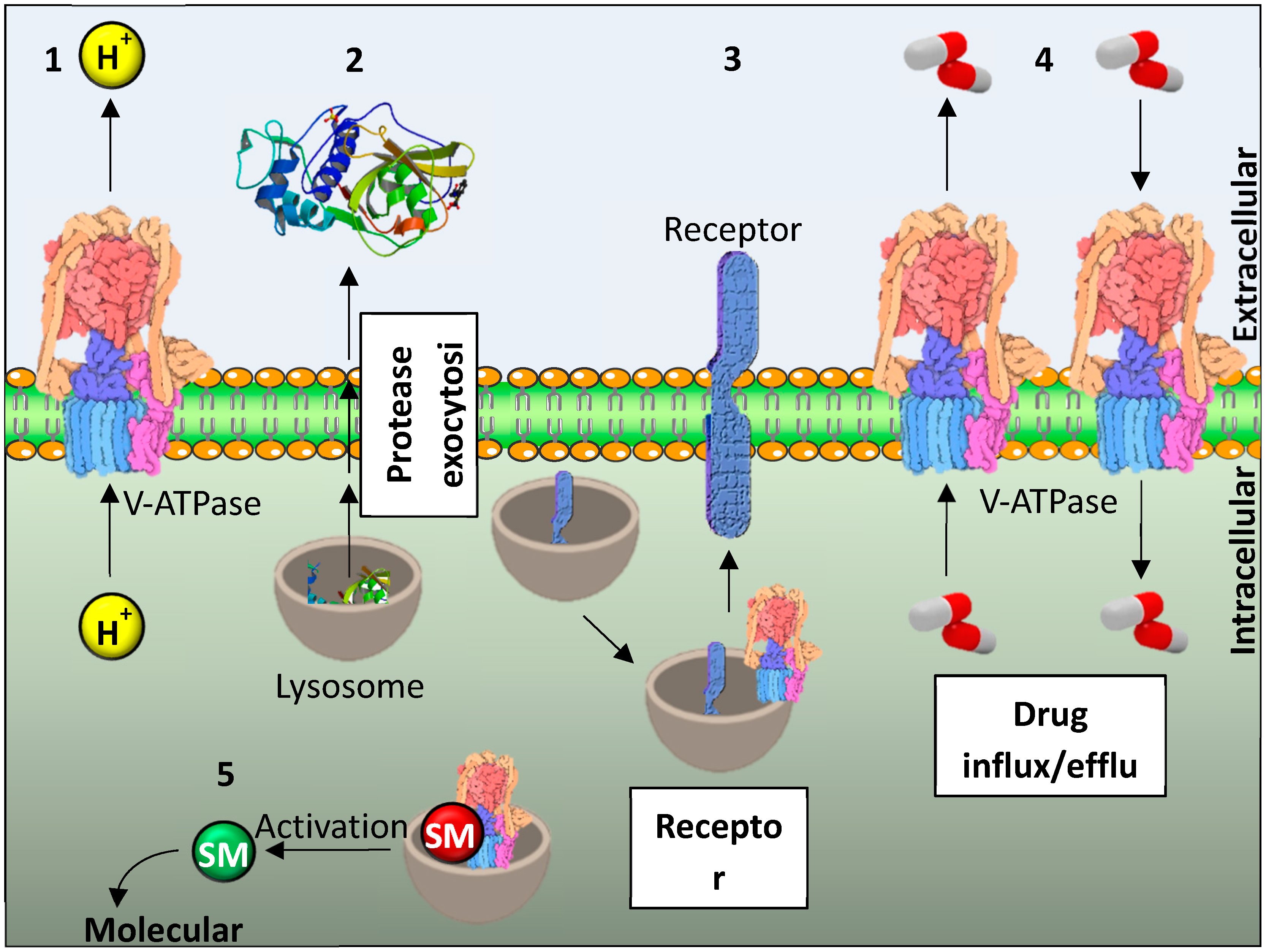{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lysosomes
3. Lysosomes, V-ATPases and Cancer
4. Resistance of Cancer Cells and Lysosomes and V-ATPase
5. Inhibition of Lysosomal Sequestration
6. Metallothioneins Regulates Lysosomal Function in Cancer Chemoresistance
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | ATP Binding Cassette |
| AIF/EndoG | Apoptosis-inducing factor /Endonuclease G |
| Akt | AKT Serine/Threonine Kinase 1 |
| AMPK | Adenylate-activated protein kinase |
| Bax | BCL2 Associated X Apoptosis Regulator |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma-extra large |
| Bid | BH3 Interacting Domain Death Agonist |
| CA-IX | Carbonic anhydrase IX |
| Cas | Carbonic anhydrases |
| CLEAR | Coordinated Lysosomal Expression and Regulation |
| DNA | Deoxyribonucleic acid |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| HCC | Hepatocellular carcinoma |
| HDAC10 | Histone deacetylase 10 |
| hENT3 | Human equilibrative nucleoside transporter-3 |
| HER2 | Human epidermal growth factor receptor 2 |
| LAMP | Lysosome-associated membrane protein |
| LIMP | Lysosomal integral membrane protein |
| LMP | Lysosomal membrane permeabilization |
| MCTs | Monocarboxylate transporters |
| MDR | Multiple drug resistance |
| MLKL | Mixed Lineage Kinase Domain-Like |
| MRP1 | Multidrug resistance-associated protein 1 |
| MTs | Metallothioneins |
| MTIF | Mitochondrial Translational Initiation Factor |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | mTOR complex-1 |
| NHE1 | Na+/H+ exchanger 1 |
| Notch | Neurogenic Locus Notch Homolog Protein |
| PDB | Protein Data Bank |
| pH | Decimal logarithm of the reciprocal of the hydrogen ion activity |
| pKa | Negative base-10 logarithm of the acid dissociation constant |
| Rab27B | Ras-related protein Rab-27B |
| Rho-GTPase Rac1 | Ras homologous GTPase Ras-related C3 botulinum toxin substrate 1 |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| RIPK3 | Receptor-interacting serine/threonine-protein kinase 3 |
| R-CSC | Rhabdomyosarcoma cancer stem cells |
| ROS | Reactive oxygen species |
| TEFB | Transcription factor EB |
| TFE3 | Transcription factor E3 |
References
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlos Rodrigo, M.A.; Jimenez Jimemez, A.M.; Haddad, Y.; Bodoor, K.; Adam, P.; Krizkova, S.; Heger, Z.; Adam, V. Metallothionein Isoforms as Double Agents—Their Roles in Carcinogenesis, Cancer Progression and Chemoresistance. Drug Resist. Update 2020, in press. [Google Scholar] [CrossRef]
- Skarkova, V.; Kralova, V.; Vitovcova, B.; Rudolf, E. Selected Aspects of Chemoresistance Mechanisms in Colorectal CarcinomaA Focus on Epithelial-to-Mesenchymal Transition, Autophagy, and Apoptosis. Cells 2019, 8, 234. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.T.; Fan, Z.Y.; Luo, G.P.; Yang, C.; Huang, Q.Y.; Fan, K.; Cheng, H.; Jin, K.Z.; Ni, Q.X.; Yu, X.J.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Update 2016, 24, 23–33. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef] [Green Version]
- Ndolo, R.A.; Jacobs, D.T.; Forrest, M.L.; Krise, J.P. Intracellular Distribution-based Anticancer Drug Targeting: Exploiting a Lysosomal Acidification Defect Associated with Cancer Cells. Mol. Cell. Pharmacol. 2010, 2, 131–136. [Google Scholar] [CrossRef]
- Ullio, C.; Brunk, U.T.; Urani, C.; Melchioretto, P.; Bonelli, G.; Baccino, F.M.; Autelli, R. Autophagy of metallothioneins prevents TNF-induced oxidative stress and toxicity in hepatoma cells. Autophagy 2015, 11, 2184–2198. [Google Scholar] [CrossRef] [Green Version]
- Appelqvist, H.; Waster, P.; Kagedal, K.; Ollinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Saftig, P.; Schroder, B.; Blanz, J. Lysosomal membrane proteins: Life between acid and neutral conditions. Biochem. Soc. Trans. 2010, 38, 1420–1423. [Google Scholar] [CrossRef] [Green Version]
- Saftig, P.; Haas, A. Turn up the lysosome. Nat. Cell Biol. 2016, 18, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Kissing, S.; Saftig, P.; Haas, A. Vacuolar ATPase in phago(lyso)some biology. Int. J. Med. Microbiol. 2018, 308, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Cotter, K.; Capecci, J.; Sennoune, S.; Huss, M.; Maier, M.; Martinez-Zaguilan, R.; Forgac, M. Activity of Plasma Membrane V-ATPases Is Critical for the Invasion of MDA-MB231 Breast Cancer Cells. J. Biol. Chem. 2015, 290, 3680–3692. [Google Scholar] [CrossRef] [Green Version]
- Halcrow, P.W.; Khan, N.; Datta, G.; Ohm, J.E.; Chen, X.; Geiger, J.D. Importance of measuring endolysosome, cytosolic, and extracellular pH in understanding the pathogenesis of and possible treatments for glioblastoma multiforme. Cancer Rep. 2019, 2, e1193. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.J.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.T.; Zou, L.H.; Xie, X.J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by mTORC1. Embo J. 2011, 30, 3242–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertollano, R. mTOR and lysosome regulation. F1000Prime Rep. 2014, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Fennelly, C.; Amaravadi, R.K. Lysosomal Biology in Cancer. Methods Mol. Biol. 2017, 1594, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Allison, A.C. Lysosomes in cancer cells. J. Clin. Pathol. Suppl. (R. Coll. Pathol.) 1974, 7, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Kallunki, T.; Olsen, O.D.; Jaattela, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Zhong, B.F.; Liu, M.; Bai, C.S.; Ruan, Y.X.; Wang, Y.Y.; Qiu, L.; Hong, Y.; Wang, X.; Li, L.F.; Li, B.H. Caspase-8 Induces Lysosome-Associated Cell Death in Cancer Cells. Mol. Ther. 2020, 28, 1078–1091. [Google Scholar] [CrossRef] [PubMed]
- Fais, S. Proton pump inhibitor-induced tumour cell death by inhibition of a detoxification mechanism. J. Intern. Med. 2010, 267, 515–525. [Google Scholar] [CrossRef]
- Daniel, C.; Bell, C.; Burton, C.; Harguindey, S.; Reshkin, S.J.; Rauch, C. The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim. Biophys. Acta-Mol. Basis Dis. 2013, 1832, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-dos-Santos, A. A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, B.; Kogel, D. Autophagy in Cancer Cell Death. Biology 2019, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K. Autophagy-induced tumor dormancy in ovarian cancer. J. Clin. Investig. 2008, 118, 3837–3840. [Google Scholar] [CrossRef] [Green Version]
- Harguindey, S.; Orive, G.; Pedraz, J.L.; Paradiso, A.; Reshkin, S.J. The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin-one single nature. Biochim. Biophys. Acta-Rev. Cancer 2005, 1756, 1–24. [Google Scholar] [CrossRef]
- Murakami, T.; Shibuya, I.; Ise, T.; Chen, Z.S.; Akiyama, S.; Nakagawa, M.; Izumi, H.; Nakamura, T.; Matsuo, K.; Yamada, Y.; et al. Elevated expression of vacuolar proton pump genes and cellular pH in cisplatin resistance. Int. J. Cancer 2001, 93, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Capecci, J.; Forgac, M. The Function of Vacuolar ATPase (V-ATPase) a Subunit Isoforms in Invasiveness of MCF10a and MCF10CA1a Human Breast Cancer Cells. J. Biol. Chem. 2013, 288, 32731–32741. [Google Scholar] [CrossRef] [Green Version]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef]
- Hinton, A.; Sennoune, S.R.; Bond, S.; Fang, M.; Reuveni, M.; Sahagian, G.G.; Jay, D.; Martinez-Zaguilan, R.; Forgac, M. Function of a Subunit Isoforms of the V-ATPase in pH Homeostasis and in Vitro Invasion of MDA-MB231 Human Breast Cancer Cells. J. Biol. Chem. 2009, 284, 16400–16408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Sayans, M.; Somoza-Martin, J.M.; Barros-Angueira, F.; Rey, J.M.G.; Garcia-Garcia, A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat. Rev. 2009, 35, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, S.; Jaunbergs, J.; Roth, M.G.; Ferguson, D.A.; De Brabander, J.K. Evaluating the potential of Vacuolar ATPase inhibitors as anticancer agents and multigram synthesis of the potent salicylihalamide analog saliphenylhalamide. Bioorg. Med. Chem. Lett. 2008, 18, 5879–5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supino, R.; Petrangolini, G.; Pratesi, G.; Tortoreto, M.; Favini, E.; Dal Bo, L.; Casalini, P.; Radaelli, E.; Croce, A.C.; Bottiroli, G.; et al. Antimetastatic effect of a small-molecule vacuolar H+-ATPase inhibitor in in vitro and in vivo preclinical studies. J. Pharmacol. Exp. Ther. 2008, 324, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Sridevi, P.; Ramirez, M.; Ludwig, K.J.; Wang, J.Y.J. β-Catenin-dependent lysosomal targeting of internalized tumor necrosis factor-α suppresses caspase-8 activation in apoptosis-resistant colon cancer cells. Mol. Biol. Cell 2013, 24, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, A.; Sormunen, R.; Westbroek, W.; Lambein, K.; Denys, H.; Sys, G.; Braems, G.; Van den Broecke, R.; Cocquyt, V.; Gespach, C.; et al. Vacuolar H plus ATPase expression and activity is required for Rab27B-dependent invasive growth and metastasis of breast cancer. Int. J. Cancer 2013, 133, 843–854. [Google Scholar] [CrossRef]
- Li, Z.H.; Fang, R.; Fang, J.; He, S.S.; Liu, T. Functional implications of Rab27 GTPases in Cancer. Cell Commun. Signal. 2018, 16, 44. [Google Scholar] [CrossRef] [Green Version]
- Schempp, C.M.; von Schwarzenberg, K.; Schreiner, L.; Kubisch, R.; Muller, R.; Wagner, E.; Vollmar, A.M. V-ATPase Inhibition Regulates Anoikis Resistance and Metastasis of Cancer Cells. Mol. Cancer Ther. 2014, 13, 926–937. [Google Scholar] [CrossRef] [Green Version]
- Kobia, F.; Duchi, S.; Deflorian, G.; Vaccari, T. Pharmacologic inhibition of vacuolar H plus ATPase reduces physiologic and oncogenic Notch signaling. Mol. Oncol. 2014, 8, 207–220. [Google Scholar] [CrossRef]
- Huang, L.J.; Lu, Q.; Han, Y.; Li, Z.; Zhang, Z.P.; Li, X.F. ABCG2/V-ATPase was associated with the drug resistance and tumor metastasis of esophageal squamous cancer cells. Diagn. Pathol. 2012, 7, 180. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Lu, S.; Huang, L.J.; Wang, T.; Wan, Y.; Zhou, C.X.; Zhang, C.H.; Zhang, Z.P.; Li, X.F. The expression of V-ATPase is associated with drug resistance and pathology of non-small-cell lung cancer. Diagn. Pathol. 2013, 8, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.F.; Chen, H.J.; Han, L.X.; Zou, X.P.; Shen, W.D. Expression and role of V1A subunit of V-ATPases in gastric cancer cells. Int. J. Clin. Oncol. 2015, 20, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Koch, R.; Radunski, U.; Corsham, S.; Cheong, N.; Inagaki, N.; Ban, N.; Wenzel, D.; Reinhardt, D.; Zapf, A.; et al. Intracellular ABC transporter A3 confers multidrug resistance in leukemia cells by lysosomal drug sequestration. Leukemia 2008, 22, 1576–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.Y.; Xie, R.; Liu, X.M.; Wen, G.R.; Jin, H.; Yu, Z.H.; Jiang, Y.X.; Zhao, Z.L.; Yang, Y.; Ji, B.; et al. Expression and functional role of vacuolar H+-ATPase in human hepatocellular carcinoma. Carcinogenesis 2012, 33, 2432–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asakura, T.; Imai, A.; Ohkubo-Uraoka, N.; Kuroda, M.; Iidaka, Y.; Uchida, K.; Shibasaki, T.; Ohkawa, K. Relationship between expression of drug-resistance factors and drug sensitivity in normal human renal proximal tubular epithelial cells in comparison with renal cell carcinoma. Oncol. Rep. 2005, 14, 601–607. [Google Scholar] [CrossRef]
- Bertolini, I.; Terrasi, A.; Martelli, C.; Gaudioso, G.; Di Cristofori, A.; Storaci, A.M.; Formica, M.; Braidotti, P.; Todoerti, K.; Ferrero, S.; et al. A GBM-like V-ATPase signature directs cell-cell tumor signaling and reprogramming via large oncosomes. EBioMedicine 2019, 41, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Couto-Vieira, J.; Nicolau-Neto, P.; Costa, E.P.; Figueira, F.F.; Simao, T.D.; Okorokova-Facanha, A.L.; Pinto, L.F.R.; Facanha, A.R. Multi-cancer V-ATPase molecular signatures: A distinctive balance of subunit C isoforms in esophageal carcinoma. EBioMedicine 2020, 51, 102581. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, S.J.; Terashima, M.; Mizunuma, N.; Slapak, C.A. Vesicular anthracycline accumulation in Doxorubicin-selected U-937 cells: Participation of lysosomes. Blood 1997, 89, 3745–3754. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.P.; Duvvuri, M.; Krise, J.P. Separate roles for the Golgi apparatus and lysosomes in the sequestration of drugs in the multidrug-resistant human leukemic cell line HL-60. J. Biol. Chem. 2003, 278, 50234–50239. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J. Biol. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, H.Y.; Jin, J.; Shu, H.Q.; Yu, B.; De Milito, A.; Lozupone, F.; Deng, Y.; Tang, N.; Yao, G.F.; Fais, S.; et al. Small interfering RNA targeting the subunit ATP6L of proton pump V-ATPase overcomes chemoresistance of breast cancer cells. Cancer Lett. 2009, 280, 110–119. [Google Scholar] [CrossRef] [PubMed]
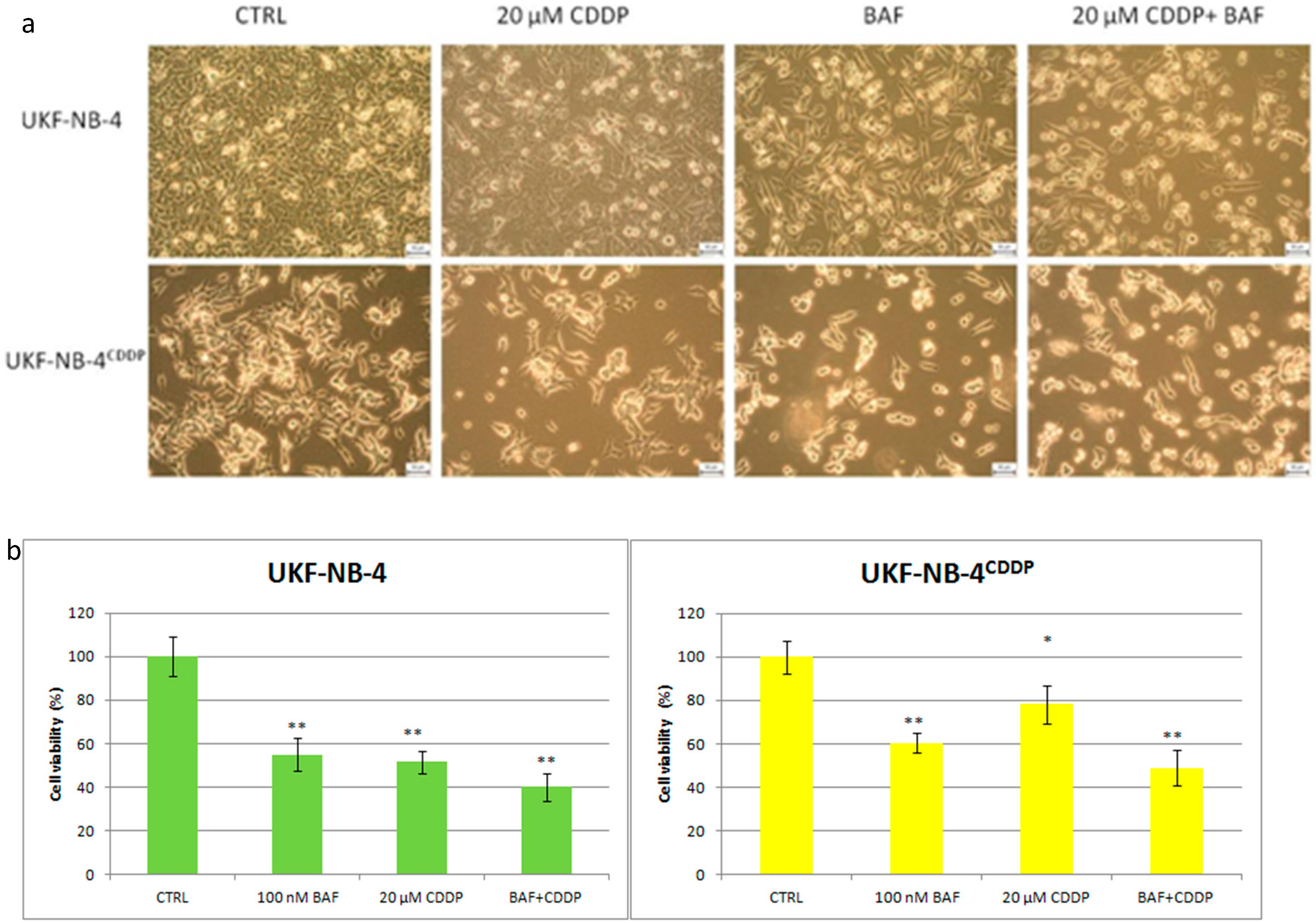
- Rodrigo, M.A.M.; Buchtelova, H.; de los Rios, V.; Casal, J.I.; Eckschlager, T.; Hrabeta, J.; Belhajova, M.; Heger, Z.; Adam, V. Proteomic Signature of Neuroblastoma Cells UKF-NB-4 Reveals Key Role of Lysosomal Sequestration and the Proteasome Complex in Acquiring Chemoresistance to Cisplatin. J. Proteome Res. 2019, 18, 1255–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piskareva, O.; Harvey, H.; Nolan, J.; Conlon, R.; Alcock, L.; Buckley, P.; Dowling, P.; O’Sullivan, F.; Bray, I.; Stallings, R.L. The development of cisplatin resistance in neuroblastoma is accompanied by epithelial to mesenchymal transition in vitro. Cancer Lett. 2015, 364, 142–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, C.; Roberg, K.; Grafstrom, R.C.; Ollinger, K. Intrinsic differences in cisplatin sensitivity of head and neck cancer cell lines: Correlation to lysosomal pH. Head Neck-J. Sci. Spec. Head Neck 2010, 32, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Katara, G.K.; Ibrahim, S.A.; Riehl, V.; Sahoo, M.; Dolan, J.; Meinke, K.W.; Pins, M.R.; Beaman, K.D. Targeting V-ATPase Isoform Restores Cisplatin Activity in Resistant Ovarian Cancer: Inhibition of Autophagy, Endosome Function, and ERK/MEK Pathway. J. Oncol. 2019, 2343876. [Google Scholar] [CrossRef] [Green Version]
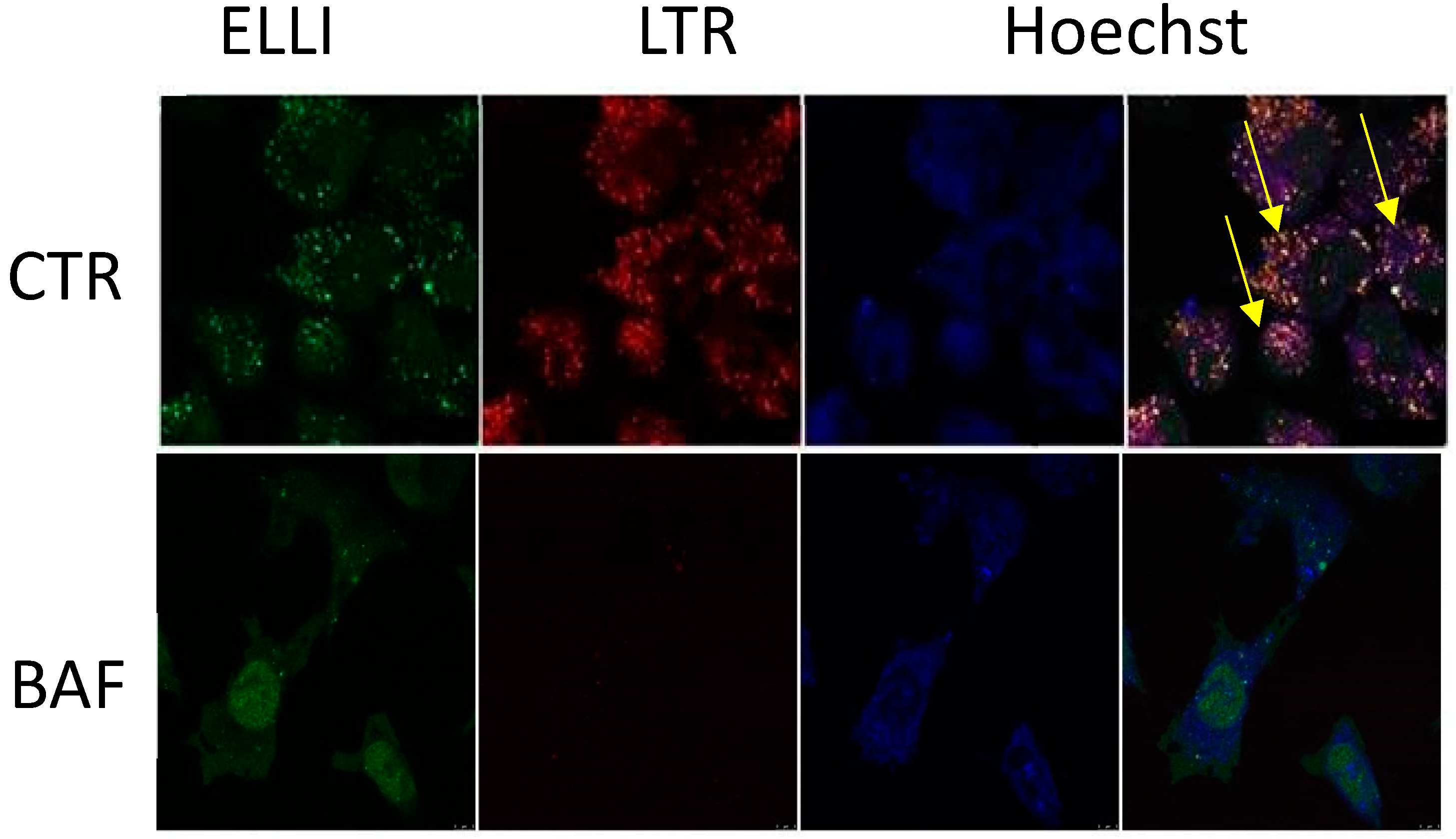
- Hrabeta, J.; Groh, T.; Khalil, M.A.; Poljakova, J.; Adam, V.; Kizek, R.; Uhlik, J.; Doktorova, H.; Cerna, T.; Frei, E.; et al. Vacuolar-ATPase-mediated intracellular sequestration of ellipticine contributes to drug resistance in neuroblastoma cells. Int. J. Oncol. 2015, 47, 971–980. [Google Scholar] [CrossRef]
- Wu, S.; Huang, L.M.; Shen, R.; Bernard-Cacciarella, M.; Zhou, P.; Hu, C.Q.; Di Benedetto, M.; Janin, A.; Bousquet, G.; Li, H.; et al. Drug resistance-related sunitinib sequestration in autophagolysosomes of endothelial cells. Int. J. Oncol. 2020, 56, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Sasazawa, Y.; Futamura, Y.; Tashiro, E.; Imoto, M. Vacuolar H+-ATPase inhibitors overcome Bcl-xL-mediated chemoresistance through restoration of a caspase-independent apoptotic pathway. Cancer Sci. 2009, 100, 1460–1467. [Google Scholar] [CrossRef]
- Salerno, M.; Avnet, S.; Bonuccelli, G.; Hosogi, S.; Granchi, D.; Baldini, N. Impairment of Lysosomal Activity as a Therapeutic Modality Targeting Cancer Stem Cells of Embryonal Rhabdomyosarcoma Cell Line RD. PLoS ONE 2014, 9, e110340. [Google Scholar] [CrossRef] [Green Version]
- Ridinger, J.; Koeneke, E.; Kolbinger, F.R.; Koerholz, K.; Mahboobi, S.; Hellweg, L.; Gunkel, N.; Miller, A.K.; Peterziel, H.; Schmezer, P.; et al. Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.W.; Schneider, L.S.; Vick, B.; Grunert, M.; Jeremias, I.; Menche, D.; Muller, R.; Vollmar, A.M.; Liebl, J. Anti-leukemic effects of the V-ATPase inhibitor Archazolid A. Oncotarget 2015, 6, 43508–43528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Shi, X.Y.; Li, Z.M.; Pan, X.H.; Li, Z.L.; Chen, Y.; Yan, S.J.; Xiao, L. Proton pump inhibitors can reverse the YAP mediated paclitaxel resistance in epithelial ovarian cancer. BMC Mol. Cell Biol. 2019, 20, 49. [Google Scholar] [CrossRef] [PubMed]
- Merk, H.; Messer, P.; Ardelt, M.A.; Lamb, D.C.; Zahler, S.; Muller, R.; Vollmar, A.M.; Pachmayr, J. Inhibition of the V-ATPase by Archazolid A: A New Strategy to Inhibit EMT. Mol. Cancer Ther. 2017, 16, 2329–2339. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.D.; Gao, D.C.; Redfern, N.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [Green Version]
- Funk, R.S.; Krise, J.P. Cationic Amphiphilic Drugs Cause a Marked Expansion of Apparent Lysosomal Volume: Implications for an Intracellular Distribution-Based Drug Interaction. Mol. Pharm. 2012, 9, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Logan, R.; Kong, A.; Krise, J.P. Evaluating the Roles of Autophagy and Lysosomal Trafficking Defects in Intracellular Distribution-Based Drug-Drug Interactions Involving Lysosomes. J. Pharm. Sci. 2013, 102, 4173–4180. [Google Scholar] [CrossRef]
- Arnaout, A.; Robertson, S.J.; Pond, G.R.; Lee, H.; Jeong, A.; Ianni, L.; Kroeger, L.; Hilton, J.; Coupland, S.; Gottlieb, C.; et al. A randomized, double-blind, window of opportunity trial evaluating the effects of chloroquine in breast cancer patients. Breast Cancer Res. Treat. 2019, 178, 327–335. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Samaras, P.; Tusup, M.; Nguyen-Kim, T.D.L.; Seifert, B.; Bachmann, H.; von Moos, R.; Knuth, A.; Pascolo, S. Phase I study of a chloroquine-gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother. Pharmacol. 2017, 80, 1005–1012. [Google Scholar] [CrossRef]
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme - A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.B.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, R.; Leone, R.; Chang, Y.Y.C.; Fecher, L.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvvuri, M.; Konkar, S.; Funk, R.S.; Krise, J.M.; Krise, J.P. A chemical strategy to manipulate the intracellular localization of drugs in resistant cancer cells. Biochemistry 2005, 44, 15743–15749. [Google Scholar] [CrossRef]
- von Schwarzenberg, K.; Lajtos, T.; Simon, L.; Muller, R.; Vereb, G.; Vollmar, A.M. V-ATPase inhibition overcomes trastuzumab resistance in breast cancer. Mol. Oncol. 2014, 8, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zheng, Y.Q.; Shi, Y.C.; Cui, Z.R. Overcoming tumor cell chemoresistance using nanoparticles: Lysosomes are beneficial for (stearoyl) gemcitabine-incorporated solid lipid nanoparticles. Int. J. Nanomed. 2018, 13, 319–336. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.-K.; Jang, M.; Song, M.-J.; Kim, D.; Kim, Y.; Jang, H.H. Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants 2019, 8, 471. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.M.L.; van Duijn, W.; Kubben, F.J.G.M.; Griffioen, G.; Lamers, C.B.H.W.; van Krieken, J.H.J.M.; van de Velde, C.J.H.; Verspaget, H.W. Prognostic Significance of Metallothionein in Human Gastrointestinal Cancer. Clinical Cancer Res. 2002, 8, 1889. [Google Scholar]
- Krizkova, S.; Ryvolova, M.; Hrabeta, J.; Adam, V.; Stiborova, M.; Eckschlager, T.; Kizek, R. Metallothioneins and zinc in cancer diagnosis and therapy. Drug Metab. Rev. 2012, 44, 287–301. [Google Scholar] [CrossRef]
- Hasumi, M.; Suzuki, K.; Matsui, H.; Koike, H.; Ito, K.; Yamanaka, H. Regulation of metallothionein and zinc transporter expression in human prostate cancer cells and tissues. Cancer Lett. 2003, 200, 187–195. [Google Scholar] [CrossRef]
- Kukic, I.; Kelleher, S.L.; Kiselyov, K. Zn2+ efflux through lysosomal exocytosis prevents Zn2+-induced toxicity. J. Cell Sci. 2014, 127, 3094–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, S.K.; Kurz, T.; Brunk, U.T. Metallothionein protects against oxidative stress-induced lysosomal destabilization. Biochem. J. 2006, 394, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.K.; Hidalgo, J.; Eddins, D.; Levin, E.D.; Aschner, M. Metallothionein in the central nervous system: Roles in protection, regeneration and cognition. NeuroToxicology 2008, 29, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, M.O.; Larsen, A.; Stoltenberg, M.; Penkowa, M. Cell death in the injured brain: Roles of metallothioneins. Prog. Histochem. Cyto. 2009, 44, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Seo, B.-R.; Koh, J.-Y. Metallothionein-3 modulates the amyloid β endocytosis of astrocytes through its effects on actin polymerization. Mol. Brain 2015, 8, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, M.l.A.M.; Buchtelova, H.; Jimenez, A.M.J.; Adam, P.; Babula, P.; Heger, Z.; Adam, V. Transcriptomic Landscape of Cisplatin-Resistant Neuroblastoma Cells. Cells 2019, 8, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-J.; Koh, J.Y. Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Mol. Brain 2010, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.H.; Lee, S.-H.; Lee, S.-J.; Kim, H.N.; Koh, J.-Y. A role of metallothionein-3 in radiation-induced autophagy in glioma cells. Sci. Rep. 2020, 10, 2015. [Google Scholar] [CrossRef]
- Petruzzelli, R.; Polishchuk, R.S. Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs. Cells 2019, 8, 1080. [Google Scholar] [CrossRef] [Green Version]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Choudhuri, S.; McKim, J.M.; Klaassen, C.D. Role of hepatic lysosomes in the degradation of metallothionein. Toxcol. Appl. Pharmacol. 1992, 115, 64–71. [Google Scholar] [CrossRef]
- Min, K.-S.; Nakatsubo, T.; Fujita, Y.; Onosaka, S.; Tanaka, K. Degradation of cadmium metallothionein in vitro by lysosomal proteases. Toxcol. Appl. Pharmacol. 1992, 113, 299–305. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hraběta, J.; Belhajová, M.; Šubrtová, H.; Merlos Rodrigo, M.A.; Heger, Z.; Eckschlager, T. Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition. Int. J. Mol. Sci. 2020, 21, 4392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124392
Hraběta J, Belhajová M, Šubrtová H, Merlos Rodrigo MA, Heger Z, Eckschlager T. Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition. International Journal of Molecular Sciences. 2020; 21(12):4392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124392
Chicago/Turabian StyleHraběta, Jan, Marie Belhajová, Hana Šubrtová, Miguel Angel Merlos Rodrigo, Zbyněk Heger, and Tomáš Eckschlager. 2020. "Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition" International Journal of Molecular Sciences 21, no. 12: 4392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124392