PGI2 Analog Attenuates Salt-Induced Renal Injury through the Inhibition of Inflammation and Rac1-MR Activation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
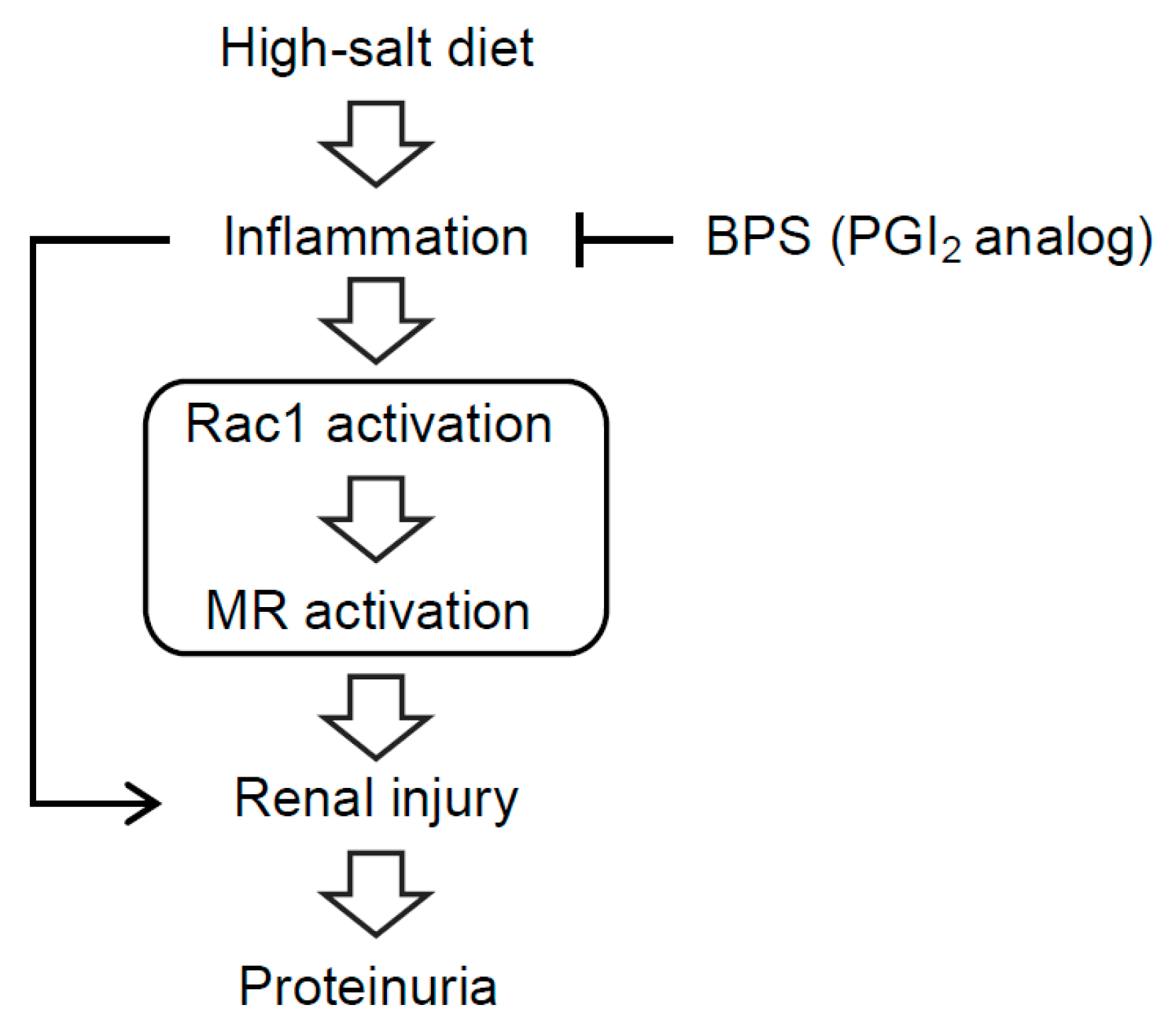
Abstract
:1. Introduction
2. Results
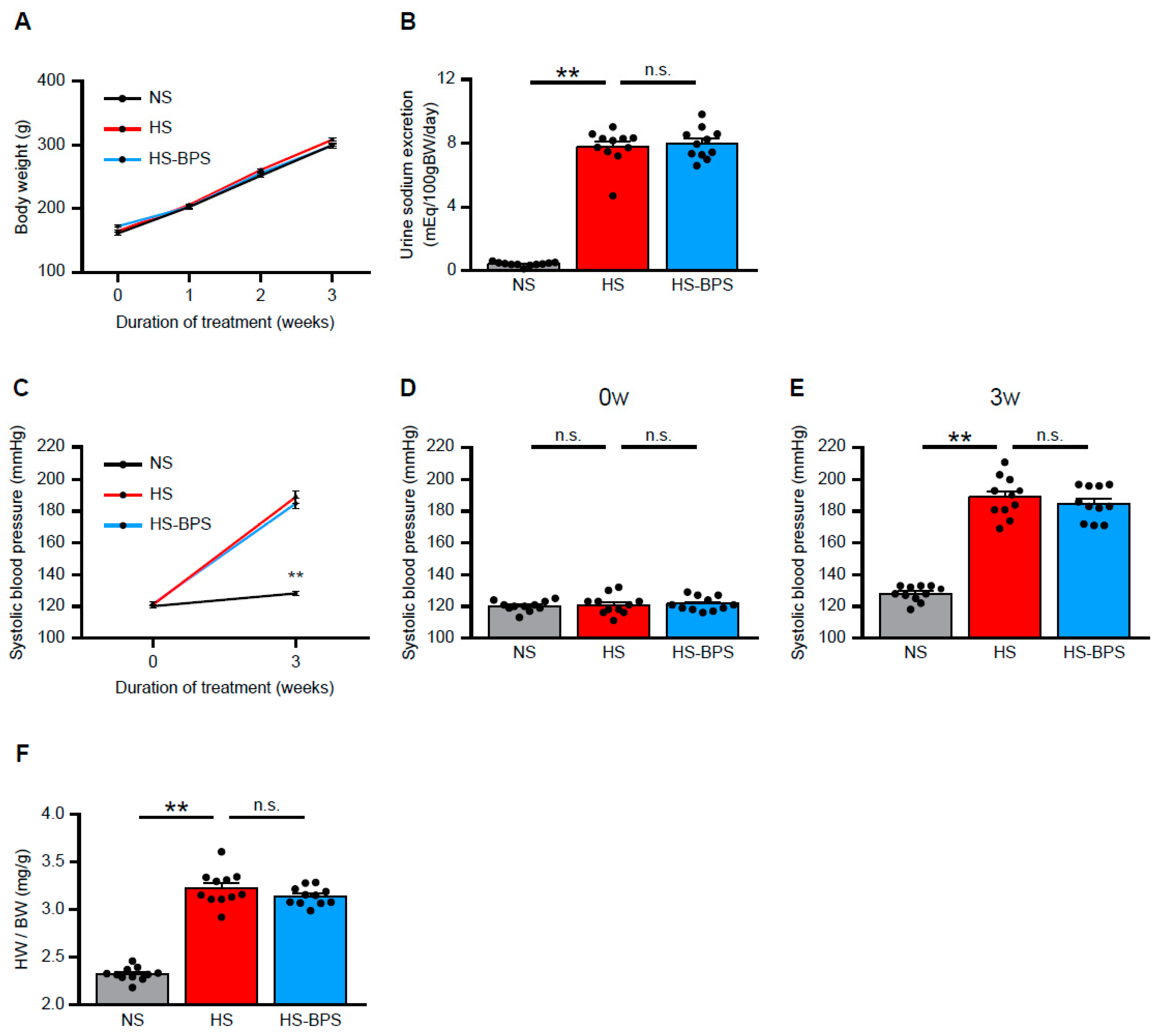
2.1. BPS Treatment Did Not Alter Blood Pressure Levels in High-Salt-Fed DS Rats
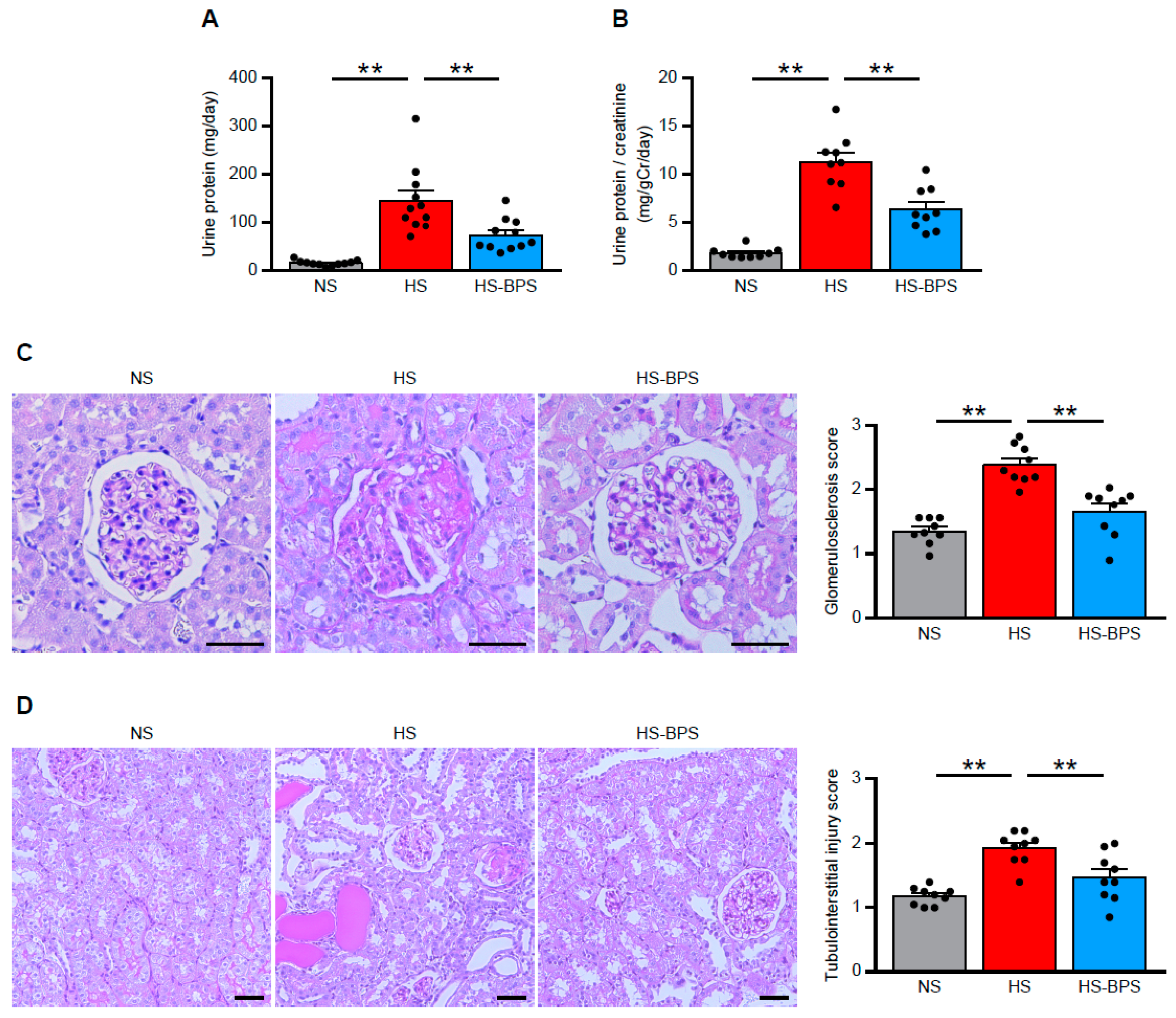
2.2. Administration of BPS Ameliorates Proteinuria and Renal Injury in High-Salt-Fed DS Rats
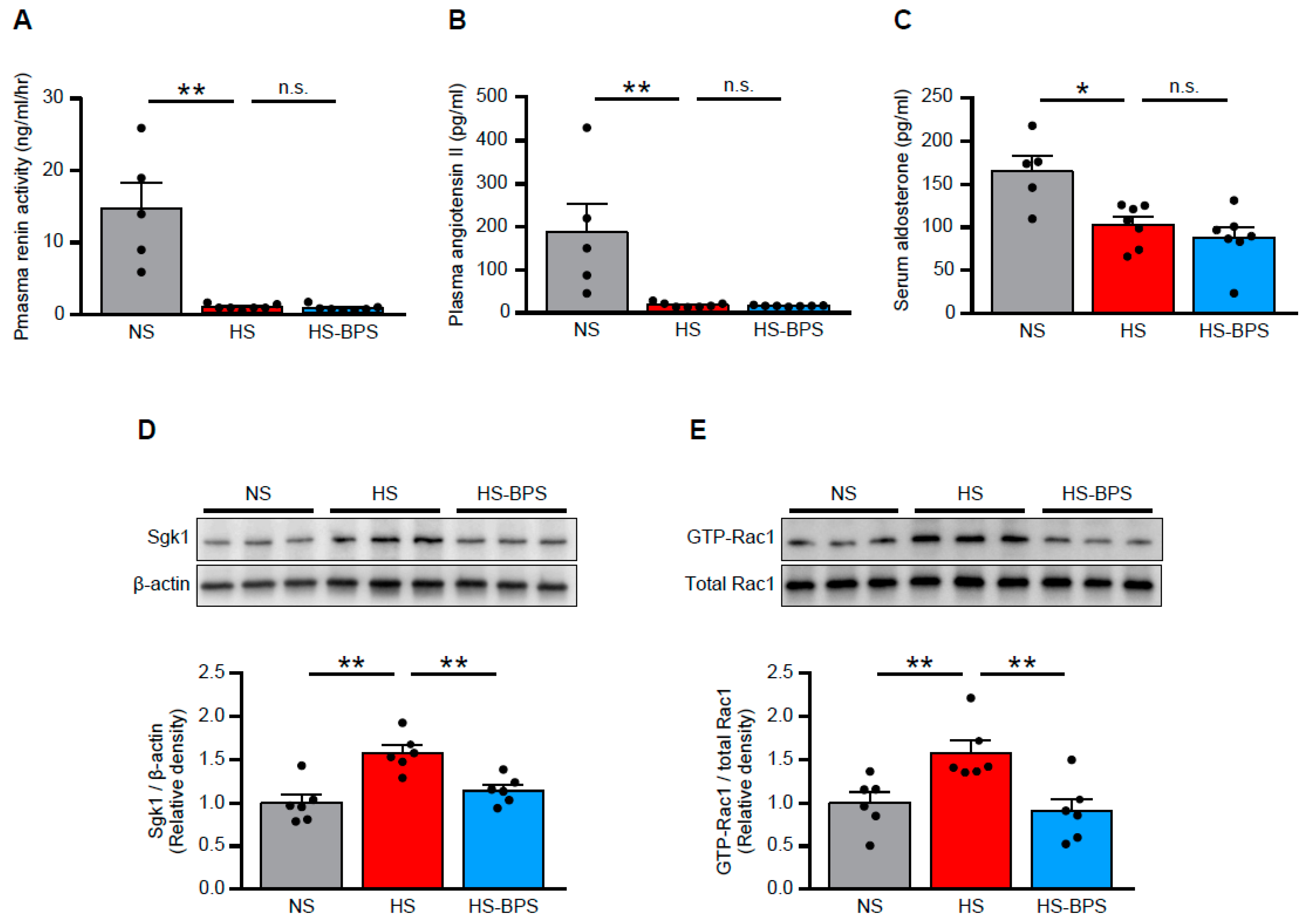
2.3. BPS Reduced Renal MR Pathway Activation via Rac1 Activity Suppression in High-Salt-Fed DS Rats
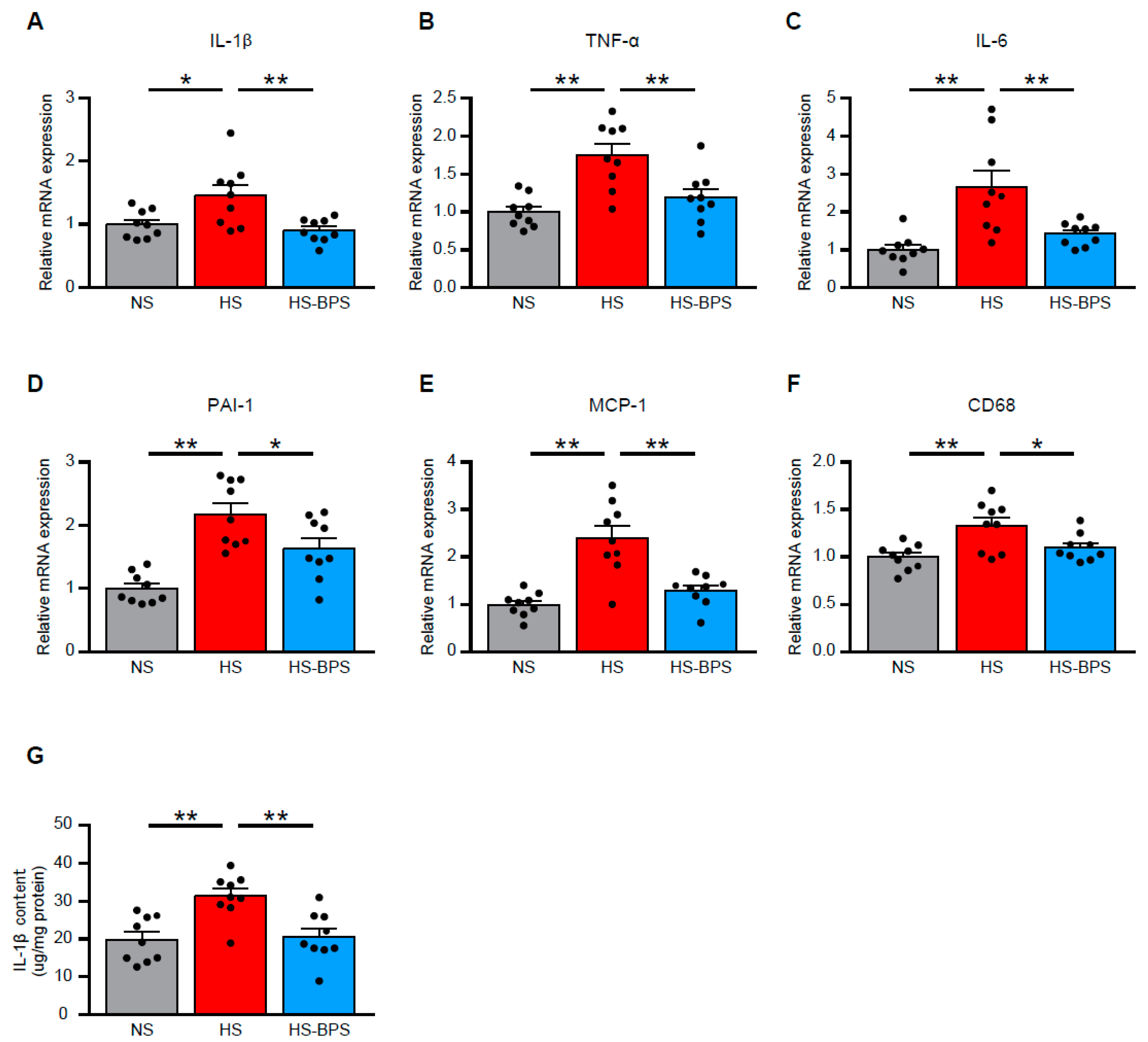
2.4. BPS Alleviates Renal Inflammation in High-Salt-Fed DS Rats
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Design
4.2. Blood Pressure Measurements in Conscious Rats
4.3. Metabolic Studies
4.4. Blood Collection and Laboratory Measurements
4.5. Renal Histology
4.6. Western Blot Analysis
4.7. Quantitative RT-PCR Analysis
4.8. IL-1β ELISA
4.9. Antibodies
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BPS | beraprost sodium |
| CKD | chronic kidney disease |
| DR rats | Dahl salt-resistant rats |
| DS rats | Dahl salt-sensitive rats |
| ELISA | enzyme-linked immunosorbent assay |
| GBM | glomerular basement membrane |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| MCP-1 | monocyte chemoattractant protein-1 |
| MR | mineralocorticoid receptor |
| PAI-1 | plasminogen activator inhibitor-1 |
| PAS | periodic acid–Schiff |
| RAAS | renin-angiotensin-aldosterone system |
| Sgk1 | serum and glucocorticoid-regulated kinase |
| TNF-α | tumor necrosis factor-α |
References
- Tian, N.; Gu, J.W.; Jordan, S.; Rose, R.A.; Hughson, M.D.; Manning, R.D., Jr. Immune suppression prevents renal damage and dysfunction and reduces arterial pressure in salt-sensitive hypertension. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1018–H1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Crowley, S.D. Inflammation in Salt-Sensitive Hypertension and Renal Damage. Curr. Hypertens. Rep. 2018, 20, 103. [Google Scholar] [CrossRef] [PubMed]
- Hirohama, D.; Fujita, T. Evaluation of the pathophysiological mechanisms of salt-sensitive hypertension. Hypertens. Res. 2019, 42, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Worou, M.E.; Liao, T.D.; D’Ambrosio, M.; Nakagawa, P.; Janic, B.; Peterson, E.L.; Rhaleb, N.E.; Carretero, O.A. Renal protective effect of N-acetyl-seryl-aspartyl-lysyl-proline in dahl salt-sensitive rats. Hypertension 2015, 66, 816–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, M.; Fujita, T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat. Rev. Nephrol. 2013, 9, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawarazaki, W.; Kurihara, H.; Tanaka, H.; Miyoshi, J.; Takai, Y.; Fujita, T. Modification of mineralocorticoid receptor function by Rac1 GTPase: Implication in proteinuric kidney disease. Nat. Med. 2008, 14, 1370–1376. [Google Scholar] [CrossRef]
- Shibata, S.; Mu, S.; Kawarazaki, H.; Muraoka, K.; Ishizawa, K.; Yoshida, S.; Kawarazaki, W.; Takeuchi, M.; Ayuzawa, N.; Miyoshi, J.; et al. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J. Clin. Investig. 2011, 121, 3233–3243. [Google Scholar] [CrossRef]
- Fujita, T. Mechanism of salt-sensitive hypertension: Focus on adrenal and sympathetic nervous systems. J. Am. Soc. Nephrol. 2014, 25, 1148–1155. [Google Scholar] [CrossRef] [Green Version]
- Nagaya, N.; Uematsu, M.; Okano, Y.; Satoh, T.; Kyotani, S.; Sakamaki, F.; Nakanishi, N.; Miyatake, K.; Kunieda, T. Effect of orally active prostacyclin analogue on survival of outpatients with primary pulmonary hypertension. J. Am. Coll. Cardiol. 1999, 34, 1188–1192. [Google Scholar] [CrossRef] [Green Version]
- Lievre, M.; Morand, S.; Besse, B.; Fiessinger, J.N.; Boissel, J.P. Oral Beraprost sodium, a prostaglandin I(2) analogue, for intermittent claudication: A double-blind, randomized, multicenter controlled trial. Beraprost et Claudication Intermittente (BERCI) Research Group. Circulation 2000, 102, 426–431. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Sasaki, R.; Sato, N.; Suzuki, M.; Tamura, M.; Matsushita, T.; Kurumatani, H. Amelioration by beraprost sodium, a prostacyclin analogue, of established renal dysfunction in rat glomerulonephritis model. Eur. J. Pharmacol. 2002, 449, 167–176. [Google Scholar] [CrossRef]
- Sato, N.; Kaneko, M.; Tamura, M.; Kurumatani, H. The prostacyclin analog beraprost sodium ameliorates characteristics of metabolic syndrome in obese Zucker (fatty) rats. Diabetes 2010, 59, 1092–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Nakashima, H.; Mochizuki, S.; Abe, Y.; Ishimura, A.; Ito, K.; Fukushima, T.; Miyake, K.; Ogahara, S.; Saito, T. Amelioration of diabetic nephropathy in OLETF rats by prostaglandin I(2) analog, beraprost sodium. Am. J. Nephrol. 2009, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, J.; Xu, Y.; Wang, Y.; Du, H.; Shao, J.; Liu, Z. The Protective Effect of Beraprost Sodium on Diabetic Nephropathy by Inhibiting Inflammation and p38 MAPK Signaling Pathway in High-Fat Diet/Streptozotocin-Induced Diabetic Rats. Int. J. Endocrinol. 2016, 2016, 1690474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenaka, M.; Machida, N.; Ida, N.; Satoh, N.; Kurumatani, H.; Yamane, Y. Effect of beraprost sodium (BPS) in a new rat partial unilateral ureteral obstruction model. Prostaglandins Leukot. Essent. Fatty Acids 2009, 80, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Y.; Chen, L.; Wang, Z.; Liu, G.; Zuo, B.; Liu, C.; Sun, D. Beraprost sodium mitigates renal interstitial fibrosis through repairing renal microvessels. J. Mol. Med. 2019, 97, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Itoh, Y.; Kubota, T.; Sendo, T.; Koyama, T.; Fujita, T.; Saeki, K.; Yuo, A.; Oishi, R. A prostacyclin analog prevents radiocontrast nephropathy via phosphorylation of cyclic AMP response element binding protein. Am. J. Pathol. 2005, 166, 1333–1342. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, K.; Nagasaka, A.; Nagata, M.; Yamamoto, K.; Imamura, S.; Oda, N.; Sawai, Y.; Hayakawa, N.; Suzuki, A.; Itoh, M. A stable prostacyclin analogue reduces high serum TNF-alpha levels in diabetic patients. Exp. Clin. Endocrinol. Diabetes 2004, 112, 390–394. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Inada, C.; Tamura, M.; Sato, N.; Yamada, M.; Itaba, S.; Okazaki, S.; Matsuura, H.; Fujii, S.; Matsuda, F.; et al. Beraprost sodium improves survival rates in anti-glomerular basement membrane glomerulonephritis and 5/6 nephrectomized chronic kidney disease rats. Eur. J. Pharmacol. 2013, 714, 325–331. [Google Scholar] [CrossRef]
- Zhu, Q.; Li, X.X.; Wang, W.; Hu, J.; Li, P.L.; Conley, S.; Li, N. Mesenchymal stem cell transplantation inhibited high salt-induced activation of the NLRP3 inflammasome in the renal medulla in Dahl S rats. Am. J. Physiol. Ren. Physiol. 2016, 310, F621–F627. [Google Scholar] [CrossRef] [Green Version]
- Uddin, S.; Lekmine, F.; Sharma, N.; Majchrzak, B.; Mayer, I.; Young, P.R.; Bokoch, G.M.; Fish, E.N.; Platanias, L.C. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J. Biol. Chem. 2000, 275, 27634–27640. [Google Scholar] [PubMed] [Green Version]
- Woo, C.H.; Eom, Y.W.; Yoo, M.H.; You, H.J.; Han, H.J.; Song, W.K.; Yoo, Y.J.; Chun, J.S.; Kim, J.H. Tumor necrosis factor-alpha generates reactive oxygen species via a cytosolic phospholipase A2-linked cascade. J. Biol. Chem. 2000, 275, 32357–32362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuringa, J.J.; Dekker, L.V.; Vellenga, E.; Kruijer, W. Sequential activation of Rac-1, SEK-1/MKK-4, and protein kinase Cdelta is required for interleukin-6-induced STAT3 Ser-727 phosphorylation and transactivation. J. Biol. Chem. 2001, 276, 27709–27715. [Google Scholar] [CrossRef] [Green Version]
- Wadei, H.M.; Textor, S.C. The role of the kidney in regulating arterial blood pressure. Nat. Rev. Nephrol. 2012, 8, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Shibata, S.; Yoshida, S.; Nagase, T.; Gotoda, T.; Fujita, T. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension 2006, 47, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawachi, H.; Fujita, T. Podocyte as the target for aldosterone: Roles of oxidative stress and Sgk1. Hypertension 2007, 49, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briet, M.; Schiffrin, E.L. Aldosterone: Effects on the kidney and cardiovascular system. Nat. Rev. Nephrol. 2010, 6, 261–273. [Google Scholar] [CrossRef]
- Shibata, S.; Ishizawa, K.; Uchida, S. Mineralocorticoid receptor as a therapeutic target in chronic kidney disease and hypertension. Hypertens. Res. 2017, 40, 221–225. [Google Scholar] [CrossRef]
- Kawarazaki, H.; Ando, K.; Nagae, A.; Fujita, M.; Matsui, H.; Fujita, T. Mineralocorticoid receptor activation contributes to salt-induced hypertension and renal injury in prepubertal Dahl salt-sensitive rats. Nephrol. Dial. Transplant. 2010, 25, 2879–2889. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, M.; Node, K.; Nakagami, H.; Liao, Y.; Grimm, M.; Takemoto, Y.; Kitakaze, M.; Liao, J.K. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J. Clin. Investig. 2001, 108, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Tanabe, S.; Maruyama, Y.; Mangmool, S.; Urayama, K.; Nagamatsu, Y.; Takagahara, S.; Turner, J.H.; Kozasa, T.; Kobayashi, H.; et al. G alpha 12/13- and reactive oxygen species-dependent activation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase by angiotensin receptor stimulation in rat neonatal cardiomyocytes. J. Biol. Chem. 2005, 280, 18434–18441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawarazaki, W.; Nagase, M.; Yoshida, S.; Takeuchi, M.; Ishizawa, K.; Ayuzawa, N.; Ueda, K.; Fujita, T. Angiotensin II- and salt-induced kidney injury through Rac1-mediated mineralocorticoid receptor activation. J. Am. Soc. Nephrol. 2012, 23, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Ishizawa, K.; Ayuzawa, N.; Ueda, K.; Takeuchi, M.; Kawarazaki, W.; Fujita, T.; Nagase, M. Local mineralocorticoid receptor activation and the role of Rac1 in obesity-related diabetic kidney disease. Nephron Exp. Nephrol. 2014, 126, 16–24. [Google Scholar] [CrossRef]
- Nagase, M.; Ayuzawa, N.; Kawarazaki, W.; Ishizawa, K.; Ueda, K.; Yoshida, S.; Fujita, T. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: Role of small GTPase Rac1. Hypertension 2012, 59, 500–506. [Google Scholar] [CrossRef] [Green Version]
- Ayuzawa, N.; Nagase, M.; Ueda, K.; Nishimoto, M.; Kawarazaki, W.; Marumo, T.; Aiba, A.; Sakurai, T.; Shindo, T.; Fujita, T. Rac1-Mediated Activation of Mineralocorticoid Receptor in Pressure Overload-Induced Cardiac Injury. Hypertension 2016, 67, 99–106. [Google Scholar] [CrossRef]
- Townsend, R.R.; Taler, S.J. Management of hypertension in chronic kidney disease. Nat. Rev. Nephrol. 2015, 11, 555–563. [Google Scholar] [CrossRef]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Perico, L.; Conti, S.; Benigni, A.; Remuzzi, G. Podocyte-actin dynamics in health and disease. Nat. Rev. Nephrol. 2016, 12, 692–710. [Google Scholar] [CrossRef]
- Ashraf, S.; Kudo, H.; Rao, J.; Kikuchi, A.; Widmeier, E.; Lawson, J.A.; Tan, W.; Hermle, T.; Warejko, J.K.; Shril, S.; et al. Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat. Commun. 2018, 9, 1960. [Google Scholar] [CrossRef] [Green Version]
- Koyama, A.; Fujita, T.; Gejyo, F.; Origasa, H.; Isono, M.; Kurumatani, H.; Okada, K.; Kanoh, H.; Kiriyama, T.; Yamada, S. Orally active prostacyclin analogue beraprost sodium in patients with chronic kidney disease: A randomized, double-blind, placebo-controlled, phase II dose finding trial. BMC Nephrol. 2015, 16, 165. [Google Scholar] [CrossRef] [Green Version]
- Nakamoto, H.; Yu, X.Q.; Kim, S.; Origasa, H.; Zheng, H.; Chen, J.; Joo, K.W.; Sritippayawan, S.; Chen, Q.; Chen, H.C.; et al. Effects of Sustained-Release Beraprost in Patients With Primary Glomerular Disease or Nephrosclerosis: CASSIOPEIR Study Results. Ther. Apher. Dial. 2020, 24, 42–55. [Google Scholar] [CrossRef]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for heart failure with preserved ejection fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, M.A.; Claggett, B.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; Gordeev, I.; et al. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial. Circulation 2015, 131, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Denus, S.; O’Meara, E.; Desai, A.S.; Claggett, B.; Lewis, E.F.; Leclair, G.; Jutras, M.; Lavoie, J.; Solomon, S.D.; Pitt, B.; et al. Spironolactone Metabolites in TOPCAT-New Insights into Regional Variation. N. Engl. J. Med. 2017, 376, 1690–1692. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, M.; Ohtsu, H.; Marumo, T.; Kawarazaki, W.; Ayuzawa, N.; Ueda, K.; Hirohama, D.; Kawakami-Mori, F.; Shibata, S.; Nagase, M.; et al. Mineralocorticoid receptor blockade suppresses dietary salt-induced ACEI/ARB-resistant albuminuria in non-diabetic hypertension: A sub-analysis of evaluate study. Hypertens. Res. 2019, 42, 514–521. [Google Scholar] [CrossRef]
- Hao, C.M.; Breyer, M.D. Physiological regulation of prostaglandins in the kidney. Annu. Rev. Physiol. 2008, 70, 357–377. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Hernanz, R.; Briones, A.M.; Salaices, M.; Alonso, M.J. New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension. Clin. Sci. 2014, 126, 111–121. [Google Scholar] [CrossRef]
- Cheng, H.F.; Harris, R.C. Renal effects of non-steroidal anti-inflammatory drugs and selective cyclooxygenase-2 inhibitors. Curr. Pharm. Des. 2005, 11, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ricciotti, E.; Scalia, R.; Tang, S.Y.; Grant, G.; Yu, Z.; Landesberg, G.; Crichton, I.; Wu, W.; Pure, E.; et al. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci. Transl. Med. 2012, 4, 132ra154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stitham, J.; Midgett, C.; Martin, K.A.; Hwa, J. Prostacyclin: An inflammatory paradox. Front. Pharmacol. 2011, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Falardeau, P.; Martineau, A. In vivo production of prostaglandin I2 in Dahl salt-sensitive and salt-resistant rats. Hypertension 1983, 5, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.Z.; Yao, B.; Wang, Y.; Yang, S.; Wang, S.; Fan, X.; Harris, R.C. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. J. Clin. Investig. 2015, 125, 4281–4294. [Google Scholar] [CrossRef] [Green Version]
- Mu, S.; Shimosawa, T.; Ogura, S.; Wang, H.; Uetake, Y.; Kawakami-Mori, F.; Marumo, T.; Yatomi, Y.; Geller, D.S.; Tanaka, H.; et al. Epigenetic modulation of the renal beta-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat. Med. 2011, 17, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Hirohama, D.; Ayuzawa, N.; Ueda, K.; Nishimoto, M.; Kawarazaki, W.; Watanabe, A.; Shimosawa, T.; Marumo, T.; Shibata, S.; Fujita, T. Aldosterone Is Essential for Angiotensin II-Induced Upregulation of Pendrin. J. Am. Soc. Nephrol. 2018, 29, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Marumo, T.; Yagi, S.; Kawarazaki, W.; Nishimoto, M.; Ayuzawa, N.; Watanabe, A.; Ueda, K.; Hirahashi, J.; Hishikawa, K.; Sakurai, H.; et al. Diabetes Induces Aberrant DNA Methylation in the Proximal Tubules of the Kidney. J. Am. Soc. Nephrol. 2015, 26, 2388–2397. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirohama, D.; Kawarazaki, W.; Nishimoto, M.; Ayuzawa, N.; Marumo, T.; Shibata, S.; Fujita, T. PGI2 Analog Attenuates Salt-Induced Renal Injury through the Inhibition of Inflammation and Rac1-MR Activation. Int. J. Mol. Sci. 2020, 21, 4433. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124433
Hirohama D, Kawarazaki W, Nishimoto M, Ayuzawa N, Marumo T, Shibata S, Fujita T. PGI2 Analog Attenuates Salt-Induced Renal Injury through the Inhibition of Inflammation and Rac1-MR Activation. International Journal of Molecular Sciences. 2020; 21(12):4433. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124433
Chicago/Turabian StyleHirohama, Daigoro, Wakako Kawarazaki, Mitsuhiro Nishimoto, Nobuhiro Ayuzawa, Takeshi Marumo, Shigeru Shibata, and Toshiro Fujita. 2020. "PGI2 Analog Attenuates Salt-Induced Renal Injury through the Inhibition of Inflammation and Rac1-MR Activation" International Journal of Molecular Sciences 21, no. 12: 4433. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21124433