Somatic Mutation Profiling in Premalignant Lesions of Vulvar Squamous Cell Carcinoma

 , ,
, ,
Abstract
:1. Introduction
2. Results
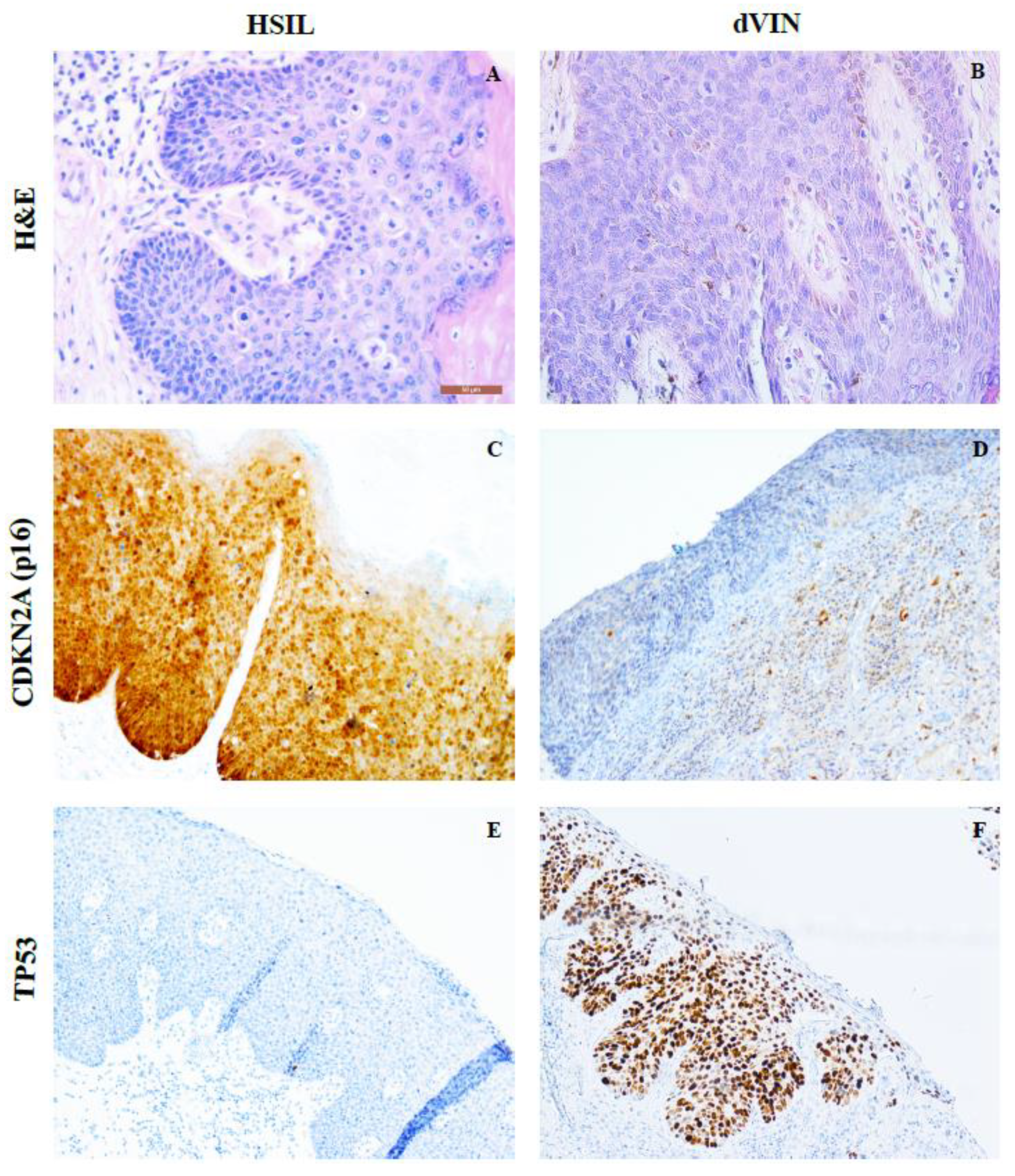
2.1. HPV Genotyping; p16 and p53 Negative Staining Results
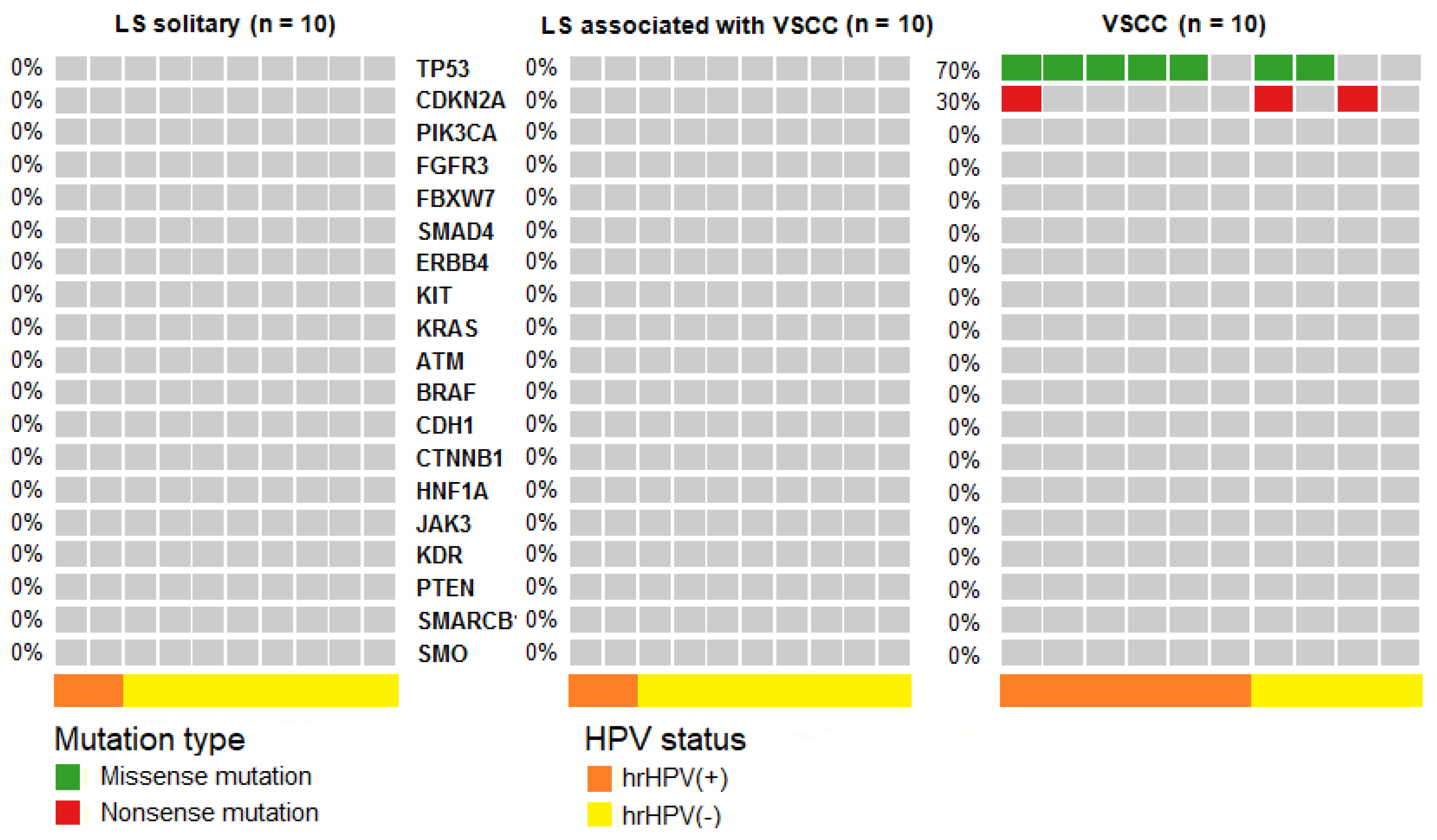
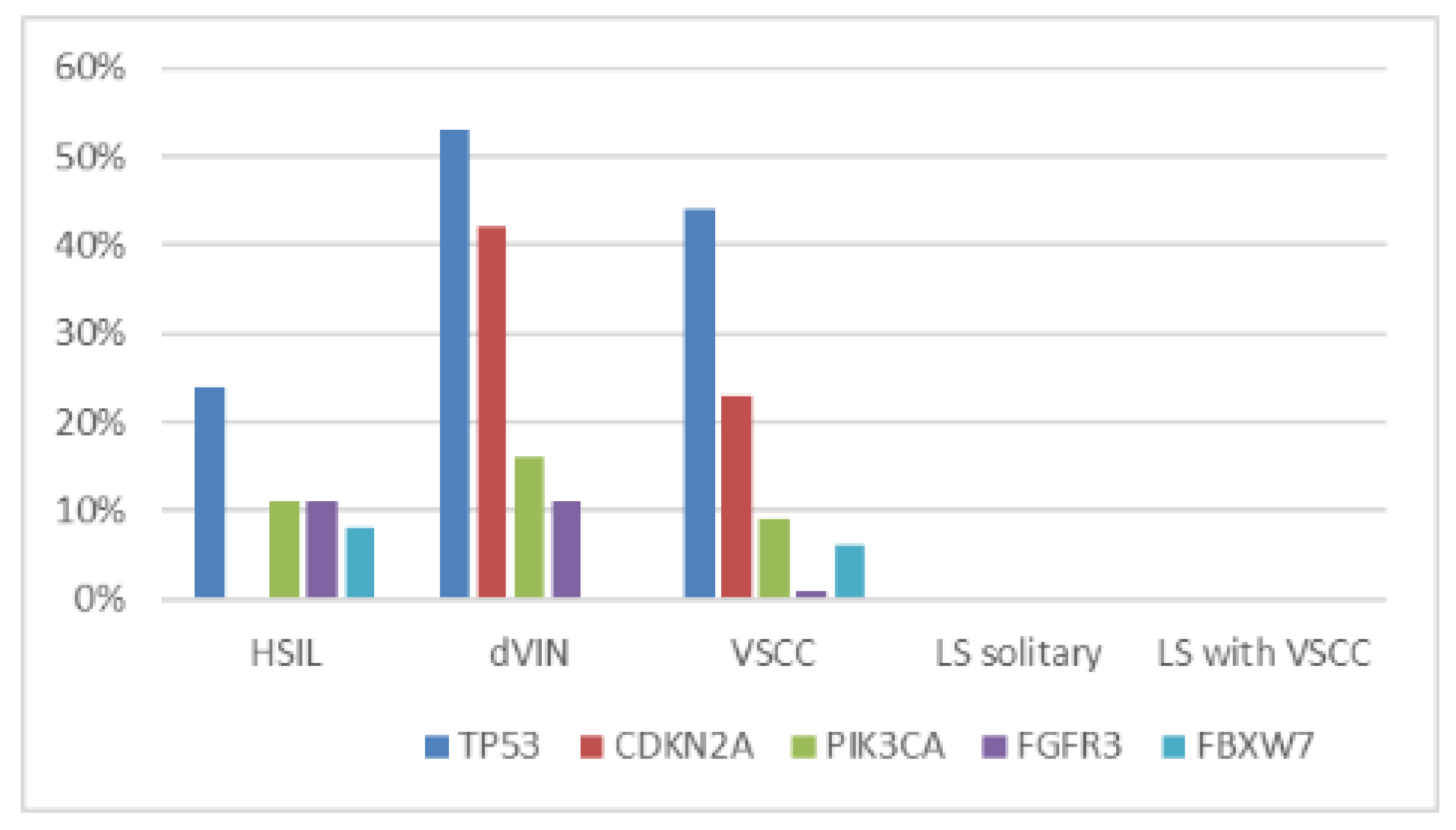
2.2. NGS Results
Pathogenic Mutations Detected in HSIL, dVIN and LS
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Isolation and HPV Genotyping
4.3. Next Generation Sequencing (NGS)
4.4. Next Generation Sequencing (NGS)
4.5. Immunohistochemistry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| dVIN | differentiated-type vulvar intraepithelial neoplasia |
| H&E | hematoxylin and eosin staining |
| HSIL | high-grade squamous intraepithelial lesion |
| HPV | human papilloma virus |
| hrHPV | high-risk HPV |
| LS | lichen sclerosus |
| VSCC | vulvar squamous cell carcinoma |
References
- International Agency for Research on Cancer: Lyon, France, 2014.
- van de Nieuwenhof, H.P.; van der Avoort, I.A.; de Hullu, J.A. Review of squamous premalignant vulvar lesions. Crit Rev. Oncol. Hematol. 2008, 68, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Hoang, L.N.; Park, K.J.; Soslow, R.A.; Murali, R. Squamous precursor lesions of the vulva: Current classification and diagnostic challenges. Pathology 2016, 48, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eva, L.J.; Ganesan, R.; Chan, K.K.; Honest, H.; Luesley, D.M. Differentiated-type vulval intraepithelial neoplasia has a high-risk association with vulval squamous cell carcinoma. Int. J. Gynecol. Cancer 2009, 19, 741–744. [Google Scholar] [CrossRef] [PubMed]
- van de Nieuwenhof, H.P.; Massuger, L.F.; van der Avoort, I.A.; Bekkers, R.L.; Casparie, M.; Abma, W.; van Kempen, L.C.; de Hullu, J.A. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur. J. Cancer 2009, 45, 851–856. [Google Scholar] [CrossRef]
- Bigby, S.M.; Eva, L.J.; Fong, K.L.; Jones, R.W. The Natural History of Vulvar Intraepithelial Neoplasia, Differentiated Type: Evidence for Progression and Diagnostic Challenges. Int. J. Gynecol. Pathol. 2016, 35, 574–584. [Google Scholar] [CrossRef]
- Judson, P.L.; Habermann, E.B.; Baxter, N.N.; Durham, S.B.; Virnig, B.A. Trends in the incidence of invasive and in situ vulvar carcinoma. Obstet. Gynecol. 2006, 107, 1018–1022. [Google Scholar] [CrossRef]
- Women and Health: Today’s Evidence Tomorrow’s Agenda; World Health Organization: Geneva, Switzerland, 2009.
- van den Einden, L.C.; de Hullu, J.A.; Massuger, L.F.; Grefte, J.M.; Bult, P.; Wiersma, A.; van Engen-van Grunsven, A.C.; Sturm, B.; Bosch, S.L.; Hollema, H.; et al. Interobserver variability and the effect of education in the histopathological diagnosis of differentiated vulvar intraepithelial neoplasia. Mod. Pathol. 2013, 26, 874–880. [Google Scholar] [CrossRef] [Green Version]
- Schuurman, M.S.; van den Einden, L.C.; Massuger, L.F.; Kiemeney, L.A.; van der Aa, M.A.; de Hullu, J.A. Trends in incidence and survival of Dutch women with vulvar squamous cell carcinoma. Eur. J. Cancer 2013, 49, 3872–3880. [Google Scholar] [CrossRef]
- Akhtar-Danesh, N.; Elit, L.; Lytwyn, A. Trends in incidence and survival of women with invasive vulvar cancer in the United States and Canada: A population-based study. Gynecol. Oncol. 2014, 134, 314–318. [Google Scholar] [CrossRef]
- Micheletti, L.; Preti, M.; Radici, G.; Boveri, S.; Di Pumpo, O.; Privitera, S.S.; Ghiringhello, B.; Benedetto, C. Vulvar Lichen Sclerosus and Neoplastic Transformation: A Retrospective Study of 976 Cases. J. Low Genit. Tract. Dis. 2016, 20, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Halonen, P.; Jakobsson, M.; Heikinheimo, O.; Riska, A.; Gissler, M.; Pukkala, E. Lichen sclerosus and risk of cancer. Int. J. Cancer 2017, 140, 1998–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Nieuwenhof, H.P.; Bulten, J.; Hollema, H.; Dommerholt, R.G.; Massuger, L.F.; van der Zee, A.G.; de Hullu, J.A.; van Kempen, L.C. Differentiated vulvar intraepithelial neoplasia is often found in lesions, previously diagnosed as lichen sclerosus, which have progressed to vulvar squamous cell carcinoma. Mod. Pathol. 2011, 24, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Neill, S.M.; Tatnall, F.M.; Cox, N.H. Guidelines for the management of lichen sclerosus. Br. J. Dermatol. 2002, 147, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Carli, P.; De Magnis, A.; Mannone, F.; Botti, E.; Taddei, G.; Cattaneo, A. Vulvar carcinoma associated with lichen sclerosus. Experience at the Florence, Italy, Vulvar Clinic. J. Reprod. Med. 2003, 48, 313–318. [Google Scholar] [PubMed]
- Dong, F.; Kojiro, S.; Borger, D.R.; Growdon, W.B.; Oliva, E. Squamous Cell Carcinoma of the Vulva: A Subclassification of 97 Cases by Clinicopathologic, Immunohistochemical, and Molecular Features (p16, p53, and EGFR). Am. J. Surg. Pathol. 2015, 39, 1045–1053. [Google Scholar] [CrossRef]
- Cheng, A.S.; Karnezis, A.N.; Jordan, S.; Singh, N.; McAlpine, J.N.; Gilks, C.B. p16 Immunostaining Allows for Accurate Subclassification of Vulvar Squamous Cell Carcinoma Into HPV-Associated and HPV-Independent Cases. Int. J. Gynecol. Pathol. 2016, 35, 385–393. [Google Scholar] [CrossRef]
- Del Pino, M.; Rodriguez-Carunchio, L.; Ordi, J. Pathways of vulvar intraepithelial neoplasia and squamous cell carcinoma. Histopathology 2013, 62, 161–175. [Google Scholar] [CrossRef]
- Trietsch, M.D.; Nooij, L.S.; Gaarenstroom, K.N.; van Poelgeest, M.I. Genetic and epigenetic changes in vulvar squamous cell carcinoma and its precursor lesions: A review of the current literature. Gynecol. Oncol. 2015, 136, 143–157. [Google Scholar] [CrossRef]
- Singh, N.; Leen, S.L.; Han, G.; Faruqi, A.; Kokka, F.; Rosenthal, A.; Jiang, X.R.; Kim, R.; McAlpine, J.N.; Gilks, C.B. Expanding the morphologic spectrum of differentiated VIN (dVIN) through detailed mapping of cases with p53 loss. Am. J. Surg. Pathol. 2015, 39, 52–60. [Google Scholar] [CrossRef]
- Pinto, A.P.; Miron, A.; Yassin, Y.; Monte, N.; Woo, T.Y.; Mehra, K.K.; Medeiros, F.; Crum, C.P. Differentiated vulvar intraepithelial neoplasia contains Tp53 mutations and is genetically linked to vulvar squamous cell carcinoma. Mod. Pathol. 2010, 23, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Zieba, S.; Kowalik, A.; Zalewski, K.; Rusetska, N.; Goryca, K.; Piascik, A.; Misiek, M.; Bakula-Zalewska, E.; Kopczynski, J.; Kowalski, K.; et al. Somatic mutation profiling of vulvar cancer: Exploring therapeutic targets. Gynecol. Oncol. 2018, 150, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer—Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Nooij, L.S.; Ter Haar, N.T.; Ruano, D.; Rakislova, N.; van Wezel, T.; Smit, V.; Trimbos, B.; Ordi, J.; van Poelgeest, M.I.E.; Bosse, T. Genomic Characterization of Vulvar (Pre)cancers Identifies Distinct Molecular Subtypes with Prognostic Significance. Clin. Cancer Res. 2017, 23, 6781–6789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murnyak, B.; Hortobagyi, T. Immunohistochemical correlates of TP53 somatic mutations in cancer. Oncotarget 2016, 7, 64910–64920. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.M.; Do, H.; Young, R.J.; Wong, S.Q.; Angel, C.; Collins, M.; Takano, E.A.; Corry, J.; Wiesenfeld, D.; Kleid, S.; et al. Differential mechanisms of CDKN2A (p16) alteration in oral tongue squamous cell carcinomas and correlation with patient outcome. Int. J. Cancer 2014, 135, 887–895. [Google Scholar] [CrossRef]
- Larque, A.B.; Conde, L.; Hakim, S.; Alos, L.; Jares, P.; Vilaseca, I.; Cardesa, A.; Nadal, A. P16(INK(4)a) overexpression is associated with CDKN2A mutation and worse prognosis in HPV-negative laryngeal squamous cell carcinomas. Virchows Arch. 2015, 466, 375–382. [Google Scholar] [CrossRef]
- Li, J.; Poi, M.J.; Tsai, M.D. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef] [Green Version]
- Woelber, L.L.P.K.; Eulenburg, C.H.; de Gregorio, N.; Klapdor, R.; Kalder, M.; Braicu, E.I.; Fuerst, S.; Klar, M.; Strauss, H.-G.; Mehlhorn, G.; et al. p53 and p16 expression profiles reveal three prognostically relevant subgroups in vulvar cancer: A TMA based study by the AGO-CaRE-translational study group. J. Clin. Oncol. 2019, 37, 5592. [Google Scholar] [CrossRef]
- Zieba, S.; Chechlinska, M.; Kowalik, A.; Kowalewska, M. Genes pathways and vulvar carcinoma—New insights from next-generation sequencing studies. Gynecol. Oncol. 2020. ahead of print. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Park, D.; Munger, K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad Sci. USA 2013, 110, 16175–16180. [Google Scholar] [CrossRef] [Green Version]
- Han, M.R.; Shin, S.; Park, H.C.; Kim, M.S.; Lee, S.H.; Jung, S.H.; Song, S.Y.; Lee, S.H.; Chung, Y.J. Mutational signatures and chromosome alteration profiles of squamous cell carcinomas of the vulva. Exp. Mol. Med. 2018, 50, e442. [Google Scholar] [CrossRef] [PubMed]
- Swarts, D.R.A.; Voorham, Q.J.M.; van Splunter, A.P.; Wilting, S.M.; Sie, D.; Pronk, D.; van Beurden, M.; Heideman, D.A.M.; Snijders, P.J.F.; Meijer, C.; et al. Molecular heterogeneity in human papillomavirus-dependent and -independent vulvar carcinogenesis. Cancer Med. 2018, 7, 4542–4553. [Google Scholar] [CrossRef] [PubMed]
- Pouwer, A.W.; Loes van den, E.; van der Linden, M.; Hehir-Kwa, J.Y.; Yu, J.; Hendriks, K.M.; Kamping, E.J.; Eijkelenboom, A.; Massuger, L.; Bulten, J.; et al. Clonal Relationship Between Lichen Sclerosus, Differentiated Vulvar Intra-epithelial Neoplasia and Non HPV-related Vulvar Squamous Cell Carcinoma. Cancer Genom. Proteom. 2020, 17, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, J.A.; Ambros, R.; Malfetano, J.; Ross, J.; Grabowski, R.; Lamb, P.; Figge, H.; Mihm, M.C., Jr. Vulvar lichen sclerosus and squamous cell carcinoma: A cohort, case control, and investigational study with historical perspective; implications for chronic inflammation and sclerosis in the development of neoplasia. Hum. Pathol. 1998, 29, 932–948. [Google Scholar] [CrossRef]
- Vanin, K.; Scurry, J.; Thorne, H.; Yuen, K.; Ramsay, R.G. Overexpression of wild-type p53 in lichen sclerosus adjacent to human papillomavirus-negative vulvar cancer. J. Invest. Dermatol. 2002, 119, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Rolfe, K.J.; MacLean, A.B.; Crow, J.C.; Benjamin, E.; Reid, W.M.; Perrett, C.W. TP53 mutations in vulval lichen sclerosus adjacent to squamous cell carcinoma of the vulva. Br. J. Cancer 2003, 89, 2249–2253. [Google Scholar] [CrossRef]
- Dasgupta, S.; Ewing-Graham, P.C.; van Kemenade, F.J.; van Doorn, H.C.; Noordhoek Hegt, V.; Koljenovic, S. Differentiated vulvar intraepithelial neoplasia (dVIN): The most helpful histological features and the utility of cytokeratins 13 and 17. Virchows Arch. 2018, 473, 739–747. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, D.; Guarch, R.; Ojer, A.; Casas, J.M.; Mendez-Meca, C.; Esteller, M.; Barba-Ramos, E.; Garcia-Bragado, F.; Puras, A. Differential hypermethylation of genes in vulvar cancer and lichen sclerosus coexisting or not with vulvar cancer. Int. J. Cancer 2011, 128, 2853–2864. [Google Scholar] [CrossRef]
- Gambichler, T.; Terras, S.; Kreuter, A.; Skrygan, M. Altered global methylation and hydroxymethylation status in vulvar lichen sclerosus: Further support for epigenetic mechanisms. Br. J. Dermatol. 2014, 170, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Abdulrahman, Z.; Kortekaas, K.E.; De Vos Van Steenwijk, P.J.; Van Der Burg, S.H.; Van Poelgeest, M.I. The immune microenvironment in vulvar (pre)cancer: Review of literature and implications for immunotherapy. Expert Opin. Biol. Ther. 2018, 18, 1223–1233. [Google Scholar] [CrossRef]
- Woelber, L.; Griebel, L.F.; Eulenburg, C.; Sehouli, J.; Jueckstock, J.; Hilpert, F.; de Gregorio, N.; Hasenburg, A.; Ignatov, A.; Hillemanns, P.; et al. Role of tumour-free margin distance for loco-regional control in vulvar cancer-a subset analysis of the Arbeitsgemeinschaft Gynakologische Onkologie CaRE-1 multicenter study. Eur. J. Cancer 2016, 69, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Te Grootenhuis, N.C.; Pouwer, A.W.; de Bock, G.H.; Hollema, H.; Bulten, J.; van der Zee, A.G.J.; de Hullu, J.A.; Oonk, M.H.M. Prognostic factors for local recurrence of squamous cell carcinoma of the vulva: A systematic review. Gynecol. Oncol. 2018, 148, 622–631. [Google Scholar] [CrossRef]
- Pleunis, N.; Leermakers, M.E.J.; van der Wurff, A.A.; Klinkhamer, P.; Ezendam, N.P.M.; Boll, D.; de Hullu, J.A.; Pijnenborg, J.M.A. Surgical margins in squamous cell carcinoma, different for the vulva? Eur. J. Surg. Oncol. 2018, 44, 1555–1561. [Google Scholar] [CrossRef]
- Yap, J.K.; Fox, R.; Leonard, S.; Ganesan, R.; Kehoe, S.T.; Dawson, C.W.; Woodman, C.B.; Luesley, D.M. Adjacent Lichen Sclerosis predicts local recurrence and second field tumour in women with vulvar squamous cell carcinoma. Gynecol. Oncol. 2016, 142, 420–426. [Google Scholar] [CrossRef]
- Leonard, S.; Pereira, M.; Fox, R.; Gordon, N.; Yap, J.; Kehoe, S.; Luesley, D.; Woodman, C.; Ganesan, R. Over-expression of DNMT3A predicts the risk of recurrent vulvar squamous cell carcinomas. Gynecol. Oncol. 2016, 143, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Koncar, R.F.; Feldman, R.; Bahassi, E.M.; Hashemi Sadraei, N. Comparative molecular profiling of HPV-induced squamous cell carcinomas. Cancer Med. 2017, 6, 1673–1685. [Google Scholar] [CrossRef]
- Janku, F.; Wheler, J.J.; Naing, A.; Falchook, G.S.; Hong, D.S.; Stepanek, V.M.; Fu, S.; Piha-Paul, S.A.; Lee, J.J.; Luthra, R.; et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013, 73, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Weberpals, J.I.; Lo, B.; Duciaume, M.M.; Spaans, J.N.; Clancy, A.A.; Dimitroulakos, J.; Goss, G.D.; Sekhon, H.S. Vulvar Squamous Cell Carcinoma (VSCC) as Two Diseases: HPV Status Identifies Distinct Mutational Profiles Including Oncogenic Fibroblast Growth Factor Receptor 3. Clin. Cancer Res. 2017, 23, 4501–4510. [Google Scholar] [CrossRef] [Green Version]
- Rosty, C.; Aubriot, M.H.; Cappellen, D.; Bourdin, J.; Cartier, I.; Thiery, J.P.; Sastre-Garau, X.; Radvanyi, F. Clinical and biological characteristics of cervical neoplasias with FGFR3 mutation. Mol. Cancer 2005, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Bersani, C.; Sivars, L.; Haeggblom, L.; DiLorenzo, S.; Mints, M.; Ahrlund-Richter, A.; Tertipis, N.; Munck-Wikland, E.; Nasman, A.; Ramqvist, T.; et al. Targeted sequencing of tonsillar and base of tongue cancer and human papillomavirus positive unknown primary of the head and neck reveals prognostic effects of mutated FGFR3. Oncotarget 2017, 8, 35339–35350. [Google Scholar] [CrossRef]
- Elkhattouti, A.; Hassan, M.; Gomez, C.R. Stromal Fibroblast in Age-Related Cancer: Role in Tumorigenesis and Potential as Novel Therapeutic Target. Front. Oncol. 2015, 5, 158. [Google Scholar] [CrossRef] [Green Version]
- Clancy, A.A.; Spaans, J.N.; Weberpals, J.I. The forgotten woman’s cancer: Vulvar squamous cell carcinoma (VSCC) and a targeted approach to therapy. Ann. Oncol. 2016, 27, 1696–1705. [Google Scholar] [CrossRef]
- Gatzka, M.V. Targeted Tumor Therapy Remixed—An Update on the Use of Small-Molecule Drugs in Combination Therapies. Cancers 2018, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic. Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Type | Mutations (% of Cases) | Polymorphisms (% of Cases) | |||
|---|---|---|---|---|---|
| TP53 | CDKN2A | TP53 P72R | KDR Q472H | KIT M541L | |
| HSIL (n = 38) | 24 | 0 | 100 | 42 | 8 |
| dVIN (n = 19) | 53 | 42 | 100 | 63 | 16 |
| solitary LS (n = 10) | 0 | 0 | 100 | 40 | 20 |
| LS associated with VSCC (n = 10) | 0 | 0 | 100 | 50 | 30 |
| VSCC (n = 10) | 70 | 30 | 100 | 50 | 30 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zięba, S.; Pouwer, A.-F.W.; Kowalik, A.; Zalewski, K.; Rusetska, N.; Bakuła-Zalewska, E.; Kopczyński, J.; Pijnenborg, J.M.A.; de Hullu, J.A.; Kowalewska, M. Somatic Mutation Profiling in Premalignant Lesions of Vulvar Squamous Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 4880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144880
Zięba S, Pouwer A-FW, Kowalik A, Zalewski K, Rusetska N, Bakuła-Zalewska E, Kopczyński J, Pijnenborg JMA, de Hullu JA, Kowalewska M. Somatic Mutation Profiling in Premalignant Lesions of Vulvar Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2020; 21(14):4880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144880
Chicago/Turabian StyleZięba, Sebastian, Anne-Floor W. Pouwer, Artur Kowalik, Kamil Zalewski, Natalia Rusetska, Elwira Bakuła-Zalewska, Janusz Kopczyński, Johanna M. A. Pijnenborg, Joanne A. de Hullu, and Magdalena Kowalewska. 2020. "Somatic Mutation Profiling in Premalignant Lesions of Vulvar Squamous Cell Carcinoma" International Journal of Molecular Sciences 21, no. 14: 4880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21144880