Drug Repositioning of the α1-Adrenergic Receptor Antagonist Naftopidil: A Potential New Anti-Cancer Drug?


Abstract
:1. Drug Repurposing: A Promising Tool for Cancer Management
2. Alpha1-Adrenergic Receptor Antagonists
3. Anti-Cancerous Properties of Naftopidil
3.1. Cytostatic Effects of Naftopidil In Vitro
3.2. Cytotoxic Effects of Naftopidil In Vitro
3.3. Anti-Cancerous Effects of Naftopidil In Vivo
3.4. Clinical Evidence of Naftopidil Anti-Cancer Effects
4. Molecular Mechanisms Involved in the Anti-Cancerous Properties of Naftopidil
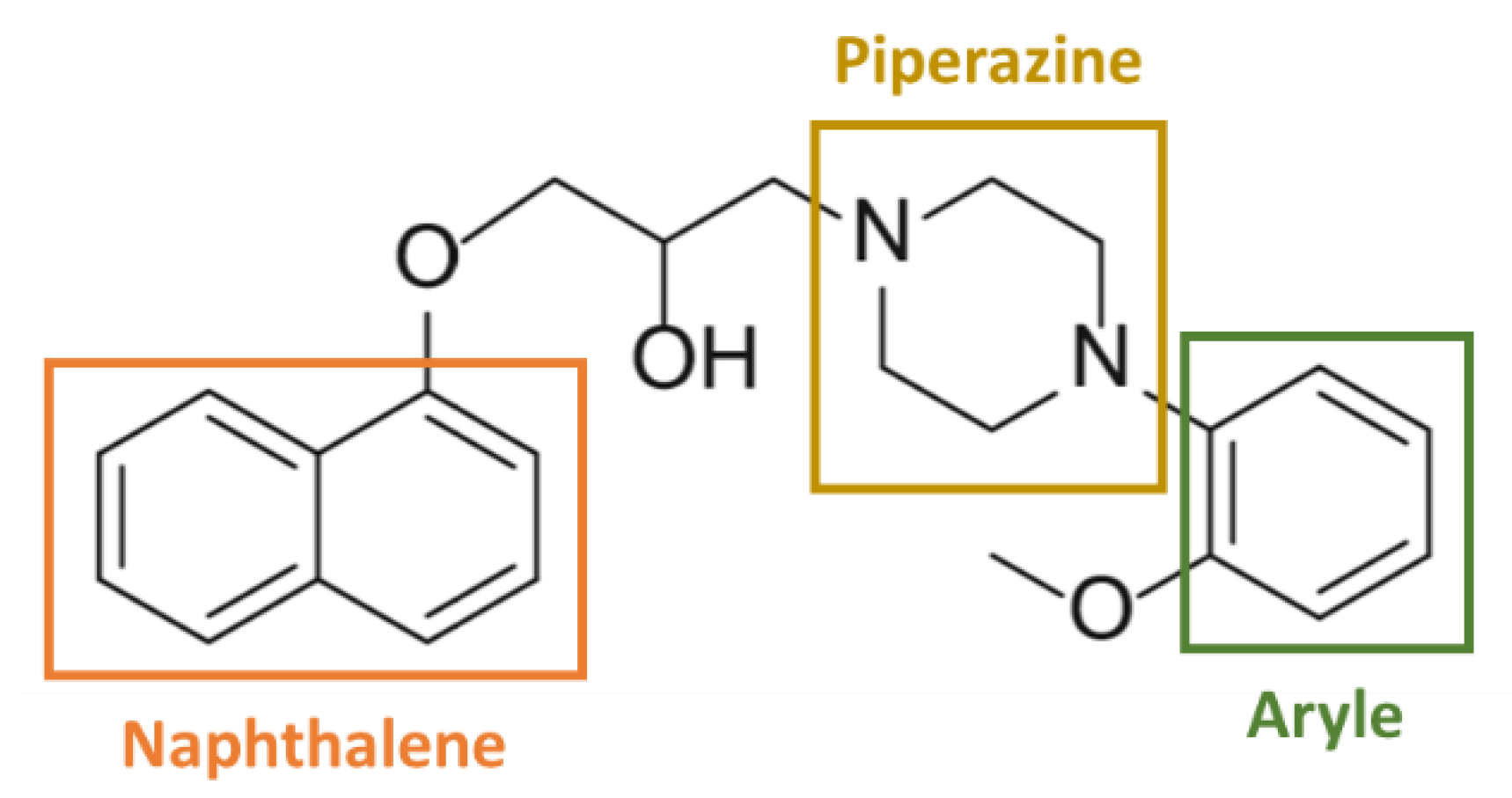
4.1. Involvement of the Chemical Structure
4.2. Alpha1-AR Independent Anti-Cancerous Action
4.3. Other Pathways Involved
4.4. Anti-Cancerous Properties of Naftopidil Analogues
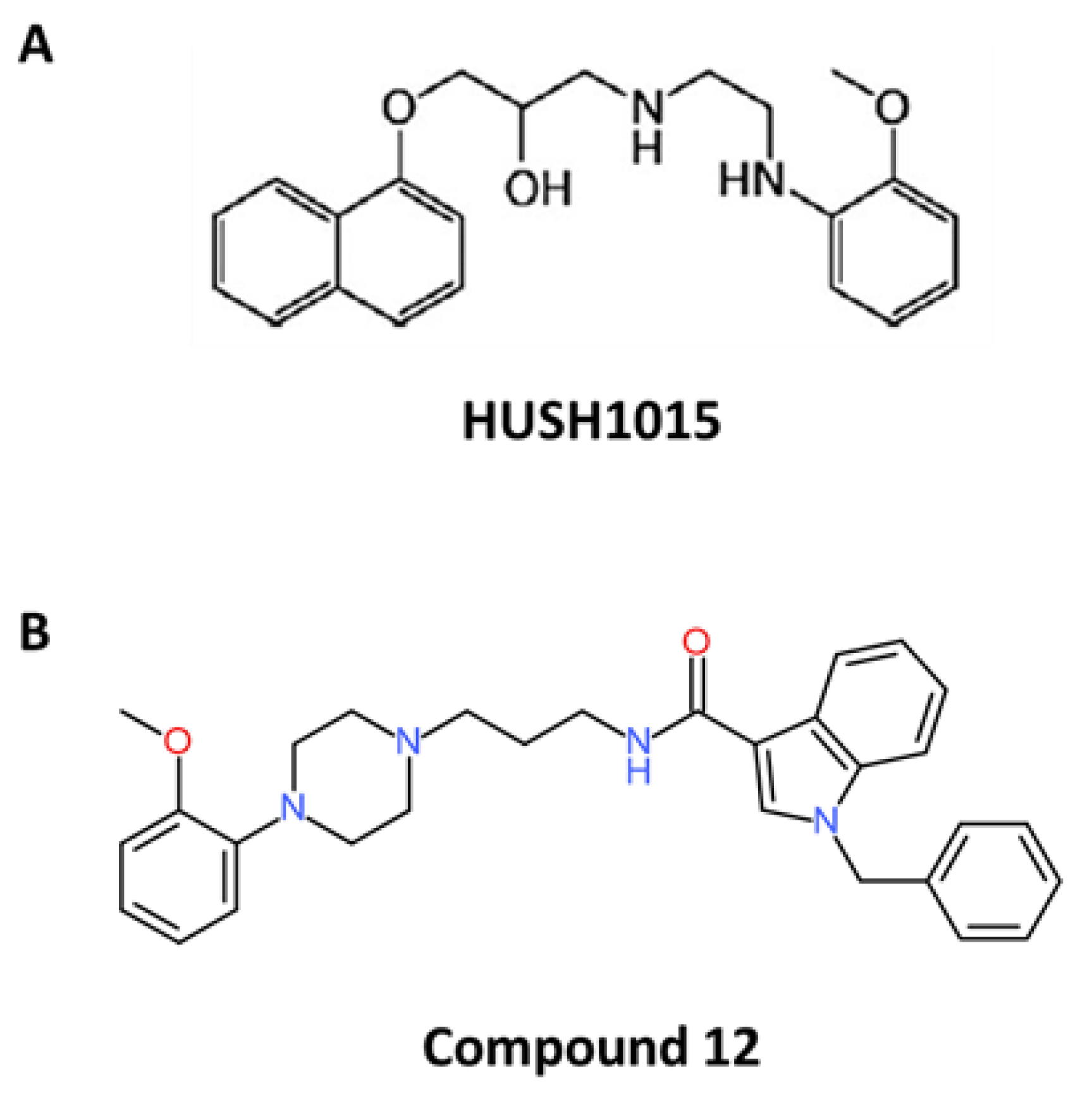
4.5. HUHS1015
4.6. Compound 12
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α1-AR | α1-Adrenergic Receptors |
| 5-ARI | 5-α Reductase Inhibitors |
| Akt | protein kinase B |
| AMID | Apoptosis-inducing factor-homologous Mitochondrion-associated Inducer of Death |
| ATF4 | Activating Transcription Factor 4 |
| AMPK | Adenosine Monophosphate-actived Protein Kinase |
| Bad | Bcl2-Associated agonist of cell death |
| Bax | Bcl-2–associated X |
| Bcl-2 | B-cell lymphoma 2 |
| Bim | Bcl-2 like 11 |
| Bcl-xL | B-cell lymphoma X long isoform |
| BPH | Benign Prostatic Hyperplasia |
| Cdk-2 | Cyclin dependent kinase-2 |
| cGMP | cyclic Guanosine Monophosphate |
| CYP2C9 | Cytochrome P450 2C9 |
| CYP2C19 | Cytochrome P450 2C19 |
| DAG | Diacylglycerol |
| DHT | Dihydrotestosterone |
| ER | Endoplasmic Reticulum |
| FDA | Food and Drug Administration |
| HDAC | Histone Deacetylase |
| Hrk | Harakiri |
| IP3 | Inositol 1,4,5-triphosphate |
| IP3-R | IP3-Receptor |
| JNK | c-Jun NH2-terminal Kinase |
| LUTS | Lower Urinary Tract Symptoms |
| Mcl-1 | Myeloid cell leukemia-1 |
| MVD | Microvessels Density |
| Noxa | Phorbol-12-myristate-13-acetate-induced protein 1 |
| PARP | Poly (ADP-Ribose) Polymerase |
| PDE5 | Phosphodiesterase type 5 |
| PDO | Patient-Derived Organoids |
| PLC | phospholipase C |
| PI3K | Phosphoinositide 3-Kinase |
| PIP2 | Phospholipid phosphatydylinositol-4,5-bisphosphate |
| PKA | Protein Kinase A |
| PKC | Protein Kinase C |
| Puma | p53 Up-regulated Modulator of Apoptosis |
| TGF-β | Tumour Growth Factor-β |
| TNF-α | Tumour Necrosis Factor-α |
| TRP | Transient Receptor Potential calcium channel |
| VGCC | Voltage-Gated Calcium Channel |
References
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Mander, K.; Finnie, J. Tumour angiogenesis, anti-angiogenic therapy and chemotherapeutic resistance. Aust. Vet. J. 2018, 96, 371–378. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharm. 2013, 34, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.C.; Ekins, S.; Williams, A.J.; Tropsha, A. A bibliometric review of drug repurposing. Drug Discov. Today 2018, 23, 661–672. [Google Scholar] [CrossRef]
- Goldstein, I.; Tseng, L.-J.; Creanga, D.; Stecher, V.; Kaminetsky, J.C. Efficacy and safety of sildenafil by age in men with erectile dysfunction. J. Sex. Med. 2016, 13, 852–859. [Google Scholar] [CrossRef]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharm. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Mallik, R.; Chowdhury, T.A. Metformin in cancer. Diabetes Res. Clin. Pract. 2018, 143, 409–419. [Google Scholar] [CrossRef]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Sauter, E.R. Breast cancer prevention: Current approaches and future directions. Eur. J. Breast Health 2018. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Banno, K.; Kunitomi, H.; Tominaga, E.; Aoki, D. Current state and outlook for drug repositioning anticipated in the field of ovarian cancer. J. Gynecol. Oncol. 2019, 30, e10. [Google Scholar] [CrossRef] [PubMed]
- Hanusova, V.; Skalova, L.; Kralova, V.; Matouskova, P. Potential anti-cancer drugs commonly used for other indications. Curr. Cancer Drug Targets 2015, 15, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Price, D.T.; Lefkowitz, R.J.; Caron, M.G.; Berkowitz, D.; Schwinn, D.A. Localization of mRNA for three distinct alpha 1-adrenergic receptor subtypes in human tissues: Implications for human alpha-adrenergic physiology. Mol. Pharm. 1994, 45, 171–175. [Google Scholar]
- Zhong, H.; Minneman, K.P. Alpha1-adrenoceptor subtypes. Eur. J. Pharm. 1999, 375, 261–276. [Google Scholar] [CrossRef]
- Docherty, J.R. The pharmacology of α1-adrenoceptor subtypes. Eur. J. Pharm. 2019, 855, 305–320. [Google Scholar] [CrossRef]
- Petitcolin, M.A.; Spitzbarth-Régrigny, E.; Bueb, J.L.; Capdeville-Atkinson, C.; Tschirhart, E. Role of G(i)-proteins in norepinephrine-mediated vasoconstriction in rat tail artery smooth muscle. Biochem. Pharm. 2001, 61, 1169–1175. [Google Scholar] [CrossRef]
- Kaplan, S.A. Current role of alpha-blockers in the treatment of benign prostatic hyperplasia. BJU Int. 2008, 102 (Suppl. S2), S3–S7. [Google Scholar] [CrossRef]
- Kim, E.H.; Larson, J.A.; Andriole, G.L. Management of benign prostatic hyperplasia. Annu. Rev. Med. 2016, 67, 137–151. [Google Scholar] [CrossRef] [Green Version]
- Asseldonk, B.V.; Barkin, J.; Elterman, D.S. Medical therapy for benign prostatic hyperplasia: A review. Can. J. Urol. 2015, 22 (Suppl. S1), S7–S17. [Google Scholar]
- Hwang, E.C.; Gandhi, S.; Jung, J.H.; Imamura, M.; Kim, M.H.; Pang, R.; Dahm, P. Naftopidil for the treatment of lower urinary tract symptoms compatible with benign prostatic hyperplasia. Cochrane Database Syst. Rev. 2018, 10, CD007360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepor, H. Pathophysiology of lower urinary tract symptoms in the aging male population. Rev. Urol. 2005, 7, 3–11. [Google Scholar] [PubMed]
- Kim, E.H.; Brockman, J.A.; Andriole, G.L. The use of 5-alpha reductase inhibitors in the treatment of benign prostatic hyperplasia. Asian J. Urol. 2018, 5, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y. Lower urinary tract symptoms/benign prostatic hypertrophy and vascular function: Role of the nitric oxide-phosphodiesterase type 5-cyclic guanosine 3’, 5’-monophosphate pathway. Int. J. Urol. Off. J. Jpn. Urol. Assoc. 2017, 24, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Tinel, H.; Stelte-Ludwig, B.; Hütter, J.; Sandner, P. Pre-clinical evidence for the use of phosphodiesterase-5 inhibitors for treating benign prostatic hyperplasia and lower urinary tract symptoms. BJU Int. 2006, 98, 1259–1263. [Google Scholar] [CrossRef]
- Schwinn, D.A.; Roehrborn, C.G. α1-Adrenoceptor subtypes and lower urinary tract symptoms: α1-Adrenoceptor subtypes & LUTS. Int. J. Urol. 2008, 15, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Kojima, Y.; Sasaki, S.; Shinoura, H.; Hayase, M.; Kubota, Y.; Hayashi, Y.; Tsujimoto, G.; Kohri, K. Change of expression levels of alpha1-adrenoceptor subtypes by administration of alpha1d-adrenoceptor-subtype-selective antagonist naftopidil in benign prostate hyperplasia patients. Prostate 2007, 67, 1285–1292. [Google Scholar] [CrossRef]
- Lepor, H. Medical treatment of benign prostatic hyperplasia. Rev. Urol. 2011, 13, 20–33. [Google Scholar] [PubMed]
- Sigala, S.; Dellabella, M.; Milanese, G.; Fornari, S.; Faccoli, S.; Palazzolo, F.; Peroni, A.; Mirabella, G.; Cunico, S.C.; Spano, P.; et al. Evidence for the presence of alpha1 adrenoceptor subtypes in the human ureter. Neurourol. Urodyn. 2005, 24, 142–148. [Google Scholar] [CrossRef]
- Malloy, B.J.; Price, D.T.; Price, R.R.; Bienstock, A.M.; Dole, M.K.; Funk, B.L.; Rudner, X.L.; Richardson, C.D.; Donatucci, C.F.; Schwinn, D.A. Alpha1-adrenergic receptor subtypes in human detrusor. J. Urol. 1998, 160, 937–943. [Google Scholar] [CrossRef]
- Hampel, C.; Dolber, P.C.; Smith, M.P.; Savic, S.L.; Th roff, J.W.; Thor, K.B.; Schwinn, D.A. Modulation of bladder alpha1-adrenergic receptor subtype expression by bladder outlet obstruction. J. Urol. 2002, 167, 1513–1521. [Google Scholar] [CrossRef]
- Michel, M.C. Potential role of a1-adrenoceptors in the aetiology of LUTS. Eur. Urol. Suppl. 2002, 5–13. [Google Scholar] [CrossRef]
- Nasu, K.; Moriyama, N.; Kawabe, K.; Tsujimoto, G.; Murai, M.; Tanaka, T.; Yano, J. Quantification and distribution of alpha 1-adrenoceptor subtype mRNAs in human prostate: Comparison of benign hypertrophied tissue and non-hypertrophied tissue. Br. J. Pharm. 1996, 119, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Sasaki, S.; Kubota, Y.; Hayase, M.; Hayashi, Y.; Shinoura, H.; Tsujimoto, G.; Kohri, K. Expression of alpha1-adrenoceptor subtype mRNA as a predictor of the efficacy of subtype selective alpha1-adrenoceptor antagonists in the management of benign prostatic hyperplasia. J. Urol. 2008, 179, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Masumori, N. Naftopidil for the treatment of urinary symptoms in patients with benign prostatic hyperplasia. Ther. Clin. Risk Manag. 2011, 7, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Walden, P.D.; Gerardi, C.; Lepor, H. Localization and expression of the alpha1A-1, alpha1B and alpha1D-adrenoceptors in hyperplastic and non-hyperplastic human prostate. J. Urol. 1999, 161, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Mobley, D.; Feibus, A.; Baum, N. Benign prostatic hyperplasia and urinary symptoms: Evaluation and treatment. Postgrad. Med. 2015, 127, 301–307. [Google Scholar] [CrossRef]
- Michel, M.C.; Grübbel, B.; Taguchi, K.; Verfürth, F.; Otto, T.; Kröpfl, D. Drugs for treatment of benign prostatic hyperplasia: Affinity comparison at cloned alpha 1-adrenoceptor subtypes and in human prostate. J. Auton. Pharm. 1996, 16, 21–28. [Google Scholar] [CrossRef]
- Take, H.; Shibata, K.; Awaji, T.; Hirasawa, A.; Ikegaki, I.; Asano, T.; Takada, T.; Tsujimoto, G. Vascular alpha1-adrenoceptor subtype selectivity and alpha1-blocker-induced orthostatic hypotension. Jpn. J. Pharm. 1998, 77, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Takei, R.; Ikegaki, I.; Shibata, K.; Tsujimoto, G.; Asano, T. Naftopidil, a novel alpha1-adrenoceptor antagonist, displays selective inhibition of canine prostatic pressure and high affinity binding to cloned human alpha1-adrenoceptors. Jpn. J. Pharm. 1999, 79, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.-J.; Cai, Y.; Yi, Y.-Z.; Huang, M.-Y.; Zhu, L.; He, F.; Liu, X.-W.; Huang, B.-Y.; Yuan, M. Pharmaceutical evaluation of naftopidil enantiomers: Rat functional assays in vitro and estrogen/androgen induced rat benign prostatic hyperplasia model in vivo. Eur. J. Pharm. 2016, 791, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Huang, J.; Jiang, R.; Yuan, M. Crystal structures, absolute configurations and molecular docking studies of naftopidil enantiomers as α1D-adrenoceptor antagonists. Acta Pharm. Sin. B 2017, 7, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Proudman, R.G.W.; Pupo, A.S.; Baker, J.G. The affinity and selectivity of α-adrenoceptor antagonists, antidepressants, and antipsychotics for the human α1A, α1B, and α1D-adrenoceptors. Pharm. Res. Perspect. 2020, 8, e00602. [Google Scholar] [CrossRef]
- Hara, N.; Mizusawa, T.; Obara, K.; Takahashi, K. The role of naftopidil in the management of benign prostatic hyperplasia. Ther. Adv. Urol. 2013, 5, 111–119. [Google Scholar] [CrossRef]
- Nishino, Y.; Masue, T.; Miwa, K.; Takahashi, Y.; Ishihara, S.; Deguchi, T. Comparison of two alpha1-adrenoceptor antagonists, naftopidil and tamsulosin hydrochloride, in the treatment of lower urinary tract symptoms with benign prostatic hyperplasia: A randomized crossover study. BJU Int. 2006, 97, 747–751. [Google Scholar] [CrossRef]
- Ukimura, O.; Kanazawa, M.; Fujihara, A.; Kamoi, K.; Okihara, K.; Miki, T. Kyoto Prefectural University of Medicine Benign Prostatic Hypertrophy Research Group Naftopidil versus tamsulosin hydrochloride for lower urinary tract symptoms associated with benign prostatic hyperplasia with special reference to the storage symptom: A prospective randomized controlled study. Int. J. Urol. Off. J. Jpn. Urol. Assoc. 2008, 15, 1049–1054. [Google Scholar] [CrossRef]
- Perumal, C.; Chowdhury, P.S.; Ananthakrishnan, N.; Nayak, P.; Gurumurthy, S. A comparison of the efficacy of naftopidil and tamsulosin hydrochloride in medical treatment of benign prostatic enlargement. Urol. Ann. 2015, 7, 74–78. [Google Scholar] [CrossRef]
- Ikemoto, I.; Kiyota, H.; Ohishi, Y.; Abe, K.; Goto, H.; Kishimoto, K.; Miki, K. Usefulness of tamsulosin hydrochloride and naftopidil in patients with urinary disturbances caused by benign prostatic hyperplasia: A comparative, randomized, two-drug crossover study. Int. J. Urol. 2003, 10, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Griwan, M.S.; Karthikeyan, Y.R.; Kumar, M.; Singh, B.J.; Singh, S.K. Comparative evaluation of naftopidil and tamsulosin in the treatment of patients with lower urinary tract symptoms with benign prostatic hyperplasia. Urol. Ann. 2014, 6, 181–186. [Google Scholar] [CrossRef]
- Farthing, M.J.; Alstead, E.M.; Abrams, S.M.; Haug, G.; Johnston, A.; Hermann, R.; Niebch, G.; Ruus, P.; Molz, K.H.; Turner, P. Pharmacokinetics of naftopidil, a novel anti-hypertensive drug, in patients with hepatic dysfunction. Postgrad. Med. J. 1994, 70, 363–366. [Google Scholar] [CrossRef]
- Himmel, H.M. Naftopidil, a novel antihypertensive drug. Cardiovasc. Drug Rev. 1994, 12, 232–247. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, X.; Zhu, L.; Zhang, X.; Fu, X.; Huang, J.; Yuan, M. Identification of human cytochrome P450 isozymes involved in the metabolism of naftopidil enantiomers in vitro: The metabolism of naftopidil enantiomers. J. Pharm. Pharm. 2014, 66, 1534–1551. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Huang, J.; Rong, Y.; Luo, C.; Guo, J.; Zhu, L.; Huang, B.; Yuan, M. Enantiospecific determination of naftopidil by RRLC-MS/MS reveals stereoselective pharmacokinetics and tissue distributions in rats. J. Pharm. Biomed. Anal. 2015, 112, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Elagamy, H.I.; Essa, E.A.; Nouh, A.; El Maghraby, G.M. Development and evaluation of rapidly dissolving buccal films of naftopidil: In vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2019, 45, 1695–1706. [Google Scholar] [CrossRef]
- Hori, Y.; Ishii, K.; Kanda, H.; Iwamoto, Y.; Nishikawa, K.; Soga, N.; Kise, H.; Arima, K.; Sugimura, Y. Naftopidil, a selective {alpha}1-adrenoceptor antagonist, suppresses human prostate tumor growth by altering interactions between tumor cells and stroma. Cancer Prev. Res. 2011, 4, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, Y.; Ishii, K.; Sasaki, T.; Kato, M.; Kanda, H.; Yamada, Y.; Arima, K.; Shiraishi, T.; Sugimura, Y. Oral naftopidil suppresses human renal-cell carcinoma by inducing G(1) cell-cycle arrest in tumor and vascular endothelial cells. Cancer Prev. Res. 2013, 6, 1000–1006. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, A.; Nagaya, H.; Kanno, T.; Nishizaki, T. Antitumor action of alpha(1)-adrenoceptor blockers on human bladder, prostate and renal cancer cells. Pharmacology 2012, 90, 242–246. [Google Scholar] [CrossRef]
- Nakagawa, Y.U.; Nagaya, H.; Miyata, T.; Wada, Y.; Oyama, T.; Gotoh, A. Piperazine-based Alpha-1 AR blocker, Naftopidil, selectively suppresses malignant human bladder cells via induction of apoptosis. Anticancer Res. 2016, 36, 1563–1570. [Google Scholar] [PubMed]
- Ishii, K.; Sugimura, Y. Identification of a new pharmacological activity of the phenylpiperazine derivative naftopidil: Tubulin-binding drug. J. Chem. Biol. 2015, 8, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Florent, R.; Weiswald, L.-B.; Lambert, B.; Brotin, E.; Abeilard, E.; Louis, M.-H.; Babin, G.; Poulain, L.; N’Diaye, M. Bim, Puma and Noxa upregulation by Naftopidil sensitizes ovarian cancer to the BH3-mimetic ABT-737 and the MEK inhibitor Trametinib. Cell Death Dis. 2020, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Ishii, K.; Ogura, Y.; Imamura, T.; Kanai, M.; Arima, K.; Sugimura, Y. Naftopidil, a selective alpha-1 adrenoceptor antagonist, inhibits growth of human prostate cancer cells by G1 cell cycle arrest. Int. J. Cancer 2008, 122, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Mikami, K.; Nagaya, H.; Gotoh, A.; Kanno, T.; Tsuchiya, A.; Nakano, T.; Nishizaki, T. Naftopidil is useful for the treatment of malignant pleural mesothelioma. Pharmacology 2014, 94, 163–169. [Google Scholar] [CrossRef]
- Masachika, E.; Kanno, T.; Nakano, T.; Gotoh, A.; Nishizaki, T. Naftopidil induces apoptosis in malignant mesothelioma cell lines independently of alpha1-adrenoceptor blocking. Anticancer Res. 2013, 33, 887–894. [Google Scholar] [PubMed]
- Yamada, D.; Nishimatsu, H.; Kumano, S.; Hirano, Y.; Suzuki, M.; Fujimura, T.; Fukuhara, H.; Enomoto, Y.; Kume, H.; Homma, Y. Reduction of prostate cancer incidence by naftopidil, an alpha1 -adrenoceptor antagonist and transforming growth factor-beta signaling inhibitor. Int. J. Urol. 2013, 20, 1220–1227. [Google Scholar] [CrossRef]
- Nakamura, A.; Matsunaga, W.; Gotoh, A. Autophagy induced by Naftopidil inhibits apoptosis of human gastric cancer cells. Anticancer Res. 2018, 38, 803–809. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Ishii, K.; Kanda, H.; Kato, M.; Miki, M.; Kajiwara, S.; Arima, K.; Shiraishi, T.; Sugimura, Y. Combination treatment with naftopidil increases the efficacy of radiotherapy in PC-3 human prostate cancer cells. J. Cancer Res. Clin. Oncol. 2017, 143, 933–939. [Google Scholar] [CrossRef]
- Ishii, K.; Matsuoka, I.; Kajiwara, S.; Sasaki, T.; Miki, M.; Kato, M.; Kanda, H.; Arima, K.; Shiraishi, T.; Sugimura, Y. Additive naftopidil treatment synergizes docetaxel-induced apoptosis in human prostate cancer cells. J. Cancer Res. Clin. Oncol. 2018, 144, 89–98. [Google Scholar] [CrossRef]
- Turanli, B.; Grøtli, M.; Boren, J.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Drug repositioning for effective prostate cancer treatment. Front. Physiol. 2018, 9, 500. [Google Scholar] [CrossRef] [Green Version]
- Besen, J.; Gan, S.D. A critical evaluation of clinical research study designs. J. Investig. Dermatol. 2014, 134, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Batty, M.; Pugh, R.; Rathinam, I.; Simmonds, J.; Walker, E.; Forbes, A.; Anoopkumar-Dukie, S.; McDermott, C.M.; Spencer, B.; Christie, D.; et al. The role of alpha1-adrenoceptor antagonists in the treatment of prostate and other cancers. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Kyprianou, N.; Benning, C.M. Suppression of human prostate cancer cell growth by alpha1-adrenoceptor antagonists doxazosin and terazosin via induction of apoptosis. Cancer Res. 2000, 60, 4550–4555. [Google Scholar] [PubMed]
- Benning, C.M.; Kyprianou, N. Quinazoline-derived α1-adrenoceptor antagonists induce prostate cancer cell apoptosis via an α1-Adrenoceptor-independent action. Cancer Res. 2002, 60, 4550–4555. [Google Scholar] [PubMed]
- Harris, A.M.; Warner, B.W.; Wilson, J.M.; Becker, A.; Rowland, R.G.; Conner, W.; Lane, M.; Kimbler, K.; Durbin, E.B.; Baron, A.T.; et al. Effect of alpha1-adrenoceptor antagonist exposure on prostate cancer incidence: An observational cohort study. J. Urol. 2007, 178, 2176–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Wang, C.L.; Sun, T.; Zhou, Z.; Niu, J.X.; Tian, X.M.; Yuan, M. Synthesis, biological evaluation and SAR of naftopidil-based arylpiperazine derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cao, Y.; Zhang, J.; Yu, S.; Zou, Y.; Chai, X.; Wu, Q.; Zhang, D.; Jiang, Y.; Sun, Q. Design, synthesis and antifungal activities of novel 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2011, 46, 3142–3148. [Google Scholar] [CrossRef] [PubMed]
- Bassetto, M.; Leyssen, P.; Neyts, J.; Yerukhimovich, M.M.; Frick, D.N.; Courtney-Smith, M.; Brancale, A. In silico identification, design and synthesis of novel piperazine-based antiviral agents targeting the hepatitis C virus helicase. Eur. J. Med. Chem. 2017, 125, 1115–1131. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-X.; Li, Z.-G.; Xie, H.-Y.; Gao, J.-R.; Zou, J.-W. Quantitative structure–activity relationship analysis of aryl alkanol piperazine derivatives with antidepressant activities. Eur. J. Med. Chem. 2009, 44, 4367–4375. [Google Scholar] [CrossRef] [PubMed]
- Fytas, C.; Zoidis, G.; Tsotinis, A.; Fytas, G.; Khan, M.A.; Akhtar, S.; Rahman, K.M.; Thurston, D.E. Novel 1-(2-aryl-2-adamantyl)piperazine derivatives with antiproliferative activity. Eur. J. Med. Chem. 2015, 93, 281–290. [Google Scholar] [CrossRef]
- Trivedi, P.; Adhikari, N.; Amin, S.A.; Bobde, Y.; Ganesh, R.; Jha, T.; Ghosh, B. Design, synthesis, biological evaluation and molecular docking study of arylcarboxamido piperidine and piperazine-based hydroxamates as potential HDAC8 inhibitors with promising anticancer activity. Eur. J. Pharm. Sci. 2019, 138, 105046. [Google Scholar] [CrossRef]
- Chen, H.; Xu, B.-B.; Sun, T.; Zhou, Z.; Ya, H.-Y.; Yuan, M. Synthesis and antitumor activity of novel arylpiperazine derivatives containing the saccharin moiety. Molecules 2017, 22, 1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; He, F.; Huang, M.; Liu, X.; Xiong, Y.; Huang, Y.; Zhu, L.; Yang, Y.; Xu, X.; Yuan, M. Novel naftopidil-related derivatives and their biological effects as alpha1-adrenoceptors antagonists and antiproliferative agents. Eur. J. Med. Chem. 2015, 96, 83–91. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Ho, C.-T.; Chang, Y.-J.; Yang, L.-X.; Wei, P.-L.; Liu, T.-Z.; Liu, J.-J. A novel microtubule-disrupting agent induces endoplasmic reticular stress-mediated cell death in human hepatocellular carcinoma cells. PLoS ONE 2015, 10, e0136340. [Google Scholar] [CrossRef] [Green Version]
- Chopra, A.; Anderson, A.; Giardina, C. Novel piperazine-based compounds inhibit microtubule dynamics and sensitize colon cancer cells to tumor necrosis factor-induced apoptosis. J. Biol. Chem. 2014, 289, 2978–2991. [Google Scholar] [CrossRef] [Green Version]
- Gayvert, K.M.; Dardenne, E.; Cheung, C.; Boland, M.R.; Lorberbaum, T.; Wanjala, J.; Chen, Y.; Rubin, M.A.; Tatonetti, N.P.; Rickman, D.S.; et al. A Computational drug repositioning approach for targeting oncogenic transcription factors. Cell Rep. 2016, 15, 2348–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.; van den Heuvel, A.P.J.; Prabhu, V.V.; Zhang, S.; El-Deiry, W.S. Targeting tumor suppressor p53 for cancer therapy: Strategies, challenges and opportunities. Curr. Drug Targets 2014, 15, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Yamaguchi, K.; Yamamoto, M.; Kurioka, R.; Kino, Y.; Matsunaga, W.; Nakao, S.; Fukuhara, H.; Tanaka, A.; Gotoh, A.; et al. Identification of HUHS190, a human naftopidil metabolite, as a novel anti-bladder cancer drug. Bioorg. Med. Chem. Lett. 2020, 30, 126744. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-J.; Zhang, Z.-H.; He, F.; Liu, X.-W.; Xu, X.-J.; Dai, L.-J.; Liu, Q.-M.; Yuan, M. Novel naftopidil derivatives containing methyl phenylacetate and their blocking effects on α1D/1A-adrenoreceptor subtypes. Bioorg. Med. Chem. Lett. 2018, 28, 547–551. [Google Scholar] [CrossRef]
- Chen, H.; Qian, Y.; Jia, H.; Yu, Y.; Zhang, H.; Shen, J.; Zhao, S. Synthesis and pharmacological evaluation of naftopidil-based arylpiperazine derivatives containing the bromophenol moiety. Pharm. Rep. 2020, PR, 1–11. [Google Scholar] [CrossRef]
- Kaku, Y.; Nagaya, H.; Tsuchiya, A.; Kanno, T.; Gotoh, A.; Tanaka, A.; Shimizu, T.; Nakao, S.; Tabata, C.; Nakano, T.; et al. Newly synthesized anticancer drug HUHS1015 is effective on malignant pleural mesothelioma. Cancer Sci. 2014, 105, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, T.; Kanno, T.; Tsuchiya, A.; Kaku, Y.; Shimizu, T.; Tanaka, A. 1-[2-(2-methoxyphenylamino) ethylamino]-3-(naphthalene-1- yloxy) propan-2-ol may be a promising anticancer drug. Molecules 2014, 19, 21462–21472. [Google Scholar] [CrossRef] [Green Version]
- Kanno, T.; Tanaka, A.; Shimizu, T.; Nakano, T.; Nishizaki, T. 1-[2-(2-methoxyphenylamino) ethylamino]-3-(naphthalene-1-yloxy) propan-2-ol as a potential anticancer drug. Pharmacology 2013, 91, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Tsuchiya, A.; Kanno, T.; Nakao, S.; Shimizu, T.; Tanaka, A.; Nishizaki, T. The newly synthesized anticancer drug HUHS1015 is useful for treatment of human gastric cancer. Cancer Chemother Pharm. 2015, 75, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Kaku, Y.; Tsuchiya, A.; Kanno, T.; Nishizaki, T. HUHS1015 induces necroptosis and caspase-independent apoptosis of MKN28 human gastric cancer cells in association with AMID accumulation in the nucleus. Anticancer Agents Med. Chem. 2015, 15, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Tsuchiya, A.; Shimizu, T.; Tanaka, A.; Nishizaki, T. HUHS1015 suppresses colonic cancer growth by inducing necrosis and apoptosis in association with mitochondrial damage. Anticancer Res. 2016, 36, 39–48. [Google Scholar] [PubMed]
- Quaglia, W.; Santoni, G.; Pigini, M.; Piergentili, A.; Gentili, F.; Buccioni, M.; Mosca, M.; Lucciarini, R.; Amantini, C.; Nabissi, M.I.; et al. Structure−activity relationships in 1,4-benzodioxan-related compounds. 8.{2-[2-(4-chlorobenzyloxy) phenoxy] ethyl}-[2-(2,6-dimethoxyphenoxy) ethyl] amine (clopenphendioxan) as a tool to highlight the involvement of α 1D- and α 1B- adrenoreceptor subtypes in the regulation of human PC-3 prostate cancer cell apoptosis and proliferation. J. Med. Chem. 2005, 48, 7750–7763. [Google Scholar] [CrossRef]
- Colciago, A.; Mornati, O.; Ferri, N.; Castelnovo, L.F.; Fumagalli, L.; Bolchi, C.; Pallavicini, M.; Valoti, E.; Negri-Cesi, P. A selective alpha1D-adrenoreceptor antagonist inhibits human prostate cancer cell proliferation and motility “in vitro”. Pharm. Res. 2016, 103, 215–226. [Google Scholar] [CrossRef]
- Thebault, S.; Roudbaraki, M.; Sydorenko, V.; Shuba, Y.; Lemonnier, L.; Slomianny, C.; Dewailly, E.; Bonnal, J.-L.; Mauroy, B.; Skryma, R.; et al. Alpha1-adrenergic receptors activate Ca(2+)-permeable cationic channels in prostate cancer epithelial cells. J. Clin. Investig. 2003, 111, 1691–1701. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.B.; Amantini, C.; Nabissi, M.; Liberati, S.; Cardinali, C.; Farfariello, V.; Tomassoni, D.; Quaglia, W.; Piergentili, A.; Bonifazi, A.; et al. Cross-talk between alpha1D-adrenoceptors and transient receptor potential vanilloid type 1 triggers prostate cancer cell proliferation. BMC Cancer 2014, 14, 921. [Google Scholar] [CrossRef] [Green Version]
- Castiglione, F.; Benigni, F.; Briganti, A.; Salonia, A.; Villa, L.; Nini, A.; Di Trapani, E.; Capitanio, U.; Hedlund, P.; Montorsi, F. Naftopidil for the treatment of benign prostate hyperplasia: A systematic review. Curr. Med. Res. Opin. 2014, 30, 719–732. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cancer Models | |||||
|---|---|---|---|---|---|
| Organs | Study | Models | Drug Effects | Observations | Authors |
| Bladder | in vitro | 253J, 5637, KK-47, T24 and UM-UC-3 | Cell viability reduction | - | Gotoh et al., 2012 [56] |
| in vitro | KK-47, 5637, T-24 | Cell viability reduction | - | Nakagawa et al., 2016 [57] | |
| in vivo | KK-47 | Tumour volume reduction | - | Nakagawa et al., 2016 [57] | |
| Cervival | in vitro | HeLa | Cell viability reduction by induction of apoptosis | Naftopidil supresses phosphorylation of Smad-2 induced by TGF-β | Yamada et al., 2013 [58] |
| Colon | in vitro | HT29 | Cell viability reduction | - | Ishii et al., 2015 [55] |
| Gastric | in vitro | HGC27 | Cell viability reduction by induction of apoptosis | Naftopidil reduces Akt phosphorylation | Nakamura et al., 2018 [59] |
| Mesothelioma | in vitro | NCI-H28, NCI-H2052, NCI-H2452, MSTO-211H | Cell viability reduction by induction of apoptosis | - | Masachika et al., 2013 [60] |
| NCI-H2052 | Cell viability reduction by induction of apoptosis | Naftopidil increases TNF-α mRNA expression and Fas-L secretion | Mikami et al., 2014 [61] | ||
| in vivo | NCI-H2052 | Tumour volume reduction | - | Mikami et al., 2014 [61] | |
| Ovarian | in vitro | IGROV1-R10 and SKOV3 | Anti-proliferative effect alone and apoptosis induction in combination with ABT-737 or Trametinib | Naftopidil increases Bim, Puma and Noxa protien expression | Florent et al., 2020 [62] |
| Prostate | in vitro | LNCaP, E9 and PrSC | Anti-proliferative effect (blockade in G0/G1 phase of the cell cycle) | Naftopidil increases p21 and p27 protein expression and reduces IL-6 secretion | Hori et al., 2011 [53] |
| LNCaP and PC-3 | Anti-proliferative effect (blockade in G0/G1 phase of the cell cycle) | Naftopidil increases p21 and p27 protein expression and reduces Akt phosphorylation | Kanda et al., 2008 [52] | ||
| DU145, LNCaP and PC-3 | Cell viability reduction | - | Gotoh et al., 2012 [56] | ||
| LNCaP | Cell viability reduction by induction of apoptosis | - | Yamada et al., 2013 [58] | ||
| LNCaP and PC-3 | Anti-proliferative effect alone and apoptosis induction in combination with Docetaxel | - | Ishii, 2018 [63] | ||
| PC-3 | Cell viability reduction enhanced with radiotherapy | Naftopidil reduces Akt phosphorylation and suppresses radiotherapy-induced MnSOD | Iwamoto et al., 2017 [64] | ||
| in vivo | E9 and PrSC | Tumour weight reduction | Naftopidil reduces Ki-67 staining and MVD | Hori et al., 2011 [53] | |
| PC-3 | Tumour volume reduction | Naftopidil reduces Ki-67 staining and MVD and increases p21 staining | Kanda et al., 2007 [52] | ||
| LNCaP and PrSC | Tumour volume reduction reduction enhanced with Docetaxel | Naftopidil reduces Ki-67 staining and tumour-bone interface and increases cleaved-caspase 3 staining | Ishii et al., 2018 [63] | ||
| PC-3 | Tumour volume reduction reduction enhanced with radiotherapy | Naftopidil reduces Ki-67 staining | Iwamoto et al., 2017 [64] | ||
| Prospective study | Naftopidil exposure, for at least 3 months, is associated with a lower incidence of prostate cancer than tamsulosin | Naftopidil treatment reduces Bcl-2 and increased p21 expression in prostate cancer cells from men | Yamada et al., 2013 [58] | ||
| Renal | in vitro | ACHN and Caki-2 | anti-proliferative effect (blockade in G0/G1 phase of the cell cycle) | Naftopidil increases p21 and reduces Cdk2 protein expression | Iwamoto et al., 2013 [54] |
| 786-O, ACHN and RCC4-VHL | cell viability reduction | - | Gotoh et al., 2012 [56] | ||
| in vivo | ACHN | Tumour weight reduction | Naftopidil increases p21 staining and reduces Ki-67 and Cdk2 staining and MVD | Iwamoto et al., 2013 [54] | |
| Patient RCC | Tumour dimensions | Naftopidil reduces MVD | Iwamoto et al., 2013 [54] | ||
| Non Cancer Models | |||||
| Endothelial | in vivo | HUVEC | Anti-proliferative effect (blockade in G0/G1 phase of the cell cycle) | Naftopidil increases p21 protein expression | Iwamoto et al., 2013 [54] |
| Cancers | Cell Lines | Drug Effects | Observations | Authors | |||
|---|---|---|---|---|---|---|---|
| Naftopidil Analogues | HUHS1015 | Bladder | in vitro | 253J, 5637, KK-47, TCCSUP, T24 and UM-UC-3 | Cell viability reduction by apoptosis induction | - | Kanno et al., 2013 [89] |
| Colorectal | in vitro | Caco-2 and CW2 | Cell viability reduction by inducing apoptosis and necrosis | HUHS1015 induces mitochondrial damage and increases Bad, Bax and Puma mRNA expression | Kaku et al., 2016 [92] | ||
| in vivo | CW2 | Tumour volume reduction and survival rate increase | - | Kaku et al., 2016 [92] | |||
| Gastric | in vitro | MKN28 and MKN45 | Cell viability reduction by induction of apoptosis and necrosis | HUHS1015 increases TNF-α mRNA and protein expression | Kaku et al., 2015 [90] | ||
| MKN28 and MKN45 | Cell viability reduction by apoptosis induction | - | Kanno et al., 2013 [89] | ||||
| in vivo | MKN45 | Tumour volume reduction and survival rate increase | - | Kaku et al., 2015 [90] | |||
| Liver | in vitro | HepG2 and HuH-7 | Cell viability reduction by apoptosis induction | - | Kanno et al., 2013 [89] | ||
| Lung | in vitro | A549, SBC-3 and Lu-65 | Cell viability reduction by apoptosis induction | - | Kanno et al., 2013 [89] | ||
| Mesothelioma | in vitro | MSTO-221H, NCHI-H28, NCI-H2052 and NCI-H2452 | Cell viability reduction by anti-proliferative effect (blockade in G0/G1 phase of the cell cycle) and induction of apoptosis and necrosis | HUHS1015 increases Puma, Noxa, Bad and HRK mRNA expression | Kaku et al., 2014 [88] | ||
| in vivo | NCI-H2052 | tumor volume reduction | - | Kaku et al., 2014 [88] | |||
| Prostate | in vitro | DU145, LNCaP and PC-3 | Cell viability reduction by apoptosis induction | - | Kanno et al., 2013 [89] | ||
| Renal | in vitro | ACHN, RCC4-VHL and 786-O | Cell viability reduction by apoptosis induction | - | Kanno et al., 2013 [89] | ||
| Compound 12 | Prostate | in vitro | PC-3, DU145, and LNCaP | Cell viability reduction by anti-proliferative effect (blockade in G0/G1 phase of the cell cycle) and induction of apoptosis | Compound 12 increases Bcl-2 and Bax mRNA expression | Huang et al., 2015 [77] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florent, R.; Poulain, L.; N'Diaye, M. Drug Repositioning of the α1-Adrenergic Receptor Antagonist Naftopidil: A Potential New Anti-Cancer Drug? Int. J. Mol. Sci. 2020, 21, 5339. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155339
Florent R, Poulain L, N'Diaye M. Drug Repositioning of the α1-Adrenergic Receptor Antagonist Naftopidil: A Potential New Anti-Cancer Drug? International Journal of Molecular Sciences. 2020; 21(15):5339. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155339
Chicago/Turabian StyleFlorent, Romane, Laurent Poulain, and Monique N'Diaye. 2020. "Drug Repositioning of the α1-Adrenergic Receptor Antagonist Naftopidil: A Potential New Anti-Cancer Drug?" International Journal of Molecular Sciences 21, no. 15: 5339. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155339