Synthesis and Pharmacological Characterization of 2-Aminoethyl Diphenylborinate (2-APB) Derivatives for Inhibition of Store-Operated Calcium Entry (SOCE) in MDA-MB-231 Breast Cancer Cells

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
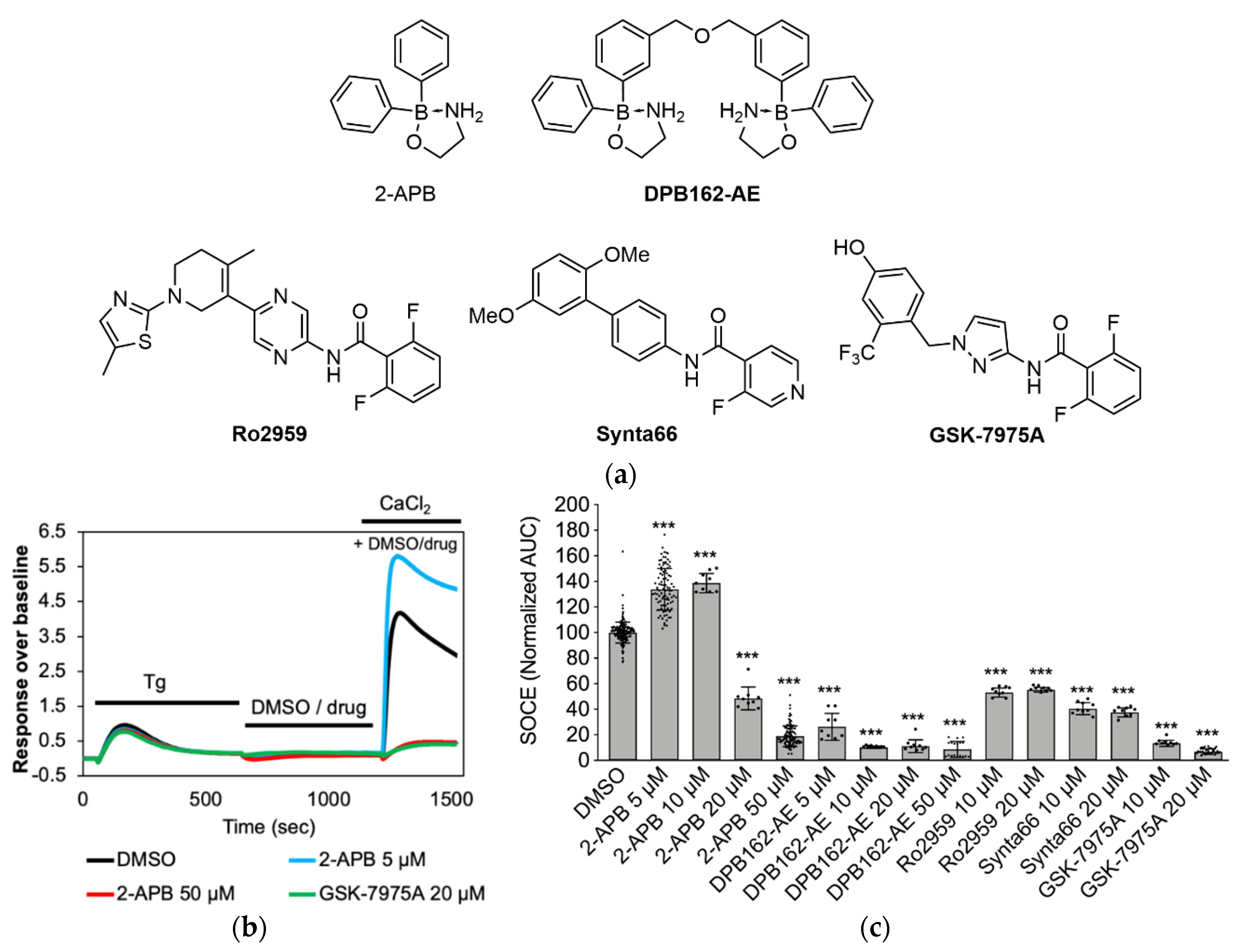
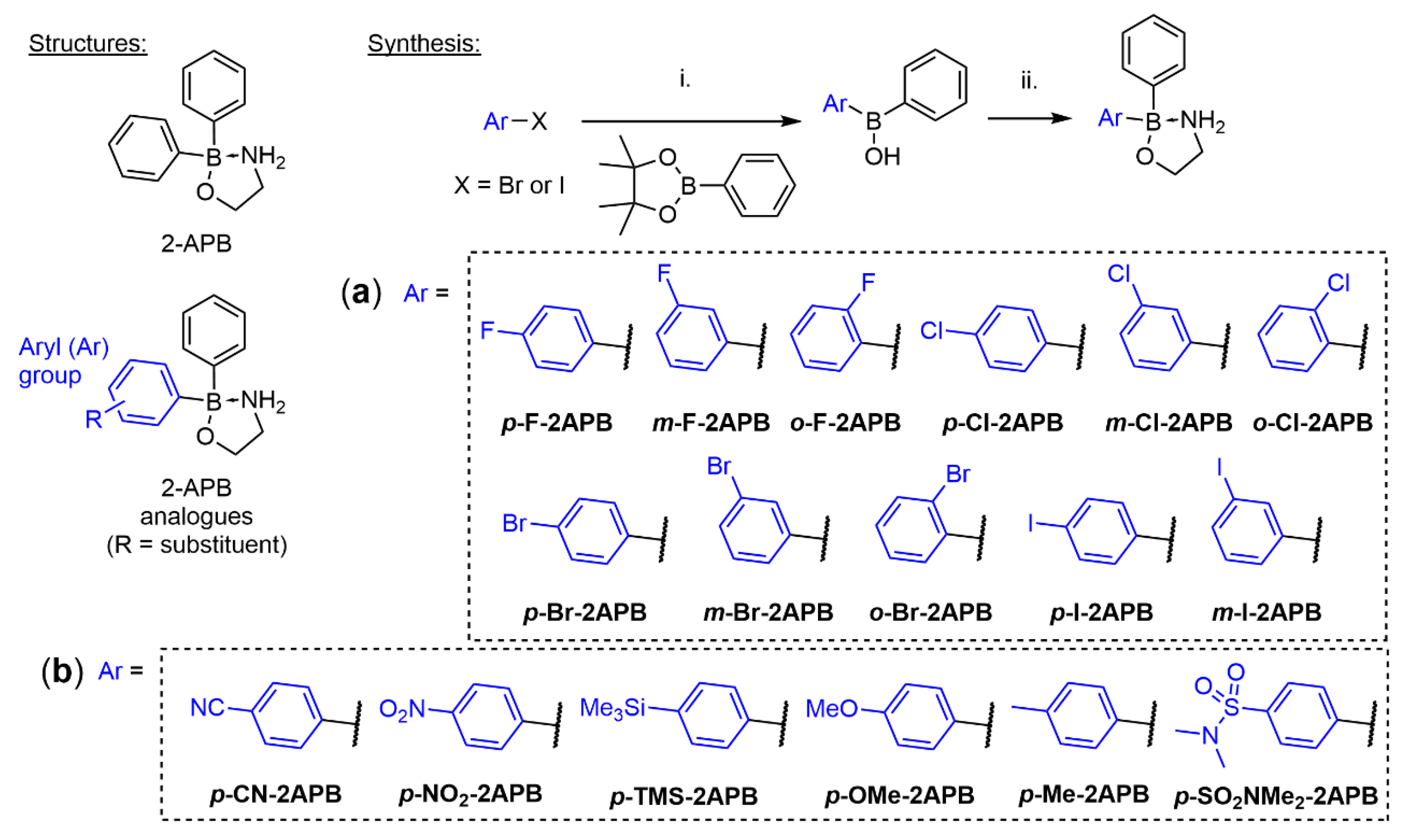
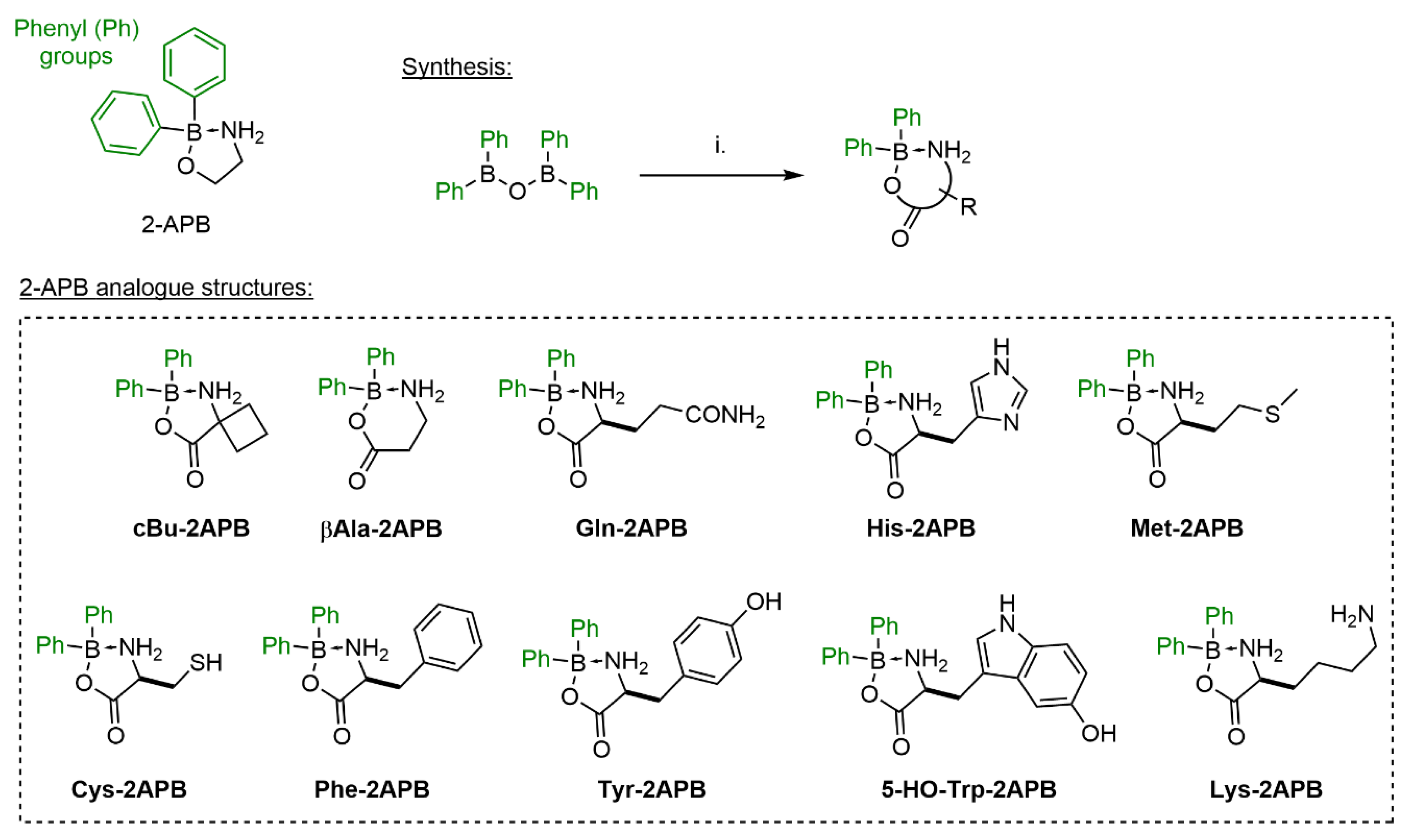
2.1. Synthesis of 2-APB Analogues
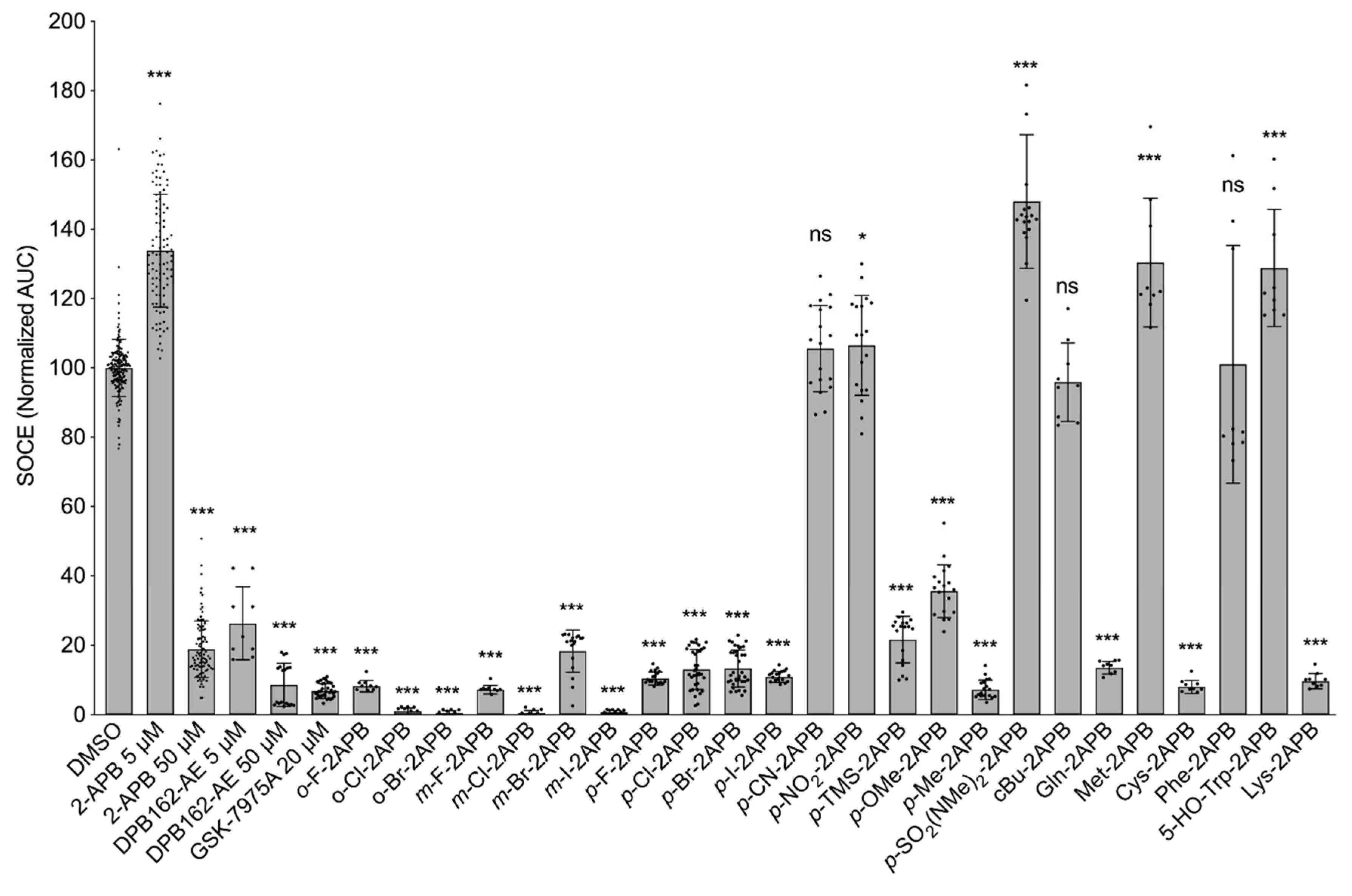
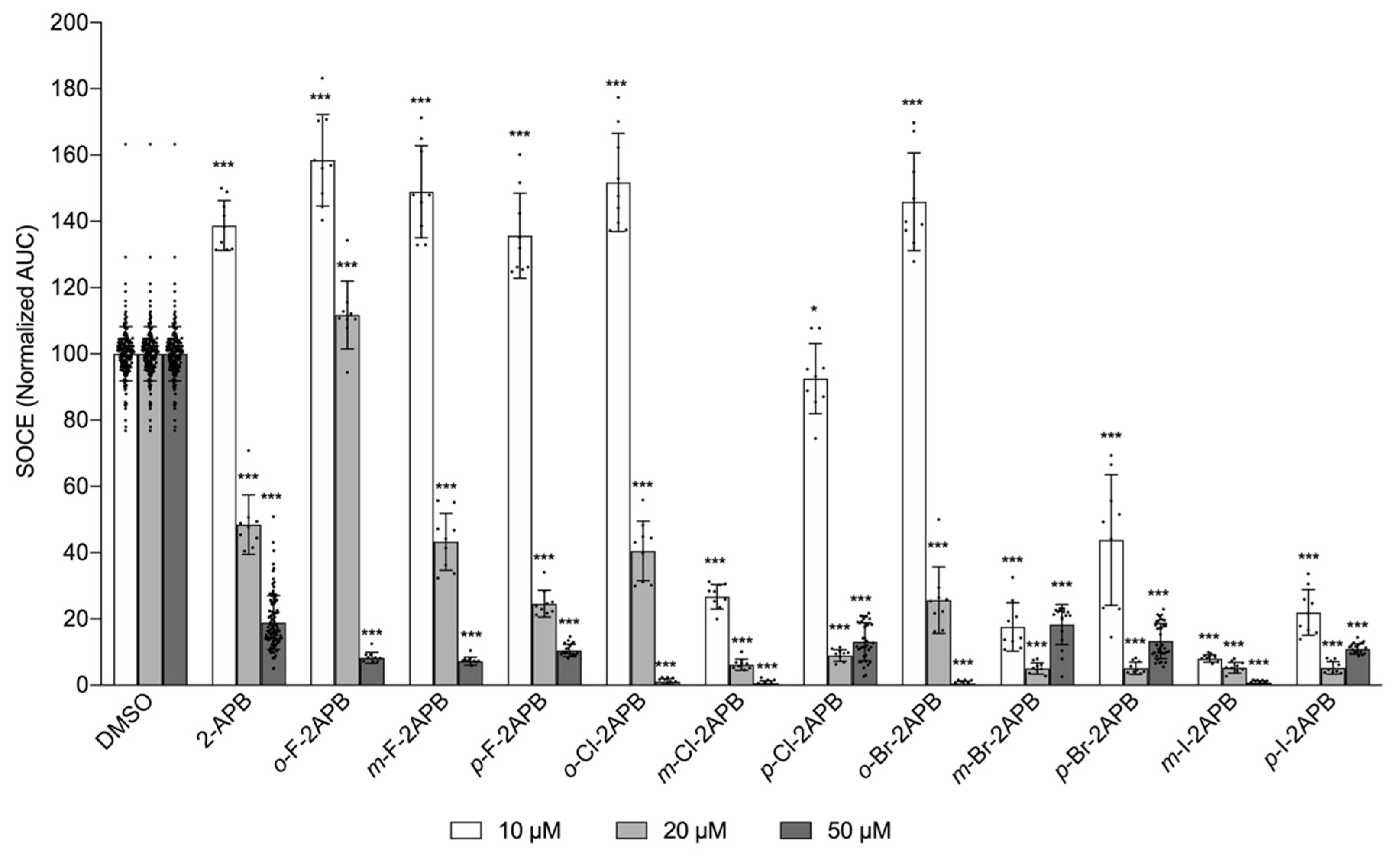
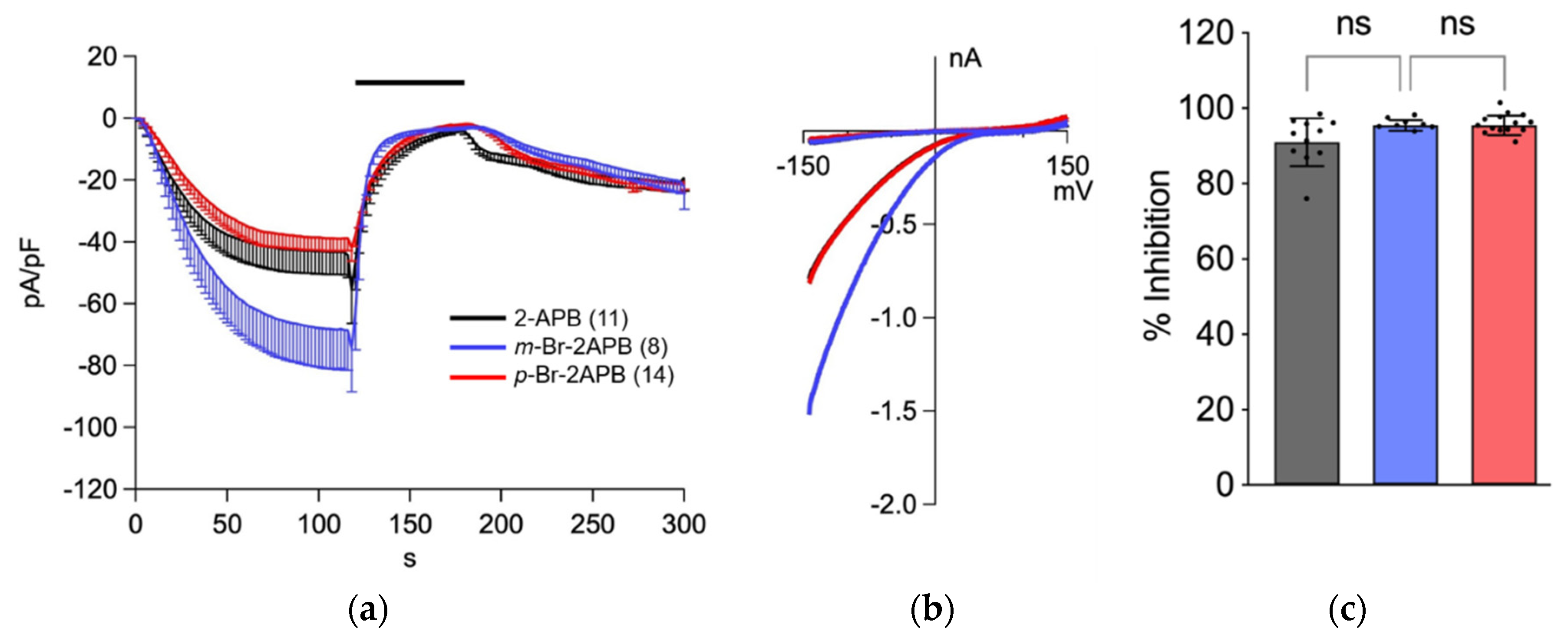
2.2. Influence of 2-APB Analogues on Store-Operated Ca2+ Entry in MDA-MB-231 Breast Cancer Cells
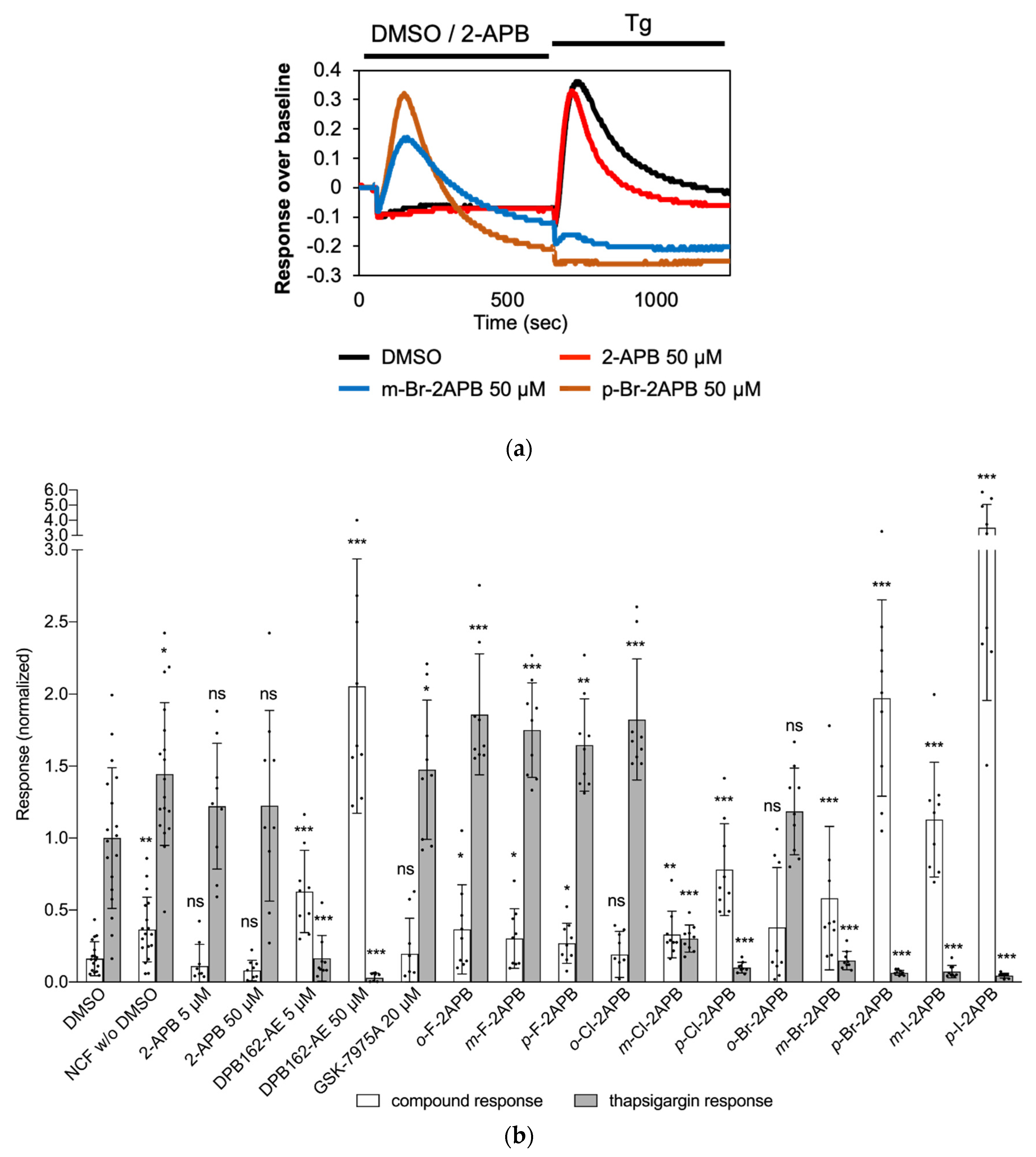
2.3. Store Depletion in MDA-MB-231 Breast Cancer Cells by 2-APB Analogues
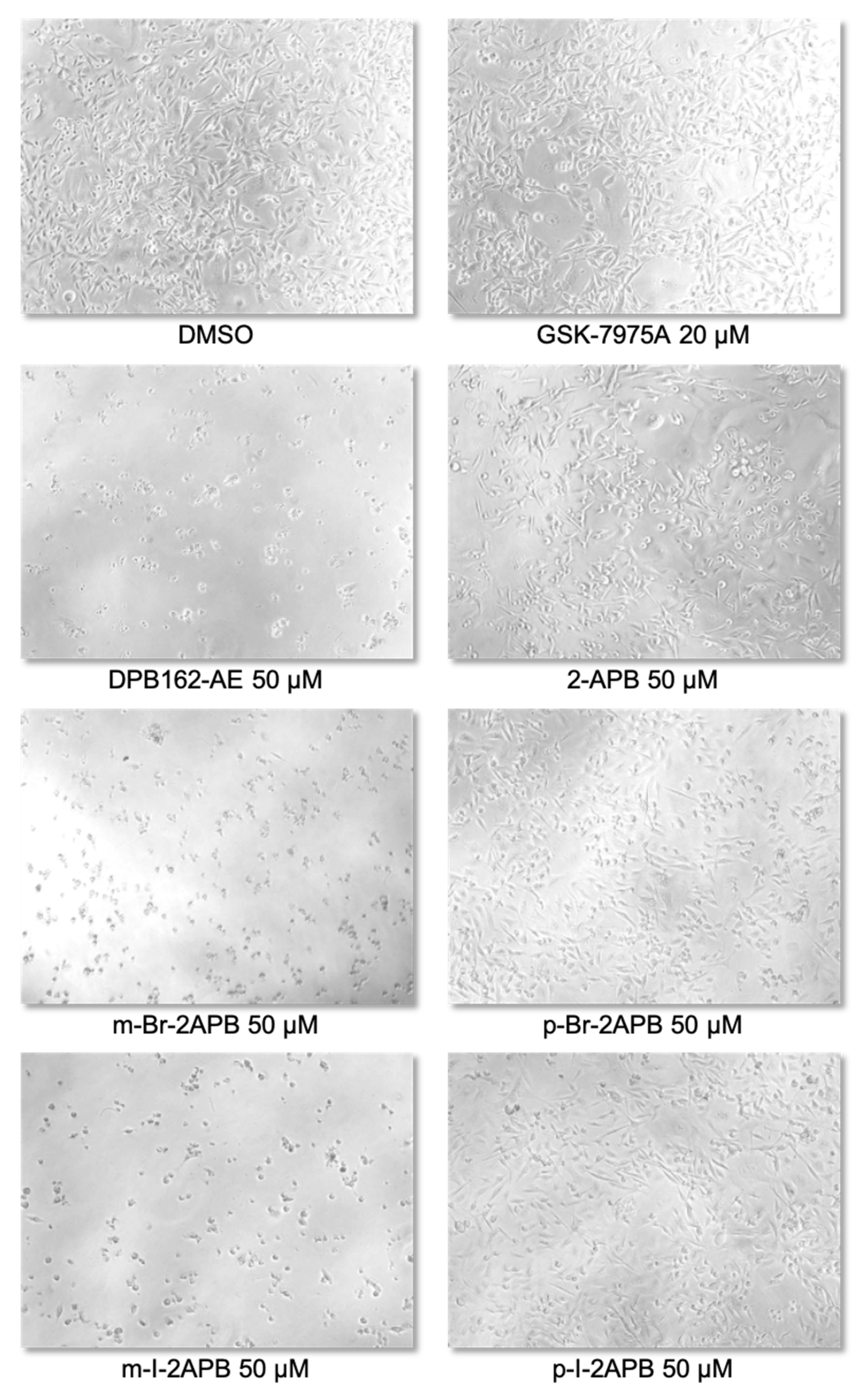
2.4. Synthetic 2-APB Analogues Effect MDA-MB-231 Cell Viability
3. Materials and Methods
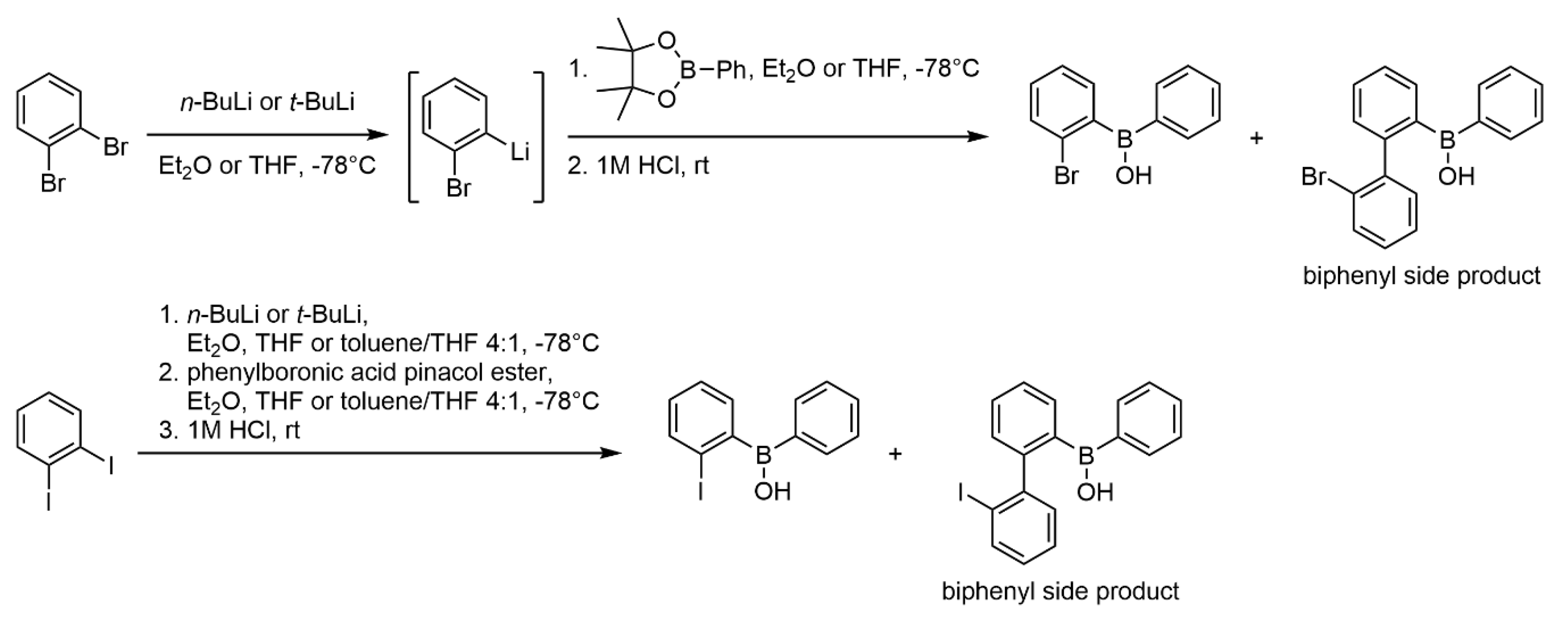
3.1. Chemical Synthesis
3.1.1. General Procedure A for the Synthesis of Mono-substituted Phenyl 2-APB Analogues
3.1.2. General Procedure B for the Synthesis of 2-APB Amino Acid Analogues
3.1.3. 2-(4-Fluorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-F-2APB)
3.1.4. 2-(3-Fluorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (m-F-2APB)
3.1.5. 2-(2-Fluorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (o-F-2APB)
3.1.6. 2-(4-Chlorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-Cl-2APB)
3.1.7. 2-(3-Chlorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (m-Cl-2APB)
3.1.8. 2-(2-Chlorophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (o-Cl-2APB)
3.1.9. 2-(4-Bromophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-Br-2APB)
3.1.10. 2-(3-Bromophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (m-Br-2APB)
3.1.11. 2-(2-Bromophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (o-Br-2APB)
3.1.12. 2-(3-Iodophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (m-I-2APB)
3.1.13. 4-(2-Phenyl-1,3,2λ4-oxazaborolidin-2-yl)benzonitrile (p-CN-2APB)
3.1.14. 2-(4-Nitrophenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-NO2-2APB)
3.1.15. 2-Phenyl-2-(4-(trimethylsilyl)phenyl)-1,3,2λ4-oxazaborolidine (p-TMS-2APB)
3.1.16. 2-(4-Methoxyphenyl)-2-phenyl-1,3,2λ4-oxazaborolidine (p-OMe-2APB)
3.1.17. N,N-Dimethyl-4-(2-phenyl-1,3,2λ4-oxazaborolidin-2-yl)benzenesulfonamide (p-SO2NMe2-2APB)
3.1.18. 6,6-Diphenyl-7-oxa-5-aza-6λ4-boraspiro[3.4]octan-8-one (cBu-2APB)
3.1.19. 2,2-Diphenyl-1,3,2λ4-oxazaborinan-6-one (βAla-2APB)
3.1.20. (S)-3-(5-Oxo-2,2-diphenyl-1,3,2λ4-oxazaborolidin-4-yl)propanamide (Gln-2APB)
3.1.21. (S)-4-((1H-Imidazol-5-yl)methyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (His-2APB)
3.1.22. (S)-4-(2-(Methylthio)ethyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Met-2APB)
3.1.23. (R)-4-(Mercaptomethyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Cys-2APB)
3.1.24. (S)-4-Benzyl-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Phe-2APB)
3.1.25. (S)-4-(4-Hydroxybenzyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Tyr-2APB)
3.1.26. (S)-4-((5-Hydroxy-1H-indol-3-yl)methyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (5-HO-Trp-2APB)
3.1.27. (S)-4-(4-Aminobutyl)-2,2-diphenyl-1,3,2λ4-oxazaborolidin-5-one (Lys-2APB)
3.2. Cell Culture
3.3. Fluorescent Ca2+ Influx Assay Using FLIPR® Tetra
3.3.1. General
3.3.2. SOCE Modulation Assay
3.3.3. ER Ca2+ Mobilization Assay (Compound Followed by Thapsigargin)
3.4. Electrophysiology
3.5. CRISPR/Cas9-Mediated Generation and Validation of STIM1 and Orai1 Knockout MDA-MB-231 Cells
3.6. Cell Viability Assay
3.7. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-APB | 2-Aminoethyl diphenylborinate |
| AUC | Area under the curve |
| CD | Current density |
| CRAC | Ca2+ release-activated Ca2+ current |
| ER | Endoplasmic reticulum |
| FLIPR | Fluorometric imaging plate reader |
| IP3 | Inositol 1,4,5-triphosphate |
| NCF | Nominal calcium-free (buffer) |
| PI(4,5)P2 | Phosphatidylinositol 4,5-bisphosphate |
| PM | Plasma membrane |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| SOCE | Store-operated calcium entry |
| STIM | Stromal interaction molecule |
| Tg | Thapsigargin |
Appendix A

References
- Prakriya, M.; Lewis, R.S. Store-operated calcium channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [Green Version]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef]
- Zheng, S.; Zhou, L.; Ma, G.; Zhang, T.; Liu, J.; Li, J.; Nguyen, N.T.; Zhang, X.; Li, W.; Nwokonko, R.; et al. Calcium store refilling and STIM activation in STIM- and Orai-deficient cell lines. Pflugers Arch. Eur. J. Physiol. 2018, 470, 1555–1567. [Google Scholar] [CrossRef] [Green Version]
- Thastrup, O.; Cullen, P.J.; Drobak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef] [Green Version]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef] [Green Version]
- Feske, S. CRAC channels and disease—From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116. [Google Scholar] [CrossRef]
- Kappel, S.; Borgstrom, A.; Stoklosa, P.; Dorr, K.; Peinelt, C. Store-operated calcium entry in disease: Beyond STIM/Orai expression levels. Semin. Cell Dev. Biol. 2019, 94, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Han, W.; Cao, W.M.; Wang, Y.; Wen, S.; Huang, Y.; Li, M.; Du, L.; Zhou, Y. Store-operated calcium entry mediated by ORAI and STIM. Compr. Physiol. 2018, 8, 981–1002. [Google Scholar] [CrossRef] [PubMed]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordonez, A.; Rosado, J.A.; Smani, T. The complex role of store operated calcium entry pathways and related proteins in the function of cardiac, skeletal and vascular smooth muscle cells. Front. Physiol. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Yoo, S.-A.; Kim, M.; Kim, W.-U. The role of calcium-calcineurin-NFAT signaling pathway in health and autoimmune diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Secondo, A.; Bagetta, G.; Amantea, D. On the role of store-operated calcium entry in acute and chronic neurodegenerative diseases. Front. Mol. Neurosci. 2018, 11, 87. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Lin, P.C.; Yeh, Y.M.; Chen, L.H.; Shen, M.R. Store-operated Ca2+ entry in tumor progression: From molecular mechanisms to clinical implications. Cancers 2019, 11, 899. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [Green Version]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Azimi, I.; Milevskiy, M.J.G.; Chalmers, S.B.; Yapa, K.; Robitaille, M.; Henry, C.; Baillie, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Orai1 and Orai3 in breast cancer molecular subtypes and the identification of Orai3 as a hypoxia sensitive gene and a regulator of hypoxia responses. Cancers 2019, 11, 208. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Store-operated Ca2+ entry in breast cancer cells: Remodeling and functional role. Int. J. Mol. Sci. 2018, 19, 4053. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting STIM and Orai proteins as an alternative approach in anticancer therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A review in the theme: Cell and molecular processes in cancer metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C457–C469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, Z.K.; Minatti, A.; Button, D.C.; Patrick, S. Small-molecule inhibitors of store-operated calcium entry. ChemMedChem 2009, 4, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Jairaman, A.; Prakriya, M. Molecular pharmacology of store-operated CRAC channels. Channels 2013, 7, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Du, L.; Zhou, Y.; Li, M. Store-operated CRAC channel inhibitors: Opportunities and challenges. Future Med. Chem. 2016, 8, 817–832. [Google Scholar] [CrossRef] [Green Version]
- Azimi, I.; Flanagan, J.U.; Stevenson, R.J.; Inserra, M.; Vetter, I.; Monteith, G.R.; Denny, W.A. Evaluation of known and novel inhibitors of Orai1-mediated store operated Ca2+ entry in MDA-MB-231 breast cancer cells using a Fluorescence Imaging Plate Reader assay. Bioorg. Med. Chem. 2017, 25, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Riva, B.; Griglio, A.; Serafini, M.; Cordero-Sanchez, C.; Aprile, S.; Di Paola, R.; Gugliandolo, E.; Alansary, D.; Biocotino, I.; Lim, D.; et al. Pyrtriazoles, a novel class of store-operated calcium entry modulators: Discovery, biological profiling, and in vivo proof-of-concept efficacy in acute pancreatitis. J. Med. Chem. 2018, 61, 9756–9783. [Google Scholar] [CrossRef]
- Djillani, A.; Doignon, I.; Luyten, T.; Lamkhioued, B.; Gangloff, S.C.; Parys, J.B.; Nusse, O.; Chomienne, C.; Dellis, O. Potentiation of the store-operated calcium entry (SOCE) induces phytohemagglutinin-activated Jurkat T cell apoptosis. Cell Calcium 2015, 58, 171–185. [Google Scholar] [CrossRef]
- Doignon, I.; Fayol, O.; Dellis, O. Improvement of the rituximab-induced cell death by potentiation of the store-operated calcium entry in mantle cell lymphoma cell lines. Oncotarget 2019, 10, 4466–4478. [Google Scholar] [CrossRef]
- Azimi, I.; Stevenson, R.J.; Zhang, X.; Meizoso-Huesca, A.; Xin, P.; Johnson, M.; Flanagan, J.U.; Chalmers, S.B.; Yoast, R.E.; Kapure, J.S.; et al. A new selective pharmacological enhancer of the Orai1 Ca2+ channel reveals roles for Orai1 in smooth and skeletal muscle functions. ACS Pharmacol. Transl. Sci. 2020, 3, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.T.; Guan, B.C.; Yang, Y.Q.; Nuttall, A.L.; Jiang, Z.G. 2-Aminoethoxydiphenyl borate blocks electrical coupling and inhibits voltage-gated K+ channels in guinea pig arteriole cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H335–H346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemonnier, L.; Prevarskaya, N.; Mazurier, J.; Shuba, Y.; Skryma, R. 2-APB inhibits volume-regulated anion channels independently from intracellular calcium signaling modulation. FEBS Lett. 2004, 556, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Bai, D.; del Corsso, C.; Srinivas, M.; Spray, D.C. Block of specific gap junction channel subtypes by 2-aminoethoxydiphenyl borate (2-APB). J. Pharmacol. Exp. Ther. 2006, 319, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilmen, J.G.; Wootton, L.L.; Godfrey, R.E.; Smart, O.S.; Michelangeli, F. Inhibition of SERCA Ca2+ pumps by 2-aminoethoxydiphenyl borate (2-APB). 2-APB reduces both Ca2+ binding and phosphoryl transfer from ATP, by interfering with the pathway leading to the Ca2+ binding sites. Eur. J. Biochem. 2002, 269, 3678–3687. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Starkov, A.A.; Fiskum, G. Cyclosporin a-insensitive permeability transition in brain mitochondria: Inhibition by 2-aminoethoxydiphenyl borate. J. Biol. Chem. 2003, 278, 27382–27389. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.T.; Venkatachalam, K.; Li, H.S.; Montell, C.; Kurosaki, T.; Patterson, R.L.; Gill, D.L. Assessment of the role of the inositol 1,4,5-trisphosphate receptor in the activation of transient receptor potential channels and store-operated Ca2+ entry channels. J. Biol. Chem. 2001, 276, 18888–18896. [Google Scholar] [CrossRef] [Green Version]
- Schindl, R.; Kahr, H.; Graz, I.; Groschner, K.; Romanin, C. Store depletion-activated CaT1 currents in rat basophilic leukemia mast cells are inhibited by 2-aminoethoxydiphenyl borate. Evidence for a regulatory component that controls activation of both CaT1 and CRAC (Ca2+ release-activated Ca2+ channel) channels. J. Biol. Chem. 2002, 277, 26950–26958. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.Z.; Zeng, F.; Boulay, G.; Grimm, C.; Harteneck, C.; Beech, D.J. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: A differential, extracellular and voltage-dependent effect. Br. J. Pharmacol. 2005, 145, 405–414. [Google Scholar] [CrossRef]
- Li, M.; Jiang, J.; Yue, L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Togashi, K.; Inada, H.; Tominaga, M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br. J. Pharmacol. 2008, 153, 1324–1330. [Google Scholar] [CrossRef]
- Callera, G.E.; He, Y.; Yogi, A.; Montezano, A.C.; Paravicini, T.; Yao, G.; Touyz, R.M. Regulation of the novel Mg2+ transporter transient receptor potential melastatin 7 (TRPM7) cation channel by bradykinin in vascular smooth muscle cells. J. Hypertens. 2009, 27, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Hermosura, M.C.; Monteilh-Zoller, M.K.; Scharenberg, A.M.; Penner, R.; Fleig, A. Dissociation of the store-operated calcium current ICRAC and the Mg-nucleotide-regulated metal ion current MagNuM. J. Physiol. 2002, 539, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Z.; Gu, Q.; Wang, C.; Colton, C.K.; Tang, J.; Kinoshita-Kawada, M.; Lee, L.Y.; Wood, J.D.; Zhu, M.X. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 2004, 279, 35741–35748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.; Montalbetti, N.; Simonin, A.; Danko, T.; Balazs, B.; Zsembery, A.; Hediger, M.A. Inhibition of the human epithelial calcium channel TRPV6 by 2-aminoethoxydiphenyl borate (2-APB). Cell Calcium 2012, 52, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kanaji, T.; Nakade, S.; Kanno, T.; Mikoshiba, K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 1997, 122, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.T.; Patterson, R.L.; van Rossum, D.B.; Birnbaumer, L.; Mikoshiba, K.; Gill, D.L. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science 2000, 287, 1647–1651. [Google Scholar] [CrossRef]
- Braun, F.J.; Broad, L.M.; Armstrong, D.L.; Putney, J.W., Jr. Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. J. Biol. Chem. 2001, 276, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Broad, L.M.; Braun, F.J.; Lievremont, J.P.; Bird, G.S.; Kurosaki, T.; Putney, J.W., Jr. Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. J. Biol. Chem. 2001, 276, 15945–15952. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. 2001, 536, 3–19. [Google Scholar] [CrossRef]
- Bakowski, D.; Glitsch, M.D.; Parekh, A.B. An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current ICRAC in RBL-1 cells. J. Physiol. 2001, 532, 55–71. [Google Scholar] [CrossRef]
- Iwasaki, H.; Mori, Y.; Hara, Y.; Uchida, K.; Zhou, H.; Mikoshiba, K. 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Recept. Channels 2001, 7, 429–439. [Google Scholar] [PubMed]
- Djillani, A.; Nusse, O.; Dellis, O. Characterization of novel store-operated calcium entry effectors. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2341–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Bird, G.S.; Putney, J.W., Jr. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 2008, 283, 19265–19273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, J.-I.; Suzuki, A.Z.; Ozaki, S.; Matsumoto, N.; Nakamura, T.; Ebisui, E.; Fleig, A.; Penner, R.; Mikoshiba, K. Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca2+ entry via STIM proteins. Cell Calcium 2010, 47, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Peinelt, C.; Lis, A.; Beck, A.; Fleig, A.; Penner, R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J. Physiol. 2008, 586, 3061–3073. [Google Scholar] [CrossRef]
- Ma, H.-T.; Venkatachalam, K.; Parys, J.B.; Gill, D.L. Modification of store-operated channel coupling and inositol trisphosphate receptor function by 2-aminoethoxydiphenyl borate in DT40 lymphocytes. J. Biol. Chem. 2002, 277, 6915–6922. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Ali, S.; Li, Y.; Yu, H.; Zhang, M.; Lu, J.; Xu, T. 2-aminoethoxydiphenyl borate potentiates CRAC current by directly dilating the pore of open Orai1. Sci. Rep. 2016, 6, 29304. [Google Scholar] [CrossRef] [Green Version]
- Hendron, E.; Wang, X.; Zhou, Y.; Cai, X.; Goto, J.I.; Mikoshiba, K.; Baba, Y.; Kurosaki, T.; Wang, Y.; Gill, D.L. Potent functional uncoupling between STIM1 and Orai1 by dimeric 2-aminodiphenyl borinate analogs. Cell Calcium 2014, 56, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Zhou, Y.; Sun, A.; Ma, G.; He, L.; Zhou, L.; Zhang, S.; Liu, J.; Zhang, S.L.; Gill, D.L.; et al. Molecular mechanisms underlying inhibition of STIM1-Orai1-mediated Ca2+ entry induced by 2-aminoethoxydiphenyl borate. Pflugers Arch. Eur. J. Physiol. 2016, 468, 2061–2074. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, X.; Zhou, Y.; Hendron, E.; Mancarella, S.; Ritchie, M.F.; Tang, X.D.; Baba, Y.; Kurosaki, T.; Mori, Y.; et al. STIM protein coupling in the activation of Orai channels. Proc. Natl. Acad. Sci. USA 2009, 106, 7391–7396. [Google Scholar] [CrossRef] [Green Version]
- Parvez, S.; Beck, A.; Peinelt, C.; Soboloff, J.; Lis, A.; Monteilh-Zoller, M.; Gill, D.L.; Fleig, A.; Penner, R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008, 22, 752–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Zhang, X.; Pathak, T.; Nwokonko, R.; Gueguinou, M.F.; Subedi, K.P.; Zhou, Y.; Ambudkar, I.S.; et al. Cross-talk between N-terminal and C-terminal domains in stromal interaction molecule 2 (STIM2) determines enhanced STIM2 sensitivity. J. Biol. Chem. 2019, 294, 6318–6332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindl, R.; Bergsmann, J.; Frischauf, I.; Derler, I.; Fahrner, M.; Muik, M.; Fritsch, R.; Groschner, K.; Romanin, C. 2-aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J. Biol. Chem. 2008, 283, 20261–20267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. 2007, 17, 794–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappel, S.; Kilch, T.; Baur, R.; Lochner, M.; Peinelt, C. The number and position of Orai3 units within heteromeric store-operated Ca2+ channels alter the pharmacology of ICRAC. Int. J. Mol. Sci. 2020, 21, 2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Iwasaki, H.; Nakamura, T.; Nakamura, K.; Maruyama, T.; Hamano, S.; Ozaki, S.; Mizutani, A.; Mikoshiba, K. 2-Aminoethyl diphenylborinate analogues: Selective inhibition for store-operated Ca2+ entry. Biochem. Biophys. Res. Commun. 2007, 352, 277–282. [Google Scholar] [CrossRef]
- Hofer, A.; Kovacs, G.; Zappatini, A.; Leuenberger, M.; Hediger, M.A.; Lochner, M. Design, synthesis and pharmacological characterization of analogs of 2-aminoethyl diphenylborinate (2-APB), a known store-operated calcium channel blocker, for inhibition of TRPV6-mediated calcium transport. Bioorg. Med. Chem. 2013, 21, 3202–3213. [Google Scholar] [CrossRef]
- Dellis, O.; Mercier, P.; Chomienne, C. The boron-oxygen core of borinate esters is responsible for the store-operated calcium entry potentiation ability. BMC Pharmacol. 2011, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Bittremieux, M.; Gerasimenko, J.V.; Schuermans, M.; Luyten, T.; Stapleton, E.; Alzayady, K.J.; De Smedt, H.; Yule, D.I.; Mikoshiba, K.; Vangheluwe, P.; et al. DPB162-AE, an inhibitor of store-operated Ca2+ entry, can deplete the endoplasmic reticulum Ca2+ store. Cell Calcium 2017, 62, 60–70. [Google Scholar] [CrossRef]
- Bittremieux, M.; La Rovere, R.M.; Schuermans, M.; Luyten, T.; Mikoshiba, K.; Vangheluwe, P.; Parys, J.B.; Bultynck, G. Extracellular and ER-stored Ca2+ contribute to BIRD-2-induced cell death in diffuse large B-cell lymphoma cells. Cell Death Discov. 2018, 4, 101. [Google Scholar] [CrossRef]
- Kukkonen, J.P.; Lund, P.E.; Akerman, K.E. 2-aminoethoxydiphenyl borate reveals heterogeneity in receptor-activated Ca2+ discharge and store-operated Ca2+ influx. Cell Calcium 2001, 30, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.J.; Azimi, I.; Flanagan, J.U.; Inserra, M.; Vetter, I.; Monteith, G.R.; Denny, W.A. An SAR study of hydroxy-trifluoromethylpyrazolines as inhibitors of Orai1-mediated store operated Ca2+ entry in MDA-MB-231 breast cancer cells using a convenient Fluorescence Imaging Plate Reader assay. Bioorg. Med. Chem. 2018, 26, 3406–3413. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor a-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Wang, X.; Shen, Q.; Yang, X.; Yu, C.; Cai, C.; Cai, G.; Meng, X.; Zou, F. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem. Biophys. Res. Commun. 2015, 458, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Seven, Ö.; Bolte, M.; Lerner, H.-W.; Wagner, M. High-yield syntheses and reactivity studies of 1,2-diborylated and 1,2,4,5-tetraborylated benzenes. Organometallics 2014, 33, 1291–1299. [Google Scholar] [CrossRef]
- Suzuki, A.Z.; Ozaki, S.; Goto, J.-I.; Mikoshiba, K. Synthesis of bisboron compounds and their strong inhibitory activity on store-operated calcium entry. Bioorg. Med. Chem. Lett. 2010, 20, 1395–1398. [Google Scholar] [CrossRef]
- Allen, D.G.; Coe, D.M.; Cooper, A.W.J.; Gore, P.M.; House, D.; Senger, S.; Sollis, S.L.; Vile, S.; Wilson, C. N-Pyrazolyl Carboxamides as CRAC Channel Inhibitors. U.S. Patent 8399436B2, 19 March 2013. [Google Scholar]
- Chen, S.; Jiang, J.; Li, H.; James, D.; Chimmanamada, D.; Borella, C.; Sun, L.; Xie, Y.; Holmqvist, M.; Mahiou, J.; et al. Compounds for Inflammation and Immune-Related Uses. U.S. Patent 7709518B2, 4 May 2010. [Google Scholar]
- Flückiger, R.; Henson, E.; Hess, G.M.; Gallop, P.M. Mass spectral and HPLC analysis of biological compounds with diphenylborinic acid. Biomed. Mass Spectrom. 1984, 11, 611–615. [Google Scholar] [CrossRef]
- Bohnert, G.; Chen, S.; Xie, Y. Vinyl-Phenyl Derivatives for Inflammation and Immune-Related Uses. U.S. Patent 7816535B2, 19 October 2010. [Google Scholar]
- Ng, S.W.; di Capite, J.; Singaravelu, K.; Parekh, A.B. Sustained activation of the tyrosine kinase Syk by antigen in mast cells requires local Ca2+ influx through Ca2+ release-activated Ca2+ channels. J. Biol. Chem. 2008, 283, 31348–31355. [Google Scholar] [CrossRef] [Green Version]
- Di Sabatino, A.; Rovedatti, L.; Kaur, R.; Spencer, J.P.; Brown, J.T.; Morisset, V.D.; Biancheri, P.; Leakey, N.A.B.; Wilde, J.I.; Scott, L.; et al. Targeting gut T cell Ca2+ release-activated Ca2+ channels inhibits T cell cytokine production and T-Box transcription factor T-Bet in inflammatory bowel disease. J. Immunol. 2009, 183, 3454–3462. [Google Scholar] [CrossRef] [Green Version]
- Derler, I.; Schindl, R.; Fritsch, R.; Heftberger, P.; Riedl, M.C.; Begg, M.; House, D.; Romanin, C. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 2013, 53, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Panicker, S.; Lau, K.-Y.; Apparsundaram, S.; Patel, V.A.; Chen, S.-L.; Soto, R.; Jung, J.K.C.; Ravindran, P.; Okuhara, D.; et al. Characterization of a novel CRAC inhibitor that potently blocks human T cell activation and effector functions. Mol. Immunol. 2013, 54, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Augustynek, B.S.; Ercan-Herbst, E.; Kandasamy, P.; Seedorf, M.; Peinelt, C.; Hediger, M.A. Ca2+/Calmodulin binding to STIM1 hydrophobic residues facilitates slow Ca2+-dependent inactivation of the Orai1 channel. Cell. Physiol. Biochem. 2020, 54, 252–270. [Google Scholar] [CrossRef] [PubMed]
- Butorac, C.; Muik, M.; Derler, I.; Stadlbauer, M.; Lunz, V.; Krizova, A.; Lindinger, S.; Schober, R.; Frischauf, I.; Bhardwaj, R.; et al. A novel STIM1-Orai1 gating interface essential for CRAC channel activation. Cell Calcium 2019, 79, 57–67. [Google Scholar] [CrossRef]
- Ercan, E.; Chung, S.-H.; Bhardwaj, R.; Seedorf, M. Di-arginine signals and the K-rich domain retain the Ca2+ sensor STIM1 in the endoplasmic reticulum. Traffic 2012, 13, 992–1003. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schild, A.; Bhardwaj, R.; Wenger, N.; Tscherrig, D.; Kandasamy, P.; Dernič, J.; Baur, R.; Peinelt, C.; Hediger, M.A.; Lochner, M. Synthesis and Pharmacological Characterization of 2-Aminoethyl Diphenylborinate (2-APB) Derivatives for Inhibition of Store-Operated Calcium Entry (SOCE) in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 5604. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165604
Schild A, Bhardwaj R, Wenger N, Tscherrig D, Kandasamy P, Dernič J, Baur R, Peinelt C, Hediger MA, Lochner M. Synthesis and Pharmacological Characterization of 2-Aminoethyl Diphenylborinate (2-APB) Derivatives for Inhibition of Store-Operated Calcium Entry (SOCE) in MDA-MB-231 Breast Cancer Cells. International Journal of Molecular Sciences. 2020; 21(16):5604. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165604
Chicago/Turabian StyleSchild, Achille, Rajesh Bhardwaj, Nicolas Wenger, Dominic Tscherrig, Palanivel Kandasamy, Jan Dernič, Roland Baur, Christine Peinelt, Matthias A. Hediger, and Martin Lochner. 2020. "Synthesis and Pharmacological Characterization of 2-Aminoethyl Diphenylborinate (2-APB) Derivatives for Inhibition of Store-Operated Calcium Entry (SOCE) in MDA-MB-231 Breast Cancer Cells" International Journal of Molecular Sciences 21, no. 16: 5604. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165604