Potential Mechanisms of Bisphenol A (BPA) Contributing to Human Disease

 and
and 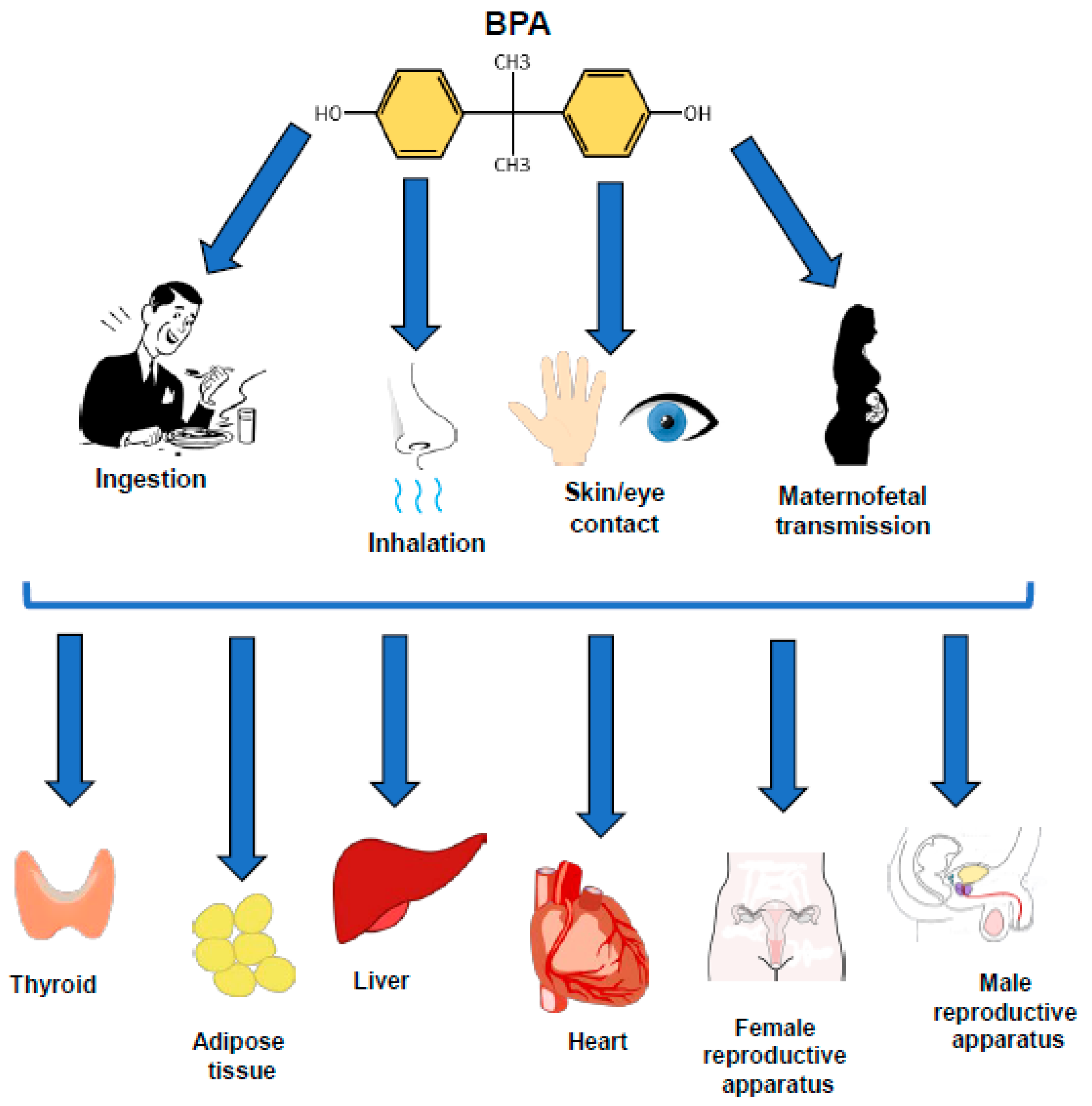{kind=link}
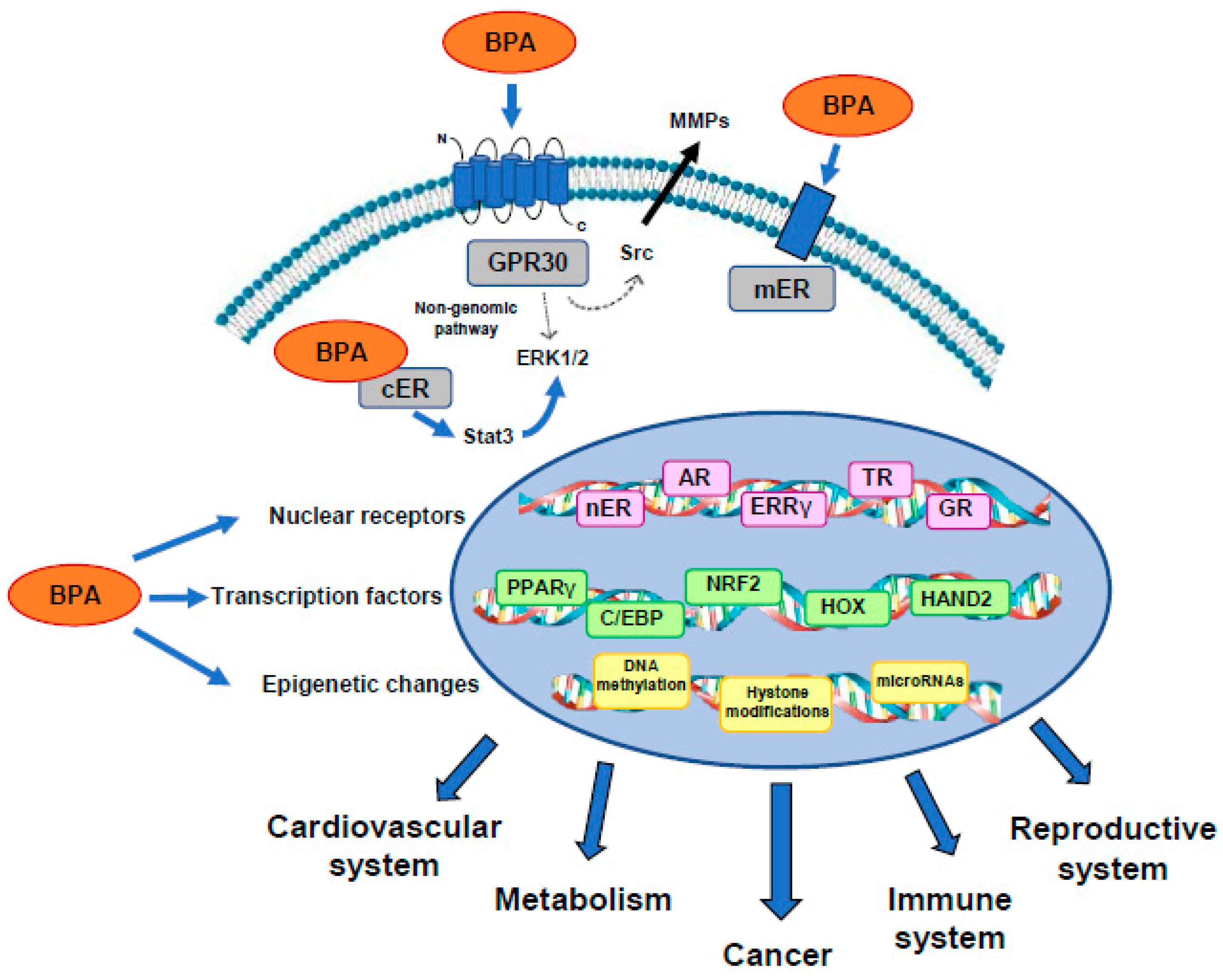{kind=link}
Abstract
:1. Introduction
2. BPA Interaction with Specific Receptors
2.1. Estrogen Receptors
2.2. GPR30
2.3. Androgen Receptor
2.4. Other Receptor Targets of BPA Action
3. BPA Regulation of Transcription Factors
3.1. PPARγ
3.2. C/EBP
3.3. NRF2
3.4. HOX
3.5. HAND2
4. BPA-Regulated Epigenetic Mechanisms
4.1. BPA-Induced Epigenetic Modifications in Metabolism
4.2. BPA-Induced Epigenetic Modifications in Cancer
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Androgen receptors |
| BPA | Bisphenol A |
| C/EBPs | CCAAT/enhancer-binding proteins |
| cER | Cytosolic estrogen receptors |
| CREB3L4 | CAMP responsive element binding protein 3 like 4 |
| Cpt1a | Carnitine palmitoyltransferase 1a |
| E2 | 17 beta estradiol |
| EGFR | Estrogen growth factor receptor |
| ER | Estrogen receptors |
| ERE | Estrogen response elements |
| ERK | Extracellular receptor kinase |
| ERRγ | Estrogen-related receptor gamma |
| EZH2 | Enhancer of Zeste homolog 2 |
| Gck | Glucokinase |
| GPR30 | G protein-coupled receptor 30 G protein-coupled receptor 30 |
| GR | Glucocorticoid receptor |
| H3 | Histone 3 |
| HAND2 | Heart- and neural crest derivatives-expressed protein 2 |
| HFD | High fat diet |
| IL | Interleukin |
| HOX | Homeobox proteins |
| IGF | Insulin-like growth factor |
| IGFR | Insulin-like growth factor receptor |
| Keap1 | Kelch-like ECH-associated protein |
| LAMP3 | Lysosomal associated membrane protein 3 |
| MAPK | Mitogen-activated protein kinase |
| mER | Membrane estrogen receptors |
| MEST | Mesoderm-specific transcript |
| NAFLD | Non-alcoholic fatty liver disease |
| nER | Nuclear estrogen receptors |
| NO | Nitric oxide |
| Nrf2 | Nuclear factor erythroid-2-related factor 2 |
| PPAR | Peroxisome proliferator-activated receptor |
| PAQR | Progestin and AdipoQ receptor family member 4 |
| PCOS | Polycystic ovary syndrome |
| PDE4D4 | Phosphodiesterase type 4 variant4 |
| PDX1 | Pancreatic and duodenal homeobox 1 |
| PI3K | Phosphoinositide 3-kinases |
| Pitx3 | Pituitary homeobox 3 |
| PKB | Protein kinase B |
| POPs | Persistent organic pollutants |
| PR | Progesterone receptor |
| Sox2 | SRY-box transcription factor 2 |
| Srebf | Sterol regulatory element-binding transcription factor 1 |
| STAT | Signal transducer and activator of transcription |
| T2D | Type 2 diabetes |
| T3 | Triiodothyronine |
| TF | Transcription factor |
| TH | Thyroid hormone |
| TR | Thyroid receptor |
| TSH | Thyroid stimulating hormone |
References
- Alharbi, O.M.L.; Basheer, A.A.; Khattab, R.A.; Ali, I. Health and environmental effects of persistent organic pollutants. J. Mol. Liq. 2018, 263, 442–453. [Google Scholar] [CrossRef]
- Ross, P.S.; Birnbaum, L.S. Integrated Human and Ecological Risk Assessment: A Case Study of Persistent Organic Pollutants (POPs) in Humans and Wildlife. Hum. Ecol. Risk Assess. 2003, 9, 303–324. [Google Scholar] [CrossRef]
- Croes, K.; Colles, A.; Koppen, G.; Govarts, E.; Bruckers, L.; Van de Mieroop, E.; Nelen, V.; Covaci, A.; Dirtu, A.C.; Thomsen, C.; et al. Persistent organic pollutants (POPs) in human milk: A biomonitoring study in rural areas of Flanders (Belgium). Chemosphere 2012, 89, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Ewers, U.; Schulz, C. Revised and new reference values for some persistent organic pollutants (POPs) in blood for human biomonitoring in environmental medicine. Int. J. Hyg. Environ. Health 2003, 206, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, Q.; Zhang, Y.; Niu, Y.; Yao, X.; Liu, H. The molecular mechanism of bisphenol A (BPA) as an endocrine disruptor by interacting with nuclear receptors: Insights from molecular dynamics (MD) simulations. PLoS ONE 2015, 10, e0120330. [Google Scholar] [CrossRef]
- Delfosse, V.; Grimaldi, M.; Pons, J.L.; Boulahtouf, A.; le Maire, A.; Cavailles, V.; Labesse, G.; Bourguet, W.; Balaguer, P. Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol A substitutes. Proc. Natl. Acad. Sci. USA 2012, 109, 14930–14935. [Google Scholar] [CrossRef] [Green Version]
- Eladak, S.; Grisin, T.; Moison, D.; Guerquin, M.J.; N’Tumba-Byn, T.; Pozzi-Gaudin, S.; Benachi, A.; Livera, G.; Rouiller-Fabre, V.; Habert, R. A new chapter in the bisphenol A story: Bisphenol S and bisphenol F are not safe alternatives to this compound. Fertil. Steril. 2015, 103, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Tyl, R.W. Abbreviated assessment of bisphenol A toxicology literature. Semin. Fetal. Neonatal. Med. 2014, 19, 195–202. [Google Scholar] [CrossRef]
- Huo, X.; Chen, D.; He, Y.; Zhu, W.; Zhou, W.; Zhang, J. Bisphenol-A and Female Infertility: A Possible Role of Gene-Environment Interactions. Int. J. Environ. Res. Public Health 2015, 12, 11101–11116. [Google Scholar] [CrossRef]
- Takahashi, Y.; Shirai, A.; Segawa, T.; Takahashi, T.; Sakakibara, K. Why does a color developing phenomenon occur on thermal paper comprising of a fluoran dye and a color developer molecule? Bull Chem. Soc. Jpn. 2002, 75, 2225–2231. [Google Scholar] [CrossRef]
- Mendum, T.; Stoler, E.; VanBenschoten, H.; Warner, J.C. Concentration of bisphenol A in thermal paper. Green Chem. Let. Rev. 2011, 4, 81–86. [Google Scholar] [CrossRef]
- Hormann, A.M.; Vom Saal, F.S.; Nagel, S.C.; Stahlhut, R.W.; Moyer, C.L.; Ellersieck, M.R.; Welshons, W.V.; Toutain, P.L.; Taylor, J.A. Holding thermal receipt paper and eating food after using hand sanitizer results in high serum bioactive and urine total levels of bisphenol A (BPA). PLoS ONE 2014, 9, e110509. [Google Scholar] [CrossRef] [PubMed]
- Ndaw, S.; Remy, A.; Jargot, D.; Robert, A. Occupational exposure of cashiers to Bisphenol A via thermal paper: Urinary biomonitoring study. Int. Arch. Occup. Environ. Health 2016, 89, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, S.; Tschudin, P.; Grob, K. Transfer of bisphenol A from thermal printer paper to the skin. Anal. Bioanal. Chem. 2010, 398, 571–576. [Google Scholar] [CrossRef]
- Liao, C.; Kannan, K. Widespread occurrence of bisphenol A in paper and paper products: Implications for human exposure. Environ. Sci. Technol. 2011, 45, 9372–9379. [Google Scholar] [CrossRef]
- Liao, C.; Liu, F.; Kannan, K. Bisphenol s, a new bisphenol analogue, in paper products and currency bills and its association with bisphenol a residues. Environ. Sci. Technol. 2012, 46, 6515–6522. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef]
- Cousins, I.T.; Staples, C.A.; Clecka, G.M.; Mackay, D. A Multimedia Assessment of the Environmental Fate of Bisphenol, A. Hum. Ecol. Risk Assess. 2002, 8, 1107–1135. [Google Scholar] [CrossRef]
- Haffner, D.; Schecter, A. Persistent Organic Pollutants (POPs): A Primer for Practicing Clinicians. Curr. Environ. Health Rep. 2014, 1, 123–131. [Google Scholar] [CrossRef]
- Corrales, J.; Kristofco, L.A.; Steele, W.B.; Yates, B.S.; Breed, C.S.; Williams, E.S.; Brooks, B.W. Global Assessment of Bisphenol A in the Environment: Review and Analysis of Its Occurrence and Bioaccumulation. Dose Response 2015, 13, 1559325815598308. [Google Scholar] [CrossRef] [Green Version]
- Valentino, R.; D’Esposito, V.; Ariemma, F.; Cimmino, I.; Beguinot, F.; Formisano, P. Bisphenol A environmental exposure and the detrimental effects on human metabolic health: Is it necessary to revise the risk assessment in vulnerable population? J. Endocrinol. Investig. 2016, 39, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodds, E.C.; Lawson, W. Synthetic, oestrogenic agents without the phenanthrene nucleus. Nature 1936, 137, 996. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Haggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Mileva, G.; Baker, S.L.; Konkle, A.T.; Bielajew, C. Bisphenol-A: Epigenetic reprogramming and effects on reproduction and behavior. Int. J. Environ. Res. Public Health 2014, 11, 7537–7561. [Google Scholar] [CrossRef]
- Almeida, S.; Raposo, A.; Almeida-González, M.; Carrascosa, C. Bisphenol A: Food Exposure and Impact on Human Health. Compr. Rev. Food. Sci. Food Saf. 2018, 17, 1503–1517. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, I.; Oriente, F.; D’Esposito, V.; Liguoro, D.; Liguoro, P.; Ambrosio, M.R.; Cabaro, S.; D’Andrea, F.; Beguinot, F.; Formisano, P.; et al. Low Dose Bisphenol-A Regulates Inflammatory Cytokines through GPR30 in Mammary Adipose Cells. J. Mol. Endocrinol. 2019, 63, 273–283. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef]
- Ziv-Gal, A.; Flaws, J.A. Evidence for bisphenol A-induced female infertility: A review (2007–2016). Fertil. Steril. 2016, 106, 827–856. [Google Scholar] [CrossRef] [Green Version]
- Ekbom, A. Growing evidence that several human cancers may originate in utero. Semin. Cancer Biol. 1998, 8, 237–244. [Google Scholar] [CrossRef]
- Margel, D.; Fleshner, N.E. Oral contraceptive use is associated with prostate cancer: An ecological study. BMJ Open 2011, 1, e000311. [Google Scholar] [CrossRef]
- Andra, S.S.; Austin, C.; Yang, J.; Patel, D.; Arora, M. Recent advances in simultaneous analysis of bisphenol A and its conjugates in human matrices: Exposure biomarker perspectives. Sci. Total Environ. 2016, 572, 770–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vom Saal, F.S.; Hughes, C. An extensive new literature concerning low-dose effects of bisphenol A shows the need for a new risk assessment. Environ. Health Perspect. 2005, 113, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu-Denoncourt, J.; Wallace, S.J.; de Solla, S.R.; Langlois, V.S. Influence of Lipophilicity on the Toxicity of Bisphenol A and Phthalates to Aquatic Organisms. Bull. Environ. Contam. Toxicol. 2016, 97, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Healy, B.F.; English, K.R.; Jagals, P.; Sly, P.D. Bisphenol A exposure pathways in early childhood: Reviewing the need for improved risk assessment models. J. Expo. Sci. Environ. Epidemiol. 2015, 25, 544–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotondo, E.; Chiarelli, F. Endocrine-Disrupting Chemicals and Insulin Resistance in Children. Biomedicines 2020, 8, 137. [Google Scholar] [CrossRef]
- Castellini, C.; Totaro, M.; Parisi, A.; D’Andrea, S.; Lucente, L.; Cordeschi, G.; Francavilla, S.; Francavilla, F.; Barbonetti, A. Bisphenol A and Male Fertility: Myths and Realities. Front. Endocrinol. (Lausanne) 2020, 11, 353. [Google Scholar] [CrossRef]
- Wehbe, Z.; Nasser, S.A.; El-Yazbi, A.; Nasreddine, S.; Eid, A.H. Estrogen and Bisphenol A in Hypertension. Curr. Hypertens. Rep. 2020, 22, 23. [Google Scholar] [CrossRef]
- Provvisiero, D.P.; Pivonello, C.; Muscogiuri, G.; Negri, M.; de Angelis, C.; Simeoli, C.; Pivonello, R.; Colao, A. Influence of Bisphenol A on Type 2 Diabetes Mellitus. Int. J. Environ. Res. Public Health 2016, 13, 989. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Huang, G.; Guo, T.L. Developmental Bisphenol A Exposure Modulates Immune-Related Diseases. Toxics 2016, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Chapin, R.E.; Adams, J.; Boekelheide, K.; Gray, L.E., Jr.; Hayward, S.W.; Lees, P.S.; McIntyre, B.S.; Portier, K.M.; Schnorr, T.M.; Selevan, S.G.; et al. NTP-CERHR expert panel report on the reproductive and developmental toxicity of bisphenol A. Birth. Defects. Res. B Dev. Reprod. Toxicol. 2008, 83, 157–395. [Google Scholar] [CrossRef]
- Nohynek, G.J.; Borgert, C.J.; Dietrich, D.; Rozman, K.K. Endocrine disruption: Fact or urban legend? Toxicol. Lett. 2013, 223, 295–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ding, Z.; Shi, Q.M.; Ge, X.; Wang, H.X.; Li, M.X.; Chen, G.; Wang, Q.; Ju, Q.; Zhang, J.P.; et al. Anti-androgenic mechanisms of Bisphenol A involve androgen receptor signaling pathway. Toxicology 2017, 387, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjold, M.; Gustafsson, J.A. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menasce, L.P.; White, G.R.; Harrison, C.J.; Boyle, J.M. Localization of the estrogen receptor locus (ESR) to chromosome 6q25.1 by FISH and a simple post-FISH banding technique. Genomics 1993, 17, 263–265. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Matthews, J.; Celius, T.; Halgren, R.; Zacharewski, T. Differential estrogen receptor binding of estrogenic substances: A species comparison. J. Steroid. Biochem. Mol. Biol. 2000, 74, 223–234. [Google Scholar] [CrossRef]
- Acconcia, F.; Pallottini, V.; Marino, M. Molecular Mechanisms of Action of BPA. Dose Response 2015, 13, 1559325815610582. [Google Scholar] [CrossRef] [Green Version]
- Angle, B.M.; Do, R.P.; Ponzi, D.; Stahlhut, R.W.; Drury, B.E.; Nagel, S.C.; Welshons, W.V.; Besch-Williford, C.L.; Palanza, P.; Parmigiani, S.; et al. Metabolic disruption in male mice due to fetal exposure to low but not high doses of bisphenol A (BPA): Evidence for effects on body weight, food intake, adipocytes, leptin, adiponectin, insulin and glucose regulation. Reprod. Toxicol. 2013, 42, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Nadal, A.; Ropero, A.B.; Laribi, O.; Maillet, M.; Fuentes, E.; Soria, B. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor alpha and estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2000, 97, 11603–11608. [Google Scholar] [CrossRef] [Green Version]
- Wetherill, Y.B.; Akingbemi, B.T.; Kanno, J.; McLachlan, J.A.; Nadal, A.; Sonnenschein, C.; Watson, C.S.; Zoeller, R.T.; Belcher, S.M. In vitro molecular mechanisms of bisphenol A action. Reprod. Toxicol. 2007, 24, 178–198. [Google Scholar] [CrossRef] [PubMed]
- Phrakonkham, P.; Viengchareun, S.; Belloir, C.; Lombes, M.; Artur, Y.; Canivenc-Lavier, M.C. Dietary xenoestrogens differentially impair 3T3-L1 preadipocyte differentiation and persistently affect leptin synthesis. J. Steroid. Biochem. Mol. Biol. 2008, 110, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Ben-Jonathan, N.; Hugo, E.R.; Brandebourg, T.D. Effects of bisphenol A on adipokine release from human adipose tissue: Implications for the metabolic syndrome. Mol. Cell. Endocrinol. 2009, 304, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, B.S.; Paranjpe, M.; DaFonte, T.; Schaeberle, C.; Soto, A.M.; Obin, M.; Greenberg, A.S. Perinatal BPA exposure alters body weight and composition in a dose specific and sex specific manner: The addition of peripubertal exposure exacerbates adverse effects in female mice. Reprod. Toxicol. 2017, 68, 130–144. [Google Scholar] [CrossRef] [Green Version]
- Ptak, A.; Gregoraszczuk, E.L. Bisphenol A induces leptin receptor expression, creating more binding sites for leptin, and activates the JAK/Stat, MAPK/ERK and PI3K/Akt signalling pathways in human ovarian cancer cell. Toxicol. Lett. 2012, 210, 332–337. [Google Scholar] [CrossRef]
- Song, H.; Zhang, T.; Yang, P.; Li, M.; Yang, Y.; Wang, Y.; Du, J.; Pan, K.; Zhang, K. Low doses of bisphenol A stimulate the proliferation of breast cancer cells via ERK1/2/ERRγ signals. Toxicol. In Vitro 2015, 30, 521–528. [Google Scholar] [CrossRef]
- Dairkee, S.H.; Luciani-Torres, M.G.; Moore, D.H.; Goodson, W.H. Bisphenol-A-induced inactivation of the p53 axis underlying deregulation of proliferation kinetics, and cell death in non-malignant human breast epithelial cells. Carcinogenesis 2013, 34, 703–712. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Laribi, O.; Ropero, A.B.; Fuentes, E.; Ripoll, C.; Soria, B.; Nadal, A. Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environ. Health Perspect. 2005, 113, 969–977. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, S.; Nakatsuka, M.; Asagiri, K.; Habara, T.; Takata, M.; Konishi, H.; Kudo, T. Bisphenol A stimulates NO synthesis through a non-genomic estrogen receptor-mediated mechanism in mouse endothelial cells. Toxicol. Lett. 2002, 135, 95–101. [Google Scholar] [CrossRef]
- O’Dowd, B.F.; Nguyen, T.; Marchese, A.; Cheng, R.; Lynch, K.R.; Heng, H.H.; Kolakowski, L.F., Jr.; George, S.R. Discovery of three novel G-protein-coupled receptor genes. Genomics 1998, 47, 310–313. [Google Scholar] [CrossRef]
- Owman, C.; Blay, P.; Nilsson, C.; Lolait, S.J. Cloning of human cDNA encoding a novel heptahelix receptor expressed in Burkitt’s lymphoma and widely distributed in brain and peripheral tissues. Biochem. Biophys. Res. Commun. 1996, 228, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Kato, C.; Kondo, S.; Korenaga, R.; Ando, J. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem. Biophys. Res. Commun. 1997, 240, 737–741. [Google Scholar] [CrossRef]
- Pietras, R.J.; Marquez-Garban, D.C. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin. Cancer Res. 2007, 13, 4672–4676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, K.J.; Liao, J.K. Non-nuclear actions of estrogen: New targets for prevention and treatment of cardiovascular disease. Mol. Interv. 2002, 2, 219–228. [Google Scholar] [CrossRef]
- Kelly, M.J.; Levin, E.R. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2001, 12, 152–156. [Google Scholar] [CrossRef]
- Levin, E.R. Cell localization, physiology, and nongenomic actions of estrogen receptors. J. Appl. Physiol. 2001, 91, 1860–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, E.R. Cellular functions of plasma membrane estrogen receptors. Steroids 2002, 67, 471–475. [Google Scholar] [CrossRef]
- Razandi, M.; Pedram, A.; Park, S.T.; Levin, E.R. Proximal events in signaling by plasma membrane estrogen receptors. J. Biol. Chem. 2003, 278, 2701–2712. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Revankar, C.M.; Mitchell, H.D.; Field, A.S.; Burai, R.; Corona, C.; Ramesh, C.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. Synthetic estrogen derivatives demonstrate the functionality of intracellular GPR30. ACS Chem. Biol. 2007, 2, 536–544. [Google Scholar] [CrossRef]
- Thomas, P.; Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J. Steroid. Biochem. Mol. Biol. 2006, 102, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Luo, J.; Moore, W.; Alkhalidy, H.; Wu, L.; Zhang, J.; Zhen, W.; Wang, Y.; Clegg, D.J.; Bin, X.; et al. GPR30 regulates diet-induced adiposity in female mice and adipogenesis in vitro. Sci. Rep. 2016, 6, 34302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Arevalo, M.; Alonso-Magdalena, P.; Rebelo Dos Santos, J.; Quesada, I.; Carneiro, E.M.; Nadal, A. Exposure to bisphenol-A during pregnancy partially mimics the effects of a high-fat diet altering glucose homeostasis and gene expression in adult male mice. PLoS ONE 2014, 9, e100214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, M.K.; Jeong, I.K.; Jung Oh, T.; Ahn, H.Y.; Kim, H.H.; Park, Y.J.; Jang, H.C.; Park, K.S. Long-term oral exposure to bisphenol A induces glucose intolerance and insulin resistance. J. Endocrinol. 2015, 226, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.O.; Arias-Pulido, H.; Kuo, D.Y.; Howard, T.; Qualls, C.R.; Lee, S.J.; Verschraegen, C.F.; Hathaway, H.J.; Joste, N.E.; Prossnitz, E.R. GPR30 predicts poor survival for ovarian cancer. Gynecol. Oncol. 2009, 114, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Terasaka, S.; Kiyama, R. Bisphenol A induces a rapid activation of Erk1/2 through GPR30 in human breast cancer cells. Environ. Pollut. 2011, 159, 212–218. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Li, Q.; Zhang, T.; Deng, Z.; Lian, J.; Jia, D.; Li, R.; Zheng, T.; Ding, X.; et al. Low concentration of BPA induces mice spermatocytes apoptosis via GPR30. Oncotarget 2017, 8, 49005–49015. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, N.; Bouskine, A.; Fenichel, P. Bisphenol A promotes testicular seminoma cell proliferation through GPER/GPR30. Int. J. Cancer 2012, 130, 241–242. [Google Scholar] [CrossRef]
- Kojima, H.; Takeuchi, S.; Sanoh, S.; Okuda, K.; Kitamura, S.; Uramaru, N.; Sugihara, K.; Yoshinari, K. Profiling of bisphenol A and eight its analogues on transcriptional activity via human nuclear receptors. Toxicology 2019, 413, 48–55. [Google Scholar] [CrossRef]
- Molina-Molina, J.M.; Amaya, E.; Grimaldi, M.; Saenz, J.M.; Real, M.; Fernandez, M.F.; Balaguer, P.; Olea, N. In vitro study on the agonistic and antagonistic activities of bisphenol-S and other bisphenol-A congeners and derivatives via nuclear receptors. Toxicol. Appl. Pharmacol. 2013, 272, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Sohoni, P.; Sumpter, J.P. Several environmental oestrogens are also anti-androgens. J. Endocrinol. 1998, 158, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Tong, W.; Branham, W.S.; Moland, C.L.; Dial, S.L.; Hong, H.; Xie, Q.; Perkins, R.; Owens, W.; Sheehan, D.M. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem. Res. Toxicol. 2003, 16, 1338–1358. [Google Scholar] [CrossRef] [PubMed]
- Perera, L.; Li, Y.; Coons, L.A.; Houtman, R.; Van Beuningen, R.; Goodwin, B.; Auerbach, S.S.; Teng, C.T. Binding of bisphenol A, bisphenol AF, and bisphenol S on the androgen receptor: Coregulator recruitment and stimulation of potential interaction sites. Toxicol. In Vitro 2017, 44, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Manfo, F.P.; Jubendradass, R.; Nantia, E.A.; Moundipa, P.F.; Mathur, P.P. Adverse effects of bisphenol A on male reproductive function. Rev. Environ. Contam. Toxicol. 2014, 228, 57–82. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Cang, X.; Liu, J. Molecular mechanism of Bisphenol A on androgen receptor antagonism. Toxicol. In Vitro 2019, 61, 104621. [Google Scholar] [CrossRef]
- Li, D.; Zhou, Z.; Qing, D.; He, Y.; Wu, T.; Miao, M.; Wang, J.; Weng, X.; Ferber, J.R.; Herrinton, L.J.; et al. Occupational exposure to bisphenol-A (BPA) and the risk of self-reported male sexual dysfunction. Hum. Reprod. 2010, 25, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Vitku, J.; Sosvorova, L.; Chlupacova, T.; Hampl, R.; Hill, M.; Sobotka, V.; Heracek, J.; Bicikova, M.; Starka, L. Differences in bisphenol A and estrogen levels in the plasma and seminal plasma of men with different degrees of infertility. Physiol. Res. 2015, 64, S303–S311. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402. [Google Scholar] [CrossRef]
- Takayanagi, S.; Tokunaga, T.; Liu, X.; Okada, H.; Matsushima, A.; Shimohigashi, Y. Endocrine disruptor bisphenol A strongly binds to human estrogen-related receptor gamma (ERRgamma) with high constitutive activity. Toxicol. Lett. 2006, 167, 95–105. [Google Scholar] [CrossRef]
- Tohme, M.; Prud’homme, S.M.; Boulahtouf, A.; Samarut, E.; Brunet, F.; Bernard, L.; Bourguet, W.; Gibert, Y.; Balaguer, P.; Laudet, V. Estrogen-related receptor gamma is an in vivo receptor of bisphenol A. FASEB J. 2014, 28, 3124–3133. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Matsushima, A.; Shimohigashi, M.; Shimohigashi, Y. A characteristic back support structure in the bisphenol A-binding pocket in the human nuclear receptor ERRγ. PLoS ONE 2014, 9, e101252. [Google Scholar] [CrossRef] [PubMed]
- Ranhotra, H.S. The mammalian orphan nuclear receptors: Orphans as cellular guardians. J. Recept. Signal Transduct. Res. 2011, 31, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Liu, N.; Weng, S.F.; Wang, H.S. Bisphenol A Increases the Migration and Invasion of Triple-Negative Breast Cancer Cells via Oestrogen-related Receptor Gamma. Basic Clin. Pharmacol. Toxicol. 2016, 119, 389–395. [Google Scholar] [CrossRef]
- Atlas, E.; Pope, L.; Wade, M.G.; Kawata, A.; Boudreau, A.; Boucher, J.G. Bisphenol A increases aP2 expression in 3T3L1 by enhancing the transcriptional activity of nuclear receptors at the promoter. Adipocyte 2014, 3, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Meeker, J.D.; Calafat, A.M.; Hauser, R. Urinary bisphenol A concentrations in relation to serum thyroid and reproductive hormone levels in men from an infertility clinic. Environ. Sci. Technol. 2010, 44, 1458–1463. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Shen, O.X.; Wang, X.R.; Zhou, L.; Zhen, S.Q.; Chen, X.D. Anti-thyroid hormone activity of bisphenol A, tetrabromobisphenol A and tetrachlorobisphenol A in an improved reporter gene assay. Toxicol. In Vitro 2009, 23, 950–954. [Google Scholar] [CrossRef]
- Gorini, F.; Bustaffa, E.; Coi, A.; Iervasi, G.; Bianchi, F. Bisphenols as Environmental Triggers of Thyroid Dysfunction: Clues and Evidence. Int. J. Environ. Res. Public Health 2020, 17, 2654. [Google Scholar] [CrossRef]
- Somm, E.; Schwitzgebel, V.M.; Toulotte, A.; Cederroth, C.R.; Combescure, C.; Nef, S.; Aubert, M.L.; Huppi, P.S. Perinatal exposure to bisphenol a alters early adipogenesis in the rat. Environ. Health Perspect. 2009, 117, 1549–1555. [Google Scholar] [CrossRef] [Green Version]
- Pu, Y.; Gingrich, J.D.; Steibel, J.P.; Veiga-Lopez, A. Sex-Specific Modulation of Fetal Adipogenesis by Gestational Bisphenol A and Bisphenol S Exposure. Endocrinology 2017, 158, 3844–3858. [Google Scholar] [CrossRef] [Green Version]
- Biasiotto, G.; Zanella, I.; Masserdotti, A.; Pedrazzani, R.; Papa, M.; Caimi, L.; Di Lorenzo, D. Municipal wastewater affects adipose deposition in male mice and increases 3T3-L1 cell differentiation. Toxicol. Appl. Pharmacol. 2016, 297, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Ariemma, F.; D’Esposito, V.; Liguoro, D.; Oriente, F.; Cabaro, S.; Liotti, A.; Cimmino, I.; Longo, M.; Beguinot, F.; Formisano, P.; et al. Low-Dose Bisphenol-A Impairs Adipogenesis and Generates Dysfunctional 3T3-L1 Adipocytes. PLoS ONE 2016, 11, e0150762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.G.; Gagne, R.; Rowan-Carroll, A.; Boudreau, A.; Yauk, C.L.; Atlas, E. Bisphenol A and Bisphenol S Induce Distinct Transcriptional Profiles in Differentiating Human Primary Preadipocytes. PLoS ONE 2016, 11, e0163318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sun, B.; Hou, M.; Pan, X.; Li, X. The environmental obesogen bisphenol A promotes adipogenesis by increasing the amount of 11beta-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int. J. Obes. (Lond.) 2013, 37, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, A.; Shidfar, F.; Hedayati, M.; Neshatbini Tehrani, A.; Farshad, A.A.; Mohammadi, S. Bisphenol A enhances adipogenic signaling pathways in human mesenchymal stem cells. Genes Environ. 2020, 42, 13. [Google Scholar] [CrossRef]
- DeBenedictis, B.; Guan, H.; Yang, K. Prenatal Exposure to Bisphenol A Disrupts Mouse Fetal Liver Maturation in a Sex-Specific Manner. J. Cell Biochem. 2016, 117, 344–350. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Zhang, Z.; Liu, H.; Jia, L.; Qin, M.; Wang, X. Exacerbating lupus nephritis following BPA exposure is associated with abnormal autophagy in MRL/lpr mice. Am. J. Transl. Res. 2020, 12, 649–659. [Google Scholar]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1428. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Donepudi, A.C.; More, V.R.; Kulkarni, S.R.; Li, L.; Guo, L.; Yan, B.; Chatterjee, T.; Weintraub, N.; Slitt, A.L. Deficiency in Nrf2 transcription factor decreases adipose tissue mass and hepatic lipid accumulation in leptin-deficient mice. Obesity 2015, 23, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Yamanaka, H.; Oguro, A.; Imaoka, S. Bisphenol A induces Nrf2-dependent drug-metabolizing enzymes through nitrosylation of Keap1. Drug Metab. Pharmacokinet. 2018, 33, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Shimpi, P.C.; More, V.R.; Paranjpe, M.; Donepudi, A.C.; Goodrich, J.M.; Dolinoy, D.C.; Rubin, B.; Slitt, A.L. Hepatic Lipid Accumulation and Nrf2 Expression following Perinatal and Peripubertal Exposure to Bisphenol A in a Mouse Model of Nonalcoholic Liver Disease. Environ. Health Perspect. 2017, 125, 087005. [Google Scholar] [CrossRef] [PubMed]
- Lappin, T.R.; Grier, D.G.; Thompson, A.; Halliday, H.L. HOX genes: Seductive science, mysterious mechanisms. Ulster. Med. J. 2006, 75, 23–31. [Google Scholar]
- Featherstone, M. HOX proteins and their co-factors in transcriptional regulation. Adv. Develop. Biol. Biochem. 2003, 13, 1–42. [Google Scholar] [CrossRef]
- Taylor, H.S.; Vanden Heuvel, G.B.; Igarashi, P. A conserved Hox axis in the mouse and human female reproductive system: Late establishment and persistent adult expression of the Hoxa cluster genes. Biol. Reprod. 1997, 57, 1338–1345. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Capecchi, M.R. Paralogous mouse Hox genes, Hoxa9, Hoxb9, and Hoxd9, function together to control development of the mammary gland in response to pregnancy. Proc. Natl. Acad. Sci. USA 1999, 96, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Taylor, H.S. Xenoestrogen exposure imprints expression of genes (Hoxa10) required for normal uterine development. FASEB J. 2007, 21, 239–246. [Google Scholar] [CrossRef]
- Hayashida, T.; Takahashi, F.; Chiba, N.; Brachtel, E.; Takahashi, M.; Godin-Heymann, N.; Gross, K.W.; Vivanco, M.; Wijendran, V.; Shioda, T.; et al. HOXB9, a gene overexpressed in breast cancer, promotes tumorigenicity and lung metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1100–1105. [Google Scholar] [CrossRef] [Green Version]
- Bodey, B.; Bodey, B., Jr.; Siegel, S.E.; Kaiser, H.E. Immunocytochemical detection of the homeobox B3, B4, and C6 gene products in breast carcinomas. Anticancer Res. 2000, 20, 3281–3286. [Google Scholar]
- Castronovo, V.; Kusaka, M.; Chariot, A.; Gielen, J.; Sobel, M. Homeobox genes: Potential candidates for the transcriptional control of the transformed and invasive phenotype. Biochem. Pharmacol. 1994, 47, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Deb, P.; Bhan, A.; Hussain, I.; Ansari, K.I.; Bobzean, S.A.; Pandita, T.K.; Perrotti, L.I.; Mandal, S.S. Endocrine disrupting chemical, bisphenol-A, induces breast cancer associated gene HOXB9 expression in vitro and in vivo. Gene 2016, 590, 234–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, I.; Bhan, A.; Ansari, K.I.; Deb, P.; Bobzean, S.A.; Perrotti, L.I.; Mandal, S.S. Bisphenol-A induces expression of HOXC6, an estrogen-regulated homeobox-containing gene associated with breast cancer. Biochim. Biophys. Acta 2015, 1849, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Davila, J.; Kannan, A.; Flaws, J.A.; Bagchi, M.K.; Bagchi, I.C. Chronic Exposure to Bisphenol A Affects Uterine Function During Early Pregnancy in Mice. Endocrinology 2016, 157, 1764–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFadden, D.G.; Barbosa, A.C.; Richardson, J.A.; Schneider, M.D.; Srivastava, D.; Olson, E.N. The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development 2005, 132, 189–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, Y.L.; Garske, K.M.; Wang, J.; Firulli, B.A.; Firulli, A.B.; Poss, K.D.; Yelon, D. Hand2 elevates cardiomyocyte production during zebrafish heart development and regeneration. Development 2014, 141, 3112–3122. [Google Scholar] [CrossRef] [Green Version]
- Moreman, J.; Lee, O.; Trznadel, M.; David, A.; Kudoh, T.; Tyler, C.R. Acute Toxicity, Teratogenic, and Estrogenic Effects of Bisphenol A and Its Alternative Replacements Bisphenol S, Bisphenol F, and Bisphenol AF in Zebrafish Embryo-Larvae. Environ. Sci. Technol. 2017, 51, 12796–12805. [Google Scholar] [CrossRef]
- Bakkers, J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Stainier, D.Y. Zebrafish genetics and vertebrate heart formation. Nat. Rev. Genet. 2001, 2, 39–48. [Google Scholar] [CrossRef]
- Lombo, M.; Gonzalez-Rojo, S.; Fernandez-Diez, C.; Herraez, M.P. Cardiogenesis impairment promoted by bisphenol A exposure is successfully counteracted by epigallocatechin gallate. Environ. Pollut. 2019, 246, 1008–1019. [Google Scholar] [CrossRef]
- Ho, S.M.; Tang, W.Y.; Belmonte de Frausto, J.; Prins, G.S. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res. 2006, 66, 5624–5632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, O.S.; Nahar, M.S.; Faulk, C.; Jones, T.R.; Liao, C.; Kannan, K.; Weinhouse, C.; Rozek, L.S.; Dolinoy, D.C. Epigenetic responses following maternal dietary exposure to physiologically relevant levels of bisphenol A. Environ. Mol. Mutagen. 2012, 53, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromer, J.G.; Zhou, Y.; Taylor, M.B.; Doherty, L.; Taylor, H.S. Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J. 2010, 24, 2273–2280. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogen Regulation of MicroRNA Expression. Curr. Genom. 2009, 10, 169–183. [Google Scholar] [CrossRef] [Green Version]
- Doherty, L.F.; Bromer, J.G.; Zhou, Y.; Aldad, T.S.; Taylor, H.S. In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: An epigenetic mechanism linking endocrine disruptors to breast cancer. Horm. Cancer 2010, 1, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Dhimolea, E.; Wadia, P.R.; Murray, T.J.; Settles, M.L.; Treitman, J.D.; Sonnenschein, C.; Shioda, T.; Soto, A.M. Prenatal exposure to BPA alters the epigenome of the rat mammary gland and increases the propensity to neoplastic development. PLoS ONE 2014, 9, e99800. [Google Scholar] [CrossRef] [Green Version]
- Veiga-Lopez, A.; Luense, L.J.; Christenson, L.K.; Padmanabhan, V. Developmental programming: Gestational bisphenol-A treatment alters trajectory of fetal ovarian gene expression. Endocrinology 2013, 154, 1873–1884. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Sartor, M.A.; Rozek, L.S.; Faulk, C.; Anderson, O.S.; Jones, T.R.; Nahar, M.S.; Dolinoy, D.C. Perinatal bisphenol A exposure promotes dose-dependent alterations of the mouse methylome. BMC Genom. 2014, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Xia, W.; Wang, D.Q.; Wan, Y.J.; Xu, B.; Chen, X.; Li, Y.Y.; Xu, S.Q. Hepatic DNA methylation modifications in early development of rats resulting from perinatal BPA exposure contribute to insulin resistance in adulthood. Diabetologia 2013, 56, 2059–2067. [Google Scholar] [CrossRef]
- Van Esterik, J.C.; Vitins, A.P.; Hodemaekers, H.M.; Kamstra, J.H.; Legler, J.; Pennings, J.L.; Steegenga, W.T.; Lute, C.; Jelinek, J.; Issa, J.P.; et al. Liver DNA methylation analysis in adult female C57BL/6JxFVB mice following perinatal exposure to bisphenol A. Toxicol. Lett. 2015, 232, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chang, H.; Xia, W.; Mao, Z.; Li, Y.; Xu, S. F0 maternal BPA exposure induced glucose intolerance of F2 generation through DNA methylation change in Gck. Toxicol. Lett. 2014, 228, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.H.; Pan, J.X.; Jin, L.Y.; Xu, H.Y.; Yu, T.T.; Ullah, K.; Rahman, T.U.; Ren, J.; Cheng, Y.; Dong, X.Y.; et al. Bisphenol A Exposure May Induce Hepatic Lipid Accumulation via Reprogramming the DNA Methylation Patterns of Genes Involved in Lipid Metabolism. Sci. Rep. 2016, 6, 31331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Ding, D.; Huang, Q.; Liu, Q.; Lu, H.; Lu, Y.; Chi, Y.; Sun, X.; Ye, G.; Zhu, H.; et al. Downregulation of miR-192 causes hepatic steatosis and lipid accumulation by inducing SREBF1: Novel mechanism for bisphenol A-triggered non-alcoholic fatty liver disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.X.; Lezmi, S. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Bastos Sales, L.; Kamstra, J.H.; Cenijn, P.H.; van Rijt, L.S.; Hamers, T.; Legler, J. Effects of endocrine disrupting chemicals on in vitro global DNA methylation and adipocyte differentiation. Toxicol. In Vitro 2013, 27, 1634–1643. [Google Scholar] [CrossRef]
- Junge, K.M.; Leppert, B.; Jahreis, S.; Wissenbach, D.K.; Feltens, R.; Grutzmann, K.; Thurmann, L.; Bauer, T.; Ishaque, N.; Schick, M.; et al. MEST mediates the impact of prenatal bisphenol A exposure on long-term body weight development. Clin. Epigenet. 2018, 10, 58. [Google Scholar] [CrossRef]
- Mao, Z.; Xia, W.; Chang, H.; Huo, W.; Li, Y.; Xu, S. Paternal BPA exposure in early life alters Igf2 epigenetic status in sperm and induces pancreatic impairment in rat offspring. Toxicol. Lett. 2015, 238, 30–38. [Google Scholar] [CrossRef]
- Wei, J.; Ding, D.; Wang, T.; Liu, Q.; Lin, Y. MiR-338 controls BPA-triggered pancreatic islet insulin secretory dysfunction from compensation to decompensation by targeting Pdx-1. FASEB J. 2017, 31, 5184–5195. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Wang, D.; Xia, W.; Pan, X.; Huo, W.; Xu, S.; Li, Y. Epigenetic disruption and glucose homeostasis changes following low-dose maternal bisphenol A exposure. Toxicol. Res. 2016, 5, 1400–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafei, A.; Ramzy, M.M.; Hegazy, A.I.; Husseny, A.K.; El-Hadary, U.G.; Taha, M.M.; Mosa, A.A. The molecular mechanisms of action of the endocrine disrupting chemical bisphenol A in the development of cancer. Gene 2018, 647, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Schaeberle, C.M.; Rubin, B.S.; Sonnenschein, C.; Soto, A.M. The male mammary gland: A target for the xenoestrogen bisphenol A. Reprod. Toxicol. 2013, 37, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durando, M.; Kass, L.; Piva, J.; Sonnenschein, C.; Soto, A.M.; Luque, E.H.; Munoz-de-Toro, M. Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ. Health Perspect. 2007, 115, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.J.; Maffini, M.V.; Ucci, A.A.; Sonnenschein, C.; Soto, A.M. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal bisphenol A exposure. Reprod. Toxicol. 2007, 23, 383–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhan, A.; Hussain, I.; Ansari, K.I.; Bobzean, S.A.; Perrotti, L.I.; Mandal, S.S. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-A and diethylstilbestrol. J. Mol. Biol. 2014, 426, 3426–3441. [Google Scholar] [CrossRef]
- Weng, Y.I.; Hsu, P.Y.; Liyanarachchi, S.; Liu, J.; Deatherage, D.E.; Huang, Y.W.; Zuo, T.; Rodriguez, B.; Lin, C.H.; Cheng, A.L.; et al. Epigenetic influences of low-dose bisphenol A in primary human breast epithelial cells. Toxicol. Appl. Pharmacol. 2010, 248, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Kanao, H.; Enomoto, T.; Kimura, T.; Fujita, M.; Nakashima, R.; Ueda, Y.; Ueno, Y.; Miyatake, T.; Yoshizaki, T.; Buzard, G.S.; et al. Overexpression of LAMP3/TSC403/DC-LAMP promotes metastasis in uterine cervical cancer. Cancer Res. 2005, 65, 8640–8645. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.Y.; Fukuda, T.; Yang, L.; Zaha, H.; Akanuma, H.; Zeng, Q.; Yoshinaga, J.; Sone, H. Effects of bisphenol A exposure on the proliferation and senescence of normal human mammary epithelial cells. Cancer Biol. Ther. 2012, 13, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Tilghman, S.L.; Bratton, M.R.; Segar, H.C.; Martin, E.C.; Rhodes, L.V.; Li, M.; McLachlan, J.A.; Wiese, T.E.; Nephew, K.P.; Burow, M.E. Endocrine disruptor regulation of microRNA expression in breast carcinoma cells. PLoS ONE 2012, 7, e32754. [Google Scholar] [CrossRef]
- Li, X.; Xie, W.; Xie, C.; Huang, C.; Zhu, J.; Liang, Z.; Deng, F.; Zhu, M.; Zhu, W.; Wu, R.; et al. Curcumin modulates miR-19/PTEN/AKT/p53 axis to suppress bisphenol A-induced MCF-7 breast cancer cell proliferation. Phytother. Res. 2014, 28, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Avissar-Whiting, M.; Veiga, K.R.; Uhl, K.M.; Maccani, M.A.; Gagne, L.A.; Moen, E.L.; Marsit, C.J. Bisphenol A exposure leads to specific microRNA alterations in placental cells. Reprod. Toxicol. 2010, 29, 401–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, G.S.; Ye, S.H.; Birch, L.; Ho, S.M.; Kannan, K. Serum bisphenol A pharmacokinetics and prostate neoplastic responses following oral and subcutaneous exposures in neonatal Sprague-Dawley rats. Reprod. Toxicol. 2011, 31, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, G.S.; Hu, W.Y.; Shi, G.B.; Hu, D.P.; Majumdar, S.; Li, G.; Huang, K.; Nelles, J.L.; Ho, S.M.; Walker, C.L.; et al. Bisphenol A promotes human prostate stem-progenitor cell self-renewal and increases in vivo carcinogenesis in human prostate epithelium. Endocrinology 2014, 155, 805–817. [Google Scholar] [CrossRef]
- Cheong, A.; Zhang, X.; Cheung, Y.Y.; Tang, W.Y.; Chen, J.; Ye, S.H.; Medvedovic, M.; Leung, Y.K.; Prins, G.S.; Ho, S.M. DNA methylome changes by estradiol benzoate and bisphenol A links early-life environmental exposures to prostate cancer risk. Epigenetics 2016, 11, 674–689. [Google Scholar] [CrossRef]
- Prins, G.S.; Ye, S.H.; Birch, L.; Zhang, X.; Cheong, A.; Lin, H.; Calderon-Gierszal, E.; Groen, J.; Hu, W.Y.; Ho, S.M.; et al. Prostate Cancer Risk and DNA Methylation Signatures in Aging Rats following Developmental BPA Exposure: A Dose-Response Analysis. Environ. Health Perspect. 2017, 125, 077007. [Google Scholar] [CrossRef]
- Renaud, L.; Huff, M.; da Silveira, W.A.; Angert, M.; Haas, M.; Hardiman, G. Genome-Wide Analysis of Low Dose Bisphenol-A (BPA) Exposure in Human Prostate Cells. Curr. Genom. 2019, 20, 260–274. [Google Scholar] [CrossRef]
- Fatma Karaman, E.; Caglayan, M.; Sancar-Bas, S.; Ozal-Coskun, C.; Arda-Pirincci, P.; Ozden, S. Global and region-specific post-transcriptional and post-translational modifications of bisphenol A in human prostate cancer cells. Environ. Pollut. 2019, 255, 113318. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Maffini, M.V.; Sonnenschein, C.; Rubin, B.S.; Soto, A.M. Bisphenol-A and the great divide: A review of controversies in the field of endocrine disruption. Endocr. Rev. 2009, 30, 75–95. [Google Scholar] [CrossRef]
- Singh, S.; Li, S.S. Epigenetic effects of environmental chemicals bisphenol A and phthalates. Int. J. Mol. Sci. 2012, 13, 10143–10153. [Google Scholar] [CrossRef]
- FitzGerald, R.; Hank, V.L.; Civitella, C.; Castoldi, A.F.; Bernasconi, G. Assessment of new information on Bisphenol S (BPS) submitted in response to the Decision 1 under REACH Regulation (EC) No. 1907/2006. EFSA 2020, 17, 1844e. [Google Scholar] [CrossRef]
- Moon, M.K. Concern about the Safety of Bisphenol A Substitutes. Diabetes Metab. J. 2019, 43, 46–48. [Google Scholar] [CrossRef] [PubMed]
- Thoene, M.; Dzika, E.; Gonkowski, S.; Wojtkiewicz, J. Bisphenol S in Food Causes Hormonal and Obesogenic Effects Comparable to or Worse than Bisphenol A: A Literature Review. Nutrients 2020, 12, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, N.; Maguer-Satta, V.; Guyot, B. Early Steps of Mammary Stem Cell Transformation by Exogenous Signals; Effects of Bisphenol Endocrine Disrupting Chemicals and Bone Morphogenetic Proteins. Cancers 2019, 11, 1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cimmino, I.; Fiory, F.; Perruolo, G.; Miele, C.; Beguinot, F.; Formisano, P.; Oriente, F. Potential Mechanisms of Bisphenol A (BPA) Contributing to Human Disease. Int. J. Mol. Sci. 2020, 21, 5761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165761
Cimmino I, Fiory F, Perruolo G, Miele C, Beguinot F, Formisano P, Oriente F. Potential Mechanisms of Bisphenol A (BPA) Contributing to Human Disease. International Journal of Molecular Sciences. 2020; 21(16):5761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165761
Chicago/Turabian StyleCimmino, Ilaria, Francesca Fiory, Giuseppe Perruolo, Claudia Miele, Francesco Beguinot, Pietro Formisano, and Francesco Oriente. 2020. "Potential Mechanisms of Bisphenol A (BPA) Contributing to Human Disease" International Journal of Molecular Sciences 21, no. 16: 5761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165761