Divergent Effect of Cigarette Smoke on Innate Immunity in Inflammatory Bowel Disease: A Nicotine-Infection Interaction


Abstract
:1. Introduction
2. Cigarette Smoke Is Detrimental in Crohn’s Disease
Factors Affecting the Severity of Symptoms in CD Smokers
3. Cigarette Smoke Is a Protective Factor in Ulcerative Colitis
Nicotine Replacment
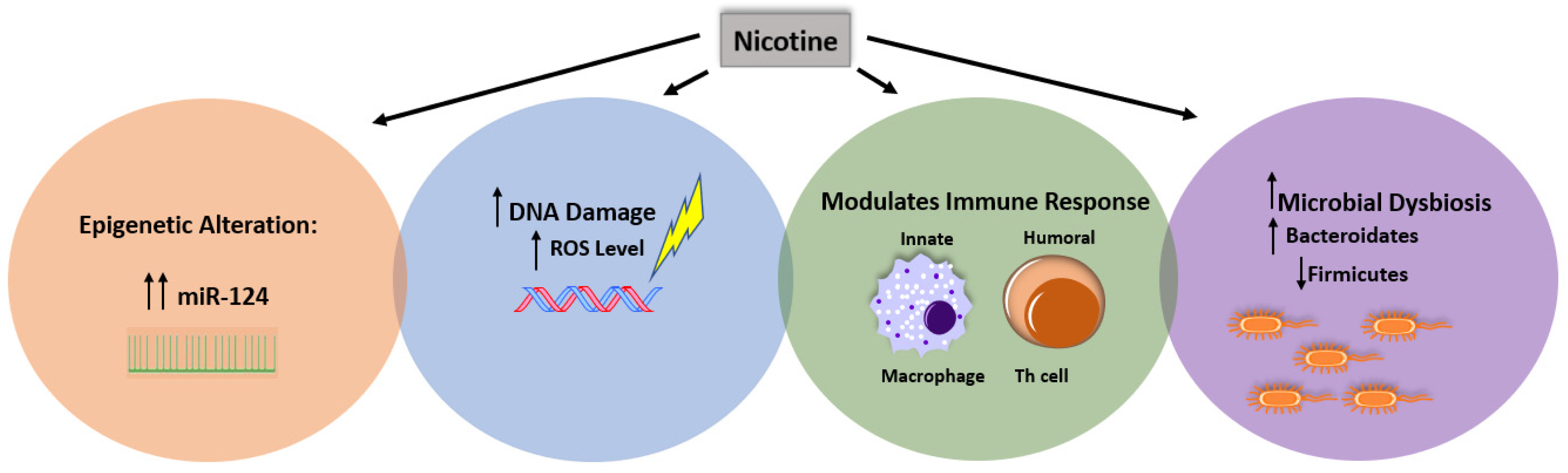
4. The Contradictory Effect of CS on IBD Subsets Is Due to Nicotine-Multifactorial Interaction
4.1. Nicotine and the Other Factors
4.2. Nicotine and Immune Response
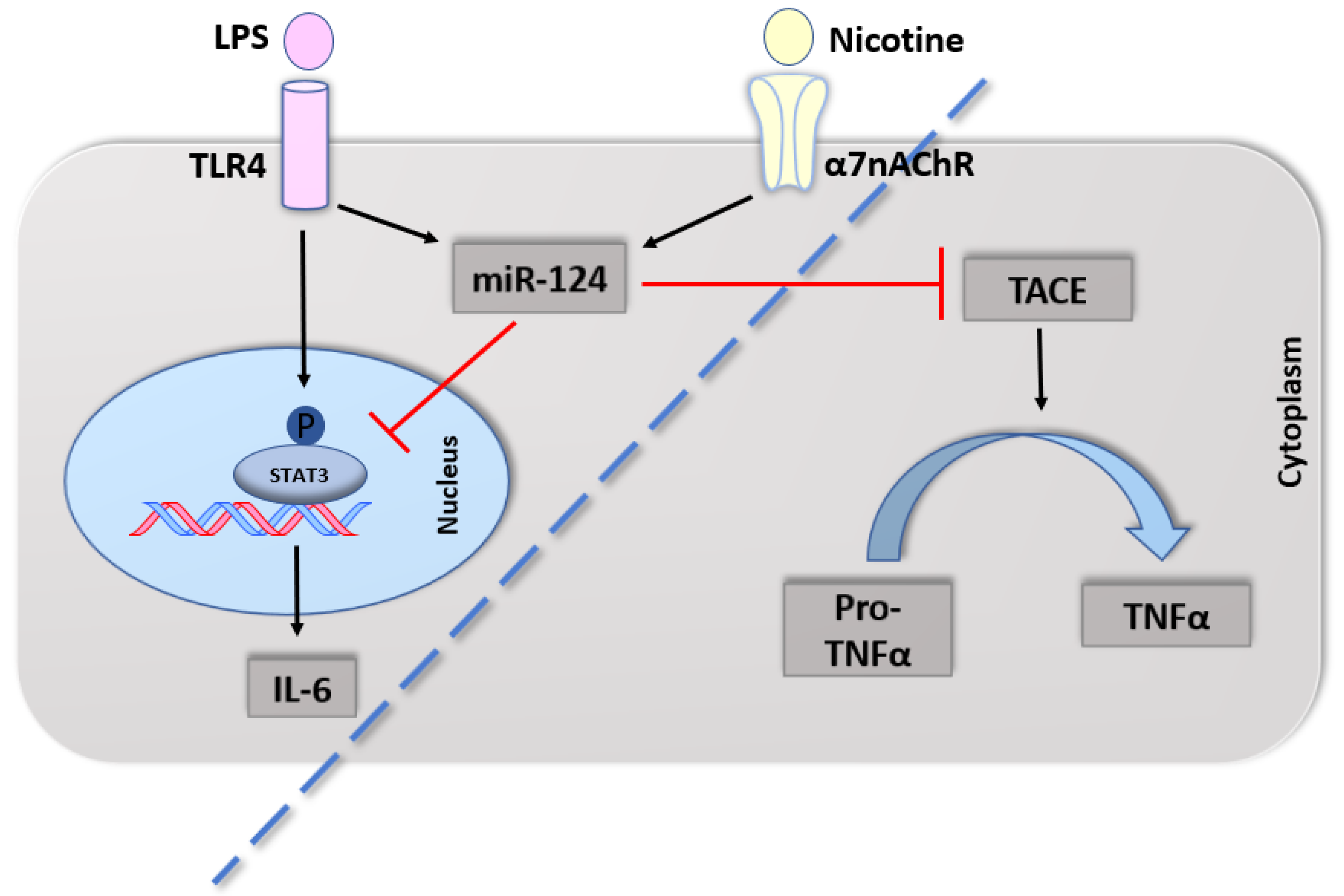
4.2.1. Cytokines
4.2.2. α7nAChR in Immune Cells
4.2.3. Microbial Dysbiosis
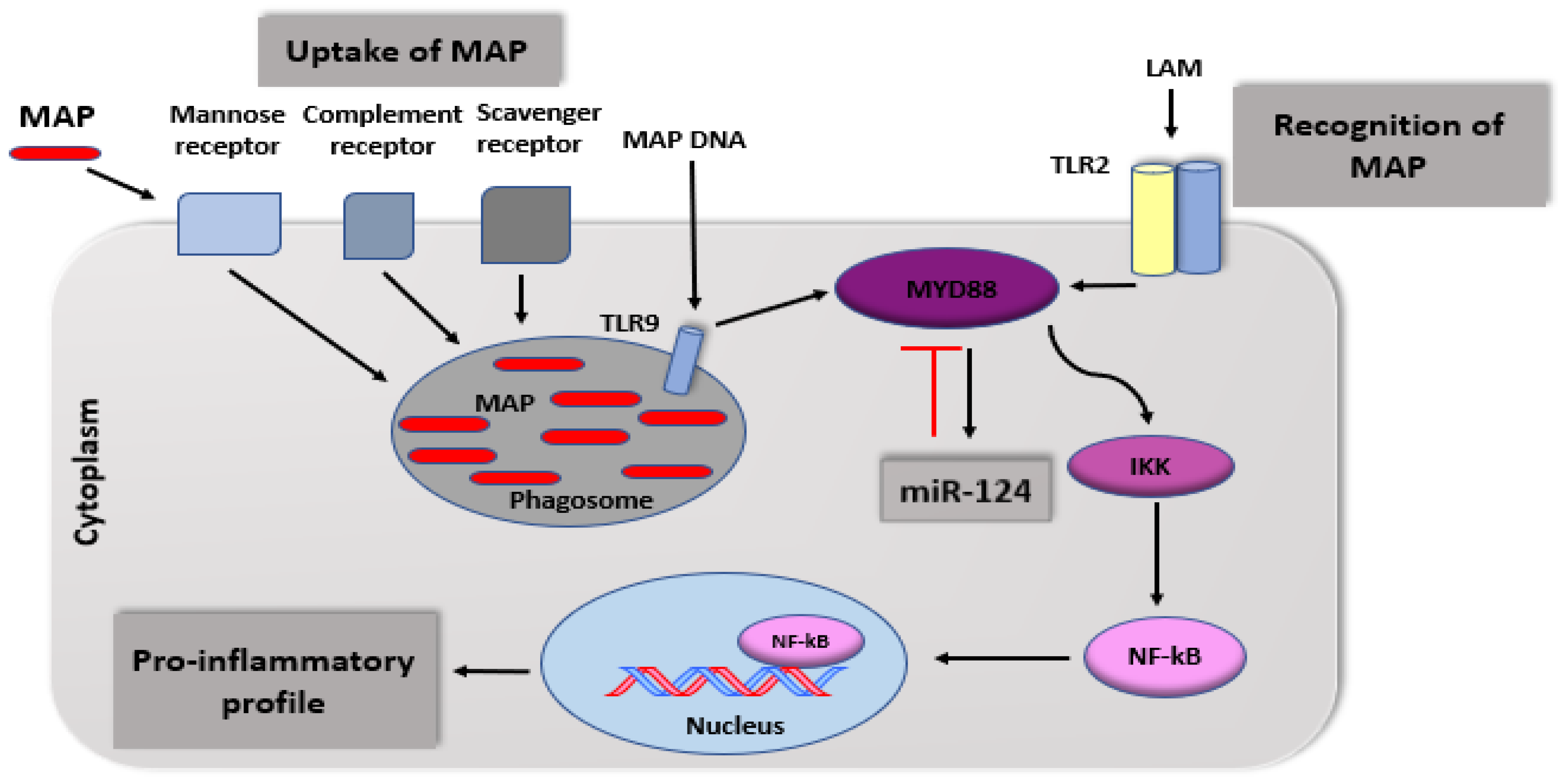
4.2.4. Epigenetic Susceptibility
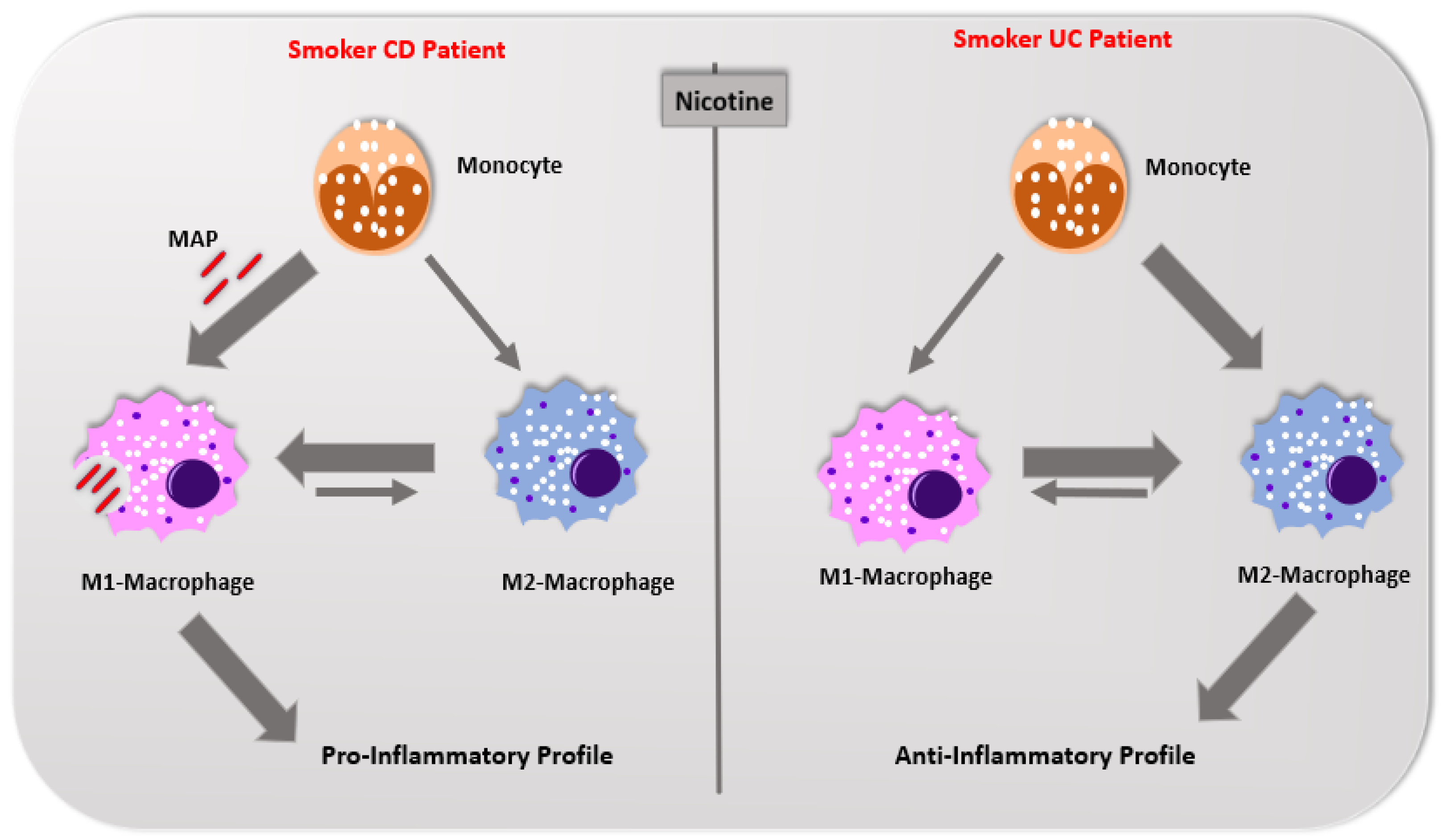
5. Differences in Immunological Response Profile between Crohn’s Disease and Ulcerative Colitis
6. Summary and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Hendrickson, B.A.; Gokhale, R.; Cho, J.H. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin. Microbiol. Rev. 2002, 15, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; Goethel, A.; Bedrani, L.; Croitoru, K. Determinants of IBD heritability: Genes, bugs, and more. Inflamm. Bowel Dis. 2018, 24, 1133–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleynen, I.; Boucher, G.; Jostins, L.; Schumm, L.P.; Zeissig, S.; Ahmad, T.; Andersen, V.; Andrews, J.M.; Annese, V.; Brand, S.; et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: A genetic association study. Lancet 2016, 387, 156–167. [Google Scholar] [CrossRef] [Green Version]
- De Hertogh, G.; Aerssens, J.; Geboes, K.P.; Geboes, K. Evidence for the involvement of infectious agents in the pathogenesis of Crohn’s disease. World J. Gastroenterol. 2008, 14, 845. [Google Scholar] [CrossRef] [Green Version]
- Naser, S.A.; Ghobrial, G.; Romero, C.; Valentine, J.F. Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn’s disease. Lancet 2004, 364, 1039–1044. [Google Scholar] [CrossRef]
- Chamberlin, W.; Graham, D.Y.; Hulten, K.; El-Zimaity, H.M.; Schwartz, M.R.; Naser, S.; Shafran, I.; El-Zaatari, F.A. Mycobacterium avium subsp. paratuberculosis as one cause of Crohn’s disease. Aliment. Pharm. Ther. 2001, 15, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Saxegaard, F.I. Isolation of Mycobacterium paratuberculosis from intestinal mucosa and mesenteric lymph nodes of goats by use of selective Dubos medium. J. Clin. Microbiol. 1985, 22, 312–313. [Google Scholar] [CrossRef] [Green Version]
- Parkes, G.C.; Whelan, K.; Lindsay, J.O. Smoking in inflammatory bowel disease: Impact on disease course and insights into the aetiology of its effect. J. Crohns Colitis 2014, 8, 717–725. [Google Scholar] [CrossRef]
- Lakatos, P.L.; Szamosi, T.; Lakatos, L. Smoking in inflammatory bowel diseases: Good, bad or ugly? World J. Gastroenterol. 2007, 13, 6134. [Google Scholar] [CrossRef] [PubMed]
- AlQasrawi, D.; Abdelli, L.S.; Naser, S.A. Mystery Solved: Why Smoke Extract Worsens Disease in Smokers with Crohn’s Disease and Not Ulcerative Colitis? Gut MAP! Microorganisms 2020, 8, 666. [Google Scholar] [CrossRef] [PubMed]
- Somerville, K.W.; Logan, R.F.; Edmond, M.; Langman, M.J. Smoking and Crohn’s disease. Br. Med. J. (Clin. Res. Ed.) 1984, 289, 954–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holdstock, G.; Savage, D.A.; Wright, R. Should patients with inflammatory bowel disease smoke? Br. Med. J. (Clin. Res. Ed.) 1984, 288, 862. [Google Scholar] [CrossRef] [Green Version]
- Timmer, A.; Sutherland, L.R.; Martin, F. Oral contraceptive use and smoking are risk factors for relapse in Crohn’s disease. Gastroenterology 1998, 114, 1143–1150. [Google Scholar] [CrossRef]
- Seksik, P.; Nion-Larmurier, I.; Sokol, H.; Beaugerie, L.; Cosnes, J. Effects of light smoking consumption on the clinical course of Crohn’s disease. Inflamm. Bowel. Dis. 2009, 15, 734–741. [Google Scholar] [CrossRef]
- Lindberg, E.; Järnerot, G.; Huitfeldt, B. Smoking in Crohn’s disease: Effect on localisation and clinical course. Gut 1992, 33, 779–782. [Google Scholar] [CrossRef] [Green Version]
- van der Heide, F.; Dijkstra, A.; Weersma, R.K.; Albersnagel, F.A.; van der Logt, E.M.; Faber, K.N.; Sluiter, W.J.; Kleibeuker, J.H.; Dijkstra, G. Effects of active and passive smoking on disease course of Crohn’s disease and ulcerative colitis. Inflamm. Bowel. Dis. 2009, 15, 1199–1207. [Google Scholar] [CrossRef]
- Aldhous, M.C.; Satsangi, J. The impact of smoking in Crohn’s disease: No smoke without fire. Frontline Gastroenterol. 2010, 1, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Picco, M.F.; Bayless, T.M. Tobacco consumption and disease duration are associated with fistulizing and stricturing behaviors in the first 8 years of Crohn’s disease. Am. J. Gastroenterol. 2003, 98, 363–368. [Google Scholar] [CrossRef]
- Louis, E.; Michel, V.; Hugot, J.P.; Reenaers, C.; Fontaine, F.; Delforge, M.; El Yafi, F.; Colombel, J.F.; Belaiche, J. Early development of stricturing or penetrating pattern in Crohn’s disease is influenced by disease location, number of flares, and smoking but not by NOD2/CARD15 genotype. Gut 2003, 52, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Mahid, S.S.; Minor, K.S.; Stromberg, A.J.; Galandiuk, S. Active and passive smoking in childhood is related to the development of inflammatory bowel disease. Inflamm. Bowel. Dis. 2007, 13, 431–438. [Google Scholar] [CrossRef]
- Samuelsson, S.M. Ulcerative Colitis in the County of Uppsala 1945–64: Clinical, Epidemiological and Socio-Medical Aspects. Ph.D. Thesis, Acta Universitatis Upsaliensis, Uppsala, Sweden, 1976; p. 19. [Google Scholar]
- Harries, A.D.; Baird, A.; Rhodes, J. Non-smoking: A feature of ulcerative colitis. Br. Med. J. (Clin. Res. Ed.) 1982, 284, 706. [Google Scholar] [CrossRef] [Green Version]
- Green, J.T.; Rhodes, J.; Ragunath, K.; Thomas, G.A.; Williams, G.T.; Mani, V.; Feyerabend, C.; Russell, M.A. Clinical status of ulcerative colitis in patients who smoke. Am. J. Gastroenterol. 1998, 93, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.; Yanai, H.; Shuster, D.; Rubin, D.T.; Hanauer, S.B. Low-dose smoking resumption in ex-smokers with refractory ulcerative colitis. J. Crohns Colitis 2012, 6, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Lakatos, P.L.; Vegh, Z.; Lovasz, B.D.; David, G.; Pandur, T.; Erdelyi, Z.; Szita, I.; Mester, G.; Balogh, M.; Szipocs, I.; et al. Is current smoking still an important environmental factor in inflammatory bowel diseases? Results from a population-based incident cohort. Inflamm. Bowel Dis. 2013, 19, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Mahid, S.S.; Minor, K.S.; Soto, R.E.; Hornung, C.A.; Galandiuk, S. Smoking and inflammatory bowel disease: A meta-analysis. Inmayo Clin. Proc. (Elsevier) 2006, 81, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Aldhous, M.C.; Drummond, H.E.; Anderson, N.; Baneshi, M.R.; Smith, L.A.; Arnott, I.D.; Satsangi, J. Smoking habit and load influence age at diagnosis and disease extent in ulcerative colitis. Am. J. Gastroenterol. 2007, 102, 589–597. [Google Scholar] [CrossRef]
- Nakarnura, Y.; Labarthe, D.R. A case-control study of ulcerative colitis with relation to smoking habits and alcohol consumption in Japan. Am. J. Epidemiol. 1994, 140, 902–911. [Google Scholar] [CrossRef]
- Lunney, P.C.; Leong, R.W. Ulcerative colitis, smoking and nicotine therapy. Aliment. Pharm. Ther. 2012, 36, 997–1008. [Google Scholar] [CrossRef]
- Pullan, R.D.; Rhodes, J.; Ganesh, S.; Mani, V.; Morris, J.S.; Williams, G.T.; Newcombe, R.G.; Russell, M.; Feyerabend, C.; Thomas, G.; et al. Transdermal nicotine for active ulcerative colitis. N. Engl. J. Med. 1994, 330, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Tremaine, W.J.; Leighton, J.A.; Lawson, G.M.; Zins, B.J.; Compton, R.F.; Mays, D.C.; Lipsky, J.J.; Batts, K.P.; Offord, K.P.; et al. Nicotine tartrate liquid enemas for mildly to moderately active left-sided ulcerative colitis unresponsive to first-line therapy: A pilot study. Aliment. Pharm. Ther. 1997, 11, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Lashner, B.A.; Hanauer, S.B.; Silverstein, M.D. Testing nicotine gum for ulcerative colitis patients. Digest. Dis. Sci. 1990, 35, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; W Ren, J.; CM Wong, C.; KK Wu, W.; X Ren, S.; Shen, J.; LY Chan, R.; H Cho, C. Effects of cigarette smoke and its active components on ulcer formation and healing in the gastrointestinal mucosa. Curr. Med. Chem. 2012, 19, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Peña, G.; Cai, B.; Ramos, L.; Vida, G.; Deitch, E.A.; Ulloa, L. Cholinergic regulatory lymphocytes re-establish neuromodulation of innate immune responses in sepsis. J. Immunol. 2011, 187, 718–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatti, M.A.; Hodgson, H.J. Peripheral blood pro-inflammatory cytokine profile in active inflammatory bowel disease (IBD) differ between smokers and non-smokers. Gut 1997, 40, 1–3. [Google Scholar]
- Wang, H.; Liao, H.; Ochani, M.; Justiniani, M.; Lin, X.; Yang, L.; Al-Abed, Y.; Wang, H.; Metz, C.; Miller, E.J.; et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004, 10, 1216–1221. [Google Scholar] [CrossRef]
- Li, X.; Naser, S.A.; Khaled, A.; Hu, H.; Li, X. When old metagenomic data meet newly sequenced genomes, a case study. PLoS ONE 2018, 13, e0198773. [Google Scholar] [CrossRef]
- Berkowitz, L.; Schultz, B.M.; Salazar, G.A.; Pardo-Roa, C.; Sebastián, V.P.; Álvarez-Lobos, M.M.; Bueno, S.M. Impact of cigarette smoking on the gastrointestinal tract inflammation: Opposing effects in Crohn’s disease and ulcerative colitis. Front. Immunol. 2018, 9, 74. [Google Scholar] [CrossRef] [Green Version]
- Bien, J.; Palagani, V.; Bozko, P. The intestinal microbiota dysbiosis and Clostridium difficile infection: Is there a relationship with inflammatory bowel disease? Ther. Adv. Gastroenter. 2013, 6, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, J.L.; Hedin, C.R.; Koutsoumpas, A.; Ng, S.C.; McCarthy, N.E.; Prescott, N.J.; Pessoa-Lopes, P.; Mathew, C.G.; Sanderson, J.; Hart, A.L.; et al. Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm. Bowel. Dis. 2012, 18, 1092–1192. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, L.; Zeitz, J.; Mwinyi, J.; Sutter-Minder, E.; Rehman, A.; Ott, S.J.; Steurer-Stey, C.; Frei, A.; Frei, P.; Scharl, M.; et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS ONE 2013, 8, e59260. [Google Scholar] [CrossRef] [PubMed]
- Jenke, A.C.; Zilbauer, M. Epigenetics in inflammatory bowel disease. Curr. Opin. Gastroen. 2012, 28, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, Q.; Gui, H.; Xu, D.P.; Yang, Y.L.; Su, D.F.; Liu, X. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. 2013, 23, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Wan, J.J.; Sun, Y.; Wu, T.; Wang, P.Y.; Du, P.; Su, D.F.; Yang, Y.; Liu, X. Nicotine protects against DSS colitis through regulating microRNA-124 and STAT3. J. Mol. Med. 2017, 95, 221–233. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, T.; Chen, W.; Chen, Y.; Li, M.; Ren, L.; Chen, J.; Cao, R.; Feng, Y.; Zhang, H.; et al. MicroRNA-124 promotes intestinal inflammation by targeting aryl hydrocarbon receptor in Crohn’s disease. J. Crohns. Colitis 2016, 10, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Wang, P.Y.; Wan, J.J.; Zhang, Y.; Wei, J.; Sun, Y.; Liu, X. MicroRNA124-IL6R mediates the effect of nicotine in inflammatory bowel disease by shifting Th1/Th2 balance toward Th1. Front. Immunol. 2020, 11, 235. [Google Scholar] [CrossRef] [Green Version]
- Naser, S.A.; Schwartz, D.; Shafran, I. Isolation of Mycobacterium avium subsp paratuberculosis from breast milk of Crohn’s disease patients. Am. J. Gastroenterol. 2000, 95, 1094. [Google Scholar] [CrossRef]
- Qasem, A.; Naser, S.A. TNFα inhibitors exacerbate Mycobacterium paratuberculosis infection in tissue culture: A rationale for poor response of patients with Crohn’s disease to current approved therapy. BMJ Open Gastroenterol. 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Qasem, A.; Naser, A.E.; Naser, S.A. The alternate effects of anti-TNFα therapeutics and their role in mycobacterial granulomatous infection in Crohn’s disease. Expert Rev. Anti-Infect. Ther. 2017, 15, 637–643. [Google Scholar] [CrossRef]
- Qasem, A.; Safavikhasraghi, M.; Naser, S.A. A single capsule formulation of RHB-104 demonstrates higher anti-microbial growth potency for effective treatment of Crohn’s disease associated with Mycobacterium avium subspecies paratuberculosis. Gut Pathog. 2016, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Qasem, A.; Abdel-Aty, A.; Abu-Suwa, H.; Naser, S.A. Oxidative stress due to Mycobacterium avium subspecies paratuberculosis (MAP) infection upregulates selenium-dependent GPx activity. Gut Pathog. 2016, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qasem, A.; Ramesh, S.; Naser, S.A. Genetic polymorphisms in tumour necrosis factor receptors (TNFRSF1A/1B) illustrate differential treatment response to TNFα inhibitors in patients with Crohn’s disease. BMJ Open Gastroenterol. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.L.; Qasem, A.; Sharp, R.C.; Abdelli, L.S.; Naser, S.A. Systematic review and meta-analysis on the association of tuberculosis in Crohn’s disease patients treated with tumor necrosis factor-α inhibitors (Anti-TNFα). World J. Gastroenterol. 2018, 24, 2764. [Google Scholar] [CrossRef] [PubMed]
- Naser, A.; Qasem, A.; Naser, S.A. Mycobacterial infection influences bone biomarker levels in patients with Crohn’s disease. Can. J. Physiol. Pharm. 2018, 96, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, R.C.; Naser, E.S.; Alcedo, K.P.; Qasem, A.; Abdelli, L.S.; Naser, S.A. Development of multiplex PCR and multi-color fluorescent in situ hybridization (m-FISH) coupled protocol for detection and imaging of multi-pathogens involved in inflammatory bowel disease. Gut Pathogens 2018, 10, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Naser, A.; Odeh, A.K.; Sharp, R.C.; Qasem, A.; Beg, S.; Naser, S.A. Polymorphisms in TNF Receptor Superfamily 1B (TNFRSF1B:rs3397) are Linked to Mycobacterium avium paratuberculosis Infection and Osteoporosis in Rheumatoid Arthritis. Microorganisms 2019, 7, 646. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Peyrin-Biroulet, L.; Eisenhut, M.; Shin, J.I. IBD immunopathogenesis: A comprehensive review of inflammatory molecules. Autoimmun. Rev. 2017, 16, 416–426. [Google Scholar] [CrossRef]
- Van Crevel, R.; Ottenhoff, T.H.; Van Der Meer, J.W. Innate immunity to Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2002, 15, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.D.; Yew, W.W.; Ganguly, D.; Davidow, A.L.; Reichman, L.B.; Dheda, K.; Rook, G.A. Smoking and tuberculosis: The epidemiological association and immunopathogenesis. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 291–298. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, S.M.; Coleman, M.M.; Chew, W.M.; Morrow, C.; McLaughlin, A.M.; Gleeson, L.E.; O’Sullivan, M.P.; Keane, J. Cigarette smoking impairs human pulmonary immunity to Mycobacterium tuberculosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Stitzel, J.A.; Bai, A.; Zambrano, C.A.; Phillips, M.; Marrack, P.; Chan, E.D. Nicotine impairs macrophage control of Mycobacterium tuberculosis. Am. J. Respir. Crit. Care Med. 2017, 57, 324–333. [Google Scholar]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Nizri, E.; Irony-Tur-Sinai, M.; Lory, O.; Orr-Urtreger, A.; Lavi, E.; Brenner, T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J. Immunol. 2009, 183, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sopori, M. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002, 2, 372–377. [Google Scholar] [CrossRef]
- Singh, S.P.; Kalra, R.; Puttfarcken, P.; Kozak, A.; Tesfaigzi, J.; Sopori, M.L. Acute and chronic nicotine exposures modulate the immune system through different pathways. Toxicol. Appl. Pharm. 2000, 164, 65–72. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Reference & Published Year | National Origin | Type of Study | Type of Enrollment | Number of Participants | Main Conclusion |
|---|---|---|---|---|---|
| Holdstock et al. (1984) [14] | UK | Case-Control Study | Retrospective | 172 IBD patients | Smoking leads to CD rather than UC |
| Somerville et al. (1984) [13] | UK | Case-Control Study | Retrospective | 82 variables | CD patients are more likely to smoke |
| Timmer et al. (1998) [15] | Canada | Cohort | Prospective | 152 CD patients | Increased rate of relapses in CD smoker patients |
| Lindberg et al. (1992) [17] | France | Case-Control Study | Retrospective | 231 CD patients | CD heavy smokers have an increased risk of surgery |
| Seksik et al. (1995–2008) [16] | France | Cohort | Prospective | 3000 CD patients | Smoking has a dose-dependent effect on CD patients |
| Van der Heide et al. (2009) [18] | Netherland | Case-Control Study | Retrospective | 820 CD patients | No unfavorable effects of active smoking on CD |
| Study Reference | National Origin | Type of Study | Type of Enrollment | Number of Participants | Main Conclusion |
|---|---|---|---|---|---|
| Samuelsson et al. (1976) [23] | Sweden | Unknown | Unknown | Unknown | Low rate of smokers among UC patients |
| Harries et al. (1982) [24] | UK | Cohort | Prospective | 230 UC patients | Low rate of smokers among UC patients |
| Nakarnura et al. (1994) [30] | Japan | Case-Control Study | Retrospective | 384 UC patients | The relationship between smoking and UC is dose-dependent |
| Green et al. (1998) [25] | UK | Cohort | Prospective | 51 UC patients | UC is a non-smoker disease |
| Aldhous et al. (2007) [29] | UK | Case-Control Study | Retrospective | 499 IBD patients | UC heavy smokers have healthier colons than UC light smokers |
| Calabrese et al. (2012) [26] | US | Case-Control Study | Retrospective | 15 UC patients | Low doses of smoking can be used as a medication in ex-smoker refractory UC patients |
| Lakatos et al. (2013) [27] | Hangiri | Cohort | Retrospective | 1420 IBD patients | Smoking prevents colectomy in UC smokers |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlQasrawi, D.; Qasem, A.; Naser, S.A. Divergent Effect of Cigarette Smoke on Innate Immunity in Inflammatory Bowel Disease: A Nicotine-Infection Interaction. Int. J. Mol. Sci. 2020, 21, 5801. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165801
AlQasrawi D, Qasem A, Naser SA. Divergent Effect of Cigarette Smoke on Innate Immunity in Inflammatory Bowel Disease: A Nicotine-Infection Interaction. International Journal of Molecular Sciences. 2020; 21(16):5801. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165801
Chicago/Turabian StyleAlQasrawi, Dania, Ahmad Qasem, and Saleh A. Naser. 2020. "Divergent Effect of Cigarette Smoke on Innate Immunity in Inflammatory Bowel Disease: A Nicotine-Infection Interaction" International Journal of Molecular Sciences 21, no. 16: 5801. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165801