Affinity Capture Enrichment versus Affinity Depletion: A Comparison of Strategies for Increasing Coverage of Low-Abundant Human Plasma Proteins


Abstract
:1. Introduction
2. Results
2.1. Head-to-Head Comparison of Plasma Proteome Coverage
2.2. Comparison of the Efficiency of the Removal of High Abundant Plasma Proteins
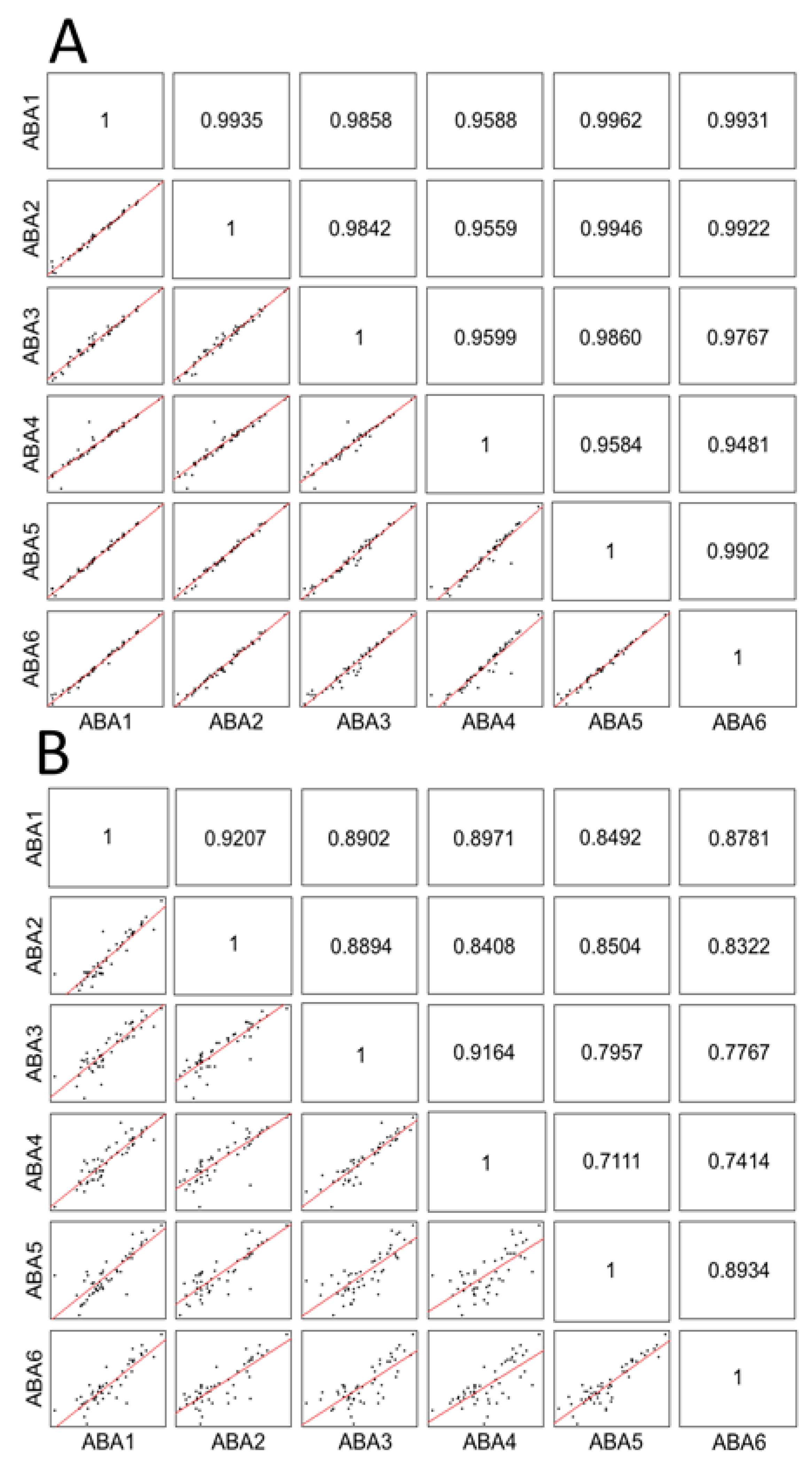
2.3. Comparison of the Reproducibility of Affinity-Based Proteomic Enrichment
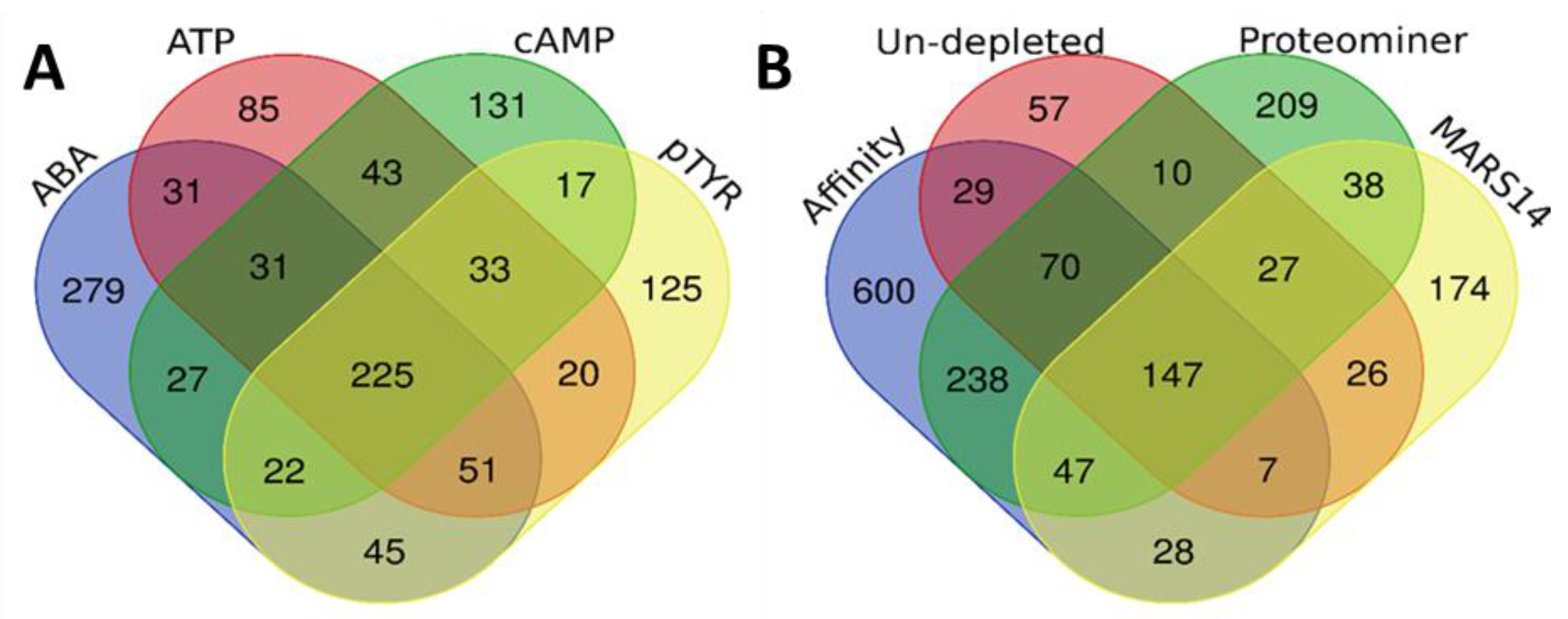
2.4. Qualitative Comparison
3. Discussion
4. Material and Methods
4.1. Plasma Samples and Reagents
4.2. High-Abundant Protein Immunodepletion, Affinity-Based Enrichment and Protein Equalization
4.3. Protein Sample Processing for Proteome Analysis
4.4. Nano-LC-MS/MS Analysis
4.5. Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | para-amino-benzamidine |
| pTYR | ortho-phospho-L-tyrosine |
| cAMP | 8-amino-hexyl-cyclic adenosine monophosphate |
| ATP | adenosine triphosphate |
| MARS14 | multiple affinity removal system human 14 |
| LAP | low-abundant protein |
| MAP | Medium-abundant protein |
| HAP | high-abundant protein |
| TEAB | tri ethyl ammonium bisphosphate |
| IAA | iodoacetamide |
| HCD | higher energy collisional dissociation |
References
- Khoonsari, P.E.; Haggmark, A.; Lonnberg, M.; Mikus, M.; Kilander, L.; Lannfelt, L.; Bergquist, J.; Ingelsson, M.; Nilsson, P.; Kultima, K. Analysis of the Cerebrospinal Fluid Proteome in Alzheimer’s Disease. PLoS ONE 2016, 11, e0150672. [Google Scholar]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Bowden, P.; Zhang, D.; Marshall, J.G. Mass spectrometry of peptides and proteins from human blood. Mass Spectrom. Rev. 2011, 30, 685–732. [Google Scholar] [PubMed]
- Tian, R.; Jiang, X.; Li, X.; Jiang, X.; Feng, S.; Xu, S.; Han, G.; Ye, M.; Zou, H. Biological fingerprinting analysis of the interactome of a kinase inhibitor in human plasma by a chemiproteomic approach. J. Chromatogr. A 2006, 1134, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Reddy, P.J.; Jain, R.; Gollapalli, K.; Moiyadi, A.; Srivastava, S. Proteomic technologies for the identification of disease biomarkers in serum: Advances and challenges ahead. Proteomics 2011, 11, 2139–2161. [Google Scholar] [CrossRef] [PubMed]
- Blume, J.E.; Manning, W.C.; Troiano, G.; Hornburg, D.; Figa, M.; Hesterberg, L.; Platt, T.L.; Zhao, X.; Cuaresma, R.A.; Everley, P.A.; et al. Rapid, deep and precise profiling of the plasma proteome with multi-nanoparticle protein corona. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Zeng, Z.; Hincapie, M.; Pitteri, S.J.; Hanash, S.; Schalkwijk, J.; Hogan, J.M.; Wang, H.; Hancock, W.S. A proteomics platform combining depletion, multi-lectin affinity chromatography (M-LAC), and isoelectric focusing to study the breast cancer proteome. Anal. Chem. 2011, 83, 4845–4854. [Google Scholar] [CrossRef] [Green Version]
- Sennels, L.; Salek, M.; Lomas, L.; Boschetti, E.; Righetti, P.G.; Rappsilber, J. Proteomic analysis of human blood serum using peptide library beads. J. Proteome Res. 2007, 6, 4055–4062. [Google Scholar] [CrossRef]
- Cao, Z.; Tang, H.Y.; Wang, H.; Liu, Q.; Speicher, D.W. Systematic comparison of fractionation methods for in-depth analysis of plasma proteomes. J. Proteome Res. 2012, 11, 3090–3100. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Valentine, S.J.; Plasencia, M.D.; Trimpin, S.; Naylor, S.; Clemmer, D.E. Mapping the human plasma proteome by SCX-LC-IMS-MS. J. Am. Soc. Mass Spectrom. 2007, 18, 1249–1264. [Google Scholar] [CrossRef] [Green Version]
- Pernemalm, M.; Lehtio, J. A novel prefractionation method combining protein and peptide isoelectric focusing in immobilized pH gradient strips. J. Proteome Res. 2013, 12, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chang, C.; Qin, P.; Cao, Q.; Tian, F.; Jiang, J.; Li, X.; Yu, W.; Zhu, Y.; He, F.; et al. Mining the human plasma proteome with three-dimensional strategies by high-resolution Quadrupole Orbitrap Mass Spectrometry. Anal. Chim. Acta 2016, 904, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Nanjappa, V.; Thomas, J.K.; Marimuthu, A.; Muthusamy, B.; Radhakrishnan, A.; Sharma, R.; Ahmad, K.A.; Balakrishnan, L.; Sahasrabuddhe, N.A.; Kumar, S.; et al. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2014, 42, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratantoni, S.A.; Piersma, S.R.; Jimenez, C.R. Comparison of the performance of two affinity depletion spin filters for quantitative proteomics of CSF: Evaluation of sensitivity and reproducibility of CSF analysis using GeLC-MS/MS and spectral counting. Proteom. Clin. Appl. 2010, 4, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.J.; Kaleta, D.T.; Petritis, B.O.; Jiang, H.; Liu, T.; Zhang, X.; Mottaz, H.M.; Varnum, S.M.; Camp, D.G.; Huang, L.; et al. Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol. Cell. Proteom. 2008, 7, 1963–1973. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, B.; Zecha, J.; Heinzlmeir, S.; Medard, G.; Lemeer, S.; Kuster, B. Evaluation of Kinase Activity Profiling Using Chemical Proteomics. ACS Chem. Biol. 2015, 10, 2743–2752. [Google Scholar] [CrossRef]
- Bantscheff, M.; Eberhard, D.; Abraham, Y.; Bastuck, S.; Boesche, M.; Hobson, S.; Mathieson, T.; Perrin, J.; Raida, M.; Rau, C.; et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. [Google Scholar] [CrossRef]
- Lemeer, S.; Zorgiebel, C.; Ruprecht, B.; Kohl, K.; Kuster, B. Comparing immobilized kinase inhibitors and covalent ATP probes for proteomic profiling of kinase expression and drug selectivity. J. Proteome Res. 2013, 12, 1723–1731. [Google Scholar] [CrossRef]
- Adachi, J.; Kishida, M.; Watanabe, S.; Hashimoto, Y.; Fukamizu, K.; Tomonaga, T. Proteome-wide discovery of unknown ATP-binding proteins and kinase inhibitor target proteins using an ATP probe. J. Proteome Res. 2014, 13, 5461–5470. [Google Scholar] [CrossRef]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kim, C.; Cheng, C.Y.; Brown, S.H.; Wu, J.; Kannan, N. Signaling through cAMP and cAMP-dependent protein kinase: Diverse strategies for drug design. Biochim. Biophys. Acta 2008, 1784, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholten, A.; Poh, M.K.; van Veen, T.A.; van, B.B.; Vos, M.A.; Heck, A.J. Analysis of the cGMP/cAMP interactome using a chemical proteomics approach in mammalian heart tissue validates sphingosine kinase type 1-interacting protein as a genuine and highly abundant AKAP. J. Proteome Res. 2006, 5, 1435–1447. [Google Scholar] [CrossRef]
- Kaupp, U.B.; Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. (Shanghai) 2008, 40, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Hofener, M.; Heinzlmeir, S.; Kuster, B.; Sewald, N. Probing SH2-domains using Inhibitor Affinity Purification (IAP). Proteome Sci. 2014, 12, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovska, E.; Svitek, M.; Holada, K.; Petrak, J. Affinity depletion versus relative protein enrichment: A side-by-side comparison of two major strategies for increasing human cerebrospinal fluid proteome coverage. Clin. Proteom. 2019, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Araujo, R.; Howard, M.; Magni, R.; Liotta, L.A.; Luchini, A. Affinity enrichment for mass spectrometry: Improving the yield of low abundance biomarkers. Expert Rev. Proteom. 2018, 15, 353–366. [Google Scholar] [CrossRef]
- Beck, H.C.; Jensen, L.O.; Gils, C.; Ilondo, A.M.M.; Frydland, M.; Hassager, C.; Moller-Helgestad, O.K.; Moller, J.E.; Rasmussen, L.M. Proteomic Discovery and Validation of the Confounding Effect of Heparin Administration on the Analysis of Candidate Cardiovascular Biomarkers. Clin. Chem. 2018, 64, 1474–1484. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Accession Number | Protein Name | [Plasma] µg/mL | % Removed | |||||
|---|---|---|---|---|---|---|---|---|
| MARS14 | Proteominer | ABA | ATP | cAMP | pTYR | |||
| P02768 | Serum Albumin | 42,000 | 99.8 | 99.5 | 98.1 | 99.8 | 99.9 | 99.4 |
| P02671 | Fibrinogen alpha chain | 3800 | 72.8 | 3.7 | 85.2 | 99.3 | 99.5 | 98.5 |
| P01009 | Alpha-1-antitrypsin | 3000 | 99.8 | 90.5 | 93.5 | 98.7 | 99.5 | 96.4 |
| P02787 | Serotransferrin | 2300 | 99.7 | 99.1 | 99.5 | 99.9 | >99.9 # | 99.8 |
| P01024 | Complement C3 | 1730 | 87.3 | 79.5 | 96.5 | 97.3 | 99.8 | 98.9 |
| P01023 | Alpha-2-macroglobulin | 1609 | 92.7 | 96.6 | 98.4 | 99.2 | 99.1 | 99.9 |
| P02647 | Apolipoprotein A-I | 1400 | 99.6 | <1 | 95.2 | 92.9 | 96.8 | 89.3 |
| P00738 | Haptoglobin | 1100 | 99.1 | 98.9 | 97.9 | 99.5 | 99.8 | 98.6 |
| P0DOX5 | Immunoglobulin gamma-1 heavy chain | 1001 | n.d. * | 95.1 | 83.4 | 95.1 | 98.1 | 90.1 |
| P01876 | Immunoglobulin heavy constant alpha 1 | 1000 | >99.9 # | 92.9 | 63.4 | 79.0 | 86.2 | 51.5 |
| P02652 | Apolipoprotein A-II | 780 | 92.7 | 51.9 | 91.3 | 92.9 | 91.6 | 61.4 |
| P02766 | Transthyretin | 770 | <1 | <1 | 94.4 | n.d. * | 99.3 | 97.5 |
| P02763 | Alpha-1-acid glycoprotein 1 | 610 | 99.7 | 99.9 | 99.9 | n.d. * | n.d. * | n.d. * |
| P01871 | Immunoglobulin heavy constant mu | 320 | 96.1 | 1.7 | <1 | <1 | <1 | <1 |
| Correlation Coefficients—50 Most Abundant Plasma Proteins | ||||||||
| Un-Depleted | MARS14 | PM | ABA | ATP | cAMP | pTYR | ||
| Concentration Range [µg/mL] | 42,000–62.36 | 42,000–47 | 42,000–57 | 42,000–47 | 42,000–41 | 42,000–15 | 42,000–27 | |
| Mean | 1.00 | 0.97 | 0.93 | 1.00 | 0.97 | 0.97 | 0.94 | |
| SD | 7.36 × 10−5 | 1.83 × 10−2 | 5.51 × 10−2 | 1.01 × 10−3 | 2.25 × 10−2 | 1.96 × 10−2 | 4.83 × 10−2 | |
| Variance | 5.42 × 10−9 | 3.35 × 10−4 | 3.04 × 10−3 | 1.03 × 10−6 | 5.06 × 10−4 | 3.83 × 10−4 | 2.33 × 10−3 | |
| Correlation Coefficients—50 Least Abundant Plasma Proteins | ||||||||
| Un-Depleted | MARS14 | PM | ABA | ATP | cAMP | pTYR | ||
| Concentration Range [µg/mL] | 1.1–0.0014 | 0.025–6.3 × 10 −6 | 0.02–0.00082 | 0.0348–0.00082 | 0.051–0.0011 | 0.13–0.00082 | 0.063–0.00082 | |
| Mean | 0.94 | 0.98 | 0.83 | 0.85 | 0.86 | 0.99 | 0.83 | |
| SD | 7.35 × 10−2 | 1.82 × 10−2 | 1.18 × 10−1 | 6.15 × 10−2 | 1.39 × 10−1 | 7.48 × 10−3 | 1.86 × 10−1 | |
| Variance | 5.40 × 10−3 | 3.31 × 10−4 | 1.40 × 10−2 | 3.78 × 10−3 | 1.93 × 10−2 | 5.59 × 10−5 | 3.47 × 10−2 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palstrøm, N.B.; Rasmussen, L.M.; Beck, H.C. Affinity Capture Enrichment versus Affinity Depletion: A Comparison of Strategies for Increasing Coverage of Low-Abundant Human Plasma Proteins. Int. J. Mol. Sci. 2020, 21, 5903. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165903
Palstrøm NB, Rasmussen LM, Beck HC. Affinity Capture Enrichment versus Affinity Depletion: A Comparison of Strategies for Increasing Coverage of Low-Abundant Human Plasma Proteins. International Journal of Molecular Sciences. 2020; 21(16):5903. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165903
Chicago/Turabian StylePalstrøm, Nicolai Bjødstrup, Lars Melholt Rasmussen, and Hans Christian Beck. 2020. "Affinity Capture Enrichment versus Affinity Depletion: A Comparison of Strategies for Increasing Coverage of Low-Abundant Human Plasma Proteins" International Journal of Molecular Sciences 21, no. 16: 5903. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165903