Advantages and Pitfalls of Capillary Electrophoresis of Pharmaceutical Compounds and Their Enantiomers in Complex Samples: Comparison of Hydrodynamically Opened and Closed Systems



Abstract
:1. Introduction
2. Results and Discussion
2.1. Comparison of Hydrodynamically Opened and Closed Separation Systems
2.1.1. Repeatability
2.1.2. Efficiency and Resolution
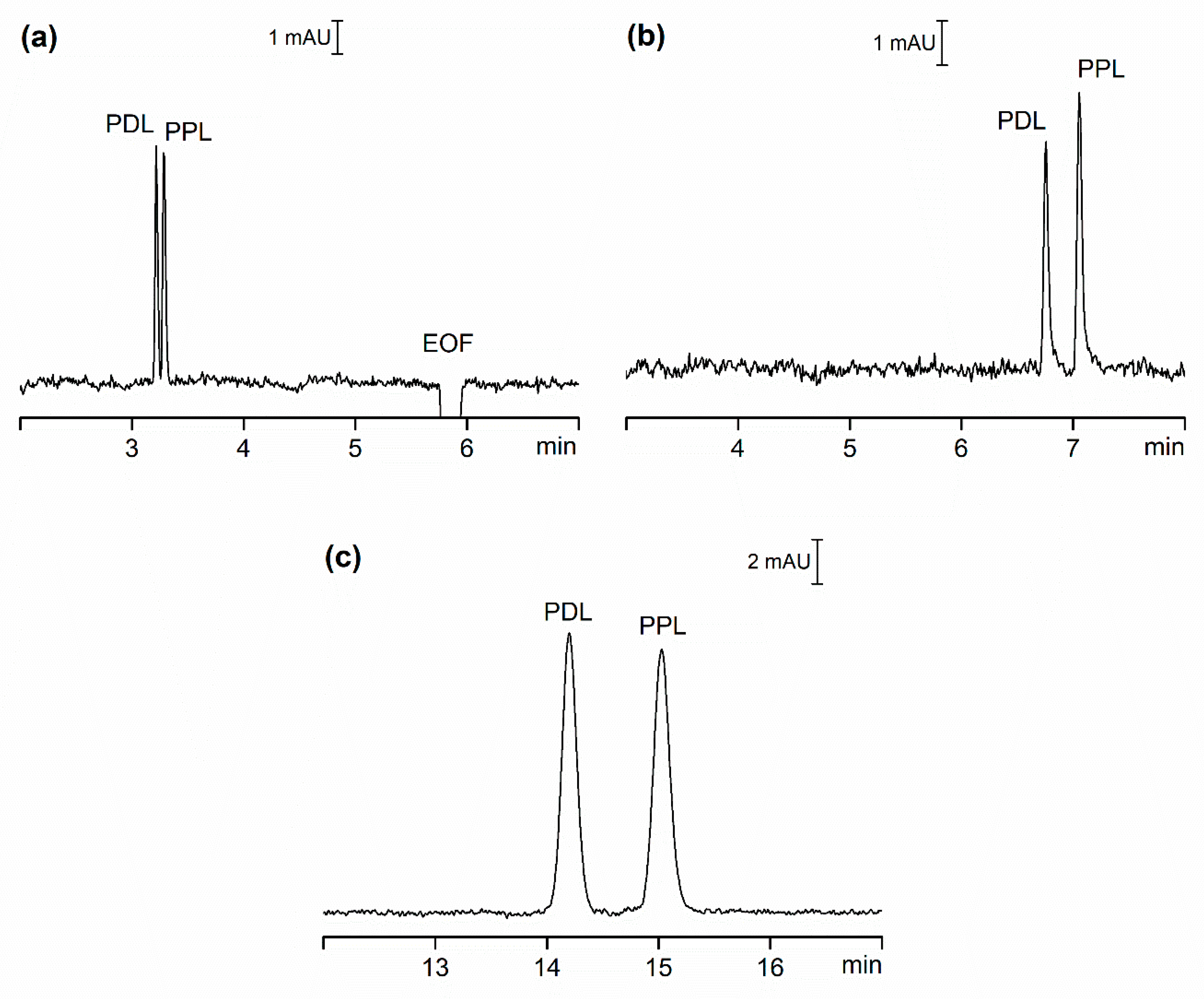
2.1.3. Sensitivity
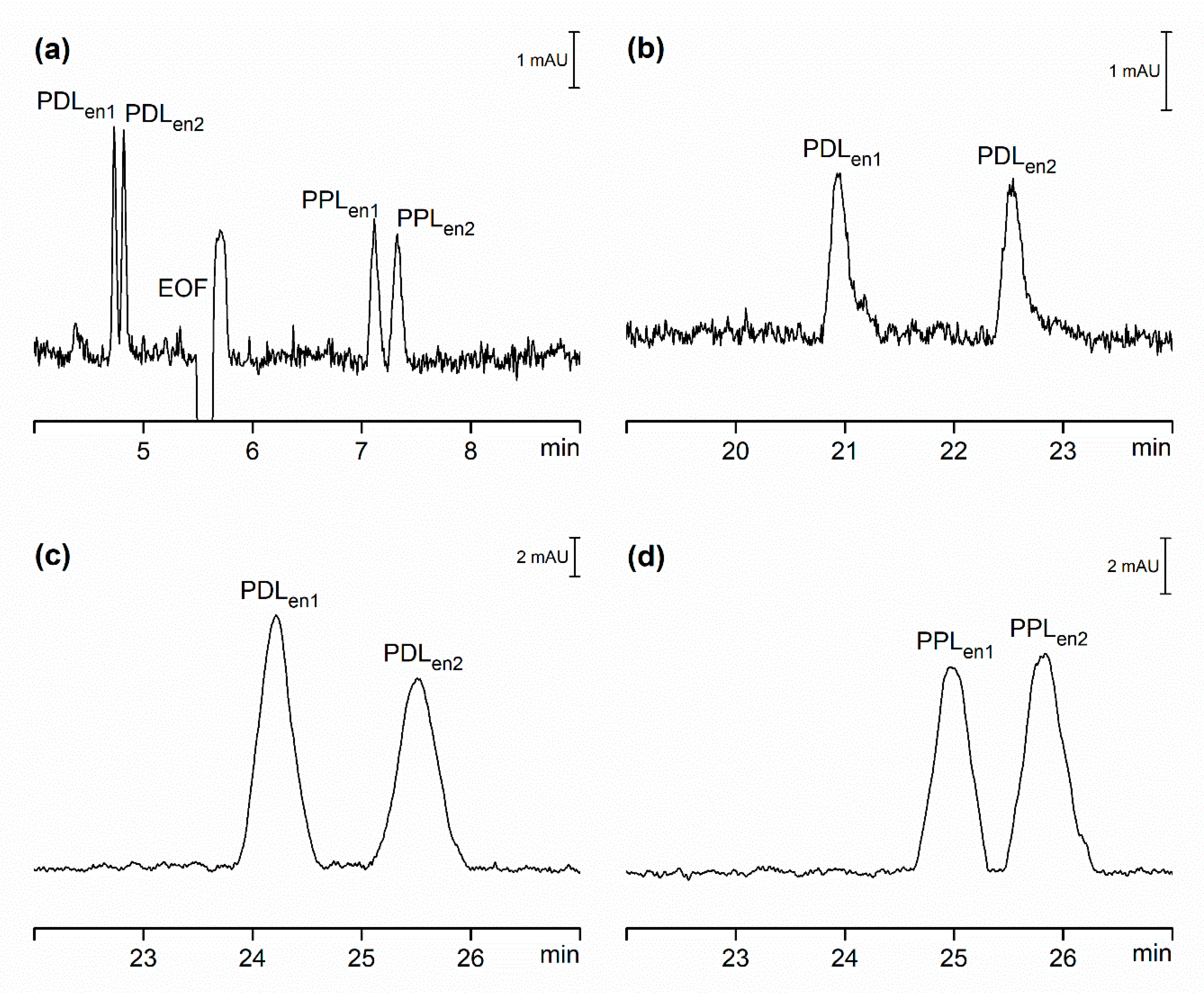
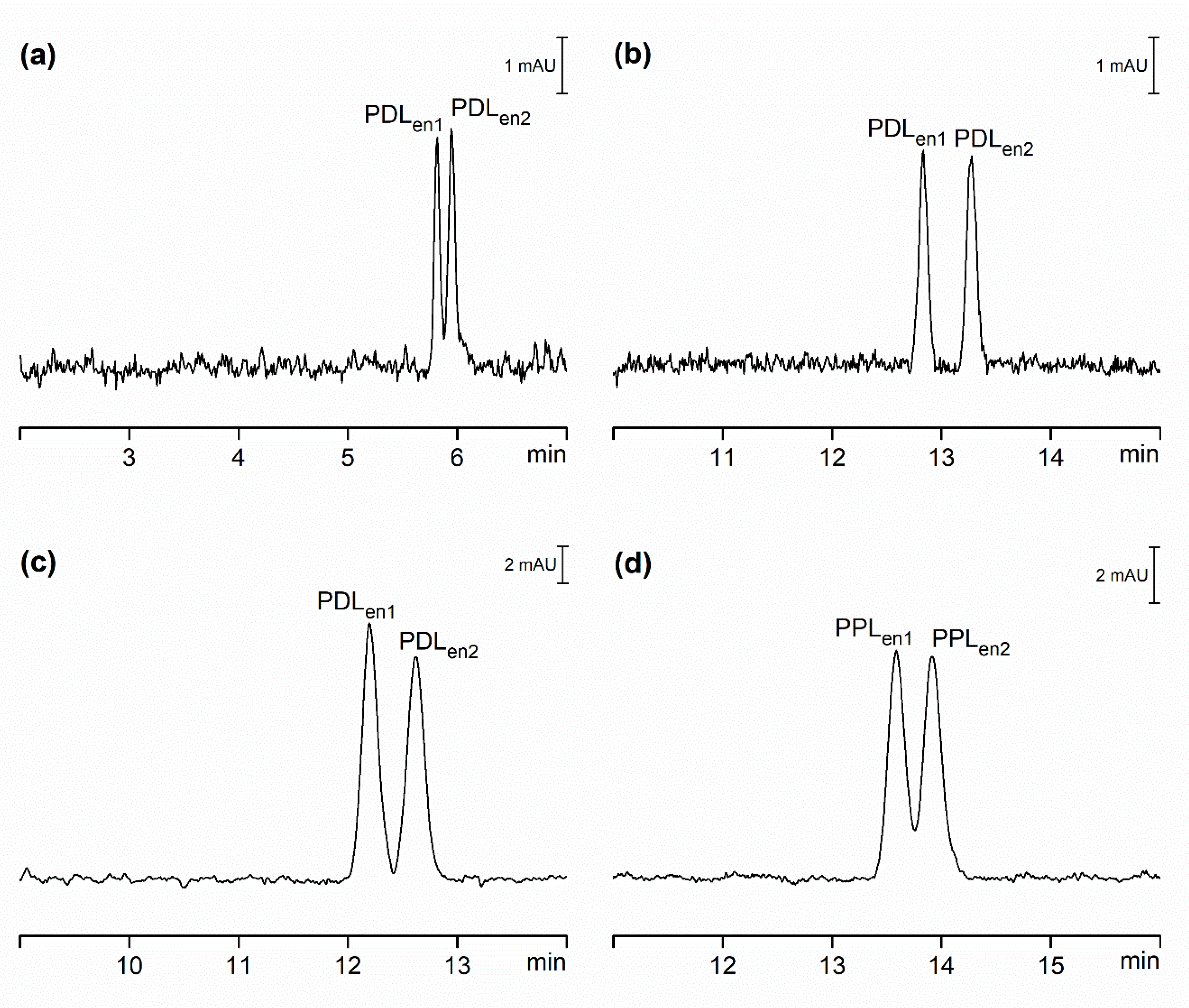
2.2. Comparison of Hydrodynamically Opened and Closed Separation Systems for Chiral Separations
2.3. Comparison of Hydrodynamically Opened and Closed Separation Systems for Analysis of Pharmaceuticals in Serum Samples
3. Materials and Methods
3.1. Instrumentation and Software
3.2. Chemicals and Reagents
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CE | Capillary electrophoresis |
| EOF | Electroosmotic flow |
| HPLC | High-performance liquid chromatography |
| i.d. | Internal diameter |
| LOD | Limit of detection |
| CSS | Hydrodynamically closed separation system with suppressed electroosmotic flow |
| OSS | Hydrodynamically opened separation system |
| µep | Electrophoretic mobility |
| HDF | Hydrodynamic flow |
| PPL | Propranolol |
| PDL | Pindolol |
| OSS-EOF | Hydrodynamically opened separation system with electroosmotic flow |
| OSS-sEOF | Hydrodynamically opened separation system with suppressed electroosmotic flow |
| MHEC | Methylhydroxyethylcelullose |
| DETA | Diethylenetriamine |
| Vinj | Injected volume |
| d | Capillary diameter |
| API | Active pharmaceutical ingredient |
| LOQ | Limit of quantitation |
| UHPLC | Ultra high-performance liquid chromatography |
| CMBCD | Carboxymethyl-β-cyclodextrin |
| µEOF | Electroosmotic mobility |
| µCD | Electrophoretic mobility of chiral selector |
| µCD,eff | Effective mobility of chiral selector |
| CEBCD | Carboxyethyl-β-cyclodextrin |
| ΔµCD | Reduction of electrophoretic mobility due to chiral selector |
| o.d. | Outer diameter |
References
- Yıldırım, S.; Erkmen, C.; Uslu, B. Novel Trends in Analytical Methods for β-Blockers: An Overview of Applications in the Last Decade. Crit. Rev. Anal. Chem. 2020, 1–39. [Google Scholar] [CrossRef] [PubMed]
- El Deeb, S.; Wätzig, H.; El-Hady, D.A.; Van De Griend, C.E.S.; Scriba, G.K.E. Recent advances in capillary electrophoretic migration techniques for pharmaceutical analysis (2013–2015). Electrophoresis 2016, 37, 1591–1608. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M. Recent advances in CE and microchip-CE in clinical applications: 2014 to mid-2017. Electrophoresis 2018, 39, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huhn, C.; Ramautar, R.; Wuhrer, M.; Somsen, G.W. Relevance and use of capillary coatings in capillary electrophoresis-mass spectrometry. Anal. Bioanal. Chem. 2010, 396, 297–314. [Google Scholar] [CrossRef]
- Gaš, B.; Kenndler, E. Dispersive phenomena in electromigration separation methods. Electrophoresis 2000, 21, 3888–3897. [Google Scholar] [CrossRef]
- Gaš, B.; Štědrý, M.; Kenndler, E. Peak broadening in capillary zone electrophoresis. Electrophoresis 1997, 18, 2123–2133. [Google Scholar] [CrossRef]
- Junaid, T.; Wu, X.; Thanukrishnan, H.; Venkataramanan, R. Therapeutic Drug Monitoring. In Clinical Pharmacy Education, Practice and Research; Thomas, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 425–436. [Google Scholar]
- Virtanen, R. Zone electrophoresis in narrow-bore tube employing potentiometric detection. A theoretical and experimental study. Acta Polytech. Scand. Chem. Technol. Ser. 1974, 123, 1–67. [Google Scholar]
- Yin, H.; Keely-Templin, C.; McManigill, D. Preparative capillary electrophoresis with wide-bore capillaries. J. Chromatogr. A 1996, 744, 45–54. [Google Scholar] [CrossRef]
- Valkó, I.E.; Porras, S.P.; Riekkola, M.-L. Capillary electrophoresis with wide-bore capillaries and non-aqueous media. J. Chromatogr. A 1998, 813, 179–186. [Google Scholar] [CrossRef]
- Kaniansky, D.; Masár, M.; Bielčı́ková, J. Electroosmotic flow suppressing additives for capillary zone electrophoresis in a hydrodynamically closed separation system. J. Chromatogr. A 1997, 792, 483–494. [Google Scholar] [CrossRef]
- Mikkers, F.; Everaerts, F.; Verheggen, T. Concentration distributions in free zone electrophoresis. J. Chromatogr. A 1979, 169, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Payán, M.; Ocaña-Gonzalez, J.A.; Fernández-Torres, R.M.; Llobera, A.; Bello-López, M.Á. Recent trends in capillary electrophoresis for complex samples analysis: A review. Electrophoresis 2018, 39, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Hajba, L.; Guttman, A. Recent advances in column coatings for capillary electrophoresis of proteins. TrAC Trends Anal. Chem. 2017, 90, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Štěpánová, S.; Kašička, V. Recent applications of capillary electromigration methods to separation and analysis of proteins. Anal. Chim. Acta 2016, 933, 23–42. [Google Scholar] [CrossRef]
- Reijenga, J.; Kenndler, E. Computational simulation of migration and dispersion in free capillary zone electrophoresis. J. Chromatogr. A 1994, 659, 403–415. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.-J.; Feng, J.-J.; Dong, W.-J.; Lu, Y.-H.; Li, Z.-H.; Riekkola, M.-L. Spermine-graft-dextran non-covalent copolymer as coating material in separation of basic proteins and neurotransmitters by capillary electrophoresis. J. Chromatogr. A 2010, 1217, 5130–5136. [Google Scholar] [CrossRef]
- Kaniansky, D.; Marak, J.; Masár, M.; Iványi, F.; Madajová, V.; Šimuničová, E.; Zelenská, V. Capillary zone electrophoresis in a hydrodynamically closed separation system with enhanced sample loadability. J. Chromatogr. A 1997, 772, 103–114. [Google Scholar] [CrossRef]
- Foret, F.; Křivánková, L.; Boček, P. Capillary Zone Electrophoresis; VCH: Weinheim, Germany, 1993. [Google Scholar]
- Lucy, C.A.; Macdonald, A.M.; Gulcev, M.D. Non-covalent capillary coatings for protein separations in capillary electrophoresis. J. Chromatogr. A 2008, 1184, 81–105. [Google Scholar] [CrossRef]
- The United States Pharmacopeial Convention. Capillary Electrophoresis. Available online: https://www.usp.org/sites/default/files/usp/document/harmonization/biotechnology/harmonization-september-2019-m859.pdf (accessed on 2 August 2020).
- Vindevogel, J.; Szücs, R.; Sandra, P. Reproducibility of migration times and propagation of error in micellar electrokinetic chromatography. J. Microcolumn Sep. 1992, 4, 399–404. [Google Scholar] [CrossRef]
- Altria, K.D. Essential peak area normalisation for quantitative impurity content determination by capillary electrophoresis. Chromatographia 1993, 35, 177–182. [Google Scholar] [CrossRef]
- International Conference on Harmonisation of Technical Requirements of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Impurities in New Drug Substances Q3A(R2). Available online: https://database.ich.org/sites/default/files/Q3A%28R2%29%20Guideline.pdf (accessed on 31 July 2020).
- International Conference on Harmonisation of Technical Requirements of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology Q2(R1). Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 31 July 2020).
- Bernardo-Bermejo, S.; Sánchez-López, E.; Castro-Puyana, M.; Marina, M.L. Chiral capillary electrophoresis. TrAC Trends Anal. Chem. 2020, 124, 115807. [Google Scholar] [CrossRef]
- Fanali, S.; Chankvetadze, B. Some thoughts about enantioseparations in capillary electrophoresis. Electrophoresis 2019, 40, 2420–2437. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.F.; Quirino, J.P. Chiral Selectors in Capillary Electrophoresis: Trends during 2017–2018. Molecules 2019, 24, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.G.; Hägele, J.S. Separation of enantiomers and positional isomers of novel psychoactive substances in solid samples by chromatographic and electrophoretic techniques—A selective review. J. Chromatogr. A 2020, 1624, 461256. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.E.; Aslan, D.; Clarke, A.; Roeraade, J.; Hagman, G. Evaluation of generic chiral liquid chromatography screens for pharmaceutical analysis. J. Chromatogr. A 2003, 1005, 83–101. [Google Scholar] [CrossRef]
- Perera, R.W.H.; Abraham, I.; Gupta, S.; Kowalska, P.; Lightsey, D.; Marathaki, C.; Singh, N.S.; Lough, W.J. Screening approach, optimisation and scale-up for chiral liquid chromatography of cathinones. J. Chromatogr. A 2012, 1269, 189–197. [Google Scholar] [CrossRef]
- Sajonz, P.; Gong, X.; Leonard, W.R.; Biba, M.; Welch, C.J. Multiparallel chiral method development screening using an 8-channel microfluidic HPLC system. Chirality 2006, 18, 803–813. [Google Scholar] [CrossRef]
- Welch, C.J.; Sajonz, P.; Biba, M.; Gouker, J.; Fairchild, J. Comparison of Multiparallel Microfluidic HPLC Instruments for High Throughput Analyses in Support of Pharmaceutical Process Research. J. Liq. Chromatogr. Relat. Technol. 2006, 29, 2185–2200. [Google Scholar] [CrossRef]
- God, R.; Gumm, H. Parallel HPLC in High-Throughput Analysis and Purification. In Analysis and Purification Methods in Combinatorial Chemistry; Yan, B., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2004; Volume 163, pp. 307–320. [Google Scholar]
- Rathore, A.S.; Horváth, C. Cyclodextrin aided separation of peptides and proteins by capillary zone electrophoresis. J. Chromatogr. A 1998, 796, 367–373. [Google Scholar] [CrossRef]
- Serno, T.; Geidobler, R.; Winter, G. Protein stabilization by cyclodextrins in the liquid and dried state. Adv. Drug Deliv. Rev. 2011, 63, 1086–1106. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Parameter | OSS-EOF | OSS-sEOF | CSS | |||
|---|---|---|---|---|---|---|
| PDL | PPL | PDL | PPL | PDL | PPL | |
| µep × 108 (m2V−1s−1) | 1.74 | 1.66 | 1.83 | 1.75 | 2.52 | 2.37 |
| RSD tm (%) | 0.3 | 0.3 | 0.4 | 0.4 | 0.5 | 0.5 |
| RSD A (%) | 2.0 | 1.4 | 3.4 | 2.4 | 2.2 | 1.6 |
| N × 10−3 (m−1) | 309 | 310 | 465 | 482 | 162 | 147 |
| LOD (µmol/L) | 1.20 | 1.10 | 1.50 | 1.40 | 0.06 | 0.07 |
| Resolution (1) | 1.47 | 3.64 | 3.23 | |||
| Required LOD | Achieved LOD (µmol/L) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| % (w/w) | mg/mL | µmol/L | OSS-EOF | OSS-sEOF | CSS | ||||
| PDL | PPL | PDL | PPL | PDL | PPL | PDL | PPL | ||
| 0.02 | 2.0 × 10−5 | 0.08 | 0.08 | 1.20 | 1.10 | 1.50 | 1.40 | 0.06 | 0.07 |
| Chiral Selector | OSS-EOF | OSS-sEOF | CSS | |||
|---|---|---|---|---|---|---|
| PDL | PPL | PDL | PPL | PDL | PPL | |
| CMBCD | Rs = 1.33 | Rs = 1.42 | Rs = 5.22 | r.m. | Rs = 2.06 | n.t. |
| CEBCD | n.t. | n.t. | n.t. | n.t. | n.t. | Rs = 1.36 |
| System | PDLen1 | PDLen2 | PPLen1 | PPLen2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| µeof | µep | µeff | µCD (1) | µeff | µCD | µep | µeff | µCD (1) | µeff | µCD | |
| OSS-EOF | +2.20 | +1.74 | +2.60 | −1.34 | +2.56 | −1.38 | +1.66 | +1.74 | −2.12 | +1.69 | −2.17 |
| OSS-sEOF | 0 | +1.83 | +0.59 | −1.24 | +0.55 | −1.28 | +1.75 | −0.37 | −2.12 | −0.42 | −2.17 |
| CSS | 0 | +2.52 | +1.18 | −1.34 | +1.12 | −1.40 | +2.37 | +1.08 (2) | −1.29 (2) | +1.05 (2) | −1.32 (2) |
| Parameter | OSS-sEOF | CSS | ||
|---|---|---|---|---|
| PDL | PPL | PDL | PPL | |
| µep × 108 (m2V−1s−1) | 1.75 | 1.66 | 2.50 | 2.36 |
| RSD tm (%) | 0.3 | 0.3 | 0.8 | 0.8 |
| RSD A (%) | 2.0 | 1.4 | 1.7 | 3.0 |
| N × 10−3 (m−1) | 392 | 410 | 151 | 124 |
| LOD (µmol/L) | 3.4 | 3.2 | 0.08 | 0.08 |
| Resolution | 3.78 | 2.73 | ||
| Chiral Selector | OSS-EOF | OSS-sEOF | CSS | |||
|---|---|---|---|---|---|---|
| PDL | PPL | PDL | PPL | PDL | PPL | |
| CMBCD | Rs = 1.16 | r.m. | Rs = 2.68 | n.t. | Rs = 1.46 | n.t. |
| CEBCD | n.t. | n.t. | n.t. | n.t. | n.t. | Rs = 1.04 |
| Chiral | Analyte | OSS-sEOF | CSS | ||
|---|---|---|---|---|---|
| Selector | µeff × 108 (m2V−1s−1) | µeff × 108 (m2V−1s−1) | |||
| Model Sample | Spiked Serum | Model Sample | Spiked Serum | ||
| CMBCD | PDLen1 | 0.59 | 0.96 | 1.18 | 2.10 |
| PDLen2 | 0.55 | 0.93 | 1.12 | 2.03 | |
| CEBCD | PPLen1 | r.m. | n.t. | 1.08 | 1.99 |
| PPLen2 | r.m. | n.t. | 1.05 | 1.94 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masár, M.; Hradski, J.; Schmid, M.G.; Szucs, R. Advantages and Pitfalls of Capillary Electrophoresis of Pharmaceutical Compounds and Their Enantiomers in Complex Samples: Comparison of Hydrodynamically Opened and Closed Systems. Int. J. Mol. Sci. 2020, 21, 6852. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186852
Masár M, Hradski J, Schmid MG, Szucs R. Advantages and Pitfalls of Capillary Electrophoresis of Pharmaceutical Compounds and Their Enantiomers in Complex Samples: Comparison of Hydrodynamically Opened and Closed Systems. International Journal of Molecular Sciences. 2020; 21(18):6852. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186852
Chicago/Turabian StyleMasár, Marián, Jasna Hradski, Martin G. Schmid, and Roman Szucs. 2020. "Advantages and Pitfalls of Capillary Electrophoresis of Pharmaceutical Compounds and Their Enantiomers in Complex Samples: Comparison of Hydrodynamically Opened and Closed Systems" International Journal of Molecular Sciences 21, no. 18: 6852. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186852