Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies


Abstract
:
1. Introduction
2. Chemotherapy Resistance
3. Molecular Biology of Osteosarcoma
3.1. Bone Regulation
3.2. The Tumour Microenvironment
3.3. Genomic Instability and Cell Cycle Regulation
3.4. Cellular Signalling Pathways
4. Osteosarcoma Therapies
4.1. Chemotherapy
4.2. Adverse Effects of Chemotherapy
4.3. Immunotherapy
4.4. Targeted Therapies
5. Mechanisms Underlying Therapy Resistance
5.1. Limitations of Drug Delivery
5.2. Decreased Intracellular Drug Accumulation and Target Specificity
5.2.1. Decreased Cellular Influx
5.2.2. Increased Cellular Efflux via ATP-Binding Transporters
5.3. Alterations to Drug Targets
5.4. Intracellular Drug Modifications
5.5. Inhibition of Apoptosis and Cell Cycle Regulation
5.5.1. B Cell Lymphoma 2
5.5.2. TP53
5.6. Alterations in DNA Repair Pathways
5.7. Signal Transduction
5.8. Autophagy
5.9. Cancer Stem Cells and Tumour Microenvironment
5.10. Bone Pathways and Treatment Resistance
5.11. Other Factors Linked to Drug Resistance in Osteosarcoma
6. Mechanisms Underlying Drug Efficacy/Synergy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harrison, D.J.; Geller, D.S.; Gill, J.; Lewis, V.O.; Gorlick, R. Current and future therapeutic approaches for osteosarcoma. Expert Rev. Anticancer Ther. 2018, 18, 39–50. [Google Scholar] [CrossRef]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottaviani, G.; Jaffe, N.; Ottaviani, G. The Epidemiology of Osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar] [PubMed]
- Hattinger, C.M.; Patrizio, M.P.; Magagnoli, F.; Luppi, S.; Serra, M. An update on emerging drugs in osteosarcoma: Towards tailored therapies? Expert Opin. Emerg. Drugs 2019, 24, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.M.; Mostoufi-Moab, S.; Stratton, K.L.; Leisenring, W.M.; Barnea, D.; Chow, E.J.; Donaldson, S.S.; Howell, R.M.; Hudson, M.M.; Mahajan, A.; et al. Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970–99: A report from the Childhood Cancer Survivor Study cohort. Lancet Oncol. 2018, 19, 1590–1601. [Google Scholar] [CrossRef]
- Robison, L.L.; Hudson, M.M. Survivors of childhood and adolescent cancer: Life-long risks and responsibilities. Nat. Rev. Cancer 2014, 14, 61–70. [Google Scholar] [CrossRef]
- Ferguson, J.L.; Turner, S.P. Bone Cancer: Diagnosis and Treatment Principles. Am. Fam. Physician 2018, 98, 205–213. [Google Scholar]
- Kager, L.; Tamamyan, G.; Bielack, S. Novel insights and therapeutic interventions for pediatric osteosarcoma. Futur. Oncol. 2017, 13, 357–368. [Google Scholar] [CrossRef]
- Chou, A.J.; Gorlick, R. Chemotherapy resistance in osteosarcoma: Current challenges and future directions. Expert Rev. Anticancer Ther. 2006, 6, 1075–1085. [Google Scholar] [CrossRef]
- Rosen, G.; Caparros, B.; Huvos, A.G.; Kosloff, C.; Nirenberg, A.; Cacavio, A.; Marcove, R.C.; Lane, J.M.; Mehta, B.; Urban, C. Preoperative chemotherapy for osteogenic sarcoma: Selection of postoperative adjuvant chemotherapy based on the response of the primary tumor to preoperative chemotherapy. Cancer 1982, 49, 1221–1230. [Google Scholar] [CrossRef]
- Link, M.P.; Goorin, A.M.; Miser, A.W.; Green, A.A.; Pratt, C.B.; Belasco, J.B.; Pritchard, J.; Malpas, J.S.; Baker, A.R.; Kirkpatrick, J.A.; et al. The Effect of Adjuvant Chemotherapy on Relapse-Free Survival in Patients with Osteosarcoma of the Extremity. N. Engl. J. Med. 1986, 314, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Stiller, C.A.; Bielack, S.; Jundt, G.; Steliarova-Foucher, E. Bone tumours in European children and adolescents, 1978–1997. Report from the Automated Childhood Cancer Information System project. Eur. J. Cancer 2006, 42, 2124–2135. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Rutherford, M.J.; Bardot, A.; Ferlay, J.; Andersson, T.M.; Myklebust, T.; Tervonen, H.; Thursfield, V.; Ransom, D.; Shack, L.; et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995–2014 (ICBP SURVMARK-2): A population-based study. Lancet Oncol. 2019, 20, 1493–1505. [Google Scholar] [CrossRef] [Green Version]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Lippert, T.H. Current Status of Methods to Assess Cancer Drug Resistance. Int. J. Med. Sci. 2011, 8, 245. [Google Scholar] [CrossRef] [Green Version]
- Giaccone, G.; Pinedo, H.M. Drug Resistance. Oncologist 1996, 1, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Dobson, S.M.; Garcia-Prat, L.; Vanner, R.J.; Wintersinger, J.; Waanders, E.; Gu, Z.; McLeod, J.; Gan, O.I.; Grandal, I.; Payne-Turner, D.; et al. Relapse-Fated Latent Diagnosis Subclones in Acute B Lineage Leukemia Are Drug Tolerant and Possess Distinct Metabolic Programs. Cancer Discov. 2020, 10, 568–587. [Google Scholar] [CrossRef] [Green Version]
- Kuijjer, M.L.; Hogendoorn, P.C.; Cleton-Jansen, A.-M. Genome-wide analyses on high-grade osteosarcoma: Making sense of a genomically most unstable tumor. Int. J. Cancer 2013, 133, 2512–2521. [Google Scholar] [CrossRef]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.B.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Singer, F.R. Paget disease: When to treat and when not to treat. Nat. Rev. Rheumatol. 2009, 5, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Heymann, M.-F.; Lézot, F.; Heymann, D.; Marie-Françoise, H.; Frédéric, L.; Dominique, H. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell. Immunol. 2019, 343, 103711. [Google Scholar] [CrossRef] [PubMed]
- Visconti, R.; Iversen, T.; Cottrell, J. A Review of Dysregulated Osteoblast and Osteoclast Coupling in Bone Disease and Failure. J. Bone Res. 2019, 7, 200. [Google Scholar]
- Théoleyre, S.; Wittrant, Y.; Tat, S.K.; Fortun, Y.; Rédini, F.; Heymann, D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004, 15, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Cell biology of the osteoclast. Exp. Hematol. 1999, 27, 1229–1241. [Google Scholar] [CrossRef]
- Kaji, H.; Sugimoto, T.; Kanatani, M.; Nishiyama, K.; Nasu, M.; Chihara, K. Insulin-like growth factor-I mediates osteoclast-like cell formation stimulated by parathyroid hormone. J. Cell. Physiol. 1997, 172, 55–62. [Google Scholar] [CrossRef]
- Pollak, M.; Sem, A.W.; Richard, M.; Tetenes, E.; Bell, R. Inhibition of Metastatic Behavior of Murine Osteosarcoma by Hypophysectomy. J. Natl. Cancer Inst. 1992, 84, 966–971. [Google Scholar] [CrossRef] [Green Version]
- Burrow, S.; Andrulis, I.L.; Pollak, M.; Bell, R.S. Expression of insulin-like growth factor receptor, IGF-1, and IGF-2 in primary and metastatic osteosarcoma. J. Surg. Oncol. 1998, 69, 21–27. [Google Scholar] [CrossRef]
- Swerdlow, A.J.; Cooke, R.; Beckers, D.; Borgström, B.; Butler, G.; Carel, J.-C.; Cianfarani, S.; Clayton, P.E.; Coste, J.; Deodati, A.; et al. Cancer Risks in Patients Treated With Growth Hormone in Childhood: The SAGhE European Cohort Study. J. Clin. Endocrinol. Metab. 2017, 102, 1661–1672. [Google Scholar] [CrossRef]
- Endo-Munoz, L.B.; Evdokiou, A.; Saunders, N.A. The role of osteoclasts and tumour-associated macrophages in osteosarcoma metastasis. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1826, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Avnet, S.; Longhi, A.; Salerno, M.; Halleen, J.M.; Perut, F.; Granchi, D.; Ferrari, S.; Bertoni, F.; Giunti, A.; Baldini, N. Increased osteoclast activity is associated with aggressiveness of osteosarcoma. Int. J. Oncol. 2008, 33, 1231–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo-Munoz, L.B.; Cumming, A.; Rickwood, D.; Wilson, D.; Cueva, C.; Ng, C.K.; Strutton, G.; Cassady, A.; Evdokiou, A.; Sommerville, S.; et al. Loss of Osteoclasts Contributes to Development of Osteosarcoma Pulmonary Metastases. Cancer Res. 2010, 70, 7063–7072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsaers, A.J.; Walkley, C.R. Cells of origin in osteosarcoma: Mesenchymal stem cells or osteoblast committed cells? Bone 2014, 62, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Navet, B.; Ando, K.; Vargas-Franco, J.W.; Brion, R.; Amiaud, J.; Mori, K.; Yagita, H.; Mueller, C.G.; Verrecchia, F.; Dumars, C.; et al. The Intrinsic and Extrinsic Implications of RANKL/RANK Signaling in Osteosarcoma: From Tumor Initiation to Lung Metastases. Cancers 2018, 10, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, X.; Isaac, C.; Greco, N.; Huard, J.; Weiss, K. Notch Signaling is Associated with ALDH Activity and an Aggressive Metastatic Phenotype in Murine Osteosarcoma Cells. Front. Oncol. 2013, 3, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Liu, X.-Z.; Choy, E.; Mankin, H.; Hornicek, F.J.; Duan, Z. Targeting hedgehog-GLI-2 pathway in osteosarcoma. J. Orthop. Res. 2013, 31, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Cai, T.-G.; Chen, Y. Wnt Pathway in Osteosarcoma, from Oncogenic to Therapeutic. J. Cell. Biochem. 2014, 115, 625–631. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Liu, G.; Rus, I.A.; Yao, S.; Chen, Y.; Akiri, G.; Grumolato, L.; Aaronson, S.A. High-Frequency Canonical Wnt Activation in Multiple Sarcoma Subtypes Drives Proliferation through a TCF/β-Catenin Target Gene, CDC25A. Cancer Cell 2011, 19, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Tetzlaff, F.; Fischer, A. Control of Blood Vessel Formation by Notch Signaling. Adv. Exp. Med. Biol. 2018, 1066, 319–338. [Google Scholar] [CrossRef]
- Lo, W.W.; Wunder, J.S.; Dickson, B.C.; Campbell, V.; McGovern, K.; Alman, B.A.; Andrulis, I. Involvement and targeted intervention of dysregulated Hedgehog signaling in osteosarcoma. Cancer 2014, 120, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.A.; Horowitz, M.; Choi, Y. Osteoimmunology: Interactions of the bone and immune system. Endocr. Rev. 2008, 29, 403–440. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef]
- Mceachron, T.A.; Triche, T.J.; Sorenson, L.; Parham, D.M.; Carpten, J.D. Profiling targetable immune checkpoints in osteosarcoma. OncoImmunology 2018, 7, e1475873. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Beird, H.C.; Livingston, J.A.; Advani, S.; Mitra, A.; Cao, S.; Reuben, A.; Ingram, D.; Wang, W.-L.; Ju, Z.; et al. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020, 11, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalski, M.N.; McCauley, L.K. Macrophages and skeletal health. Pharmacol. Ther. 2017, 174, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Condeelis, J. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; Cleton-Jansen, A.-M.; et al. Tumor-Infiltrating Macrophages Are Associated with Metastasis Suppression in High-Grade Osteosarcoma: A Rationale for Treatment with Macrophage Activating Agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [Green Version]
- Dumars, C.; Ngyuen, J.-M.; Gaultier, A.; Lanel, R.; Corradini, N.; Gouin, F.; Heymann, D.; Heymann, M.-F. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget 2016, 7, 78343–78354. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Xian, M.; Xiang, S.; Xiang, D.; Shao, X.; Wang, J.; Cao, J.; Yang, X.; Yang, B.; Ying, M.; et al. All-Trans Retinoic Acid Prevents Osteosarcoma Metastasis by Inhibiting M2 Polarization of Tumor-Associated Macrophages. Cancer Immunol. Res. 2017, 5, 547–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Shi, H.; Liu, F. CD163 + M2-type tumor-associated macrophage support the suppression of tumor-infiltrating T cells in osteosarcoma. Int. Immunopharmacol. 2016, 34, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Fritzsching, B.; Fellenberg, J.; Moskovszky, L.; Sápi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendrõi, M.; Bosch, A.L.; et al. CD8+/FOXP3+-ratio in osteosarcoma microenvironment separates survivors from non-survivors: A multicenter validated retrospective study. Oncoimmunology 2015, 4, e990800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biller, B.; Guth, A.; Burton, J.H.; Dow, S. Decreased ratio of CD8+ T cells to regulatory T cells associated with decreased survival in dogs with osteosarcoma. J. Vet. Intern. Med. 2010, 24, 1118–1123. [Google Scholar] [CrossRef]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.-M.; Marie, B.; Larousserie, F.; Entz-Werlé, N.; De Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. OncoImmunology 2017, 6, e1331193. [Google Scholar] [CrossRef]
- Sottnik, J.L.; U’Ren, L.W.; Thamm, D.H.; Withrow, S.J.; Dow, S. Chronic bacterial osteomyelitis suppression of tumor growth requires innate immune responses. Cancer Immunol. Immunother. 2010, 59, 367–378. [Google Scholar] [CrossRef]
- Culp, W.T.N.; Olea-Popelka, F.; Sefton, J.; Aldridge, C.F.; Withrow, S.J.; Lafferty, M.H.; Rebhun, R.B.; Kent, M.S.; Ehrhart, N. Evaluation of outcome and prognostic factors for dogs living greater than one year after diagnosis of osteosarcoma: 90 cases (1997–2008). J. Am. Vet. Med Assoc. 2014, 245, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Jeys, L.M.; Grimer, R.J.; Carter, S.R.; Tillman, R.M.; Abudu, A. Post Operative Infection and Increased Survival in Osteosarcoma Patients: Are They Associated? Ann. Surg. Oncol. 2007, 14, 2887–2895. [Google Scholar] [CrossRef]
- Baud’huin, M.; Renault, R.; Charrier, C.; Riet, A.; Moreau, A.; Brion, R.; Gouin, F.; Duplomb, L.; Heymann, D. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in RANKL-induced osteoclastogenesis. J. Pathol. 2010, 221, 77–86. [Google Scholar]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef]
- Roodman, G.D.; Kurihara, N.; Ohsaki, Y.; Kukita, A.; Hosking, D.; Demulder, A.; Smith, J.F.; Singer, F.R. Interleukin 6. A potential autocrine/paracrine factor in Paget’s disease of bone. J. Clin. Investig. 1992, 89, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.A.; Roodman, G.D. The role of immune cells and inflammatory cytokines in Paget’s disease and multiple myeloma. Immunol. Rev. 2005, 208, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Huang, Y.; Sun, Y.; Zhang, J.; Yao, Y.; Shen, Z.; Xiang, D.; He, A. Prognostic value of inflammation-based scores in patients with osteosarcoma. Sci. Rep. 2016, 6, 39862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Carey, L.A.; Sharpless, N.E. PARP and cancer—If it’s broke, don’t fix it. N. Engl. J. Med. 2011, 364, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Bae, J.S.; Kim, K.M.; Moon, Y.J.; Park, S.-H.; Ha, S.H.; Hussein, U.K.; Zhang, Z.; Park, H.S.; Park, B.-H.; et al. The PARP inhibitor olaparib potentiates the effect of the DNA damaging agent doxorubicin in osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 107. [Google Scholar] [CrossRef]
- Kováč, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kováčová, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef]
- Holme, H.; Gulati, A.; Brough, R.; Fleuren, E.D.G.; Bajrami, I.; Campbell, J.; Chong, I.Y.; Costa-Cabral, S.; Elliott, R.; Fenton, T.; et al. Chemosensitivity profiling of osteosarcoma tumour cell lines identifies a model of BRCAness. Sci. Rep. 2018, 8, 10614. [Google Scholar] [CrossRef] [Green Version]
- Hattinger, C.M.; Michelacci, F.; Sella, F.; Magagnoli, G.; Benini, S.; Gambarotti, M.; Palmerini, E.; Picci, P.; Serra, M.; Ferrari, S. Excision repair cross-complementation group 1 protein expression predicts survival in patients with high-grade, non-metastatic osteosarcoma treated with neoadjuvant chemotherapy. Histopathology 2015, 67, 338–347. [Google Scholar] [CrossRef]
- Shah, M.A.; Schwartz, G.K. Cell cycle-mediated drug resistance: An emerging concept in cancer therapy. Clin. Cancer Res. 2001, 7, 2168–2181. [Google Scholar]
- Ferrara, R.; Simionato, F.; Ciccarese, C.; Grego, E.; Cingarlini, S.; Iacovelli, R.; Bria, E.; Tortora, G.; Melisi, D. The development of PARP as a successful target for cancer therapy. Expert Rev. Anticancer Ther. 2018, 18, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Rao, P.H.; Favis, R.; Lu, X.-Y.; Elowitz, M.B.; Barany, F.; Ladanyi, M.; Gorlick, R.; Levine, A.J. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc. Natl. Acad. Sci. USA 2003, 100, 11547–11552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarity, B.S.; Otto, G.M.; Rahrmann, E.P.; Rathe, S.K.; Wolf, N.K.; Weg, M.T.; Manlove, L.A.; LaRue, R.S.; Temiz, N.A.; Molyneux, S.D.; et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat. Genet. 2015, 47, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Yeager, M.; Mai, P.L.; Gastier-Foster, J.M.; Gorlick, R.; Khanna, C.; Patiño-García, A.; Sierrasesúmaga, L.; Lecanda, F.; Andrulis, I.L.; et al. Germline TP53 Variants and Susceptibility to Osteosarcoma. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell. Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Tarkkanen, M.; Karhu, R.; Kallioniemi, A.; Elomaa, I.; Kivioja, A.H.; Nevalainen, J.; Böhling, T.; Karaharju, E.; Hyytinen, E.; Knuutila, S. Gains and losses of DNA sequences in osteosarcomas by comparative genomic hybridization. Cancer Res. 1995, 55, 1334–1338. [Google Scholar] [PubMed]
- Tarkkanen, M.; Elomaa, I.; Blomqvist, C.; Kivioja, A.H.; Kellokumpu-Lehtinen, P.; Valle, J.; Knuutila, S. DNA sequence copy number increase at 8q: A potential new prognostic marker in high-grade osteosarcoma. Int. J. Cancer 1999, 84, 114–121. [Google Scholar] [CrossRef]
- Hansen, M.F.; Koufos, A.; Gallie, B.L.; Phillips, R.A.; Fodstad, O.; Brögger, A.; Gedde-Dahl, T.; Cavenee, W.K. Osteosarcoma and retinoblastoma: A shared chromosomal mechanism revealing recessive predisposition. Proc. Natl. Acad. Sci. USA 1985, 82, 6216–6220. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Jin, W.; Liu, H.; Wang, L.L. RECQ DNA Helicases and Osteosarcoma. Adv. Exp. Med. Biol. 2014, 804, 129–145. [Google Scholar] [CrossRef]
- Ji, J.; Quindipan, C.; Parham, D.; Shen, L.; Ruble, D.; Bootwalla, M.; Maglinte, D.T.; Gai, X.; Saitta, S.C.; Biegel, J.A.; et al. Inherited germline ATRX mutation in two brothers with ATR-X syndrome and osteosarcoma. Am. J. Med Genet. Part A 2017, 173, 1390–1395. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Gorlick, R.; Huvos, A.G.; Heller, G.; Aledo, A.; Beardsley, G.P.; Healey, J.H.; Meyers, P.A. Expression of HER2/erbB-2 correlates with survival in osteosarcoma. J. Clin. Oncol. 1999, 17, 2781–2788. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.P.M.; Thomas, D.G.; Giordano, T.J.; Baker, L.H.; McDonagh, K.T. Cell surface expression of epidermal growth factor receptor and Her-2 with nuclear expression of Her-4 in primary osteosarcoma. Cancer Res. 2004, 64, 2047–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ma, Y.-H.; Sun, Z.; Rui, Y.-J.; Yin, Q.-D.; Song, S.; Wei, X.-M.; Liu, J.; Liu, X.-G.; Hu, K. Effect of c-erbB2 overexpression on prognosis in osteosarcoma: Evidence from eight studies. Tumor Biol. 2014, 35, 8939–8943. [Google Scholar] [CrossRef] [PubMed]
- Akatsuka, T.; Wada, T.; Kokai, Y.; Kawaguchi, S.; Isu, K.; Yamashiro, K.; Yamashita, T.; Sawada, N.; Yamawaki, S.; Ishii, S. ErbB2 expression is correlated with increased survival of patients with osteosarcoma. Cancer 2002, 94, 1397–1404. [Google Scholar] [CrossRef]
- Somers, G.R.; Ho, M.; Zielenska, M.; Squire, J.A.; Thorner, P.S. HER2 Amplification and Overexpression Is Not Present in Pediatric Osteosarcoma: A Tissue Microarray Study. Pediatr. Dev. Pathol. 2005, 8, 525–532. [Google Scholar] [CrossRef]
- Anninga, J.K.; Van De Vijver, M.J.; Cleton-Jansen, A.; Kristel, P.; Taminiau, A.; Nooij, M.; Egeler, R.; Hogendoorn, P.C. Overexpression of the HER-2 oncogene does not play a role in high-grade osteosarcomas. Eur. J. Cancer 2004, 40, 963–970. [Google Scholar] [CrossRef]
- Wen, Y.H.; Koeppen, H.; Garcia, R.; Chiriboga, L.; Tarlow, B.; Peters, B.; Eigenbrot, C.; Yee, H.; Steiner, G.; Greco, M.A. Epidermal growth factor receptor in osteosarcoma: Expression and mutational analysis. Hum. Pathol. 2007, 38, 1184–1191. [Google Scholar] [CrossRef]
- Kersting, C.; Gebert, C.; Agelopoulos, K.; Schmidt, H.; Van Diest, P.J.; Juergens, H.; Winkelmann, W.; Kevric, M.; Gosheger, G.; Brandt, B.; et al. Epidermal Growth Factor Receptor Expression in High-Grade Osteosarcomas Is Associated with a Good Clinical Outcome. Clin. Cancer Res. 2007, 13, 2998–3005. [Google Scholar] [CrossRef] [Green Version]
- MacEwen, E.G.; Pastor, J.; Kutzke, J.; Tsan, R.; Kurzman, I.D.; Thamm, D.H.; Wilson, M.; Radinsky, R. IGF-1 receptor contributes to the malignant phenotype in human and canine osteosarcoma. J. Cell. Biochem. 2004, 92, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Kaya, M.; Wada, T.; Akatsuka, T.; Kawaguchi, S.; Nagoya, S.; Shindoh, M.; Higashino, F.; Mezawa, F.; Okada, F.; Ishii, S. Vascular endothelial growth factor expression in untreated osteosarcoma is predictive of pulmonary metastasis and poor prognosis. Clin. Cancer Res. 2000, 6, 572–577. [Google Scholar] [PubMed]
- Lee, Y.H.; Tokunaga, T.; Oshika, Y.; Suto, R.; Yanagisawa, K.; Tomisawa, M.; Fukuda, H.; Nakano, H.; Abe, S.; Tateishi, A.; et al. Cell-retained isoforms of vascular endothelial growth factor (VEGF) are correlated with poor prognosis in osteosarcoma. Eur. J. Cancer 1999, 35, 1089–1093. [Google Scholar] [CrossRef]
- Sulzbacher, I.; Birner, P.; Trieb, K.; Träxler, M.; Lang, S.; Chott, A. Expression of Platelet-Derived Growth Factor-AA is Associated with Tumor Progression in Osteosarcoma. Mod. Pathol. 2003, 16, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Cicenas, J.; Zalyte, E.; Bairoch, A.; Gaudet, P. Kinases and Cancer. Cancers 2018, 10, 63. [Google Scholar] [CrossRef] [Green Version]
- Pignochino, Y.; Grignani, G.; Cavalloni, G.; Motta, M.; Tapparo, M.; Bruno, S.; Bottos, A.; Gammaitoni, L.; Migliardi, G.; Camussi, G.; et al. Sorafenib blocks tumour growth, angiogenesis and metastatic potential in preclinical models of osteosarcoma through a mechanism potentially involving the inhibition of ERK1/2, MCL-1 and ezrin pathways. Mol. Cancer 2009, 8, 118. [Google Scholar] [CrossRef] [Green Version]
- Bishop, M.W.; Janeway, K. Emerging concepts for PI3K/mTOR inhibition as a potential treatment for osteosarcoma. F1000Research 2016, 5, 1590. [Google Scholar] [CrossRef]
- Basu-Roy, U.; Bayin, N.S.; Rattanakorn, K.; Han, E.; Placantonakis, D.; Mansukhani, A.; Basilico, C. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat. Commun. 2015, 6, 6411. [Google Scholar] [CrossRef] [Green Version]
- Basu-Roy, U.; Seo, E.; Ramanathapuram, L.; Rapp, T.B.; Perry, J.A.; Orkin, S.H.; Mansukhani, A.; Basilico, C. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene 2012, 31, 2270–2282. [Google Scholar] [CrossRef] [Green Version]
- Verrecchia, F.; Redini, F. Transforming Growth Factor-beta Signaling Plays a Pivotal Role in the Interplay Between Osteosarcoma Cells and Their Microenvironment. Front. Oncol. 2018, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- LaMora, A.; Talbot, J.; Bougras, G.; Amiaud, J.; LeDuc, M.; Chesneau, J.; Taurelle, J.; Stresing, V.; Le Deley, M.-C.; Heymann, M.F.; et al. Overexpression of Smad7 Blocks Primary Tumor Growth and Lung Metastasis Development in Osteosarcoma. Clin. Cancer Res. 2014, 20, 5097–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blattmann, C.; Oertel, S.; Schulz-Ertner, D.; Rieken, S.; Haufe, S.; Ewerbeck, V.; Unterberg, A.; Karapanagiotou-Schenkel, I.; Combs, S.E.; Nikoghosyan, A.; et al. Non-randomized therapy trial to determine the safety and efficacy of heavy ion radiotherapy in patients with non-resectable osteosarcoma. BMC Cancer 2010, 10, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rang, H.; Ritter, J.; Flower, R.; Henderson, G. Rang and Dale’s Pharmacology, 8th ed.; Elsevier, Churchill, Livingstone: Edinburgh, UK, 2016. [Google Scholar]
- Meyers, P.A.; Heller, G.; Healey, J.; Huvos, A.; Lane, J.; Marcove, R.; Applewhite, A.; Vlamis, V.; Rosen, G. Chemotherapy for nonmetastatic osteogenic sarcoma: The Memorial Sloan-Kettering experience. J. Clin. Oncol. 1992, 10, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: The Addition of Muramyl Tripeptide to Chemotherapy Improves Overall Survival—A Report From the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef]
- Kang, J.-M.; Ju, H.Y.; Joo, J.; Sung, J.Y.; Park, S.Y.; Kim, J.H.; Kang, H.G.; Kwon, M.; Park, M.; Park, H.J.; et al. Histologic Response and Toxicity following Interval-Compressed Four-Drug Therapy Given Preoperatively in Children and Young Adults with Osteosarcoma: A Retrospective Study. Oncology 2020, 98, 81–90. [Google Scholar] [CrossRef]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Perez, E.A.; Shang, X.; Burlingame, S.M.; Okcu, M.F.; Ge, N.; Russell, H.V.; Egler, R.A.; David, R.D.; Vasudevan, S.A.; Yang, J.; et al. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Catarina, A.; Nuno, J.; Simões, S. Combination Chemotherapy in Cancer: Principles, Evaluation and Drug Delivery Strategies. In Current Cancer Treatment—Novel Beyond Conventional Approaches; IntechOpen: London, UK, 2011; pp. 693–711. [Google Scholar]
- DeVita, V.T.; Young, R.C.; Canellos, G.P. Combination versus single agent chemotherapy: A review of the basis for selection of drug treatment of cancer. Cancer 1975, 35, 98–110. [Google Scholar] [CrossRef]
- Ferrari, S.; Serra, M. An update on chemotherapy for osteosarcoma. Expert Opin. Pharmacother. 2015, 16, 2727–2736. [Google Scholar] [CrossRef]
- Isakoff, M.S.; Bielack, S.S.; Meltzer, P.; Gorlick, R. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J. Clin. Oncol. 2015, 33, 3029–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, J.S.; Bielack, S.S.; Marina, N.; Smeland, S.; Jovic, G.; Hook, J.M.; Krailo, M.; Anninga, J.; Butterfass-Bahloul, T.; Böhling, T.; et al. EURAMOS-1, an international randomised study for osteosarcoma: Results from pre-randomisation treatment. Ann. Oncol. 2015, 26, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Anninga, J.K.; Gelderblom, H.; Fiocco, M.; Kroep, J.; Taminiau, A.H.; Hogendoorn, P.C.; Egeler, R.M. Chemotherapeutic adjuvant treatment for osteosarcoma: Where do we stand? Eur. J. Cancer 2011, 47, 2431–2445. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Ruggieri, P.; Cefalo, G.; Tamburini, A.; Capanna, R.; Fagioli, F.; Comandone, A.; Bertulli, R.M.; Bisogno, G.; Palmerini, E.; et al. Neoadjuvant Chemotherapy With Methotrexate, Cisplatin, and Doxorubicin With or Without Ifosfamide in Nonmetastatic Osteosarcoma of the Extremity: An Italian Sarcoma Group Trial ISG/OS-1. J. Clin. Oncol. 2012, 30, 2112–2118. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, S.; Smeland, S.; Mercuri, M.; Bertoni, F.; Longhi, A.; Ruggieri, P.; Alvegård, T.A.; Picci, P.; Capanna, R.; Bernini, G.; et al. Neoadjuvant Chemotherapy With High-Dose Ifosfamide, High-Dose Methotrexate, Cisplatin, and Doxorubicin for Patients With Localized Osteosarcoma of the Extremity: A Joint Study by the Italian and Scandinavian Sarcoma Groups. J. Clin. Oncol. 2005, 23, 8845–8852. [Google Scholar] [CrossRef]
- Le Deley, M.-C.; Guinebretière, J.-M.; Gentet, J.-C.; Pacquement, H.; Pichon, F.; Marec-Berard, P.; Entz-Werlé, N.; Schmitt, C.; Brugiéres, L.; Vanel, D.; et al. SFOP OS94: A randomised trial comparing preoperative high-dose methotrexate plus doxorubicin to high-dose methotrexate plus etoposide and ifosfamide in osteosarcoma patients. Eur. J. Cancer 2007, 43, 752–761. [Google Scholar] [CrossRef]
- Marina, N.M.; Smeland, S.; Bielack, S.S.; Bernstein, M.; Jovic, G.; Krailo, M.D.; Hook, J.M.; Arndt, C.; Berg, H.V.D.; Brennan, B.; et al. Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): An open-label, international, randomised controlled trial. Lancet Oncol. 2016, 17, 1396–1408. [Google Scholar] [CrossRef] [Green Version]
- Bielack, S.S.; Smeland, S.; Whelan, J.S.; Marina, N.; Jovic, G.; Hook, J.M.; Krailo, M.D.; Gebhardt, M.; Pápai, Z.; Meyer, J.; et al. Methotrexate, Doxorubicin, and Cisplatin (MAP) Plus Maintenance Pegylated Interferon Alfa-2b Versus MAP Alone in Patients With Resectable High-Grade Osteosarcoma and Good Histologic Response to Preoperative MAP: First Results of the EURAMOS-1 Good Response Randomized Controlled Trial. J. Clin. Oncol. 2015, 33, 2279–2287. [Google Scholar] [CrossRef]
- Daw, N.C.; Neel, M.D.; Rao, B.N.; Billups, C.A.; Wu, J.; Jenkins, J.J.; Quintana, J.; Luchtman-Jones, L.; Villarroel, M.; Santana, V.M. Frontline treatment of localized osteosarcoma without methotrexate. Cancer 2011, 117, 2770–2778. [Google Scholar] [CrossRef] [Green Version]
- Marec-Berard, P.; Laurence, V.; Occean, B.-V.; Ray-Coquard, I.; Linassier, C.; Corradini, N.; Collard, O.; Chaigneau, L.; Cupissol, D.; Kerbrat, P.; et al. Methotrexate-Etoposide-Ifosfamide Compared with Doxorubicin-Cisplatin-Ifosfamide Chemotherapy in Osteosarcoma Treatment, Patients Aged 18–25 Years. J. Adolesc. Young Adult Oncol. 2020, 9, 172–182. [Google Scholar] [CrossRef]
- Assi, H.; Missenard, G.; Terrier, P.; Le Pechoux, C.; Bonvalot, S.; Vanel, D.; Meric, J.; Tursz, T.; Lecesne, A. Intensive induction chemotherapy without methotrexate in adult patients with localized osteosarcoma: Results of the Institut Gustave-Roussy phase II trial. Curr. Oncol. 2010, 17, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piperno-Neumann, S.; Ray-Coquard, I.; Occean, B.V.; Laurence, V.; Cupissol, D.; Perrin, C.; Penel, N.; Bompas, E.; Rios, M.; Le Cesne, A.; et al. Results of API-AI based regimen in osteosarcoma adult patients included in the French OS2006/Sarcome-09 study. Int J Cancer 2020, 146, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.G.; Bielack, S.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brennan, B.; et al. Bone sarcomas: ESMO-PaedCan-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv79–iv95. [Google Scholar] [CrossRef] [PubMed]
- Lagmay, J.P.; Krailo, M.D.; Dang, H.; Kim, A.; Hawkins, D.S.; Beaty, O.; Widemann, B.C.; Zwerdling, T.; Bomgaars, L.; Langevin, A.-M.; et al. Outcome of Patients With Recurrent Osteosarcoma Enrolled in Seven Phase II Trials Through Children’s Cancer Group, Pediatric Oncology Group, and Children’s Oncology Group: Learning From the Past to Move Forward. J. Clin. Oncol. 2016, 34, 3031–3038. [Google Scholar] [CrossRef] [PubMed]
- Fagioli, F.; Aglietta, M.; Tienghi, A.; Ferrari, S.; Del Prever, A.B.; Vassallo, E.; Palmero, A.; Biasin, E.; Bacci, G.; Picci, P.; et al. High-Dose Chemotherapy in the Treatment of Relapsed Osteosarcoma: An Italian Sarcoma Group Study. J. Clin. Oncol. 2002, 20, 2150–2156. [Google Scholar] [CrossRef]
- Massimo, B.; Giovanni, G.; Stefano, F.; Eleonora, B.; Adalberto, B.D.P.; Sandra, A.; Francesco, S.; Massimo, A.; Franca, F. Phase 2 trial of two courses of cyclophosphamide and etoposide for relapsed high-risk osteosarcoma patients. Cancer 2009, 115, 2980–2987. [Google Scholar] [CrossRef]
- Pratt, C.B.; Horowitz, M.E.; Meyer, W.H.; Etcubanas, E.; Thompson, E.I.; Douglass, E.C.; Wilimas, J.A.; Hayes, F.A.; Green, A.A. Phase II trial of ifosfamide in children with malignant solid tumors. Cancer Treat. Rep. 1987, 71, 131–135. [Google Scholar]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for Soft Tissue Sarcoma: Current Status and Future Perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef] [Green Version]
- Laverdière, C.; Kolb, E.A.; Supko, J.G.; Gorlick, R.; Meyers, P.; Maki, R.G.; Wexler, L.; Demetri, G.D.; Healey, J.H.; Huvos, A.G.; et al. Phase II study of ecteinascidin 743 in heavily pretreated patients with recurrent osteosarcoma. Cancer 2003, 98, 832–840. [Google Scholar] [CrossRef]
- Synold, T.W.; Dussault, I.; Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef]
- Scotlandi, K.; Perdichizzi, S.; Manara, M.C.; Serra, M.; Benini, S.; Cerisano, V.; Strammiello, R.; Mercuri, M.; Reverter-Branchat, G.; Faircloth, G.; et al. Effectiveness of Ecteinascidin-743 against drug-sensitive and -resistant bone tumor cells. Clin. Cancer Res. 2002, 8, 3893–3903. [Google Scholar] [PubMed]
- Gastaud, L.; Saada-Bouzid, E.; Le Morvan, V.; Pourquier, P.; Ianessi, A.; Thariat, J.; Italiano, A.; Thyss, A. Major Efficacy of Trabectedin in 2 Metastatic Osteosarcoma Patients with Wild-Type Asp1104 ERCC5 Tumor Status. Onkologie 2013, 36, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, N.; Frei, E.; Traggis, D.; Bishop, Y. Adjuvant Methotrexate and Citrovorum-Factor Treatment of Osteogenic Sarcoma. N. Engl. J. Med. 1974, 291, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Krailo, M.; Ertel, I.; Makley, J.; Fryer, C.J.H.; Baum, E.; Weetman, R.; Yunis, E.; Barnes, L.; Bleyer, W.A.; Hammond, G.D. A randomized study comparing high-dose methotrexate with moderate-dose methotrexate as components of adjuvant chemotherapy in childhood nonmetastatic osteosarcoma: A report from the childrens cancer study group. Med. Pediatr. Oncol. 1987, 15, 69–77. [Google Scholar] [CrossRef]
- Working Party on Bone Sarcoma. A trial of chemotherapy in patients with osteosarcoma. Br. J. Cancer 1986, 53, 513–518. [Google Scholar] [CrossRef]
- Houghton, P.J.; Kurmasheva, R.T.; Kolb, E.A.; Gorlick, R.; Maris, J.M.; Wu, J.; Tong, Z.; Arnold, M.A.; Chatterjee, M.; Williams, T.M.; et al. Initial testing (stage 1) of the tubulin binding agent nanoparticle albumin-bound (nab) paclitaxel (Abraxane(®)) by the Pediatric Preclinical Testing Program (PPTP). Pediatr. Blood Cancer 2015, 62, 1214–1221. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Kolb, E.A.; Gorlick, R.; Wu, J.; Kurmasheva, R.T.; Houghton, P.J.; Smith, M. Initial testing (stage 1) of the anti-microtubule agents cabazitaxel and docetaxel, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2015, 62, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- Palmerini, E.; Jones, R.L.; Marchesi, E.; Paioli, A.; Cesari, M.; Longhi, A.; Meazza, C.; Coccoli, L.; Fagioli, F.; Asaftei, S.D.; et al. Gemcitabine and docetaxel in relapsed and unresectable high-grade osteosarcoma and spindle cell sarcoma of bone. BMC Cancer 2016, 16, 280. [Google Scholar] [CrossRef] [Green Version]
- Preston, J.N.; Trivedi, M.V. Eribulin: A Novel Cytotoxic Chemotherapy Agent. Ann. Pharmacother. 2012, 46, 802–811. [Google Scholar] [CrossRef]
- Kolb, E.A.; Gorlick, R.; Reynolds, C.P.; Kang, M.H.; Carol, H.; Lock, R.; Keir, S.T.; Maris, J.M.; Billups, C.A.; Desjardins, C.; et al. Initial testing (stage 1) of eribulin, a novel tubulin binding agent, by the pediatric preclinical testing program. Pediatr. Blood Cancer 2013, 60, 1325–1332. [Google Scholar] [CrossRef] [Green Version]
- Sampson, V.B.; Vetter, N.S.; Zhang, W.; Patil, P.U.; Mason, R.W.; George, E.; Gorlick, R.; Kolb, E.A. Integrating mechanisms of response and resistance against the tubulin binding agent Eribulin in preclinical models of osteosarcoma. Oncotarget 2016, 7, 86594–86607. [Google Scholar] [CrossRef] [Green Version]
- Isakoff, M.S.; Goldsby, R.; Villaluna, D.; Krailo, M.D.; Hingorani, P.; Collier, A.; Morris, C.D.; Kolb, E.A.; Doski, J.J.; Womer, R.B.; et al. A phase II study of eribulin in recurrent or refractory osteosarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2019, 66, e27524. [Google Scholar] [CrossRef] [PubMed]
- Kager, L.; Zoubek, A.; Pötschger, U.; Kastner, U.; Flege, S.; Kempf-Bielack, B.; Branscheid, D.; Kotz, R.; Salzer-Kuntschik, M.; Winkelmann, W.; et al. Primary Metastatic Osteosarcoma: Presentation and Outcome of Patients Treated on Neoadjuvant Cooperative Osteosarcoma Study Group Protocols. J. Clin. Oncol. 2003, 21, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Basile, P.; Greengard, E.; Weigel, B.; Spector, L. Prognostic Factors for Development of Subsequent Metastases in Localized Osteosarcoma: A Systematic Review and Identification of Literature Gaps. Sarcoma 2020, 2020, 7431549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeland, S.; Bielack, S.S.; Whelan, J.; Bernstein, M.; Hogendoorn, P.C.; Krailo, M.D.; Gorlick, R.; Janeway, K.A.; Ingleby, F.C.; Anninga, J.; et al. Survival and prognosis with osteosarcoma: Outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur. J. Cancer 2019, 109, 36–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.N.; Grell, K.; Nersting, J.; Abrahamsson, J.; Lund, B.; Kanerva, J.; Jónsson, Ó.; Vaitkevičienė, G.; Pruunsild, K.; Hjalgrim, L.L.; et al. DNA-thioguanine nucleotide concentration and relapse-free survival during maintenance therapy of childhood acute lymphoblastic leukaemia (NOPHO ALL2008): A prospective substudy of a phase 3 trial. Lancet Oncol. 2017, 18, 515–524. [Google Scholar] [CrossRef]
- Bisogno, G.; De Salvo, G.L.; Bergeron, C.; Melcón, S.G.; Merks, J.H.; Kelsey, A.; Martelli, H.; Minard-Colin, V.; Orbach, D.; Glosli, H.; et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 1566–1575. [Google Scholar] [CrossRef]
- Lancia, C.; Anninga, J.; Spitoni, C.; Sydes, M.; Whelan, J.; Hogendoorn, P.C.; Gelderblom, H.; Fiocco, M. Method to measure the mismatch between target and achieved received dose intensity of chemotherapy in cancer trials: A retrospective analysis of the MRC BO06 trial in osteosarcoma. BMJ Open 2019, 9, e022980. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.J.; Dubois, S.G.; Fligor, B.; Li, X.; Goorin, A.; Grier, H.E. Ototoxicity in children treated for osteosarcoma. Pediatr. Blood Cancer 2009, 52, 387–391. [Google Scholar] [CrossRef]
- Li, Y.; Womer, R.; Silber, J. Predicting cisplatin ototoxicity in children: The influence of age and the cumulative dose. Eur. J. Cancer 2004, 40, 2445–2451. [Google Scholar] [CrossRef]
- Harned, T.M.; Kalous, O.; Neuwelt, A.; Loera, J.; Ji, L.; Iovine, P.; Sposto, R.; Neuwelt, E.A.; Reynolds, C.P. Sodium Thiosulfate Administered Six Hours after Cisplatin Does Not Compromise Antineuroblastoma Activity. Clin. Cancer Res. 2008, 14, 533–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, P.; Maibach, R.; Childs, M.; Rajput, K.; Roebuck, D.; Sullivan, M.J.; Laithier, V.; Ronghe, M.; Dall’Igna, P.; Hiyama, E.; et al. Sodium Thiosulfate for Protection from Cisplatin-Induced Hearing Loss. N. Engl. J. Med. 2018, 378, 2376–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freyer, D.R.; Brock, P.; Knight, K.; Reaman, G.; Cabral, S.; Robinson, P.D.; Sung, L. Interventions for cisplatin-induced hearing loss in children and adolescents with cancer. Lancet Child Adolesc. Health 2019, 3, 578–584. [Google Scholar] [CrossRef]
- Viglietta, V.; Shi, F.; Hu, Q.-Y.; Ren, Y.; Keilty, J.; Wolff, H.; McCarthy, R.; Kropp, J.; Weber, P.; Soglia, J. Phase 1 study to evaluate safety, tolerability and pharmacokinetics of a novel intra-tympanic administered thiosulfate to prevent cisplatin-induced hearing loss in cancer patients. Investig. New Drugs 2020, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, K.; Grier, H.E. Sequelae of osteosarcoma medical therapy: A review of rare acute toxicities and late effects. Lancet Oncol. 2010, 11, 670–678. [Google Scholar] [CrossRef]
- Widemann, B.C.; Schwartz, S.; Jayaprakash, N.; Christensen, R.; Pui, C.H.; Chauhan, N.; Daugherty, C.; King, T.R.; Rush, J.E.; Howard, S.C. Efficacy of glucarpidase (carboxypeptidase g2) in patients with acute kidney injury after high-dose methotrexate therapy. Pharmacotherapy 2014, 34, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, S.; Palmerini, E.; Staals, E.; Abate, M.; Longhi, A.; Cesari, M.; Balladelli, A.; Pratelli, L.; Bacci, G. Sex- and Age-Related Chemotherapy Toxicity in Patients with Non-Metastatic Osteosarcoma. J. Chemother. 2009, 21, 205–210. [Google Scholar] [CrossRef]
- Negrao, M.V.; da Silva Rocha, L.S.; da Motta Girardi, D.; Feher, O. Perioperative chemotherapy with and without high-dose methotrexate in adult osteosarcoma. Anticancer Drugs 2017, 28, 915–921. [Google Scholar] [CrossRef]
- Xu, M.; Xu, S.; Yu, X. Clinical analysis of osteosarcoma patients treated with high-dose methotrexate-free neoadjuvant chemotherapy. Curr. Oncol. 2014, 21, e678–e684. [Google Scholar] [CrossRef] [Green Version]
- Skinner, R.; Pearson, A.; English, M.; Price, L.; Wyllie, R.; Coulthard, M.; Craft, A. Cisplatin dose rate as a risk factor for nephrotoxicity in children. Br. J. Cancer 1998, 77, 1677–1682. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, P.C.K.; Hadj-Aïssa, A.; Schell, M.; Dubourg, L.; Brunat-Mentigny, M.; Cochat, P. Long-term nephrotoxicity of cisplatin, ifosfamide, and methotrexate in osteosarcoma. Pediatr. Nephrol. 1998, 12, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, O.; Fawaz, O.; Rey, A.; Niaudet, P.; Ridola, V.; Orbach, D.; Bergeron, C.; Thomassin-Defachelles, A.-S.; Gentet, J.-C.; Schmitt, C.; et al. Long-Term Evaluation of Ifosfamide-Related Nephrotoxicity in Children. J. Clin. Oncol. 2009, 27, 5350–5355. [Google Scholar] [CrossRef] [PubMed]
- Berrak, S.G.; Pearson, M.; Berberoğlu, S.; Ilhan, I.E.; Jaffe, N. High-dose ifosfamide in relapsed pediatric osteosarcoma: Therapeutic effects and renal toxicity. Pediatr. Blood Cancer 2005, 44, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.A.; Morgenstern, B.; Hawkins, U.; Wilson, D.; Liedtke, R.; Miser, J. Renal function following combination chemotherapy with ifosfamide and cisplatin in patients with osteogenic sarcoma. Med. Pediatr. Oncol. 1999, 32, 93–96. [Google Scholar] [CrossRef]
- Schwartz, C.L.; Wexler, L.H.; Krailo, M.D.; Teot, L.A.; Devidas, M.; Steinherz, L.J.; Goorin, A.M.; Gebhardt, M.C.; Healey, J.H.; Sato, J.K.; et al. Intensified Chemotherapy With Dexrazoxane Cardioprotection in Newly Diagnosed Nonmetastatic Osteosarcoma: A Report From the Children’s Oncology Group. Pediatr. Blood Cancer 2016, 63, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulrooney, D.A.; Yeazel, M.W.; Kawashima, T.; Mertens, A.C.; Mitby, P.; Stovall, M.; Donaldson, S.S.; Green, D.M.; Sklar, C.A.; Robison, L.L.; et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: Retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ 2009, 339, b4606. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.R.; Vijarnsorn, C.; Potts, J.; Milner, R.; Sandor, G.G.S.; Fryer, C. Anthracycline induced cardiac toxicity in pediatric Ewing sarcoma: A longitudinal study. Pediatr. Blood Cancer 2013, 60, 842–848. [Google Scholar] [CrossRef]
- Smith, M.; Ungerleider, R.S.; Horowitz, M.E.; Simon, R. Influence of Doxorubicin Dose Intensity on Response and Outcome for Patients With Osteogenic Sarcoma and Ewing’s Sarcoma. J. Natl. Cancer Inst. 1991, 83, 1460–1470. [Google Scholar] [CrossRef]
- Bacci, G.; Picci, P.; Ferrari, S.; Casadei, R.; Del Prever, A.B.; Tienghi, A.; Mancini, A. Influence of Adriamycin Dose in the Outcome of Patients with Osteosarcoma Treated with Multidrug Neoadjuvant Chemotherapy: Results of Two Sequential Studies. J. Chemother. 1993, 5, 237–246. [Google Scholar] [CrossRef]
- Paulides, M.; Kremers, A.; Stöhr, W.; Bielack, S.; Jürgens, H.; Treuner, J.; Beck, J.; Langer, T.; German Late Effects Working Group in the Society of Pediatric Oncology and Haematology (GPOH). Prospective longitudinal evaluation of doxorubicin-induced cardiomyopathy in sarcoma patients: A report of the late effects surveillance system (LESS). Pediatr. Blood Cancer 2006, 46, 489–495. [Google Scholar] [CrossRef]
- Geidel, S.; Garn, M.; Grävinghoff, L.; Hausdorf, G.; Morf, G.; Bielack, S.; Knop, J.; Winkler, K. Cardiomyopathy after osteosarcoma treatment: A contribution to the cardiotoxicity of adriamycin. Klin. Padiatr. 1991, 203, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Longhi, A.; Ferrari, S.; Bacci, G.; Specchia, S. Long-term follow-up of patients with doxorubicin-induced cardiac toxicity after chemotherapy for osteosarcoma. Anticancer Drugs 2007, 18, 737–744. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, P.A. Anthracycline cardiotoxicity: An update on mechanisms, monitoring and prevention. Heart 2018, 104, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Yan, T.; Jendrny, C.; Nemecek, A.; Vincetic, M.; Gödtel-Armbrust, U.; Wojnowski, L. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both topoisomerase II isoforms. BMC Cancer 2014, 14, 842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, N. Pharmacological strategies to overcome treatment resistance in acute myeloid leukemia: Increasing leukemic drug exposure by targeting the resistance factor SAMHD1 and the toxicity factor Top2β. Expert Opin. Drug Discov. 2020, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Gerber, M.C.; Ewer, M.S.; Bianchine, J.R.; Gams, R.A. Delayed administration of dexrazoxane provides cardioprotection for patients with advanced breast cancer treated with doxorubicin-containing therapy. J. Clin. Oncol. 1997, 15, 1333–1340. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Scully, R.E.; Lipsitz, S.R.; Sallan, S.E.; Silverman, L.B.; Miller, T.L.; Barry, E.V.; Asselin, B.L.; Athale, U.; Clavell, L.A.; et al. Assessment of dexrazoxane as a cardioprotectant in doxorubicin-treated children with high-risk acute lymphoblastic leukaemia: Long-term follow-up of a prospective, randomised, multicentre trial. Lancet Oncol. 2010, 11, 950–961. [Google Scholar] [CrossRef] [Green Version]
- Kopp, L.; Womer, R.B.; Schwartz, C.L.; Ebb, D.H.; Franco, V.I.; Hall, D.; Barkauskas, D.A.; Krailo, M.D.; Grier, H.E.; Meyers, P.A.; et al. Effects of dexrazoxane on doxorubicin-related cardiotoxicity and second malignant neoplasms in children with osteosarcoma: A report from the Children’s Oncology Group. Cardiooncology 2019, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strander, H.; Bauer, H.C.F.; Brosjö, O.; Kreicbergs, A.; Lindholm, J.; Nilsonne, U.; Silfverswärd, C.; Szamosi, A. Adjuvant interferon treatment in human osteosarcoma. Cancer Treat. Res. 1993, 62, 29–32. [Google Scholar] [PubMed]
- Strander, H.; Bauer, H.C.; Brosjö, O.; Fernberg, J.O.; Kreicbergs, A.; Nilsonne, U.; Silfverswärd, C.; Signomklao, T.; Söderlund, V. Long-Term Adjuvant Interferon Treatment of Human Osteosarcoma: A pilot study. Acta Oncol. 1995, 34, 877–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, C.R.; Smeland, S.; Bauer, H.C.F.; Sæter, G.; Strander, H. Interferon-α as the only adjuvant treatment in high-grade osteosarcoma: Long term results of the Karolinska Hospital series. Acta Oncol. 2005, 44, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Whelan, J.; Patterson, D.; Perisoglou, M.; Bielack, S.; Marina, N.; Smeland, S.; Bernstein, M. The role of interferons in the treatment of osteosarcoma. Pediatr. Blood Cancer 2010, 54, 350–354. [Google Scholar] [CrossRef]
- Kaufman, H.L. Combination Immunotherapy for Melanoma. JAMA Oncol. 2015, 1, 387–388. [Google Scholar] [CrossRef] [Green Version]
- Arndt, C.A.; Koshkina, N.V.; Inwards, C.Y.; Hawkins, U.S.; Krailo, M.D.; Villaluna, D.; Anderson, P.M.; Goorin, A.M.; Blakely, M.L.; Bernstein, M.; et al. Inhaled granulocyte-macrophage colony stimulating factor for first pulmonary recurrence of osteosarcoma: Effects on disease-free survival and immunomodulation. a report from the Children’s Oncology Group. Clin. Cancer Res. 2010, 16, 4024–4030. [Google Scholar] [CrossRef] [Green Version]
- Meyers, P. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Rev. Anticancer Ther. 2009, 9, 1035–1049. [Google Scholar] [CrossRef]
- Anderson, P.; Meyers, P.; Kleinerman, E.; Venkatakrishnan, K.; Hughes, D.; Herzog, C.; Huh, W.; Sutphin, R.; Vyas, Y.M.; Shen, V.; et al. Mifamurtide in metastatic and recurrent osteosarcoma: A patient access study with pharmacokinetic, pharmacodynamic, and safety assessments. Pediatr. Blood Cancer 2014, 61, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Hunsberger, S.; Freidlin, B.; Smith, M.A. Complexities in Interpretation of Osteosarcoma Clinical Trial Results. J. Clin. Oncol. 2008, 26, 3103–3104. [Google Scholar] [CrossRef]
- Kager, L.; Pötschger, U.; Bielack, S. Review of mifamurtide in the treatment of patients with osteosarcoma. Ther. Clin. Risk Manag. 2010, 6, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.S.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Shimizu, T.; Fuchimoto, Y.; Okita, H.; Fukuda, K.; Kitagawa, Y.; Ueno, S.; Kuroda, T. A curative treatment strategy using tumor debulking surgery combined with immune checkpoint inhibitors for advanced pediatric solid tumors: An in vivo study using a murine model of osteosarcoma. J. Pediatr. Surg. 2018, 53, 2460–2464. [Google Scholar] [CrossRef]
- Osipov, A.; Saung, M.T.; Zheng, L.; Murphy, A. Small molecule immunomodulation: The tumor microenvironment and overcoming immune escape. J. Immunother. Cancer 2019, 7, 224. [Google Scholar] [CrossRef] [Green Version]
- Laoui, D.; Van Overmeire, E.; De Baetselier, P.; Van Ginderachter, J.A.; Raes, G. Functional Relationship between Tumor-Associated Macrophages and Macrophage Colony-Stimulating Factor as Contributors to Cancer Progression. Front. Immunol. 2014, 5, 489. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Lu, Y.; Li, R.; Jiang, Y.; Zheng, Y.; Qian, J.; Bi, E.; Zheng, C.; Hou, J.; Wang, S.; et al. Therapeutic effects of CSF1R-blocking antibodies in multiple myeloma. Leukemia 2018, 32, 176–183. [Google Scholar] [CrossRef]
- Goulart, M.; Pluhar, G.E.; Ohlfest, J.R. Identification of Myeloid Derived Suppressor Cells in Dogs with Naturally Occurring Cancer. PLoS ONE 2012, 7, e33274. [Google Scholar] [CrossRef] [Green Version]
- Ségaliny, A.I.; Mohamadi, A.; Dizier, B.; Lokajczyk, A.; Brion, R.; Lanel, R.; Amiaud, J.; Charrier, C.; Boisson-Vidal, C.; Heymann, D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int. J. Cancer 2014, 137, 73–85. [Google Scholar] [CrossRef]
- Kubota, Y.; Takubo, K.; Shimizu, T.; Ohno, H.; Kishi, K.; Shibuya, M.; Saya, H.; Suda, T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med. 2009, 206, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- London, C.; Mathie, T.; Stingle, N.; Clifford, C.; Haney, S.; Klein, M.K.; Beaver, L.; Vickery, K.; Vail, D.M.; Hershey, B.; et al. Preliminary evidence for biologic activity of toceranib phosphate (Palladia(®)) in solid tumours. Vet. Comp. Oncol. 2012, 10, 194–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Hajjami, H.M.-E.; et al. T cell–induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Eissler, N.; Le Blanc, K.; Johnsen, J.I.; Kogner, P.; Kiessling, R. Targeting Suppressive Myeloid Cells Potentiates Checkpoint Inhibitors to Control Spontaneous Neuroblastoma. Clin. Cancer Res. 2016, 22, 3849–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-C.; Wu, C.; Hung, R.-L. Response of refractory osteosarcoma to thalidomide and celecoxib. Lancet Oncol. 2005, 6, 997–999. [Google Scholar] [CrossRef]
- D’Amato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, G.-J.; Lin, H.-H.; Xu, Q.-T.; Wang, M.-W. Thalidomide inhibits growth of tumors through COX-2 degradation independent of antiangiogenesis. Vasc. Pharmacol. 2005, 43, 112–119. [Google Scholar] [CrossRef]
- Dickens, D.; Kozielski, R.; Khan, J.; Forus, A.; Cripe, T.P. Cyclooxygenase-2 Expression in Pediatric Sarcomas. Pediatr. Dev. Pathol. 2002, 5, 356–364. [Google Scholar] [CrossRef]
- Lemos, S.; Sampaio-Marques, B.; Ludovico, P.; Gaivão, I.; Palmeira, C.; Martins, G.; Peixoto, F.; Pinto-Leite, R.; Oliveira, P. Elucidating the mechanisms of action of parecoxib in the MG-63 osteosarcoma cell line. Anticancer Drugs 2020, 31, 507–517. [Google Scholar] [CrossRef]
- Liu, J.; Wu, J.; Zhou, L.; Pan, C.; Zhou, Y.; Du, W.; Chen, J.-M.; Zhu, X.; Shen, J.; Chen, S.; et al. ZD6474, a new treatment strategy for human osteosarcoma, and its potential synergistic effect with celecoxib. Oncotarget 2015, 6, 21341–21352. [Google Scholar] [CrossRef]
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H.; Cutolo, M. Glucocorticoids and chronic inflammation. Rheumatology 2016, 55 (Suppl. 2), ii6–ii14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Xia, J.; Wang, S.-Q.; Wei, Y.-B.; Wu, J.-G.; Chen, F.-Y.; Huang, G.-Y.; Shi, J.-S. Pathway enrichment analysis of human osteosarcoma U-2 OS bone cells expose to dexamethasone. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2631–2639. [Google Scholar] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Baselga, J.; Kim, S.-B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.M.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, S.M.; Miles, D.; Kim, S.-B.; Im, Y.-H.; Im, S.-A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef]
- Ebb, D.H.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II Trial of Trastuzumab in Combination With Cytotoxic Chemotherapy for Treatment of Metastatic Osteosarcoma With Human Epidermal Growth Factor Receptor 2 Overexpression: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 2545–2551. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, S.E.; Geisinger, K.R.; King, T.S.; Sciarrotta, J.; Ward, W.G.; Gold, S.H.; Bos, G.D. Clinicopathologic analysis of HER-2/neu immunoexpression among various histologic subtypes and grades of osteosarcoma. Mod. Pathol. 2001, 14, 1277–1283. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.; Hingorani, P.; Roth, M.; Gorlick, R. HER2-Targeted Therapy in Osteosarcoma. Adv. Exp. Med. Biol. 2020, 1257, 55–66. [Google Scholar] [CrossRef]
- Raben, D.; Helfrich, B.A.; Chan, D.; Johnson, G.; Bunn, P.A. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, alone and in combination with radiation and chemotherapy as a new therapeutic strategy in non-small cell lung cancer. Semin. Oncol. 2002, 29 (Suppl. 4), 37–46. [Google Scholar] [CrossRef]
- Lee, J.A.; Ko, Y.; Kim, D.H.; Lim, J.S.; Kong, C.-B.; Cho, W.H.; Jeon, D.-G.; Lee, S.-Y.; Koh, J.S. Epidermal Growth Factor Receptor: Is It a Feasible Target for the Treatment of Osteosarcoma? Cancer Res. Treat. 2012, 44, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Sevelda, F.; Mayr, L.; Kubista, B.; Lötsch, D.; Van Schoonhoven, S.; Windhager, R.; Pirker, C.; Micksche, M.; Berger, W. EGFR is not a major driver for osteosarcoma cell growth in vitro but contributes to starvation and chemotherapy resistance. J. Exp. Clin. Cancer Res. 2015, 34, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, J.; Yin, M.; Sun, Z.; Kang, X.; Liu, D.; Jiang, K.; Xu, J.; Zhao, F.; Guo, Q.; Zheng, W. SPC24 promotes osteosarcoma progression by increasing EGFR/MAPK signaling. Oncotarget 2017, 8, 105276–105283. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Ramos, M.; Zamudio-Cuevas, Y.; Medina-Luna, D.; Martínez-Flores, K.; Martínez-Nava, G.; Fernández-Torres, J.; López-Reyes, A.; Solca, F. Afatinib is active in osteosarcoma in osteosarcoma cell lines. J. Cancer Res. Clin. Oncol. 2020, 146, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Felip, E.; Cobo, M.; Lu, S.; Syrigos, K.; Lee, K.H.; Goker, E.; Georgoulias, V.; Li, W.; Isla, D.; et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): An open-label randomised controlled phase 3 trial. Lancet Oncol. 2015, 16, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudier, B.; Worden, F.; Kudo, M. Sorafenib: Key lessons from over 10 years of experience. Expert Rev. Anticancer Ther. 2019, 19, 177–189. [Google Scholar] [CrossRef]
- Kunz, P.; Fellenberg, J.; Moskovszky, L.; Sápi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendrõi, M.; Ruef, P.; et al. Improved Survival in Osteosarcoma Patients with Atypical Low Vascularization. Ann. Surg. Oncol. 2015, 22, 489–496. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Dileo, P.; Asaftei, S.D.; D’Ambrosio, L.; Pignochino, Y.; Mercuri, M.; Picci, P.; Fagioli, F.; Casali, P.G.; et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: An Italian Sarcoma Group study. Ann. Oncol. 2012, 23, 508–516. [Google Scholar] [CrossRef]
- Pignochino, Y.; Dell’Aglio, C.; Basiricò, M.; Capozzi, F.; Soster, M.; Marchio’, S.; Bruno, S.; Gammaitoni, L.; Sangiolo, D.; Torchiaro, E.; et al. The Combination of Sorafenib and Everolimus Abrogates mTORC1 and mTORC2 Upregulation in Osteosarcoma Preclinical Models. Clin. Cancer Res. 2013, 19, 2117–2131. [Google Scholar] [CrossRef] [Green Version]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.M.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Mross, K.; Frost, A.; Steinbild, S.; Hedbom, S.; Büchert, M.; Fasol, U.; Unger, C.; Krätzschmar, J.; Heinig, R.; Boix, O.; et al. A Phase I Dose-Escalation Study of Regorafenib (BAY 73-4506), an Inhibitor of Oncogenic, Angiogenic, and Stromal Kinases, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2012, 18, 2658–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Davis, L.E.; Bolejack, V.; Ryan, C.W.; Ganjoo, K.N.; Loggers, E.T.; Chawla, S.; Agulnik, M.; Livingston, M.B.; Reed, D.; Keedy, V.; et al. Randomized Double-Blind Phase II Study of Regorafenib in Patients With Metastatic Osteosarcoma. J. Clin. Oncol. 2019, 37, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Woude, G.F.V. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef]
- Italiano, A.; Mir, O.; Mathoulin-Pelissier, S.; Penel, N.; Piperno-Neumann, S.; Bompas, E.; Chevreau, C.; Duffaud, F.; Entz-Werlé, N.; Saada, E.; et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 446–455. [Google Scholar] [CrossRef]
- Ohba, T.; Cates, J.M.M.; Cole, H.A.; Slosky, D.A.; Haro, H.; Ichikawa, J.; Ando, T.; Schwartz, H.S.; Schoenecker, J.G. Pleiotropic effects of bisphosphonates on osteosarcoma. Bone 2014, 63, 110–120. [Google Scholar] [CrossRef]
- Clezardin, P. Mechanisms of action of bisphosphonates in oncology: A scientific concept evolving from antiresorptive to anticancer activities. BoneKEy Rep. 2013, 2, 267. [Google Scholar] [CrossRef] [Green Version]
- Liao, D.; Zhong, L.; Yin, J.; Zeng, C.; Wang, X.; Huang, X.; Chen, J.; Zhang, H.; Zhang, R.; Guan, X.-Y.; et al. Author Correction: Chromosomal translocation-derived aberrant Rab22a drives metastasis of osteosarcoma. Nat. Cell Biol. 2020, 22, 907. [Google Scholar] [CrossRef]
- Heymann, M.-F.; Lezot, F.; Heymann, D. Bisphosphonates in common pediatric and adult bone sarcomas. Bone 2020, 139, 115523. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Le Deley, M.C.; Rédini, F.; Pacquement, H.; Marec-Bérard, P.; Petit, P.; Brisse, H.; Lervat, C.; Gentet, J.C.; Entz-Werlé, N.; et al. Zoledronate in combination with chemotherapy and surgery to treat osteosarcoma (OS2006): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 1070–1080. [Google Scholar] [CrossRef]
- Kolb, E.A.; Kamara, D.; Zhang, W.; Lin, J.; Hingorani, P.; Baker, L.; Houghton, P.; Gorlick, R. R1507, a fully human monoclonal antibody targeting IGF-1R, is effective alone and in combination with rapamycin in inhibiting growth of osteosarcoma xenografts. Pediatr. Blood Cancer 2010, 55, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappo, A.S.; Vassal, G.; Crowley, J.J.; Bolejack, V.; Hogendoorn, P.C.W.; Chugh, R.; Ladanyi, M.; Grippo, J.F.; Dall, G.; Staddon, A.P.; et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: Results of a Sarcoma Alliance. Cancer 2014, 120, 2448–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gvozdenovic, A.; Boro, A.; Born, W.; Muff, R.; Fuchs, B. A bispecific antibody targeting IGF-IR and EGFR has tumor and metastasis suppressive activity in an orthotopic xenograft osteosarcoma mouse model. Am. J. Cancer Res. 2017, 7, 1435–1449. [Google Scholar]
- Rousseau, J.; Escriou, V.; Lamoureux, F.; Brion, R.; Chesneau, J.; Battaglia, S.; Amiaud, J.; Scherman, D.; Heymann, D.; Rédini, F.; et al. Formulated siRNAs targeting Rankl prevent osteolysis and enhance chemotherapeutic response in osteosarcoma models. J. Bone Miner. Res. 2011, 26, 2452–2462. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.G.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Engert, F.; Kovac, M.; Baumhoer, D.; Nathrath, M.; Fulda, S. Osteosarcoma cells with genetic signatures of BRCAness are susceptible to the PARP inhibitor talazoparib alone or in combination with chemotherapeutics. Oncotarget 2017, 8, 48794–48806. [Google Scholar] [CrossRef] [Green Version]
- Rose, A.A.N.; Biondini, M.; Curiel, R.; Siegel, P.M. Targeting GPNMB with glembatumumab vedotin: Current developments and future opportunities for the treatment of cancer. Pharmacol. Ther. 2017, 179, 127–141. [Google Scholar] [CrossRef]
- Roth, M.; Barris, D.M.; Piperdi, S.; Kuo, V.; Everts, S.; Geller, D.; Houghton, P.; Kolb, E.A.; Hawthorne, T.; Gill, J.; et al. Targeting Glycoprotein NMB With Antibody-Drug Conjugate, Glembatumumab Vedotin, for the Treatment of Osteosarcoma. Pediatr. Blood Cancer 2016, 63, 32–38. [Google Scholar] [CrossRef]
- Yardley, D.A.; Weaver, R.; Melisko, M.E.; Saleh, M.N.; Arena, F.P.; Forero, A.; Cigler, T.; Stopeck, A.; Citrin, D.; Oliff, I.; et al. EMERGE: A Randomized Phase II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Advanced Glycoprotein NMB-Expressing Breast Cancer. J. Clin. Oncol. 2015, 33, 1609–1619. [Google Scholar] [CrossRef]
- Kolb, E.A.; Gorlick, R.; Billups, C.A.; Hawthorne, T.; Kurmasheva, R.T.; Houghton, P.J.; Smith, M. Initial testing (stage 1) of glembatumumab vedotin (CDX-011) by the pediatric preclinical testing program. Pediatr. Blood Cancer 2014, 61, 1816–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, L.; Malempati, S.; Krailo, M.; Gao, Y.; Buxton, A.; Weigel, B.J.; Hawthorne, T.; Crowley, E.; Moscow, J.A.; Reid, J.M.; et al. Phase II trial of the glycoprotein non-metastatic B-targeted antibody–drug conjugate, glembatumumab vedotin (CDX-011), in recurrent osteosarcoma AOST1521: A report from the Children’s Oncology Group. Eur. J. Cancer 2019, 121, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, W.; Zhang, H.; Gao, P.; Fan, B.; Huang, C.; Fu, J.; Chen, G.; Shi, L.; Zhu, H.; et al. Individualized chemotherapy for osteosarcoma and identification of gene mutations in osteosarcoma. Tumor Boil. 2015, 36, 2427–2435. [Google Scholar] [CrossRef]
- Anderson, P.M.; Bielack, S.S.; Gorlick, R.; Skubitz, K.; Daw, N.C.; Herzog, C.E.; Monge, O.R.; Lassaletta, A.; Boldrini, E.; Pápai, Z.; et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma. Pediatr. Blood Cancer 2016, 63, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Eilber, F.R.; Rosen, G. Adjuvant chemotherapy for osteosarcoma. Semin. Oncol. 1989, 16, 312–322. [Google Scholar] [PubMed]
- Cores, E.P. Doxorubicin in disseminated osteosarcoma. JAMA 1972, 221, 1132–1138. [Google Scholar] [CrossRef]
- Cortes, E.P.; Holland, J.F.; Wang, J.J.; Sinks, L.F.; Blom, J.; Senn, H.; Bank, A.; Glidewell, O. Amputation and Adriamycin in Primary Osteosarcoma. N. Engl. J. Med. 1974, 291, 998–1000. [Google Scholar] [CrossRef]
- Goorin, A.M.; Perez-Atayde, A.; Gebhardt, M.; Andersen, J.W.; Wilkinson, R.H.; Delorey, M.J.; Watts, H.; Link, M.; Jaffe, N.; Frei, E., 3rd. Weekly high-dose methotrexate and doxorubicin for osteosarcoma: The Dana-Farber Cancer Institute/the Children’s Hospital--study III. J. Clin. Oncol. 1987, 5, 1178–1184. [Google Scholar] [CrossRef]
- Rosen, G.; Nirenberg, A.; Caparros, B.; Juergens, H.; Kosloff, C.; Mehta, B.M.; Marcove, R.C.; Huvos, A.G. Osteogenic sarcoma: Eight-percent, three-year, disease-free survival with combination chemotherapy (T-7). Natl. Cancer Inst. Monogr. 1981, 56, 213–220. [Google Scholar]
- Winkler, K.; Beron, G.; Kotz, R.; Salzer-Kuntschik, M.; Beck, J.; Beck, W.; Brandeis, W.; Ebell, W.; Erttmann, R.; Göbel, U. Neoadjuvant chemotherapy for osteogenic sarcoma: Results of a Cooperative German/Austrian study. J. Clin. Oncol. 1984, 2, 617–624. [Google Scholar] [CrossRef]
- Graf, N.; Winkler, K.; Betlemovic, M.; Fuchs, N.; Bode, U. Methotrexate pharmacokinetics and prognosis in osteosarcoma. J. Clin. Oncol. 1994, 12, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Lancia, C.; Anninga, J.K.; Sydes, M.; Spitoni, C.; Whelan, J.S.; Hogendoorn, P.C.; Gelderblom, H.; Fiocco, M. A novel method to address the association between received dose intensity and survival outcome: Benefits of approaching treatment intensification at a more individualised level in a trial of the European Osteosarcoma Intergroup. Cancer Chemother. Pharmacol. 2019, 83, 951–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramwell, V.H.; Burgers, M.; Sneath, R.; Souhami, R.; Van Oosterom, A.T.; Voûte, P.A.; Rouesse, J.; Spooner, D.; Craft, A.W.; Somers, R. A comparison of two short intensive adjuvant chemotherapy regimens in operable osteosarcoma of limbs in children and young adults: The first study of the European Osteosarcoma Intergroup. J. Clin. Oncol. 1992, 10, 1579–1591. [Google Scholar] [CrossRef] [PubMed]
- Souhami, R.L.; Craft, A.W.; Van Der Eijken, J.W.; Nooij, M.; Spooner, D.; Bramwell, V.H.; Wierzbicki, R.; Malcolm, A.J.; Kirkpatrick, A.; Uscinska, B.M.; et al. Randomised trial of two regimens of chemotherapy in operable osteosarcoma: A study of the European Osteosarcoma Intergroup. Lancet 1997, 350, 911–917. [Google Scholar] [CrossRef]
- Li, S.; Sun, W.; Wang, H.; Zuo, N.; Hua, Y.; Cai, Z. Research progress on the multidrug resistance mechanisms of osteosarcoma chemotherapy and reversal. Tumor Biol. 2015, 36, 1329–1338. [Google Scholar] [CrossRef]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenetics 2019, 11, 1–24. [Google Scholar] [CrossRef]
- Chen, R.; Wang, G.; Zheng, Y.; Hua, Y.; Cai, Z. Drug resistance-related microRNAs in osteosarcoma: Translating basic evidence into therapeutic strategies. J. Cell. Mol. Med. 2019, 23, 2280–2292. [Google Scholar] [CrossRef] [Green Version]
- Tunggal, J.K.; Cowan, D.S.; Shaikh, H.; Tannock, I.F. Penetration of anticancer drugs through solid tissue: A factor that limits the effectiveness of chemotherapy for solid tumors. Clin. Cancer Res. 1999, 5, 1583–1586. [Google Scholar]
- West, G.W.; Weichselbaum, R.; Little, J.B. Limited penetration of methotrexate into human osteosarcoma spheroids as a proposed model for solid tumor resistance to adjuvant chemotherapy. Cancer Res. 1980, 40, 3665–3668. [Google Scholar]
- Wang, S.-Y.; Hu, H.-Z.; Qing, X.-C.; Zhang, Z.-C.; Shao, Z.-W. Recent advances of drug delivery nanocarriers in osteosarcoma treatment. J. Cancer 2020, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Avnet, S.; Lemma, S.; Cortini, M.; Pellegrini, P.; Perut, F.; Zini, N.; Kusuzaki, K.; Chano, T.; Grisendi, G.; Dominici, M.; et al. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget 2016, 7, 63408–63423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Kolb, E.A.; Qin, J.; Chou, A.J.; Sowers, R.; Hoang, B.; Healey, J.H.; Huvos, A.G.; Meyers, P.; Gorlick, R. The Folate Receptor Is Frequently Overexpressed in Osteosarcoma Samples and Plays a Role in the Uptake of the Physiologic Substrate 5-Methyltetrahydrofolate. Clin. Cancer Res. 2007, 13, 2557–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Healey, J.H.; Meyers, P.A.; Ladanyi, M.; Huvos, A.G.; Bertino, J.R.; Gorlick, R. Mechanisms of methotrexate resistance in osteosarcoma. Clin. Cancer Res. 1999, 5, 621–627. [Google Scholar] [PubMed]
- Dixon, K.H.; Lanpher, B.C.; Chiu, J.; Kelley, K.; Cowan, K.H. A novel cDNA restores reduced folate carrier activity and methotrexate sensitivity to transport deficient cells. J. Biol. Chem. 1994, 269, 17–20. [Google Scholar] [PubMed]
- Da Costa, M.E.M.; Marchais, A.; Gomez-Brouchet, A.; Job, B.; Assoun, N.; Daudigeos-Dubus, E.; Fromigué, O.; Santos, C.; Geoerger, B.; Gaspar, N.; et al. In-Vitro and In-Vivo Establishment and Characterization of Bioluminescent Orthotopic Chemotherapy-Resistant Human Osteosarcoma Models in NSG Mice. Cancers 2019, 11, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patiño-García, A.; Zalacaín, M.; Marrodán, L.; San-Julian, M.; Sierrasesúmaga, L. Methotrexate in Pediatric Osteosarcoma: Response and Toxicity in Relation to Genetic Polymorphisms and Dihydrofolate Reductase and Reduced Folate Carrier 1 Expression. J. Pediatr. 2009, 154, 688–693. [Google Scholar] [CrossRef]
- Ifergan, I.; Meller, I.; Issakov, J.; Assaraf, Y.G. Reduced folate carrier protein expression in osteosarcoma. Cancer 2003, 98, 1958–1966. [Google Scholar] [CrossRef]
- Flintoff, W.F.; Sadlish, H.; Gorlick, R.; Yang, R.; Williams, F.M. Functional analysis of altered reduced folate carrier sequence changes identified in osteosarcomas. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2004, 1690, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Serra, M.; Reverter-Branchat, G.; Maurici, D.; Benini, S.; Shen, J.-N.; Chano, T.; Hattinger, C.M.; Manara, M.C.; Pasello, M.; Scotlandi, K.; et al. Analysis of dihydrofolate reductase and reduced folate carrier gene status in relation to methotrexate resistance in osteosarcoma cells. Ann. Oncol. 2004, 15, 151–160. [Google Scholar] [CrossRef]
- Yang, R.; Sowers, R.; Mazza, B.; Healey, J.H.; Huvos, A.; Grier, H.; Bernstein, M.; Beardsley, G.P.; Krailo, M.; Devidas, M.; et al. Sequence alterations in the reduced folate carrier are observed in osteosarcoma tumor samples. Clin. Cancer Res. 2003, 9, 837–844. [Google Scholar]
- Yang, R.; Qin, J.; Hoang, B.H.; Healey, J.H.; Gorlick, R. Polymorphisms and Methylation of the Reduced Folate Carrier in Osteosarcoma. Clin. Orthop. Relat. Res. 2008, 466, 2046–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trippett, T.; Meyers, P.; Gorlick, R.; Steinherz, P.; Wollner, N.; Bertino, J.R. High dose trimetrexate with leucovorin protection in recurrent childhood malignancies: A phase II trial. J. Clin. Oncol. (ASCO Annu. Meet. Abstr.) 1999, 9, 889. [Google Scholar]
- Sirotnak, F.; DeGraw, J.; Moccio, D.; Samuels, L.; Goutas, L. New folate analogs of the 10-deaza-aminopterin series Basis for structural design and biochemical and pharmacologic properties. Cancer Chemother. Pharmacol. 1984, 12, 18–25. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.A.; Pro, B.; Pinter-Brown, L.; Bartlett, N.L.; Popplewell, L.; Coiffier, B.; Lechowicz, M.J.; Savage, K.J.; Shustov, A.R.; Gisselbrecht, C.; et al. Pralatrexate in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results From the Pivotal PROPEL Study. J. Clin. Oncol. 2011, 29, 1182–1189. [Google Scholar] [CrossRef]
- Cole, P.; Smith, A.; Kamen, B. Osteosarcoma cells, resistant to methotrexate due to nucleoside and nucleobase salvage, are sensitive to nucleoside analogs. Cancer Chemother. Pharmacol. 2002, 50, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Fruci, D.; Cho, W.C.; Romania, P.; Nobili, V.; Locatelli, F.; Alisi, A. Drug Transporters and Multiple Drug Resistance in the Most Common Pediatric Solid Tumors. Curr. Drug Metab. 2016, 17, 308–316. [Google Scholar] [CrossRef]
- Wunder, J.S.; Bull, S.B.; Aneliunas, V.; Lee, P.D.; Davis, A.; Beauchamp, C.P.; Conrad, E.U.; Grimer, R.J.; Healey, J.H.; Rock, M.J.; et al. MDR1 Gene Expression and Outcome in Osteosarcoma: A Prospective, Multicenter Study. J. Clin. Oncol. 2000, 18, 2685–2694. [Google Scholar] [CrossRef]
- Rajkumar, T.; Yamuna, M. Multiple pathways are involved in drug resistance to doxorubicin in an osteosarcoma cell line. Anticancer Drugs 2008, 19, 257–265. [Google Scholar] [CrossRef]
- Chan, H.S.L.; Haddad, G.; DeBoer, G.; Ling, V.; Grogan, T.M. P-glycoprotein Expression: Critical Determinant in the Response to Osteosarcoma Chemotherapy. J. Natl. Cancer Inst. 1997, 89, 1706–1715. [Google Scholar] [CrossRef] [Green Version]
- Baldini, N.; Scotlandi, K.; Barbanti-Bròdano, G.; Manara, M.C.; Maurici, D.; Bacci, G.; Bertoni, F.; Picci, P.; Sottili, S.; Campanacci, M.; et al. Expression of P-Glycoprotein in High-Grade Osteosarcomas in Relation to Clinical Outcome. N. Engl. J. Med. 1995, 333, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Pasello, M.; Manara, M.C.; Scotlandi, K.; Ferrari, S.; Bertoni, F.; Mercuri, M.; Alvegard, T.A.; Picci, P.; Bacci, G.; et al. May P-glycoprotein status be used to stratify high-grade osteosarcoma patients? Results from the Italian/Scandinavian Sarcoma Group 1 treatment protocol. Int. J. Oncol. 2006, 29, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Kan, W.-S.; Zhao, Z.-G.; Ding, F.; Liu, M.; Ma, D.-Z.; Zheng, C.-K. Association between P-Glycoprotein expression and response to chemotherapy in patients with osteosarcoma: A systematic and meta-analysis. J. Cancer Res. Ther. 2014, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liao, Y.; Shen, J.K.; Feng, Y.; Choy, E.; Cote, G.M.; Harmon, D.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. Evaluation of P-glycoprotein (Pgp) expression in human osteosarcoma by high-throughput tissue microarray. J. Orthop. Res. 2016, 34, 1606–1612. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.; Gorlick, R.; Teot, L.; Krailo, M.; Chen, Z.; Goorin, A.; Grier, H.E.; Bernstein, M.L.; Meyers, P. Multiple Drug Resistance in Osteogenic Sarcoma: INT0133 From the Children’s Oncology Group. J. Clin. Oncol. 2007, 25, 2057–2062. [Google Scholar] [CrossRef]
- Ferrari, S.; Bertoni, F.; Zanella, L.; Setola, E.; Bacchini, P.; Alberghini, M.; Versari, M.; Bacci, G. Evaluation of P-glycoprotein, HER-2/ErbB-2, p53, and Bcl-2 in primary tumor and metachronous lung metastases in patients with high-grade osteosarcoma. Cancer 2004, 100, 1936–1942. [Google Scholar] [CrossRef]
- Wu, D.; Liu, L.; Yan, X.; Wang, C.; Wang, Y.; Han, K.; Lin, S.; Gan, Z.; Min, D. Pleiotrophin promotes chemoresistance to doxorubicin in osteosarcoma by upregulating P-glycoprotein. Oncotarget 2017, 8, 63857–63870. [Google Scholar] [CrossRef]
- Windsor, R.E.; Strauss, S.J.; Kallis, C.; Wood, N.W.; Whelan, J.S. Germline genetic polymorphisms may influence chemotherapy response and disease outcome in osteosarcoma. Cancer 2012, 118, 1856–1867. [Google Scholar] [CrossRef]
- Yang, X.; Yang, P.; Shen, J.; Osaka, E.; Choy, E.; Cote, G.M.; Harmon, D.; Zhang, Z.; Mankin, H.; Hornicek, F.J.; et al. Prevention of multidrug resistance (MDR) in osteosarcoma by NSC23925. Br. J. Cancer 2014, 110, 2896–2904. [Google Scholar] [CrossRef]
- Lu, Y.; Li, F.; Xu, T.; Sun, J. Tetrandrine prevents multidrug resistance in the osteosarcoma cell line, U-2OS, by preventing Pgp overexpression through the inhibition of NF-κB signaling. Int. J. Mol. Med. 2017, 39, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, Z.; Zhang, Q.; Bernstein, K.D.A.; Lozano-Calderon, S.; Choy, E.; Hornicek, F.J.; Duan, Z. Targeting ABCB1 (MDR1) in multi-drug resistant osteosarcoma cells using the CRISPR-Cas9 system to reverse drug resistance. Oncotarget 2016, 7, 83502–83513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanelli, M.; Hattinger, C.M.; Vella, S.; Tavanti, E.; Michelacci, F.; Gudeman, B.; Barnett, D.; Picci, P.; Serra, M. Targeting ABCB1 and ABCC1 with their Specific Inhibitor CBT-1® can Overcome Drug Resistance in Osteosarcoma. Curr. Cancer Drug Targets 2016, 16, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K.W. Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits MDR1 activation. Proc. Natl. Acad. Sci. USA 2000, 97, 6775–6779. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.; Lasthaus, C.; Guérin, E.; Marcellin, L.; Pencreach, E.; Gaub, M.-P.; Guenot, D.; Entz-Werlé, N. Role of Topoisomerases in Pediatric High Grade Osteosarcomas: TOP2A Gene Is One of the Unique Molecular Biomarkers of Chemoresponse. Cancers 2013, 5, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Gorlick, R.; Göker, E.; Trippett, T.; Waltham, M.; Banerjee, D.; Bertino, J.R. Intrinsic and Acquired Resistance to Methotrexate in Acute Leukemia. N. Engl. J. Med. 1996, 335, 1041–1048. [Google Scholar] [CrossRef]
- Goker, E.; Waltham, M.; Kheradpour, A.; Trippett, T.; Mazumdar, M.; Elisseyeff, Y.; Schnieders, B.; Steinherz, P.; Tan, C.; Berman, E. Amplification of the dihydrofolate reductase gene is a mechanism of acquired resistance to methotrexate in patients with acute lymphoblastic leukemia and is correlated with p53 gene mutations. Blood 1995, 86, 677–684. [Google Scholar] [CrossRef]
- Li, W.; Fan, J.; Hochhauser, D.; Banerjee, D.; Zieliński, Z.; Almasan, A.; Yin, Y.; Kelly, R.; Wahl, G.M.; Bertino, J.R. Lack of functional retinoblastoma protein mediates increased resistance to antimetabolites in human sarcoma cell lines. Proc. Natl. Acad. Sci. USA 1995, 92, 10436–10440. [Google Scholar] [CrossRef] [Green Version]
- Sowers, R.; Toguchida, J.; Qin, J.; Meyers, P.; Healey, J.H.; Huvos, A.; Banerjee, D.; Bertino, J.R.; Gorlick, R. mRNA expression levels of E2F transcription factors correlate with dihydrofolate reductase, reduced folate carrier, and thymidylate synthase mRNA expression in osteosarcoma. Mol. Cancer Ther. 2003, 2, 535–541. [Google Scholar] [PubMed]
- Hattinger, C.M.; Stoico, G.; Michelacci, F.; Pasello, M.; Scionti, I.; Remondini, D.; Castellani, G.C.; Fanelli, M.; Scotlandi, K.; Picci, P.; et al. Mechanisms of gene amplification and evidence of coamplification in drug-resistant human osteosarcoma cell lines. Genes Chromosom. Cancer 2009, 48, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Hattinger, C.M. Genomic imbalances associated with methotrexate resistance in human osteosarcoma cell lines detected by comparative genomic hybridization-based techniques. Eur. J. Cell Biol. 2003, 82, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Wang, Y.; Titmus, M.A.; Botchkina, G.; Formentini, A.; Kornmann, M.; Ju, J. Molecular mechanism of chemoresistance by miR-215 in osteosarcoma and colon cancer cells. Mol. Cancer 2010, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tew, K.D. Glutathione-Associated Enzymes In Anticancer Drug Resistance. Cancer Res. 2016, 76, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Shoieb, A.; Hahn, K. Detection and significance of glutathione-S-transferase pi in osteogenic tumors of dogs. Int. J. Oncol. 1997, 10, 635–639. [Google Scholar] [CrossRef]
- Uozaki, H.; Horiuchi, H.; Ishida, T.; Iijima, T.; Imamura, T.; Machinami, R. Overexpression of resistance-related proteins (metallothioneins, glutathione-S-transferase pi, heat shock protein 27, and lung resistance-related protein) in osteosarcoma. Cancer 1997, 79, 2336–2344. [Google Scholar] [CrossRef]
- Pasello, M.; Michelacci, F.; Scionti, I.; Hattinger, C.M.; Zuntini, M.; Caccuri, A.M.; Scotlandi, K.; Picci, P.; Serra, M. Overcoming Glutathione S-Transferase P1-Related Cisplatin Resistance in Osteosarcoma. Cancer Res. 2008, 68, 6661–6668. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Mills, L.; Worth, L.L. Expression of human glutathione S-transferase P1 mediates the chemosensitivity of osteosarcoma cells. Mol. Cancer Ther. 2007, 6, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Bruheim, S.; Bruland, Ø.S.; Breistol, K.; Maelandsmo, G.M.; Fodstad, O. Human osteosarcoma xenografts and their sensitivity to chemotherapy. Pathol. Oncol. Res. 2004, 10, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-L.; Mao, N.-F.; Sun, J.-Y.; Shi, Z.-C.; Wang, B.; Sun, Y.-J. Predictive potential of glutathione S-transferase polymorphisms for prognosis of osteosarcoma patients on chemotherapy. Asian Pac. J. Cancer Prev. 2012, 13, 2705–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.M.; Li, X.H.; Bao, C.F. Glutathione S-transferase P1 and DNA polymorphisms influence response to chemotherapy and prognosis of bone tumors. Asian Pac. J. Cancer Prev. 2012, 13, 5883–5886. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yi, Z.; Ling, M.; Shi, J.; Qiu, Y.; Yang, S. Predictive potential of ABCB1, ABCC3, and GSTP1 gene polymorphisms on osteosarcoma survival after chemotherapy. Tumor Biol. 2014, 35, 9897–9904. [Google Scholar] [CrossRef]
- Rhee, M.S.; Wang, Y.; Nair, M.G.; Galivan, J. Acquisition of resistance to antifolates caused by enhanced gamma-glutamyl hydrolase activity. Cancer Res. 1993, 53, 2227–2230. [Google Scholar]
- Bertino, J.R.; Goker, E.; Gorlick, R.; Li, W.W.; Banerjee, D. Resistance Mechanisms to Methotrexate in Tumors. Stem Cells 1996, 14, 5–9. [Google Scholar] [CrossRef]
- Bacci, G.; Ferrari, S.; Delepine, N.; Bertoni, F.; Picci, P.; Mercuri, M.; Bacchini, P.; Del Prever, A.B.; Tienghi, A.; Comandone, A.; et al. Predictive factors of histologic response to primary chemotherapy in osteosarcoma of the extremity: Study of 272 patients preoperatively treated with high-dose methotrexate, doxorubicin, and cisplatin. J. Clin. Oncol. 1998, 16, 658–663. [Google Scholar] [CrossRef]
- Yu, W.; Min, D.; Lin, F.; Zheng, S.; Tang, L.; He, A.; Hu, H.; Shen, Z. SKA 1 induces de novo MTX-resistance in osteosarcoma through inhibiting FPGS transcription. FEBS J. 2019, 286, 2399–2414. [Google Scholar] [CrossRef]
- DeChant, M.J.; Ewerbeck, V.; Mau, H. Identification of drug-regulated genes in osteosarcoma cells. Int. J. Cancer 2003, 105, 636–643. [Google Scholar] [CrossRef]
- Reed, J.C. Double identity for proteins of the Bcl-2 family. Nature 1997, 387, 773–776. [Google Scholar] [CrossRef]
- Wu, X.; Cai, Z.-D.; Lou, L.-M.; Zhu, Y.-B. Expressions of p53, c-MYC, BCL-2 and apoptotic index in human osteosarcoma and their correlations with prognosis of patients. Cancer Epidemiol. 2012, 36, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Nedelcu, T.; Kubista, B.; Koller, A.; Sulzbacher, I.; Mosberger, I.; Arrich, F.; Trieb, K.; Kotz, R.; Toma, C.D. Livin and Bcl-2 expression in high-grade osteosarcoma. J. Cancer Res. Clin. Oncol. 2008, 134, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Kaseta, M.-K.A.; Khaldi, L.; Gomatos, I.P.; Tzagarakis, G.P.; Alevizos, L.; Leandros, E.; Papagelopoulos, P.J.; Soucacos, P.N. Prognostic value of bax, bcl-2, and p53 staining in primary osteosarcoma. J. Surg. Oncol. 2008, 97, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, C.; Zeng, B.-F.; Wu, X.-S.; Gao, T.-T.; Oda, Y. Enhanced chemosensitivity of drug-resistant osteosarcoma cells by lentivirus-mediated Bcl-2 silencing. Biochem. Biophys. Res. Commun. 2009, 390, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Eliseev, R.A.; Dong, Y.-F.; Sampson, E.; Zuscik, M.J.; Schwarz, E.M.; O’Keefe, R.J.; Rosier, R.N.; Drissi, H. Runx2-mediated activation of the Bax gene increases osteosarcoma cell sensitivity to apoptosis. Oncogene 2008, 27, 3605–3614. [Google Scholar] [CrossRef] [Green Version]
- Hemann, M.T.; Lowe, S.W. The p53–Bcl-2 connection. Cell Death Differ. 2006, 13, 1256–1259. [Google Scholar] [CrossRef]
- Patatsos, K.; Shekhar, T.M.; Hawkins, C.J. Pre-clinical evaluation of proteasome inhibitors for canine and human osteosarcoma. Vet. Comp. Oncol. 2018, 16, 544–553. [Google Scholar] [CrossRef]
- Lampson, B.L.; Davids, M.S. The Development and Current Use of BCL-2 Inhibitors for the Treatment of Chronic Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2017, 12, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Goto, A.; Kanda, H.; Ishikawa, Y.; Matsumoto, S.; Kawaguchi, N.; Machinami, R.; Kato, Y.; Kitagawa, T. Association of Loss of Heterozygosity at the p53 Locus with Chemoresistance in Osteosarcomas. Jpn. J. Cancer Res. 1998, 89, 539–547. [Google Scholar] [CrossRef]
- Rossner, P.; Gammon, M.D.; Zhang, Y.-J.; Terry, M.B.; Hibshoosh, H.; Memeo, L.; Mansukhani, M.; Long, C.-M.; Garbowski, G.; Agrawal, M.; et al. Mutations in p53, p53 protein overexpression and breast cancer survival. J. Cell. Mol. Med. 2009, 13, 3847–3857. [Google Scholar] [CrossRef]
- Pakos, E.E.; Kyzas, P.A.; Ioannidis, J.P.A. Prognostic Significance of TP53 Tumor Suppressor Gene Expression and Mutations in Human Osteosarcoma. Clin. Cancer Res. 2004, 10, 6208–6214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, N.; Tsuchiya, H.; Tomita, K. De novo deletions of p53 gene and wild-type p53 correlate with acquired cisplatin-resistance in human osteosarcoma OST cell line. Anticancer Res. 1999, 19, 5131–5137. [Google Scholar] [PubMed]
- Tsuchiya, H.; Mori, Y.; Ueda, Y.; Okada, G.; Tomita, K. Sensitization and caffeine potentiation of cisplatin cytotoxicity resulting from introduction of wild-type p53 gene in human osteosarcoma. Anticancer Res. 2000, 20, 235–242. [Google Scholar] [PubMed]
- Song, B.; Wang, Y.; Xi, Y.; Kudo, K.; Bruheim, S.; Botchkina, G.I.; Gavin, E.; Formentini, A.; Kornmann, M.; Fodstad, O.; et al. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene 2009, 28, 4065–4074. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Bertino, J.R. Modulation of Cisplatinum Cytotoxicity by p53: Effect of p53-Mediated Apoptosis and DNA Repair. Mol. Pharmacol. 1999, 56, 966–972. [Google Scholar] [CrossRef]
- Wong, R.P.; Tsang, W.P.; Chau, P.Y.; Co, N.N.; Tsang, T.Y.; Kwok, T.T. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Mol. Cancer Ther. 2007, 6, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Wiegering, A.; Matthes, N.; Mühling, B.; Koospal, M.; Quenzer, A.; Peter, S.; Germer, C.-T.; Linnebacher, M.; Otto, C. Reactivating p53 and Inducing Tumor Apoptosis (RITA) Enhances the Response of RITA-Sensitive Colorectal Cancer Cells to Chemotherapeutic Agents 5-Fluorouracil and Oxaliplatin. Neoplasia 2017, 19, 301–309. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Patrizio, M.P.; Luppi, S.; Magagnoli, F.; Picci, P.; Serra, M. Current understanding of pharmacogenetic implications of DNA damaging drugs used in osteosarcoma treatment. Expert Opin. Drug Metab. Toxicol. 2019, 15, 299–311. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Vella, S.; Tavanti, E.; Fanelli, M.; Picci, P.; Serra, M. Pharmacogenomics of second-line drugs used for treatment of unresponsive or relapsed osteosarcoma patients. Pharmacogenomics 2016, 17, 2097–2114. [Google Scholar] [CrossRef] [Green Version]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Nathrath, M.; Kremer, M.; Letzel, H.; Remberger, K.; Höfler, H.; Ulle, T. Expression of genes of potential importance in the response to chemotherapy in osteosarcoma patients. Klin. Padiatr. 2002, 214, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Caronia, D.; Patiño-García, A.; Milne, R.L.; Zalacain-Díez, M.; Pita, G.; Alonso, M.R.; Moreno, L.T.; Sierrasesumaga-Ariznabarreta, L.; Benítez, J.; González-Neira, A.; et al. Common variations in ERCC2 are associated with response to cisplatin chemotherapy and clinical outcome in osteosarcoma patients. Pharm. J. 2009, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Deng, C.; Tian, Y.; Ma, X. Meta-analysis showing that ERCC1 polymorphism is predictive of osteosarcoma prognosis. Oncotarget 2017, 8, 62769–62779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Eitan, L.N.; Alrabadi, N.; Sultan, I.; Al Shatti, M.; Zihlif, M. The effect of ERCC1 and ERCC2 gene polymorphysims on response to cisplatin based therapy in osteosarcoma patients. BMC Med. Genet. 2018, 19, 112. [Google Scholar] [CrossRef]
- Ji, W.-P.; He, N.-B. Investigation on the DNA repaired gene polymorphisms and response to chemotherapy and overall survival of osteosarcoma. Int. J. Clin. Exp. Pathol. 2015, 8, 894–899. [Google Scholar]
- Fishel, M.L.; Kelley, M.R. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol. Asp. Med. 2007, 28, 375–395. [Google Scholar] [CrossRef]
- Yang, J.; Yang, D.; Cogdell, D.; Du, X.; Li, H.; Pang, Y.; Sun, Y.; Hu, L.; Sun, B.; Trent, J.; et al. APEX1 gene amplification and its protein overexpression in osteosarcoma: Correlation with recurrence, metastasis, and survival. Technol. Cancer Res. Treat. 2010, 9, 161–169. [Google Scholar] [CrossRef]
- Wang, D.; Zhong, Z.-Y.; Li, M.-X.; Xiang, D.-B.; Li, Z.-P. Vector-based Ape1 small interfering RNA enhances the sensitivity of human osteosarcoma cells to endostatin in vivo. Cancer Sci. 2007, 98, 1993–2001. [Google Scholar] [CrossRef]
- Dai, N.; Qing, Y.; Cun, Y.; Zhong, Z.; Li, C.; Zhang, S.; Shan, J.; Yang, X.; Dai, X.; Cheng, Y.; et al. miR-513a-5p regulates radiosensitivity of osteosarcoma by targeting human apurinic/apyrimidinic endonuclease. Oncotarget 2018, 9, 25414–25426. [Google Scholar] [CrossRef]
- Liang, W.; Li, C.; Li, M.; Wang, N.; Zhong, Z. MicroRNA-765 sensitizes osteosarcoma cells to cisplatin via downregulating APE1 expression. OncoTargets Ther. 2019, 12, 7203–7214. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, M.; Tavanti, E.; Patrizio, M.P.; Vella, S.; Fernandez-Ramos, A.; Magagnoli, F.; Luppi, S.; Hattinger, C.M.; Serra, M. Cisplatin Resistance in Osteosarcoma: In vitro Validation of Candidate DNA Repair-Related Therapeutic Targets and Drugs for Tailored Treatments. Front. Oncol. 2020, 10, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Zhu, Y.; Deng, Z.; Long, H.; Zhang, S.; Chen, X. VEGF and EMMPRIN expression correlates with survival of patients with osteosarcoma. Surg. Oncol. 2011, 20, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, N.; Dai, R.; Xu, J.; Feng, H. Transferrin receptor-1 and VEGF are prognostic factors for osteosarcoma. J. Orthop. Surg. Res. 2019, 14, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, J.M.; Courtright, J.; Houghton, P.J.; Morton, C.L.; Gorlick, R.; Kolb, E.A.; Lock, R.B.; Tajbakhsh, M.; Reynolds, C.P.; Keir, S.T.; et al. Initial testing of the VEGFR inhibitor AZD2171 by the pediatric preclinical testing program. Pediatr. Blood Cancer 2008, 50, 581–587. [Google Scholar] [CrossRef]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [Green Version]
- Gupte, A.; Baker, E.K.; Wan, S.-S.; Stewart, E.; Loh, A.; Shelat, A.; Gould, C.M.; Chalk, A.M.; Taylor, S.; Lackovic, K.; et al. Systematic screening identifies dual PI3K and mTOR inhibition as a conserved therapeutic vulnerability in osteosarcoma. Clin. Cancer Res. 2015, 21, 3216–3229. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.E.; Bekarev, M.; Kim, M.; Lin, J.; Piperdi, S.; Gorlick, R.; Geller, D.S. Cell surface receptor expression patterns in osteosarcoma. Cancer 2012, 118, 740–749. [Google Scholar] [CrossRef]
- Wang, Y.; Han, X.-D.; Qiu, Y.; Xiong, J.; Yü, Y.; Wang, B.; Zhu, Z.-Z.; Qian, B.-P.; Chen, Y.-X.; Wang, S.F.; et al. Increased expression of insulin-like growth factor-1 receptor is correlated with tumor metastasis and prognosis in patients with osteosarcoma. J. Surg. Oncol. 2012, 105, 235–243. [Google Scholar] [CrossRef]
- Luk, F.; Yu, Y.; Walsh, W.; Yang, J.-L. IGF1R-Targeted Therapy and Its Enhancement of Doxorubicin Chemosensitivity in Human Osteosarcoma Cell Lines. Cancer Investig. 2011, 29, 521–532. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Cheh, C.-W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Ni, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D. HMGB1 Promotes Drug Resistance in Osteosarcoma. Cancer Res. 2012, 72, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.J.; Cao, L.; Tang, D.; Ni, J. Targeting HMGB1-mediated autophagy as a novel therapeutic strategy for osteosarcoma. Autophagy 2012, 8, 275–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Gao, R.; Wang, J.; Yuan, W.; Wang, C.; Zhou, X. High-mobility group nucleosome-binding domain 5 increases drug resistance in osteosarcoma through upregulating autophagy. Tumor Biol. 2014, 35, 6357–6363. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, W.; Li, Y.; Yang, D.; Li, X.; Shen, C.; Liu, Y.; Ke, X.; Guo, S.; Guo, Z. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 201. [Google Scholar] [CrossRef]
- Liu, B.; Ma, W.; Jha, R.K.; Gurung, K. Cancer stem cells in osteosarcoma: Recent progress and perspective. Acta Oncol. 2011, 50, 1142–1150. [Google Scholar] [CrossRef]
- Di Fiore, R.; Santulli, A.; Ferrante, R.D.; Giuliano, M.; De Blasio, A.; Messina, C.; Pirozzi, G.; Tirino, V.; Tesoriere, G.; Vento, R. Identification and expansion of human osteosarcoma-cancer-stem cells by long-term 3-aminobenzamide treatment. J. Cell. Physiol. 2009, 219, 301–313. [Google Scholar] [CrossRef]
- Martins-Neves, S.R.; Lopes, Á.O.; Carmo, A.D.; Paiva, A.; Simões, P.C.; Abrunhosa, A.; Gomes, C. Therapeutic implications of an enriched cancer stem-like cell population in a human osteosarcoma cell line. BMC Cancer 2012, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Honoki, K.; Fujii, H.; Tsujiuchi, T.; Kido, A.; Yoshitani, K.; Takakura, Y. Sphere-forming stem-like cell populations with drug resistance in human sarcoma cell lines. Int. J. Oncol. 2009, 34, 1381–1386. [Google Scholar] [CrossRef] [Green Version]
- Honoki, K.; Fujii, H.; Kubo, A.; Kido, A.; Mori, T.; Tanaka, Y.; Tsujiuchi, T. Possible involvement of stem-like populations with elevated ALDH1 in sarcomas for chemotherapeutic drug resistance. Oncol. Rep. 2010, 24, 501–505. [Google Scholar] [CrossRef]
- Chan, C.-H.; Morrow, J.K.; Li, C.-F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Wang, C.; Cui, Y.; Han, X.; Zhou, Y.; Bai, J.; Li, R. S-phase kinase-associated protein 2 is involved in epithelial-mesenchymal transition in methotrexate-resistant osteosarcoma cells. Int. J. Oncol. 2018, 52, 1841–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Guo, Y.; Yang, Y.; Kan, J.; Dai, S.; Helian, M.; Li, B.; Xu, J.; Liu, C. Nitidine chloride suppresses epithelial-to-mesenchymal transition in osteosarcoma cell migration and invasion through Akt/GSK-3β/Snail signaling pathway. Oncol. Rep. 2016, 36, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Santucci, R.; Levêque, D.; Kemmel, V.; Lutz, P.; Gérout, A.-C.; N’Guyen, A.; Lescoute, A.; Schneider, F.; Bergerat, J.-P.; Herbrecht, R. Severe intoxication with methotrexate possibly associated with concomitant use of proton pump inhibitors. Anticancer Res. 2010, 30, 963–965. [Google Scholar] [PubMed]
- Ratti, C.; Botti, L.; Cancila, V.; Galvan, S.; Torselli, I.; Garofalo, C.; Manara, M.C.; Bongiovanni, L.; Valenti, C.; Burocchi, A.; et al. Trabectedin Overrides Osteosarcoma Differentiative Block and Reprograms the Tumor Immune Environment Enabling Effective Combination with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 5149–5161. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Tian, S.; Yu, C.; Chen, M.; Sun, C. TRIM37 promoted the growth and migration of the pancreatic cancer cells. Tumor Biol. 2016, 37, 2629–2634. [Google Scholar] [CrossRef]
- Tao, Y.; Xin, M.; Cheng, H.; Huang, Z.; Hu, T.; Zhang, T.; Wang, J. TRIM37 promotes tumor cell proliferation and drug resistance in pediatric osteosarcoma. Oncol. Lett. 2017, 14, 6365–6372. [Google Scholar] [CrossRef]
- Kolb, E.A.; Gorlick, R.; Keir, S.T.; Maris, J.M.; Lock, R.B.; Carol, H.; Kurmasheva, R.T.; Reynolds, C.P.; Kang, M.H.; Wu, J.; et al. Initial testing (stage 1) by the pediatric preclinical testing program of RO4929097, a γ-secretase inhibitor targeting notch signaling. Pediatr. Blood Cancer 2012, 58, 815–818. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.M.; Johnson, S.A.; Sims, N.A.; Trivett, M.K.; Slavin, J.L.; Rubin, B.P.; Waring, P.; McArthur, G.A.; Walkley, C.R.; Holloway, A.J.; et al. Terminal osteoblast differentiation, mediated by runx2 and p27KIP1, is disrupted in osteosarcoma. J. Cell Biol. 2004, 167, 925–934. [Google Scholar] [CrossRef]
- Calzolari, A.; Oliviero, I.; Deaglio, S.; Mariani, G.; Biffoni, M.; Sposi, N.M.; Malavasi, F.; Peschle, C.; Testa, U. Transferrin receptor 2 is frequently expressed in human cancer cell lines. Blood Cells Mol. Dis. 2007, 39, 82–91. [Google Scholar] [CrossRef]
- Bhuvaneshwar, K.; Harris, M.; Gusev, Y.; Madhavan, S.; Iyer, R.; Vilboux, T.; Deeken, J.F.; Yang, E.; Shankar, S. Genome sequencing analysis of blood cells identifies germline haplotypes strongly associated with drug resistance in osteosarcoma patients. BMC Cancer 2019, 19, 357. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, H.; Duan, Z.; Zou, J.X.; Chen, H.; He, W.; Wang, J. Estrogen-Related Receptor Alpha Confers Methotrexate Resistance via Attenuation of Reactive Oxygen Species Production and P53 Mediated Apoptosis in Osteosarcoma Cells. BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, K.; Li, Y.; Guo, R.; Zhang, K.; Zhong, G.; He, Q. Oestrogen-related receptor alpha mediates chemotherapy resistance of osteosarcoma cells via regulation of ABCB1. J. Cell. Mol. Med. 2019, 23, 2115–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, Y.; Zhang, X.; Shen, J.; Xu, R.; Liu, Y.; Yu, X. Circular RNA hsa_circ_0000073 contributes to osteosarcoma cell proliferation, migration, invasion and methotrexate resistance by sponging miR-145-5p and miR-151-3p and upregulating NRAS. Aging 2020, 12, 14157–14173. [Google Scholar] [CrossRef]
- Yang, X.-R.; Xiong, Y.; Duan, H.; Gong, R.-R. Identification of genes associated with methotrexate resistance in methotrexate-resistant osteosarcoma cell lines. J. Orthop. Surg. Res. 2015, 10, 136. [Google Scholar] [CrossRef] [Green Version]
- Rudd, S.G.; Tsesmetzis, N.; Sanjiv, K.; Paulin, C.B.; Sandhow, L.; Kutzner, J.; Hed Myrberg, I.; Bunten, S.S.; Axelsson, H.; Zhang, S.M.; et al. Ribonucleotide reductase inhibitors suppress SAMHD1 ara-CTPase activity enhancing cytarabine efficacy. EMBO Mol. Med. 2020, 12, e10419. [Google Scholar] [CrossRef]
- Xu, J.-F.; Pan, X.-H.; Zhang, S.-J.; Zhao, C.; Qiu, B.-S.; Gu, H.-F.; Hong, J.-F.; Cao, L.; Chen, Y.; Xia, B.; et al. CD47 blockade inhibits tumor progression human osteosarcoma in xenograft models. Oncotarget 2015, 6, 23662–23670. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, S.; Yerneni, K.; Theruvath, J.L.; Graef, C.M.; Nejadnik, H.; Lenkov, O.; Pisani, L.; Rosenberg, J.; Mitra, S.; Cordero, A.S.; et al. Nanoparticle enhanced MRI can monitor macrophage response to CD47 mAb immunotherapy in osteosarcoma. Cell Death Dis. 2019, 10, 36. [Google Scholar] [CrossRef]
- Fucikova, J.; Kralikova, P.; Skallová-Fialová, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spísek, R. Human Tumor Cells Killed by Anthracyclines Induce a Tumor-Specific Immune Response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, S.; Aghighi, M.; Yerneni, K.; Theruvath, J.L.; Daldrup-Link, H.E. Improving the efficacy of osteosarcoma therapy: Combining drugs that turn cancer cell ‘don’t eat me’ signals off and ‘eat me’ signals on. Mol. Oncol. 2019, 13, 2049–2061. [Google Scholar] [CrossRef] [Green Version]
- Asano, N.; Takeshima, H.; Yamashita, S.; Takamatsu, H.; Hattori, N.; Kubo, T.; Yoshida, A.; Kobayashi, E.; Nakayama, R.; Matsumoto, M.; et al. Epigenetic reprogramming underlies efficacy of DNA demethylation therapy in osteosarcomas. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]




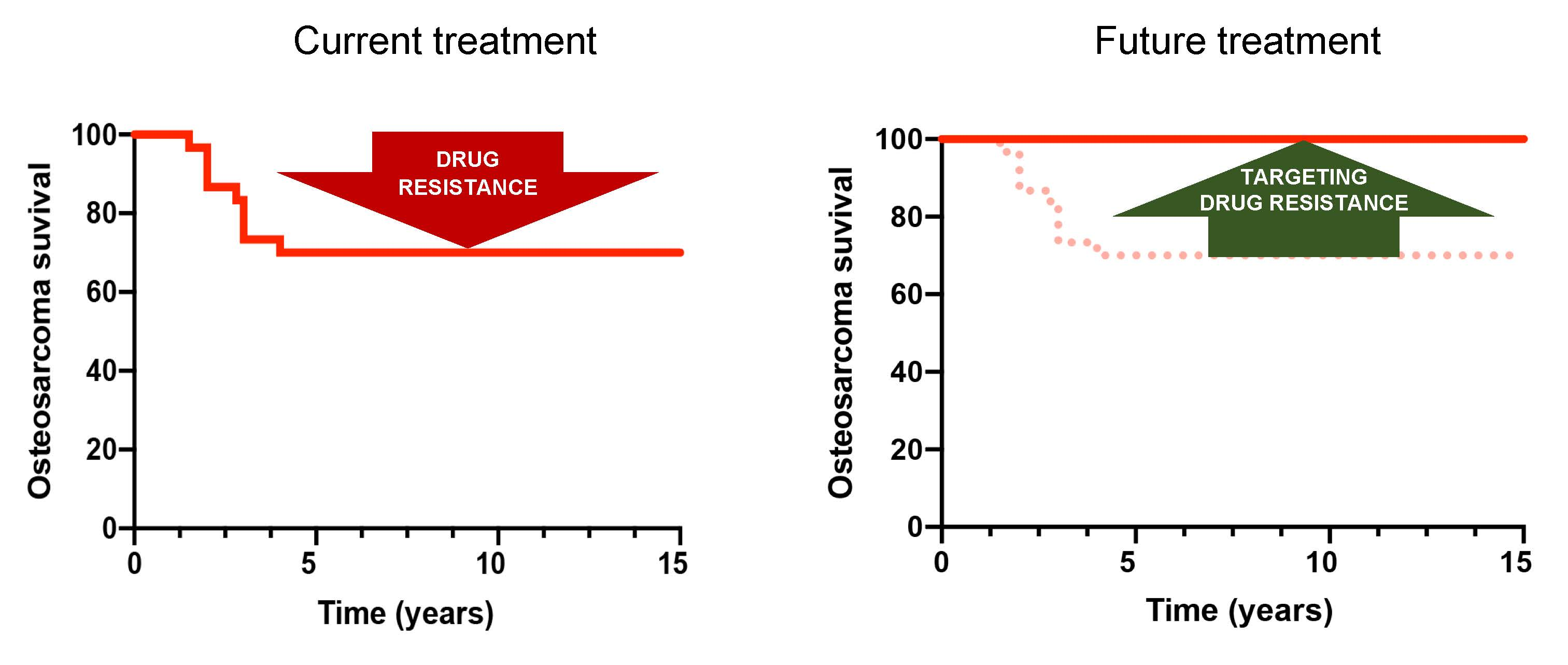{kind=link}
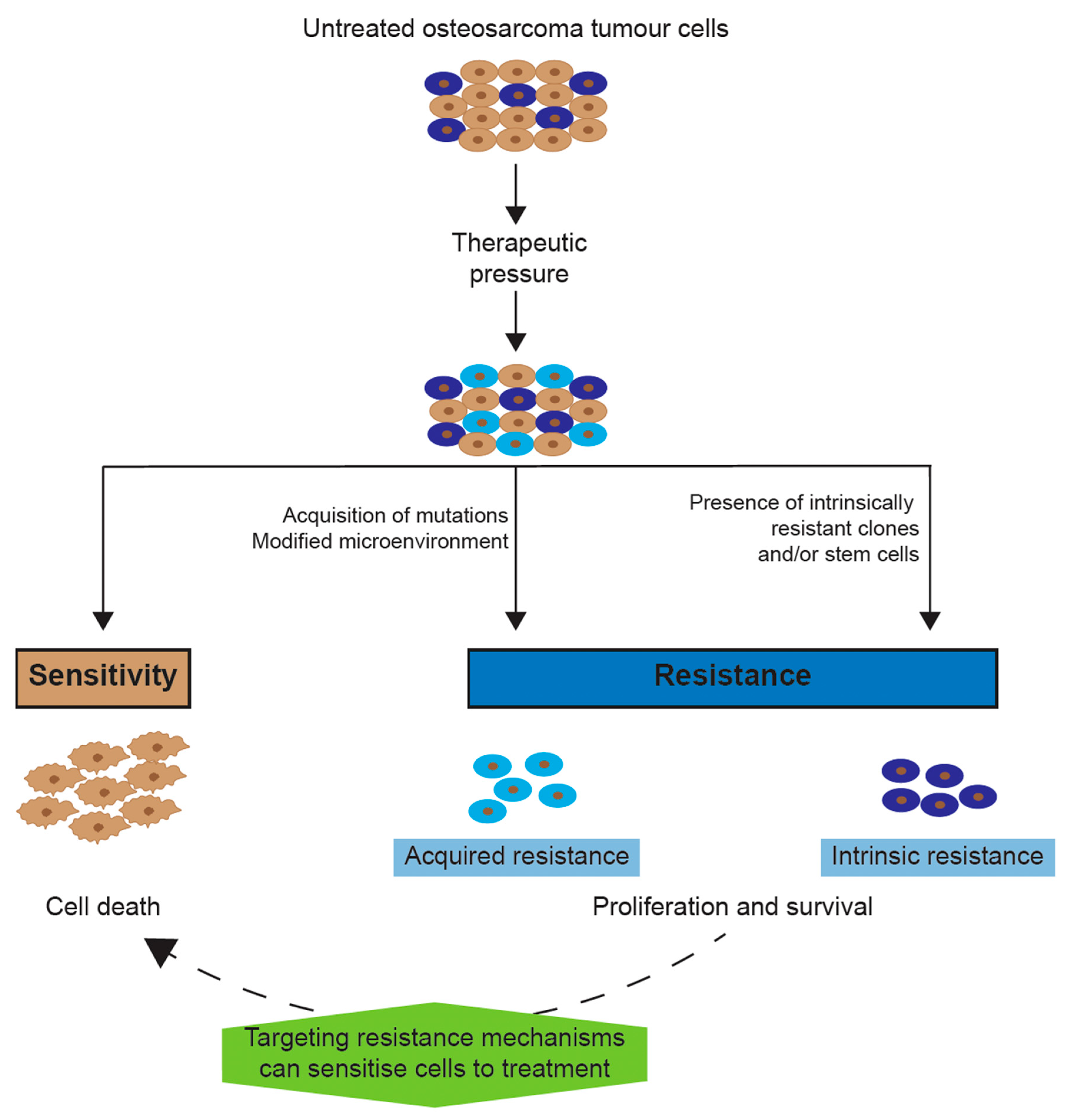{kind=link}
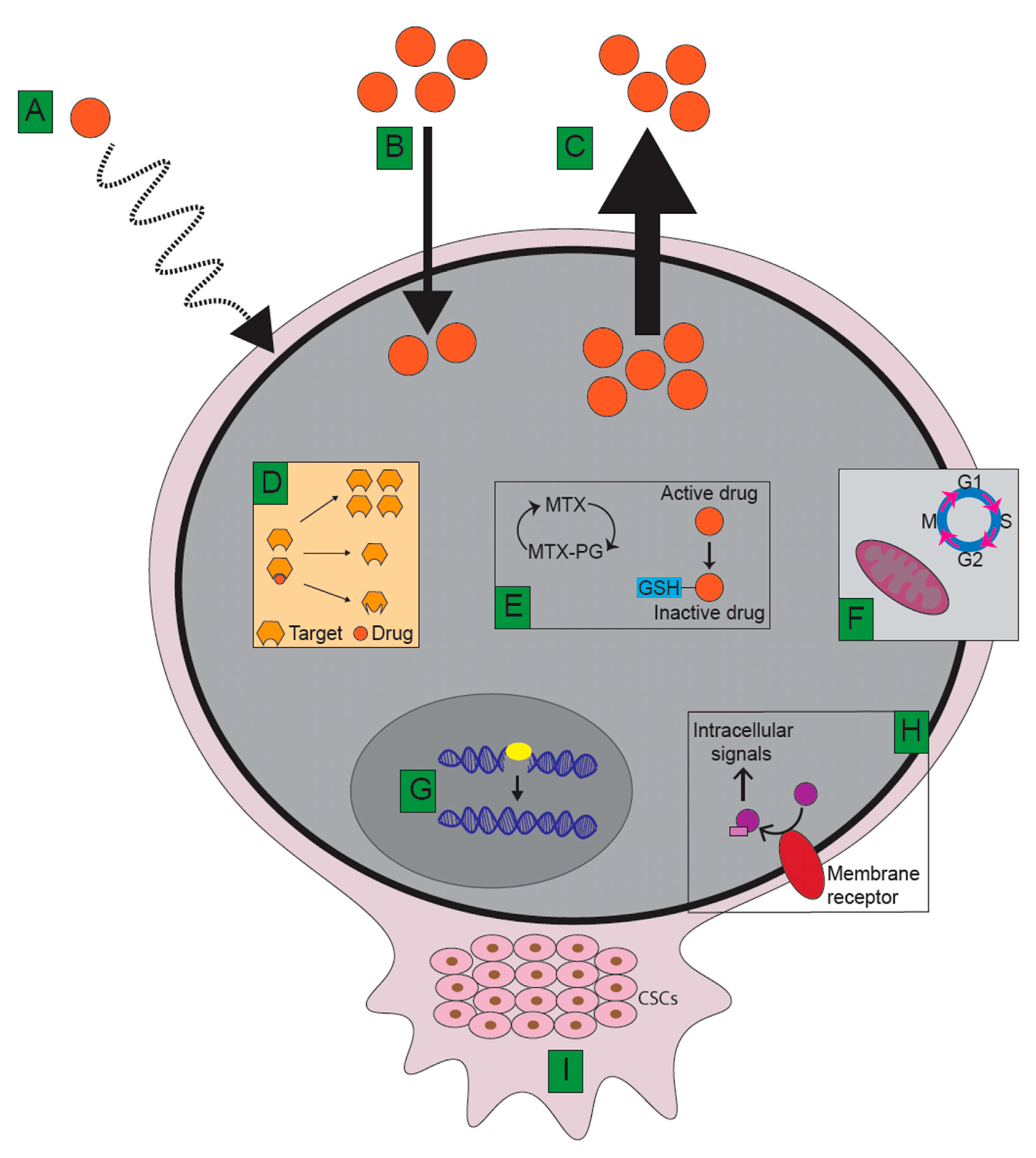{kind=link}
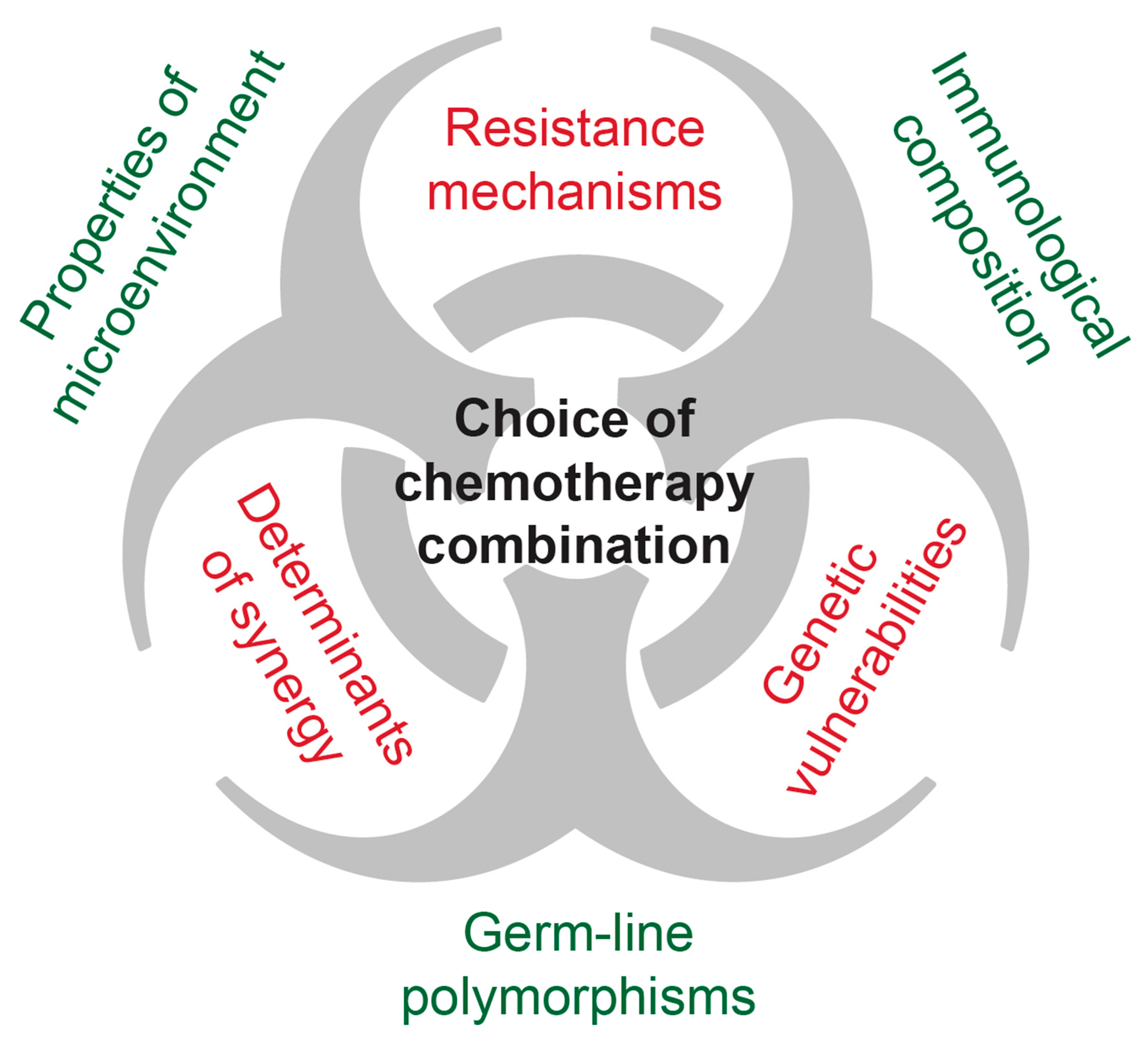{kind=link}
| Targetable Elements | Function(s) | Evidence Found in Osteosarcoma Tissue Samples | Clinical Results in Human Trials |
|---|---|---|---|
| Unspecific immune modulators | Antiproliferative and proapoptotic effects in cancer cells | Inhibit tumour progression in preclinical models [197,198,200] | Pooled IFNα added to standard treatment yielded better outcome in a nonrandomized trial [194] |
| IFN-α-2b together with MAP did not improve outcome in a large randomized trial [130] | |||
| Inhalation of aerosolized GM-CSF did not hinder disease progression nor improve outcome [199] | |||
| L-MTP-PE showed some effects on survival (but interactions with ifosfamide in a factorial study design) [116,201] | |||
| CTLA-4 | Negative regulator of cytotoxic T-cells | Osteosarcomas are enriched in CTLA-4 pathways [52] | Ipilimumab showed partial responses or stable disease in a small number of paediatric patients [204] |
| PD-1 | Negative regulator of cytotoxic T-cells | Patients positive for PD-1 ligand had worse event-free survival, and osteosarcomas are enriched in PD-1 signalling [52] | Nivolumab showed some efficacy in adults [205] |
| Pembrolizumab had limited efficacy in adults [206] | |||
| ErbB/Her receptors (including Her1, Her2, Her3, Her4, and EGFR) | Involved in differentiation, proliferation, and cell cycle control | Overexpression of Her2/HER2 and EGFR in patient samples [93,94] | Trastuzumab (anti-Her2) in combination with MAP did not reduce metastatic disease nor improve overall outcome [229] |
| Receptor-associated and cytoplasmic protein kinases, including MAPK, BRAF, VEGFRs, PDGFR, and IGF-1R | Transmembrane signalling and intracellular transduction | Overexpression of several tyrosine kinases and their receptors in patients linked to poor response to therapy and outcome [29,103,104,105,107] | The broad tyrosine kinase inhibitor sorafenib led to a 46% progression-free survival after 4 months [241] |
| Sorafenib plus the mTOR inhibitor everolimus did not improve 6-month progression-free survival [242] | |||
| Regorafenib, a multikinase inhibitor with broader activity than sorafenib, led to reduced disease progression [245,246] | |||
| Robatumumab, a monoclonal anti-IGF1R antibody, had little to no effect on clinical outcomes [266] | |||
| Cabozantinib (VEGFR2 and MET inhibitor) yielded partial response and inhibited disease progression in some patients [248] | |||
| GpNMB | Involved in tissue repair, adhesion, and growth | gpNMB mRNA found to be overexpressed in primary patient samples [261] | Glembatumumab vedotin (gpNMB antibody coupled to a microtubule inhibitor) did not have significant clinical efficacy [264] |
| Small Rho GTPases | Promote osteoclast activity | Increased RhoA activity found in some metastatic osteosarcoma patients and found in osteosarcoma mice with metastatic disease [251] | Zoledronate (a bisphosphonate that inhibits Rho) did not improve outcome when combined with traditional therapy [253] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lilienthal, I.; Herold, N. Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies. Int. J. Mol. Sci. 2020, 21, 6885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186885
Lilienthal I, Herold N. Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies. International Journal of Molecular Sciences. 2020; 21(18):6885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186885
Chicago/Turabian StyleLilienthal, Ingrid, and Nikolas Herold. 2020. "Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies" International Journal of Molecular Sciences 21, no. 18: 6885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186885