Cellular and Molecular Mechanisms of Environmental Pollutants on Hematopoiesis

{kind=link}
{kind=link}
Abstract
:1. Hematopoiesis Overview
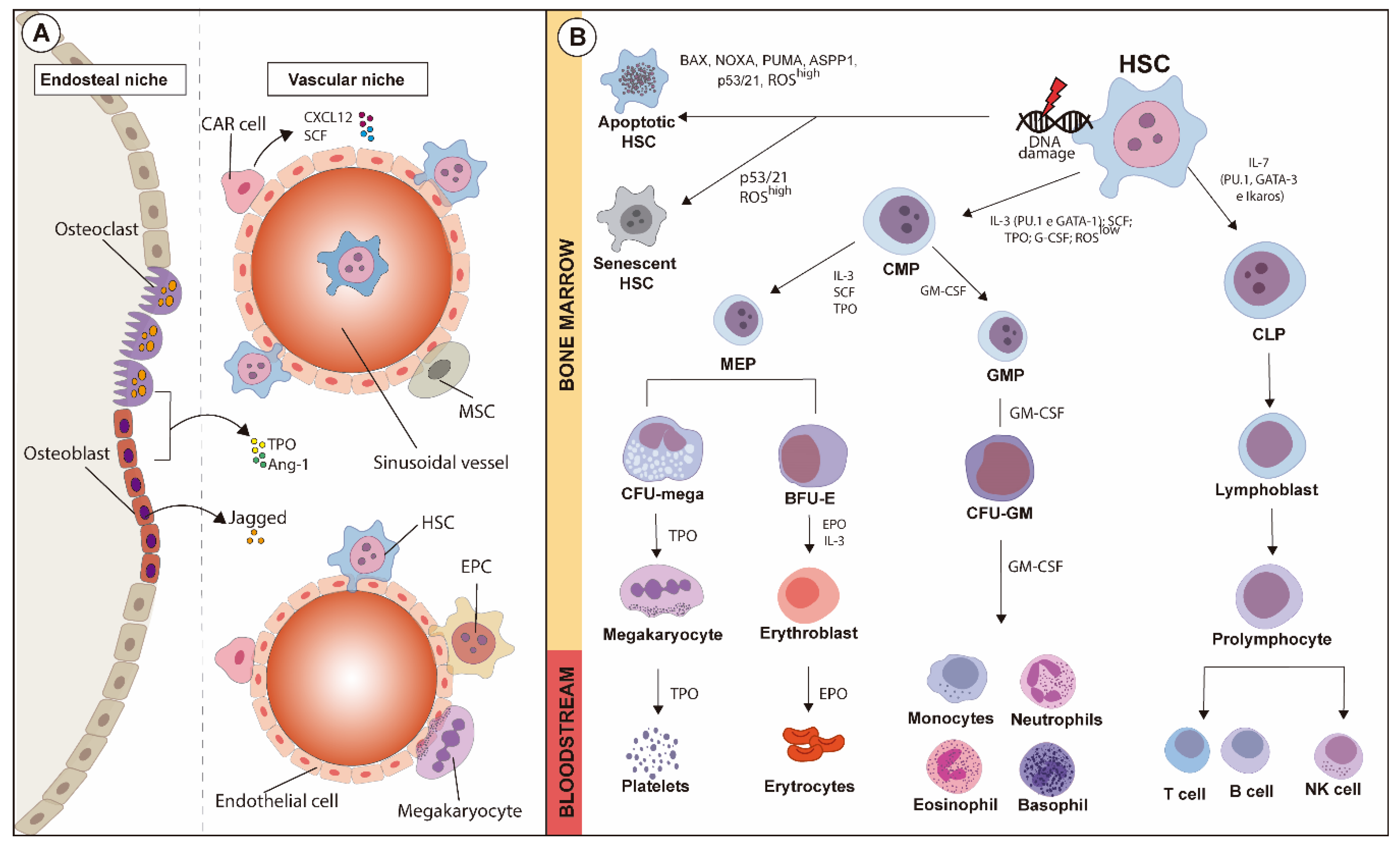
1.1. Hematopoiesis and Hematopoietic Hierarchy
1.2. HSC Quiescence
1.3. HSC Niches
1.4. HSC Mobilization
1.5. DNA Damage, ROS Generation and Hypoxia in the Control of HSC
2. Cellular and Molecular Mechanisms of Toxicity on Hematopoiesis
2.1. Benzene and Its Metabolites
2.2. Engineered Nanoparticles
2.3. Incidental Environmental Nanoparticles and Particulate Matter
2.4. Dioxins and Polychlorinated Biphenyls (PCBs)
2.5. Heavy Metals
2.5.1. Lead (Pb)
2.5.2. Cadmium (Cd)
2.5.3. Arsenic (As)
2.5.4. Mercury (Hg)
3. Conclusions
Funding
Conflicts of Interest
References
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Cheshier, S.H.; Morrison, S.J.; Liao, X.; Weissman, I.L. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3120–3125. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.J.; Jamieson, C.H.; Weissman, I.L. Stems Cells and the Pathways to Aging and Cancer. Cell 2008, 132, 681–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, E.M.; Warr, M.R.; Passegué, E. Cell cycle regulation in hematopoietic stem cells. J. Exp. Med. 2011, 208, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Kondo, M. Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol. Rev. 2010, 238, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Pang, S.H.M.; De Graaf, C.A.; Hilton, D.J.; Huntington, N.D.; Carotta, S.; Wu, L.; Nutt, S.L. PU.1 Is Required for the Developmental Progression of Multipotent Progenitors to Common Lymphoid Progenitors. Front. Immunol. 2018, 9, 1264. [Google Scholar] [CrossRef]
- Onder, L.; Narang, P.; Scandella, E.; Chai, Q.; Iolyeva, M.; Hoorweg, K.; Halin, C.; Richie, E.; Kaye, P.M.; Westermann, J.; et al. IL-7–producing stromal cells are critical for lymph node remodeling. Blood 2012, 120, 4675–4683. [Google Scholar] [CrossRef]
- Zhang, Q.; Iida, R.; Yokota, T.; Kincade, P.W. Early events in lymphopoiesis. Curr. Opin. Hematol. 2013, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Zheng, Z.; Cheng, T. New paradigms on hematopoietic stem cell differentiation. Protein Cell 2019, 11, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socolovsky, M.; Nam, H.-S.; Fleming, M.D.; Haase, V.H.; Brugnara, C.; Lodish, H.F. Ineffective erythropoiesis in Stat5a−/−5b−/− mice due to decreased survival of early erythroblasts. Blood 2001, 98, 3261–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crispino, J.D.; Weiss, M.J. Erythro-megakaryocytic transcription factors associated with hereditary anemia. Blood 2014, 123, 3080–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Tesio, M.; Tang, Y.; Müdder, K.; Saini, M.; Von Paleske, L.; MacIntyre, E.; Pasparakis, M.; Waisman, A.; Trumpp, A. Hematopoietic stem cell quiescence and function are controlled by the CYLD-TRAF2-p38MAPK pathway. J. Cell Biol. 2015, 209, 2091. [Google Scholar] [CrossRef]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent Hematopoietic Stem Cells Accumulate DNA Damage during Aging that Is Repaired upon Entry into Cell Cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef]
- Yamada, T.; Park, C.S.; Burns, A.; Nakada, D.; Lacorazza, H.D. The Cytosolic Protein G0S2 Maintains Quiescence in Hematopoietic Stem Cells. PLoS ONE 2012, 7, e38280. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Haylock, D.N.; Williams, B.; Johnston, H.M.; Liu, M.C.; Rutherford, K.E.; Whitty, G.A.; Simmons, P.J.; Bertoncello, I.; Nilsson, S.K. Hemopoietic Stem Cells with Higher Hemopoietic Potential Reside at the Bone Marrow Endosteum. Stem Cells 2007, 25, 1062–1069. [Google Scholar] [CrossRef]
- Wei, Q.; Frenette, P.S. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018, 48, 632–648. [Google Scholar] [CrossRef] [Green Version]
- Abkowitz, J.L.; Robinson, A.E.; Kale, S.; Long, M.W.; Chen, J. Mobilization of hematopoietic stem cells during homeostasis and after cytokine exposure. Blood 2003, 102, 1249–1253. [Google Scholar] [CrossRef] [Green Version]
- Yannaki, E.; Karponi, G.; Zervou, F.; Constantinou, V.; Bouinta, A.; Tachynopoulou, V.; Kotta, K.; Jonlin, E.; Papayannopoulou, T.; Anagnostopoulos, A.; et al. Hematopoietic Stem Cell Mobilization for Gene Therapy: Superior Mobilization by the Combination of Granulocyte–Colony Stimulating Factor Plus Plerixafor in Patients with β-Thalassemia Major. Hum. Gene Ther. 2013, 24, 852–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro-Spinetti, E.; Taichman, R.S.; Balduino, A. The Bone Marrow Endosteal Niche: How Far from the Surface? J. Cell. Biochem. 2014, 116, 6–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levesque, J.-P.; Helwani, F.M.; Winkler, I.G. The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia 2010, 24, 1979–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilly, A.J.; Johnson, W.E.; Bunce, C. The Haematopoietic Stem Cell Niche: New Insights into the Mechanisms Regulating Haematopoietic Stem Cell Behaviour. Stem Cells Int. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, J.M.; Nolan, D.J.; Vertes, E.L.; Varnum-Finney, B.; Kobayashi, H.; Hooper, A.T.; Seandel, M.; Shido, K.; White, I.A.; Kobayashi, M.; et al. Endothelial Cells Are Essential for the Self-Renewal and Repopulation of Notch-Dependent Hematopoietic Stem Cells. Cell Stem Cell 2010, 6, 251–264. [Google Scholar] [CrossRef] [Green Version]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The Essential Functions of Adipo-osteogenic Progenitors as the Hematopoietic Stem and Progenitor Cell Niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Bailey, A.S.; Jiang, S.; Liu, B.; Goldman, D.C.; Fleming, W.H. Endothelial cells mediate the regeneration of hematopoietic stem cells. Stem Cell Res. 2010, 4, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Psaila, B.; Lyden, D.; Roberts, I. Megakaryocytes, malignancy and bone marrow vascular niches. J. Thromb. Haemost. 2012, 10, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Pontikoglou, C.; Deschaseaux, F.; Sensebé, L.; Papadaki, H.A. Bone Marrow Mesenchymal Stem Cells: Biological Properties and Their Role in Hematopoiesis and Hematopoietic Stem Cell Transplantation. Stem Cell Rev. Rep. 2011, 7, 569–589. [Google Scholar] [CrossRef]
- Parekkadan, B.; Milwid, J.M. Mesenchymal Stem Cells as Therapeutics. Annu. Rev. Biomed. Eng. 2010, 12, 87–117. [Google Scholar] [CrossRef] [Green Version]
- Le Blanc, K.; Samuelsson, H.; Gustafsson, B.; Remberger, M.; Sundberg, B.; Arvidson, J.; Ljungman, P.; Lönnies, H.; Nava, S.; Ringdén, O. Transplantation of mesenchymal stem cells to enhance engraftment of hematopoietic stem cells. Leukemia 2007, 21, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Lazar-Karsten, P.; Dorn, I.; Meyer, G.; Lindner, U.; Driller, B.; Schlenke, P. The influence of extracellular matrix proteins and mesenchymal stem cells on erythropoietic cell maturation. Vox Sang. 2010, 101, 65–76. [Google Scholar] [CrossRef]
- Yin, T.; Li, L. The stem cell niches in bone. J. Clin. Investig. 2006, 116, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; Van Der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of Putative Progenitor Endothelial Cells for Angiogenesis. Science 1997, 275, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Dimmeler, S. Endothelial progenitor cells: Characterization and role in vascular biology. Circ. Res. 2004, 95, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Slukvin, I.I.; Kumar, A. The mesenchymoangioblast, mesodermal precursor for mesenchymal and endothelial cells. Cell. Mol. Life Sci. 2018, 75, 3507–3520. [Google Scholar] [CrossRef]
- Yang, J.; Ii, M.; Kamei, N.; Alev, C.; Kwon, S.-M.; Kawamoto, A.; Akimaru, H.; Masuda, H.; Sawa, Y.; Asahara, T. CD34+ Cells Represent Highly Functional Endothelial Progenitor Cells in Murine Bone Marrow. PLoS ONE 2011, 6, e20219. [Google Scholar] [CrossRef]
- Moccia, F.; Guerra, G. Ca2+ Signalling in Endothelial Progenitor Cells: Friend or Foe? J. Cell. Physiol. 2015, 231, 314–327. [Google Scholar] [CrossRef]
- Marquez, L.A.; Turner, A.R.; Sridharan, S.; Ratajczak, M.Z.; Janowska-Wieczorek, A. The Ins and Outs of Hematopoietic Stem Cells: Studies to Improve Transplantation Outcomes. Stem Cell Rev. Rep. 2010, 7, 590–607. [Google Scholar] [CrossRef] [Green Version]
- Schuettpelz, L.G.; Link, D.C. Regulation of Hematopoietic Stem Cell Activity by Inflammation. Front. Immunol. 2013, 4, 204. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Zhai, Q.; Qiu, L.; Meng, H.; Zou, D.; Wang, Y.; Li, Q.; Yu, Z.; Han, J.; Zhou, B. Degradation of BM SDF-1 by MMP-9: The role in G-CSF-induced hematopoietic stem/progenitor cell mobilization. Bone Marrow Transplant. 2008, 42, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Heissig, B.; Hattori, K.; Dias, S.; Friedrich, M.; Ferris, B.; Hackett, N.R.; Crystal, R.G.; Besmer, P.; Lyden, D.; Moore, M.A.; et al. Recruitment of Stem and Progenitor Cells from the Bone Marrow Niche Requires MMP-9 Mediated Release of Kit-Ligand. Cell 2002, 109, 625–637. [Google Scholar] [CrossRef] [Green Version]
- Lapidot, T.; Kollet, O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2mnull mice. Leukemia 2002, 16, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Giralt, S.A.; Costa, L.; Schriber, J.; DiPersio, J.; Maziarz, R.; Mccarty, J.; Shaughnessy, P.; Snyder, E.; Bensinger, W.; Copelan, E.; et al. Optimizing Autologous Stem Cell Mobilization Strategies to Improve Patient Outcomes: Consensus Guidelines and Recommendations. Biol. Blood Marrow Transplant. 2014, 20, 295–308. [Google Scholar] [CrossRef] [Green Version]
- Levesque, J.-P.; Takamatsu, Y.; Nilsson, S.K.; Haylock, D.; Simmons, P.J. Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood 2001, 98, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Bautz, F.; Denzlinger, C.; Kanz, L.; Möhle, R. Chemotaxis and transendothelial migration of CD34+hematopoietic progenitor cells induced by the inflammatory mediator leukotriene D4 are mediated by the 7-transmembrane receptor CysLT1. Blood 2001, 97, 3433–3440. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.W.; Kim, G.-Y.; Mun, Y.-C.; Ahn, J.-Y.; Seong, C.-M.; Kim, J.-H. Leukotriene B4 pathway regulates the fate of the hematopoietic stem cells. Exp. Mol. Med. 2005, 37, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M. Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, I.D.; Spatti, M.; Hastreiter, A.; Santin, J.R.; Fock, R.A.; Gil, C.D.; Oliani, S.M.; Perretti, M.; Farsky, S.H.P. Annexin A1 Is a Physiological Modulator of Neutrophil Maturation and Recirculation Acting on the CXCR4/CXCL12 Pathway. J. Cell Physiol. 2016, 231, 2418–2427. [Google Scholar] [CrossRef]
- Ponomaryov, T.; Peled, A.; Petit, I.; Taichman, R.S.; Habler, L.; Sandbank, J.; Arenzana-Seisdedos, F.; Magérus-Chatinet, A.; Caruz, A.; Fujii, N.; et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J. Clin. Investig. 2000, 106, 1331–1339. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xiao, H.; Zhang, X.; Wang, C.; Huang, H. The role of telomeres and telomerase in hematologic malignancies and hematopoietic stem cell transplantation. J. Hematol. Oncol. 2014, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.; Passegué, E. Hematopoietic Stem Cell Quiescence Promotes Error-Prone DNA Repair and Mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.H.; Bassing, C.H.; Teitell, M.A. Regulation of cell differentiation by the DNA damage response. Trends Cell Biol. 2011, 21, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Pilzecker, B.; Buoninfante, O.A.; Berk, P.V.D.; Lancini, C.; Song, J.-Y.; Citterio, E.; Jacobs, H. DNA damage tolerance in hematopoietic stem and progenitor cells in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E6875–E6883. [Google Scholar] [CrossRef] [Green Version]
- Pilzecker, B.; Buoninfante, O.A.; Jacobs, H. DNA damage tolerance in stem cells, ageing, mutagenesis, disease and cancer therapy. Nucleic Acids Res. 2019, 47, 7163–7181. [Google Scholar] [CrossRef] [Green Version]
- Insinga, A.; Cicalese, A.; Faretta, M.; Gallo, B.; Albano, L.; Ronzoni, S.; Furia, L.; Viale, A.; Pelicci, P.G. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc. Natl. Acad. Sci. USA 2013, 110, 3931–3936. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; Nitta, E.; Suda, T. Aspp1 Preserves Hematopoietic Stem Cell Pool Integrity and Prevents Malignant Transformation. Cell Stem Cell 2015, 17, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, E.C.; Sykes, S.M.; McMahon, S.B.; E Murphy, M. The p53 family and programmed cell death. Oncogene 2008, 27, 6507–6521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Peng, L.; Rao, Q.; Xing, H.; Huai, L.; Yu, P.; Chen, Y.; Wang, C.; Wang, M.; Mi, Y.; et al. Oncogene iASPP enhances self-renewal of hematopoietic stem cells and facilitates their resistance to chemotherapy and irradiation. FASEB J. 2014, 28, 2816–2827. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Fang, Y.; Cai, J.; Li, X.; Xu, F.; Yuan, N.; Zhang, S.; Wang, J. ROS functions as an upstream trigger for autophagy to drive hematopoietic stem cell differentiation. Hematology 2016, 21, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsini, M.; Chateauvieux, S.; Rhim, J.; Gaigneaux, A.; Cheillan, D.; Christov, C.; Dicato, M.; Morceau, F.; Han, B.W. Sphingolipid-mediated inflammatory signaling leading to autophagy inhibition converts erythropoiesis to myelopoiesis in human hematopoietic stem/progenitor cells. Cell Death Differ. 2018, 26, 1796–1812. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Yuan, N.; Wang, Z.; Cao, Y.; Fang, Y.; Li, X.; Xu, F.; Song, L.; Wang, J.; Zhang, H.; et al. Autophagy confers DNA damage repair pathways to protect the hematopoietic system from nuclear radiation injury. Sci. Rep. 2015, 5, 12362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koschade, S.E.; Brandts, C.H. Selective Autophagy in Normal and Malignant Hematopoiesis. J. Mol. Biol. 2020, 432, 261–282. [Google Scholar] [CrossRef]
- Mortensen, M.; Watson, A.S.; Simon, A.K. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 2011, 7, 1069–1070. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar] [CrossRef]
- Sohal, R.S. Role of oxidative stress and protein oxidation in the aging process. Free. Radic. Biol. Med. 2002, 33, 37–44. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxidative Med. Cell. Longev. 2016, 2016, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, M.E.; Valle, N.R.-D.; Chandra, J. Redox Control of Leukemia: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 1349–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillar, J.R.; Germon, Z.P.; De Iuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef] [Green Version]
- Samimi, A.; Khodayar, M.J.; Alidadi, H.; Khodadi, E. The Dual Role of ROS in Hematological Malignancies: Stem Cell Protection and Cancer Cell Metastasis. Stem Cell Rev. Rep. 2020, 16, 262–275. [Google Scholar] [CrossRef]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.-J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive Oxygen Species Regulate Hematopoietic Stem Cell Self-Renewal, Migration and Development, As Well As Their Bone Marrow Microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.-Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive oxygen species in haematopoiesis: Leukaemic cells take a walk on the wild side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.P. Oxidative Stress, Antioxidant Defenses, and Damage Removal, Repair, and Replacement Systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef]
- Lü, J.-M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2009, 14, 840–860. [Google Scholar] [CrossRef] [PubMed]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1α Level Is Essential for Hematopoietic Stem Cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinkovic, D.; Zhang, X.; Yalcin, S.; Luciano, J.P.; Brugnara, C.; Huber, T.; Ghaffari, S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J. Clin. Investig. 2007, 117, 2133–2144. [Google Scholar] [CrossRef] [Green Version]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Massard-Guilbaud, G.; Mathis, C.F. A Brief Introduction to the History of Pollution: From Local to Global. In Microbial Ecotoxicology; Cravo-Laureau, C., Cagnon, C., Lauga, B., Duran, R., Eds.; Springer: Cham, Switzerland, 2017; pp. 3–15. [Google Scholar] [CrossRef]
- Ferguson, L.; Taylor, J.; Davies, M.; Shrubsole, C.; Symonds, P.; Dimitroulopoulou, S. Exposure to indoor air pollution across socio-economic groups in high-income countries: A scoping review of the literature and a modelling methodology. Environ. Int. 2020, 143, 105748. [Google Scholar] [CrossRef] [PubMed]
- Hopke, P.K.; Dai, Q.; Li, L.; Feng, Y. Global review of recent source apportionments for airborne particulate matter. Sci. Total. Environ. 2020, 740, 140091. [Google Scholar] [CrossRef]
- Snyder, R. Leukemia and Benzene. Int. J. Environ. Res. Public Health 2012, 9, 2875–2893. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; He, X.; Bi, Y.; Ma, Q. Stem Cell and Benzene-Induced Malignancy and Hematotoxicity. Chem. Res. Toxicol. 2012, 25, 1303–1315. [Google Scholar] [CrossRef]
- IARC monographs on the evaluation of carcinogenic risks to humans. Food Chem. Toxicol. 1989, 27, 549. [CrossRef]
- Hunter, W.J.; Aresini, G.; Haigh, R.; Papadopoulos, P.; Von Der Hude, W. Occupational exposure limits for chemicals in the European Union. Occup. Environ. Med. 1997, 54, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Rich, A.L.; Orimoloye, H. Elevated Atmospheric Levels of Benzene and Benzene-Related Compounds from Unconventional Shale Extraction and Processing: Human Health Concern for Residential Communities. Environ. Health Insights 2016, 10, 75–82. [Google Scholar] [CrossRef]
- Hanninen, O.; Knol, A.B.; Jantunen, M.J.; Lim, T.-A.; Conrad, A.; Rappolder, M.; Carrer, P.; Fanetti, A.-C.; Kim, R.; Buekers, J.; et al. Environmental Burden of Disease in Europe: Assessing Nine Risk Factors in Six Countries. Environ. Health Perspect. 2014, 122, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Fontes, T.; Manso, M.C.; Prata, J.C.; Carvalho, M.; Silva, C.; Barros, N. Exposure to BTEX in buses: The influence of vehicle fuel type. Environ. Pollut. 2019, 255, 113100. [Google Scholar] [CrossRef]
- Moreno, T.; Pacitto, A.; Fernández, A.; Amato, F.; Marco, E.; Grimalt, J.O.; Buonanno, G.; Querol, X. Vehicle interior air quality conditions when travelling by taxi. Environ. Res. 2019, 172, 529–542. [Google Scholar] [CrossRef]
- Johnson, E.S.; Langård, S.; Lin, Y.-S. A critique of benzene exposure in the general population. Sci. Total Environ. 2007, 374, 183–198. [Google Scholar] [CrossRef]
- Stabbert, R.; Dempsey, R.; Diekmann, J.; Euchenhofer, C.; Hagemeister, T.; Haussmann, H.-J.; Knorr, A.; Mueller, B.P.; Pospisil, P.; Reininghaus, W.; et al. Studies on the contributions of smoke constituents, individually and in mixtures, in a range of in vitro bioactivity assays. Toxicol. In Vitro 2017, 42, 222–246. [Google Scholar] [CrossRef]
- Snyder, R. Xenobiotic Metabolism and the Mechanism(s) of Benzene Toxicity. Drug Metab. Rev. 2004, 36, 531–547. [Google Scholar] [CrossRef]
- Smith, M.T. Advances in understanding benzene health effects and susceptibility. Annu. Rev. Public Health 2010, 31, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Valentine, J.L.; Lee, S.S.; Seaton, M.J.; Asgharian, B.; Farris, G.; Corton, J.C.; Gonzales, F.J.; Medinsky, M.A. Reduction of benzene metabolism and toxicity in mice that lack CYP2E1 expression. Toxicol. Appl. Pharmacol. 1996, 141, 205–213. [Google Scholar] [CrossRef]
- Bauer, A.K.; Faiola, B.; Abernethy, D.J.; Marchan, R.; Pluta, L.J.; Wong, V.A.; Roberts, K.; Jaiswal, A.K.; Gonzalez, F.J.; Butterworth, B.E.; et al. Genetic susceptibility to benzene-induced toxicity: Role of NADPH: Quinone oxidoreductase-1. Cancer Res. 2003, 63, 929–935. [Google Scholar]
- Rothman, N.; Smith, M.T.; Hayes, R.B.; Traver, R.D.; Hoener, B.; Campleman, S.; Li, G.L.; Dosemeci, M.; Linet, M.; Zhang, L.; et al. Benzene poisoning, a risk factor for hematological malignancy, is associated with the NQO1 609C-->T mutation and rapid fractional excretion of chlorzoxazone. Cancer Res. 1997, 57, 2839–2842. [Google Scholar] [PubMed]
- Han, W.; Wang, S.; Li, M.; Jiang, L.; Wang, X.; Xie, K.-Q. The protective effect of diallyl trisulfide on cytopenia induced by benzene through modulating benzene metabolism. Food Chem. Toxicol. 2018, 112, 393–399. [Google Scholar] [CrossRef]
- Snyder, R.; Witz, G.; Goldstein, B.D. The toxicology of benzene. Environ. Health Perspect. 1993, 100, 293–306. [Google Scholar] [CrossRef]
- Aksoy, M. Hematotoxicity and Carcinogenicity of Benzene. Environ. Health Perspect. 1989, 82, 193. [Google Scholar] [CrossRef]
- Lovreglio, P.; Doria, D.; Fracasso, M.E.; Barbieri, A.; Sabatini, L.; Drago, I.; Violante, F.S.; Soleo, L. DNA damage and repair capacity in workers exposed to low concentrations of benzene. Environ. Mol. Mutagen. 2015, 57, 151–158. [Google Scholar] [CrossRef]
- Thomas, R.; Hubbard, A.E.; McHale, C.M.; Zhang, L.; Rappaport, S.M.; Lan, Q.; Rothman, N.; Vermeulen, R.; Guyton, K.Z.; Jinot, J.; et al. Characterization of Changes in Gene Expression and Biochemical Pathways at Low Levels of Benzene Exposure. PLoS ONE 2014, 9, e91828. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Phadnis-Moghe, A.S.; Crawford, R.B.; Kaminski, N.E. Aryl hydrocarbon receptor activation by 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs human B lymphopoiesis. Toxicology 2016, 378, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jiang, S.; Chen, Y.; Ma, R.; Chen, J.; Qian, S.; Shi, Y.; Han, Y.; Zhang, S.; Yu, K. Benzene metabolite hydroquinone induces apoptosis of bone marrow mononuclear cells through inhibition of beta-catenin signaling. Toxicol. In Vitro 2018, 46, 361–369. [Google Scholar] [CrossRef]
- Schnatter, A.R.; Glass, D.C.; Tang, G.; Irons, R.D.; Rushton, L. Myelodysplastic Syndrome and Benzene Exposure Among Petroleum Workers: An International Pooled Analysis. J. Natl. Cancer Inst. 2012, 104, 1724–1737. [Google Scholar] [CrossRef] [Green Version]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.; Wang, Y.-H. Environmental and chemotherapeutic agents induce breakage at genes involved in leukemia-causing gene rearrangements in human hematopoietic stem/progenitor cells. Mutat. Res. Mol. Mech. Mutagen. 2015, 779, 86–95. [Google Scholar] [CrossRef]
- Binato, R.; Meyer, C.; Macedo-Silva, M.L.; Garcia, D.; De Figueiredo, A.F.; Hofmann, J.; Vieira, T.P.; Abdelhay, E.; Marschalek, R. Analyzing acute leukemia patients with complex MLL rearrangements by a sequential LDI-PCR approach. Cancer Lett. 2013, 338, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Tarlé, S.A.; Hajra, A.; Claxton, D.F.; Marlton, P.; Freedman, M.; Siciliano, M.J.; Collins, F.S. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science 1993, 261, 1041–1044. [Google Scholar] [CrossRef]
- Qian, S.; Han, Y.; Shi, Y.; Xu, W.; Zhu, Y.; Jiang, S.; Chen, Y.; Yu, Z.; Zhang, S.; Yang, Y.; et al. Benzene induces haematotoxicity by promoting deacetylation and autophagy. J. Cell. Mol. Med. 2018, 23, 1022–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, W.; Guo, X.; Ren, J.; Gao, A. lncRNAVNN3 mediated benzene-induced hematotoxicity through promoting autophagy and apoptosis. Ecotoxicol. Environ. Saf. 2019, 185, 109672. [Google Scholar] [CrossRef] [PubMed]
- Kaleağasıoğlu, F.; Ali, D.M.; Berger, M.R. Multiple Facets of Autophagy and the Emerging Role of Alkylphosphocholines as Autophagy Modulators. Front. Pharmacol. 2020, 11, 547. [Google Scholar] [CrossRef]
- Forristal, C.E.; Winkler, I.G.; Nowlan, B.; Barbier, V.; Walkinshaw, G.; Levesque, J.-P. Pharmacologic stabilization of HIF-1α increases hematopoietic stem cell quiescence in vivo and accelerates blood recovery after severe irradiation. Blood 2013, 121, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Zhang, J.; Yin, L.-H.; Pu, Y. Involvement of hypoxia-inducible factor-1 α (HIF-1α) in inhibition of benzene on mouse hematopoietic system. J. Toxicol. Environ. Health Part A 2016, 79, 402–406. [Google Scholar] [CrossRef]
- Man, Z.; Meng, X.; Sun, F.; Pu, Y.; Xu, K.; Sun, R.; Zhang, J.; Yin, L.-H.; Pu, Y. Global Identification of HIF-1α Target Genes in Benzene Poisoning Mouse Bone Marrow Cells. Int. J. Environ. Res. Public Health 2018, 15, 2531. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Meng, X.; Pu, Y.; Sun, F.; Man, Z.; Zhang, J.; Yin, L.; Pu, Y. Overexpression of HIF-1a could partially protect K562 cells from 1,4-benzoquinone induced toxicity by inhibiting ROS, apoptosis and enhancing glycolysis. Toxicol. In Vitro 2019, 55, 18–23. [Google Scholar] [CrossRef]
- Koh, D.-H.; Jeon, H.-K.; Lee, S.-G.; Ryu, H.-W. The relationship between low-level benzene exposure and blood cell counts in Korean workers. Occup. Environ. Med. 2015, 72, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Sun, Q.; Jiang, Y.; Zhang, X.; Chen, L.; Xie, C.; Qin, F.; Chen, Y.; Lv, H.; Chen, W.; et al. MiR-146a affects the alteration in myeloid differentiation induced by hydroquinone in human CD34+hematopoietic progenitor cells and HL-60 cells. Toxicol. Res. 2016, 5, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froom, P.; Dyerassi, L.; Cassel, A.; Aghai, E. Erythropoietin-independent colonies of red blood cells and leukocytosis in a worker exposed to low levels of benzene. Scand. J. Work. Environ. Health 1994, 20, 306–308. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Chen, Y.; Yuan, W.; Qin, F.; Zhang, Q.; Deng, N.; Liu, X.; Ma, X.; Zhang, X.; Zhang, B.; et al. Down-regulation of miRNA-451a and miRNA-486-5p involved in benzene-induced inhibition on erythroid cell differentiation in vitro and in vivo. Arch. Toxicol. 2017, 92, 259–272. [Google Scholar] [CrossRef]
- Doré, L.C.; Amigo, J.D.; Dos Santos, C.O.; Zhang, Z.; Gai, X.; Tobias, J.W.; Yu, D.; Klein, A.M.; Dorman, C.; Wu, W.; et al. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc. Natl. Acad. Sci. USA 2008, 105, 3333–3338. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Simmini, S.; Abreu-Goodger, C.; Bartonicek, N.; Di Giacomo, M.; Bilbao-Cortes, D.; Horos, R.; Von Lindern, M.; Enright, A.J.; O’Carroll, D. The miR-144/451 locus is required for erythroid homeostasis. J. Exp. Med. 2010, 207, 1351–1358. [Google Scholar] [CrossRef]
- Wang, L.-S.; Li, L.; Li, L.; Chu, S.; Shiang, K.-D.; Li, M.; Sun, H.-Y.; Xu, J.; Xiao, F.-J.; Sun, G.; et al. MicroRNA-486 regulates normal erythropoiesis and enhances growth and modulates drug response in CML progenitors. Blood 2015, 125, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Zhang, J.; Xiong, M.; Wei, H.; Tan, K.; Yin, L.-H.; Pu, Y. Altered Expression of Genes in Signaling Pathways Regulating Proliferation of Hematopoietic Stem and Progenitor Cells in Mice with Subchronic Benzene Exposure. Int. J. Environ. Res. Public Health 2015, 12, 9298–9313. [Google Scholar] [CrossRef] [Green Version]
- Tishevskaya, N.; Bolotov, A.A.; Lebedeva, Y.E. Dynamics of Erythropoiesis in Erythroblastic Islands in the Bone Marrow in Experimental Benzene-Induced Anemia. Bull. Exp. Biol. Med. 2016, 161, 384–387. [Google Scholar] [CrossRef]
- Vogel, C.F.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef] [PubMed]
- Heluany, C.S.; Kupa, L.D.V.K.; Viana, M.N.; Fernandes, C.M.; Farsky, S. Hydroquinone exposure worsens the symptomatology of rheumatoid arthritis. Chem. Interactions 2018, 291, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Heluany, C.S.; Kupa, L.D.V.K.; Viana, M.N.; Fernandes, C.M.; Silveira, E.L.V.; Farsky, S. In vivo exposure to hydroquinone during the early phase of collagen-induced arthritis aggravates the disease. Toxicology 2018, 408, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.-I.; Hirabayashi, Y.; Kawasaki, Y.; Kodama, Y.; Kaneko, T.; Kanno, J.; Kim, D.-Y.; Fujii-Kuriyama, Y.; Inoue, T. Aryl hydrocarbon receptor mediates benzene-induced hematotoxicity. Toxicol. Sci. 2002, 70, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Yoon, B.-I.; Li, G.-X.; Kanno, J.; Inoue, T. Mechanism of Benzene-Induced Hematotoxicity and Leukemogenicity: Current Review with Implication of Microarray Analyses. Toxicol. Pathol. 2004, 32, 12–16. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Inoue, T. Benzene-induced bone-marrow toxicity: A hematopoietic stem-cell-specific, aryl hydrocarbon receptor-mediated adverse effect. Chem. Interact. 2010, 184, 252–258. [Google Scholar] [CrossRef]
- Nakahama, T.; Kimura, A.; Nguyen, N.T.; Chinen, I.; Hanieh, H.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 14222–14227. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Nakahama, T.; Kishimoto, T. Aryl hydrocarbon receptor and experimental autoimmune arthritis. Semin. Immunopathol. 2013, 35, 637–644. [Google Scholar] [CrossRef]
- Adachi, M.; Okamoto, S.; Chujyo, S.; Arakawa, T.; Yokoyama, M.; Yamada, K.; Hayashi, A.; Akita, K.; Takeno, M.; Itoh, S.; et al. Cigarette Smoke Condensate Extracts Induce IL-1-Beta Production from Rheumatoid Arthritis Patient-Derived Synoviocytes, but Not Osteoarthritis Patient-Derived Synoviocytes, Through Aryl Hydrocarbon Receptor-Dependent NF-Kappa-B Activation and Novel NF-Kappa-B Sites. J. Interf. Cytokine Res. 2013, 33, 297–307. [Google Scholar] [CrossRef]
- Talbot, J.; Peres, R.S.; Pinto, L.G.; Oliveira, R.D.R.; De Lima, K.A.; Donate, P.B.; Silva, J.R.; Ryffel, B.; Cunha, T.M.; Alves-Filho, J.C.; et al. Smoking-induced aggravation of experimental arthritis is dependent of aryl hydrocarbon receptor activation in Th17 cells. Arthritis Res. 2018, 20, 119. [Google Scholar] [CrossRef] [Green Version]
- Kazantseva, M.G.; Highton, J.; Stamp, L.K.; Hessian, P.A. Dendritic cells provide a potential link between smoking and inflammation in rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, 208. [Google Scholar] [CrossRef] [Green Version]
- Colombo, G.; Clerici, M.; Giustarini, D.; Portinaro, N.M.; Aldini, G.; Rossi, R.; Milzani, A.D.G.; Dalle-Donne, I. Pathophysiology of tobacco smoke exposure: Recent insights from comparative and redox proteomics. Mass Spectrom. Rev. 2013, 33, 183–218. [Google Scholar] [CrossRef] [PubMed]
- Osman, N.M.; Sexton, D.W.; Saleem, I.Y. Toxicological assessment of nanoparticle interactions with the pulmonary system. Nanotoxicology 2019, 14, 21–58. [Google Scholar] [CrossRef]
- Miller, M.R.; Poland, C.A. Nanotoxicology: The Need for a Human Touch? Small 2020, 2001516. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, A.; Costa, B.; Ferreira, M.V.; Miguéis, D.; Louros, J.M.S.; Durazzo, A.; Lucarini, M.; Eder, P.; Chaud, M.; Morsink, M.; et al. Nanotoxicology and Nanosafety: Safety-By-Design and Testing at a Glance. Int. J. Environ. Res. Public Health 2020, 17, 4657. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Liu, Y.; Li, R.; Wu, W.; Fawcett, J.P.; Gu, J. Absorption, distribution, metabolism and excretion of the biomaterials used in Nanocarrier drug delivery systems. Adv. Drug Deliv. Rev. 2019, 143, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Porter, A.E.; Ryan, M.P.; Schwander, S.; Chung, K.F.; Tetley, T.D.; Zhang, J.J.; Georgopoulos, P.G. Modeling In Vivo Interactions of Engineered Nanoparticles in the Pulmonary Alveolar Lining Fluid. Nanomaterials 2015, 5, 1223–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, L.; Sheykhzade, M.; Jensen, K.A.; Saber, A.T.; Jacobsen, N.R.; Vogel, U.B.; Wallin, H.; Loft, S.; Møller, P. Modest effect on plaque progression and vasodilatory function in atherosclerosis-prone mice exposed to nanosized TiO2. Part. Fibre Toxicol. 2011, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Spigoni, V.; Cito, M.; Alinovi, R.; Pinelli, S.; Passeri, G.; Zavaroni, I.; Goldoni, M.; Campanini, M.; Aliatis, I.; Mutti, A.; et al. Effects of TiO2 and Co3O4 Nanoparticles on Circulating Angiogenic Cells. PLoS ONE 2015, 10, e0119310. [Google Scholar] [CrossRef]
- Rafii, S.; Lyden, D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat. Med. 2003, 9, 702–712. [Google Scholar] [CrossRef]
- Kwon, J.-T.; Kim, D.-S.; Minai-Tehrani, A.; Hwang, S.-K.; Chang, S.-H.; Lee, E.-S.; Xu, C.-X.; Lim, H.T.; Kim, J.-E.; Yoon, B.-I.; et al. Inhaled fluorescent magnetic nanoparticles induced extramedullary hematopoiesis in the spleen of mice. J. Occup. Health 2009, 51, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Simkó, M.; Mattsson, M.-O. Risks from accidental exposures to engineered nanoparticles and neurological health effects: A critical review. Part. Fibre Toxicol. 2010, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newell, K.; Kartsonaki, C.; Lam, K.B.H.; Kurmi, O.P. Cardiorespiratory health effects of particulate ambient air pollution exposure in low-income and middle-income countries: A systematic review and meta-analysis. Lancet Planet. Health 2017, 1, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Annesi-Maesano, I. The air of Europe: Where are we going? Eur. Respir. Rev. 2017, 26, 170024. [Google Scholar] [CrossRef] [Green Version]
- Mir, R.H.; Sawhney, G.; Pottoo, F.H.; Mohi-Ud-Din, R.; Madishetti, S.; Jachak, S.M.; Ahmed, Z.; Masoodi, M.H. Role of environmental pollutants in Alzheimer’s disease: A review. Environ. Sci. Pollut. Res. 2020, 1–19. [Google Scholar] [CrossRef]
- Nakane, H. Translocation of particles deposited in the respiratory system: A systematic review and statistical analysis. Environ. Health Prev. Med. 2011, 17, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.-F.; Xu, Y.-H.; Shi, M.-H.; Lian, Y.-X. The impact of PM2.5 on the human respiratory system. J. Thorac. Dis. 2016, 8, 69–74. [Google Scholar]
- Rychlik, K.A.; Secrest, J.R.; Lau, C.; Pulczinski, J.; Zamora, M.L.; Leal, J.; Langley, R.; Myatt, L.G.; Raju, M.; Chang, R.C.-A.; et al. In utero ultrafine particulate matter exposure causes offspring pulmonary immunosuppression. Proc. Natl. Acad. Sci. USA 2019, 116, 3443–3448. [Google Scholar] [CrossRef] [Green Version]
- Loxham, M.; Nieuwenhuijsen, M. Health effects of particulate matter air pollution in underground railway systems – a critical review of the evidence. Part. Fibre Toxicol. 2019, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Li, X.; Wang, T.; Wang, B.; Li, C.; Zeng, G. A review on airborne microorganisms in particulate matters: Composition, characteristics and influence factors. Environ. Int. 2018, 113, 74–90. [Google Scholar] [CrossRef]
- Wei, M.; Xu, C.; Xu, X.; Zhu, C.; Li, J.; Lv, G. Size distribution of bioaerosols from biomass burning emissions: Characteristics of bacterial and fungal communities in submicron (PM1.0) and fine (PM2.5) particles. Ecotoxicol. Environ. Saf. 2019, 171, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Gao, D.; Liao, F.; Zhou, F.; Wang, X. The health effects of ambient PM2.5 and potential mechanisms. Ecotoxicol. Environ. Saf. 2016, 128, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Di, Q.; Wang, Y.; Zanobetti, A.; Wang, Y.; Koutrakis, P.; Choirat, C.; Dominici, F.; Schwartz, J.D. Air Pollution and Mortality in the Medicare Population. N. Engl. J. Med. 2017, 376, 2513–2522. [Google Scholar] [CrossRef] [PubMed]
- Abu Awad, Y.; Di, Q.; Wang, Y.; Choirat, C.; Coull, B.A.; Zanobetti, A.; Schwartz, J. Change in PM2.5 exposure and mortality among Medicare recipients. Environ. Epidemiol. 2019, 3, e054. [Google Scholar] [CrossRef]
- Papadogeorgou, G.; Kioumourtzoglou, M.-A.; Braun, D.; Zanobetti, A. Low Levels of Air Pollution and Health: Effect Estimates, Methodological Challenges, and Future Directions. Curr. Environ. Health Rep. 2019, 6, 105–115. [Google Scholar] [CrossRef]
- Gestro, M.; Condemi, V.; Bardi, L.; Tomaino, L.; Roveda, E.; Bruschetta, A.; Solimene, U.; Esposito, F. Short-term air pollution exposure is a risk factor for acute coronary syndromes in an urban area with low annual pollution rates: Results from a retrospective observational study (2011–2015). Arch. Cardiovasc. Dis. 2020, 113, 308–320. [Google Scholar] [CrossRef]
- Yang, J.; Kim, E.K.; Park, H.J.; McDowell, A.; Kim, Y.-K. The impact of bacteria-derived ultrafine dust particles on pulmonary diseases. Exp. Mol. Med. 2020, 52, 338–347. [Google Scholar] [CrossRef]
- Tan, W.C.; Qiu, D.; Liam, B.L.; Ng, T.P.; Lee, S.H.; Van Eeden, S.F.; D’Yachkova, Y.; Hogg, J.C. The Human Bone Marrow Response to Acute Air Pollution Caused by Forest Fires. Am. J. Respir. Crit. Care Med. 2000, 161, 1213–1217. [Google Scholar] [CrossRef]
- Goto, Y.; Ishii, H.; Hogg, J.C.; Shih, C.-H.; Yatera, K.; Vincent, R.; Van Eeden, S.F. Particulate Matter Air Pollution Stimulates Monocyte Release from the Bone Marrow. Am. J. Respir. Crit. Care Med. 2004, 170, 891–897. [Google Scholar] [CrossRef]
- Van Eeden, S.F.; Tan, W.C.; Suwa, T.; Mukae, H.; Terashima, T.; Fujii, T.; Qui, D.; Vincent, R.; Hogg, J.C. Cytokines Involved in the Systemic Inflammatory Response Induced by Exposure to Particulate Matter Air Pollutants (PM10). Am. J. Respir. Crit. Care Med. 2001, 164, 826–830. [Google Scholar] [CrossRef]
- Van Eeden, S.F.; Hogg, J.C. Systemic Inflammatory Response Induced by Particulate Matter Air Pollution: The Importance of Bone-Marrow Stimulation. J. Toxicol. Environ. Health Part A 2002, 65, 1597–1613. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Bard, R.L.; Burnett, R.T.; Shin, H.H.; Vette, A.; Croghan, C.; Phillips, M.; Rodes, C.; Thornburg, J.; Williams, R. Differences in blood pressure and vascular responses associated with ambient fine particulate matter exposures measured at the personal versus community level. Occup. Environ. Med. 2010, 68, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Burnett, R.T.; Kwong, J.C.; Villeneuve, P.J.; Goldberg, M.S.; Brook, R.D.; Van Donkelaar, A.; Jerrett, M.; Martin, R.V.; Kopp, A.; et al. Spatial Association Between Ambient Fine Particulate Matter and Incident Hypertension. Circulation 2013, 129, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampfrath, T.; Maiseyeu, A.; Ying, Z.; Shah, Z.; Deiuliis, J.A.; Xu, X.; Kherada, N.; Brook, R.D.; Reddy, K.M.; Padture, N.P.; et al. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ. Res. 2011, 108, 716–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.-Y.; Wang, J.-S.; Chao, M.-W. Causation by Diesel Exhaust Particles of Endothelial Dysfunctions in Cytotoxicity, Pro-inflammation, Permeability, and Apoptosis Induced by ROS Generation. Cardiovasc. Toxicol. 2016, 17, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Seki, T.; Naito, Y.; Tachibana, S.; Hirabayashi, N.; Nakasaka, T.; Ohara, N.; Kobayashi, H. Tracheal instillation of diesel exhaust particles component causes blood and pulmonary neutrophilia and enhances myocardial oxidative stress in mice. J. Toxicol. Sci. 2008, 33, 609–620. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, T.E.; Hellmann, J.; Wheat, L.; Haberzettl, P.; Lee, J.; Conklin, D.J.; Bhatnagar, A.; Pope, C.A. Episodic exposure to fine particulate air pollution decreases circulating levels of endothelial progenitor cells. Circ. Res. 2010, 107, 200–203. [Google Scholar] [CrossRef] [Green Version]
- Haberzettl, P.; Conklin, D.J.; Abplanalp, W.T.; Bhatnagar, A.; O’Toole, T.E. Inhalation of Fine Particulate Matter Impairs Endothelial Progenitor Cell Function Via Pulmonary Oxidative Stress. Arter. Thromb. Vasc. Biol. 2017, 38, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elmagd, M.; Alghamdi, M.A.; Shamy, M.; Khoder, M.; Costa, M.; Assidi, M.; Kadam, R.; Alsehli, H.; Gari, M.; Pushparaj, P.N.; et al. Evaluation of the Effects of Airborne Particulate Matter on Bone Marrow-Mesenchymal Stem Cells (BM-MSCs): Cellular, Molecular and Systems Biological Approaches. Int. J. Environ. Res. Public Health 2017, 14, 440. [Google Scholar] [CrossRef] [Green Version]
- Bhattarai, G.; Lee, J.B.; Kim, M.-H.; Ham, S.; So, H.-S.; Oh, S.; Sim, H.-J.; Lee, J.-C.; Song, M.; Kook, S.-H. Maternal exposure to fine particulate matter during pregnancy induces progressive senescence of hematopoietic stem cells under preferential impairment of the bone marrow microenvironment and aids development of myeloproliferative disease. Leukemia 2019, 34, 1481–1484. [Google Scholar] [CrossRef] [Green Version]
- Breivik, K.; Alcock, R.; Li, Y.-F.; Bailey, R.E.; Fiedler, H.; Pacyna, J.M. Primary sources of selected POPs: Regional and global scale emission inventories. Environ. Pollut. 2004, 128, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Holt, E.; Weber, R.; Stevenson, G.; Gaus, C. Polychlorinated Dibenzo-p-Dioxins and Dibenzofurans (PCDD/Fs) Impurities in Pesticides: A Neglected Source of Contemporary Relevance. Environ. Sci. Technol. 2010, 44, 5409–5415. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances & Disease Registry (ATSDR). Toxicological Profile for Polychlorinated Biphenyls (PCBs); US Department of Health and Human Services: Atlanta, GA, USA, 2000.
- Lallas, P.L. The Stockholm Convention on Persistent Organic Pollutants. Am. J. Int. Law 2001, 95, 692. [Google Scholar] [CrossRef]
- Klocke, C.; Lein, P.J. Evidence Implicating Non-Dioxin-Like Congeners as the Key Mediators of Polychlorinated Biphenyl (PCB) Developmental Neurotoxicity. Int. J. Mol. Sci. 2020, 21, 1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogenboom, R.L.; Malisch, R.; Van Leeuwen, S.P.; Vanderperren, H.; Hove, H.; Fernandes, A.; Schächtele, A.; Rose, M. Congener patterns of polychlorinated dibenzo-p-dioxins, dibenzofurans and biphenyls as a useful aid to source identification during a contamination incident in the food chain. Sci. Total. Environ. 2020, 746, 141098. [Google Scholar] [CrossRef] [PubMed]
- US EPA (Environmental Protection Agency). Recommended Toxicity Equivalence Factors (TEFs) for Human Health Risk Assessments of 2,3,7,8-Tetrachlorodibenzo-p-dioxin and Dioxin-Like Compounds; Risk Assessment Forum: Washington, DC, USA, 2010.
- Paustenbach, D.J.; Wenning, R.J.; Lau, V.; Harrington, N.W.; Rennix, D.K.; Parsons, A.H. Recent developments on the hazards posed by 2,3,7,8-tetrachlorodibenzo-p-dioxin in soil: Implications for setting risk-based cleanup levels at residential and industrial sites. J. Toxicol. Environ. Health Part A 1992, 36, 103–149. [Google Scholar] [CrossRef]
- Rowlands, J.C.; Gustafsson, J.-Å. Aryl Hydrocarbon Receptor-Mediated Signal Transduction. Crit. Rev. Toxicol. 1997, 27, 109–134. [Google Scholar] [CrossRef]
- Mandal, P.K. Dioxin: A review of its environmental effects and its aryl hydrocarbon receptor biology. J. Comp. Physiol. B 2005, 175, 221–230. [Google Scholar] [CrossRef]
- Cox, M.B.; Miller, C.A. Cooperation of heat shock protein 90 and p23 in aryl hydrocarbon receptor signaling. Cell Stress Chaperones 2004, 9, 4–20. [Google Scholar] [CrossRef]
- Safe, S.; Han, H.; Goldsby, J.; Mohankumar, K.; Chapkin, R.S. Aryl hydrocarbon receptor (AhR) ligands as selective AhR modulators: Genomic studies. Curr. Opin. Toxicol. 2018, 11-12, 10–20. [Google Scholar] [CrossRef]
- Beischlag, T.V.; Morales, J.L.; Hollingshead, B.D.; Perdew, G.H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef] [Green Version]
- Tomita, S.; Jiang, H.-B.; Ueno, T.; Takagi, S.; Tohi, K.; Maekawa, S.-I.; Miyatake, A.; Furukawa, A.; Gonzalez, F.J.; Takeda, J.; et al. T cell-specific disruption of arylhydrocarbon receptor nuclear translocator (Arnt) gene causes resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced thymic involution. J. Immunol. 2003, 171, 4113–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitot, H.C.; Goldsworthy, T.; Campbell, H.A.; Poland, A. Quantitative evaluation of the promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from diethylnitrosamine. Cancer Res. 1980, 40, 3616–3620. [Google Scholar] [PubMed]
- Singh, K.P.; Bennett, J.A.; Casado, F.L.; Walrath, J.L.; Welle, S.L.; Gasiewicz, T.A. Loss of Aryl Hydrocarbon Receptor Promotes Gene Changes Associated with Premature Hematopoietic Stem Cell Exhaustion and Development of a Myeloproliferative Disorder in Aging Mice. Stem Cells Dev. 2013, 23, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.A.; Singh, K.P.; Welle, S.L.; Boule, L.A.; Lawrence, B.P.; Gasiewicz, T.A. Conditional deletion of Ahr alters gene expression profiles in hematopoietic stem cells. PLoS ONE 2018, 13, e0206407. [Google Scholar] [CrossRef]
- Unnisa, Z.; Singh, K.P.; Henry, E.C.; Donegan, C.L.; Bennett, J.A.; Gasiewicz, T.A. Aryl Hydrocarbon Receptor Deficiency in an Exon 3 Deletion Mouse Model Promotes Hematopoietic Stem Cell Proliferation and Impacts Endosteal Niche Cells. Stem Cells Int. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Angelos, M.G.; Ruh, P.N.; Webber, B.R.; Blum, R.H.; Ryan, C.D.; Bendzick, L.; Shim, S.; Yingst, A.M.; Tufa, D.M.; Verneris, M.R.; et al. Aryl hydrocarbon receptor inhibition promotes hematolymphoid development from human pluripotent stem cells. Blood 2017, 129, 3428–3439. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.; Zulick, E.; Skvir, N.J.; Vanuytsel, K.; Morrison, T.A.; Naing, Z.H.; Wang, Z.; Dai, Y.; Chui, D.H.K.; Steinberg, M.H.; et al. Notch and Aryl Hydrocarbon Receptor Signaling Impact Definitive Hematopoiesis from Human Pluripotent Stem Cells. Stem Cells 2018, 36, 1004–1019. [Google Scholar] [CrossRef] [Green Version]
- Bertazzi, P.A.; Pesatori, A.C.; Consonni, D.; Tironi, A.; Landi, M.T.; Zocchetti, C. Cancer incidence in a population accidentally exposed to 2,3,7,8-tetrachlorodibenzo-para-dioxin. Epidemiology 1993, 4, 398–406. [Google Scholar] [CrossRef]
- Hosnijeh, F.S.; Boers, D.; Portengen, L.; Bueno-De-Mesquita, H.B.; Heederik, D.J.; Vermeulen, R.C.H. Plasma Cytokine Concentrations in Workers Exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Front. Oncol. 2012, 2, 37. [Google Scholar] [CrossRef] [Green Version]
- Fracchiolla, N.S.; Annaloro, C.; Guidotti, F.; Fattizzo, B.; Cortelezzi, A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) role in hematopoiesis and in hematologic diseases: A critical review. Toxicology 2016, 374, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Ahrenhoerster, L.S.; Tate, E.R.; Lakatos, P.A.; Wang, X.; Laiosa, M.D. Developmental exposure to 2,3,7,8 tetrachlorodibenzo-p-dioxin attenuates capacity of hematopoietic stem cells to undergo lymphocyte differentiation. Toxicol. Appl. Pharmacol. 2014, 277, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baccarelli, A.; Mocarelli, P.; Patterson, D.G.; Bonzini, M.; Pesatori, A.C.; Caporaso, N.; Landi, M.T. Immunologic effects of dioxin: New results from Seveso and comparison with other studies. Environ. Health Perspect. 2002, 110, 1169–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesatori, A.C.; Consonni, D.; Rubagotti, M.; Grillo, P.; Bertazzi, P.A. Cancer incidence in the population exposed to dioxin after the “Seveso accident”: Twenty years of follow-up. Environ. Health 2009, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Thurmond, T.S.; Gasiewicz, T.A. A single dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin produces a time- and dose-dependent alteration in the murine bone marrow B-lymphocyte maturation profile. Toxicol. Sci. 2000, 58, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, B.; Chaudhry, A.; Yewdell, W.T.; Angeletti, D.; Yen, W.-F.; Wheatley, A.K.; Bradfield, C.A.; McDermott, A.B.; Yewdell, J.W.; Rudensky, A.Y.; et al. The aryl hydrocarbon receptor controls cell-fate decisions in B cells. J. Exp. Med. 2016, 214, 197–208. [Google Scholar] [CrossRef]
- Sakai, R.; Kajiume, T.; Inoue, H.; Kanno, R.; Miyazaki, M.; Ninomiya, Y.; Kanno, M. TCDD treatment eliminates the long-term reconstitution activity of hematopoietic stem cells. Toxicol. Sci. 2003, 72, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.P.; Casado, F.L.; Opanashuk, L.A.; Gasiewicz, T.A. The aryl hydrocarbon receptor has a normal function in the regulation of hematopoietic and other stem/progenitor cell populations. Biochem. Pharmacol. 2009, 77, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Casado, F.L.; Singh, K.P.; Gasiewicz, T.A. Aryl hydrocarbon receptor activation in hematopoietic stem/progenitor cells alters cell function and pathway-specific gene modulation reflecting changes in cellular trafficking and migration. Mol. Pharmacol. 2011, 80, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Jensen, B.A.; Leeman, R.J.; Schlezinger, J.J.; Sherr, D.H. Aryl hydrocarbon receptor (AhR) agonists suppress interleukin-6 expression by bone marrow stromal cells: An immunotoxicology study. Environ. Health 2003, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Laiosa, M.D.; Tate, E.R. Fetal Hematopoietic Stem Cells Are the Canaries in the Coal Mine That Portend Later Life Immune Deficiency. Endocrinology 2015, 156, 3458–3465. [Google Scholar] [CrossRef] [PubMed]
- Suvarapu, L.N.; Baek, S.-O. Determination of heavy metals in the ambient atmosphere. Toxicol. Ind. Health 2016, 33, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Omrane, F.; Gargouri, I.; Khadhraoui, M.; Elleuch, B.; Zmirou-Navier, D. Risk assessment of occupational exposure to heavy metal mixtures: A study protocol. BMC Public Health 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchounwou, P.B.; Yedjou, C.G.; Patlolla, A.K.; Sutton, D.J. Heavy metals toxicity and the environment. In Molecular, Clinical and Environmental Toxicology; Luch, A., Ed.; Springer: Basel, Switzerland, 2012; pp. 133–164. [Google Scholar] [CrossRef] [Green Version]
- Qing, Y.; Yang, J.; Zhu, Y.; Li, Y.; Ma, W.; Zhang, C.; Li, X.; Wu, M.; Wang, H.; Kauffman, A.E.; et al. Cancer risk and disease burden of dietary cadmium exposure changes in Shanghai residents from 1988 to 2018. Sci. Total. Environ. 2020, 734, 139411. [Google Scholar] [CrossRef]
- Golzadeh, N.; Barst, B.D.; Basu, N.; Baker, J.M.; Auger, J.C.; McKinney, M.A. Evaluating the concentrations of total mercury, methylmercury, selenium, and selenium:mercury molar ratios in traditional foods of the Bigstone Cree in Alberta, Canada. Chemosphere 2020, 250, 126285. [Google Scholar] [CrossRef]
- Ren, J.; Cui, J.; Chen, Q.; Zhou, N.; Zhou, Z.; Zhang, G.-H.; Wu, W.; Yang, H.; Cao, J. Low-level lead exposure is associated with aberrant sperm quality and reproductive hormone levels in Chinese male individuals: Results from the MARHCS study low-level lead exposure is associated with aberrant sperm quality. Chemosphere 2020, 244, 125402. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; KyunPark, S. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimers Dis. 2020, 1–28. [Google Scholar] [CrossRef]
- Manjarres-Suarez, A.; Olivero-Verbel, J. Hematological parameters and hair mercury levels in adolescents from the Colombian Caribbean. Environ. Sci. Pollut. Res. 2020, 27, 14216–14227. [Google Scholar] [CrossRef]
- Vianna, A.D.S.; De Matos, E.P.; De Jesus, I.M.; Asmus, C.I.R.F.; Câmara, V.D.M. Human exposure to mercury and its hematological effects: A systematic review. Cad. Saúde Pública 2019, 35, e00091618. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Huo, X.; Zhang, Y.; Xiao, Z.; Zhang, Y.; Xu, X. Lead exposure is associated with risk of impaired coagulation in preschool children from an e-waste recycling area. Environ. Sci. Pollut. Res. 2018, 25, 20670–20679. [Google Scholar] [CrossRef]
- Li, Q.; Yang, Z.; Zhang, P.; Zhao, Y.; Yu, Y.; Xue, P.; Shao, Y.; Li, Q.; Jia, X.; Zhang, Q.; et al. Mercury impact on hematopoietic stem cells is regulated by IFNgamma-dependent bone marrow-resident macrophages in mice. Toxicol. Lett. 2018, 295, 54–63. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, X.; Sun, D.; Cao, J.; Zhang, Y.; Huo, X. Alteration of the number and percentage of innate immune cells in preschool children from an e-waste recycling area. Ecotoxicol. Environ. Saf. 2017, 145, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Dobrakowski, M.; Boroń, M.; Czuba, Z.P.; Birkner, E.; Chwalba, A.; Hudziec, E.; Kasperczyk, S. Blood morphology and the levels of selected cytokines related to hematopoiesis in occupational short-term exposure to lead. Toxicol. Appl. Pharmacol. 2016, 305, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Heuvel, R.V.D.; Leppens, H.; Schoeters, G. Lead and catechol hematotoxicity in vitro using human and murine hematopoietic progenitor cells. Cell Biol. Toxicol. 1999, 15, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, P.; Yu, X.; Zhao, Y.; Li, Q.; Zhang, Y.; Yang, Z.; Xie, Y.; Xue, P.; Sun, S.; et al. Lead Transiently Promotes Granulocyte-Macrophage Progenitor Differentiation and Subsequently Suppresses Common Myeloid Progenitor Differentiation. Toxicol. Sci. 2017, 160, 268–283. [Google Scholar] [CrossRef]
- Kurotaki, D.; Yamamoto, M.; Nishiyama, A.; Uno, K.; Ban, T.; Ichino, M.; Sasaki, H.; Matsunaga, S.; Yoshinari, M.; Ryo, A.; et al. IRF8 inhibits C/EBPα activity to restrain mononuclear phagocyte progenitors from differentiating into neutrophils. Nat. Commun. 2014, 5, 4978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, T.; Zhao, Y.; Zhang, P.; Shao, Y.; He, J.; Xue, P.; Zheng, W.; Qu, W.; Jia, X.; Zhou, Z.; et al. Lead impairs the development of innate lymphoid cells by impeding the differentiation of their progenitors. Toxicol. Sci. 2020, 176, 410–422. [Google Scholar] [CrossRef]
- Diefenbach, A.; Colonna, M.; Koyasu, S. Development, differentiation and diversity of innate lymphoid cells. Immunity 2014, 41, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; Velde, A.A.T.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Atreya, I.; Kindermann, M.; Wirtz, S. Innate lymphoid cells in intestinal cancer development. Semin. Immunol. 2019, 41, 101267. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, M.; Akhtar, M.J.; Verma, S.K.; Kumar, A.; Siddiqui, M.K. Environmental lead exposure as a risk for childhood aplastic anemia. Biosci. Trends 2011, 5, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrot, T.S.; Staessen, J.A.; Roels, H.A.; Munters, E.; Cuypers, A.; Richart, T.; Ruttens, A.; Smeets, K.; Clijsters, H.; Vangronsveld, J. Cadmium exposure in the population: From health risks to strategies of prevention. BioMetals 2010, 23, 769–782. [Google Scholar] [CrossRef] [Green Version]
- Nogawa, K.; Suwazono, Y.; Nishijo, M.; Sakurai, M.; Ishizaki, M.; Morikawa, Y.; Watanabe, Y.; Kido, T.; Nakagawa, H. Increase of lifetime cadmium intake dose-dependently increased all cause of mortality in female inhabitants of the cadmium-polluted Jinzu River basin, Toyama, Japan. Environ. Res. 2018, 164, 379–384. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, X.; Sun, S.; Li, Q.; Xie, Y.; Li, Q.; Zhao, Y.; Pei, J.; Zhang, W.; Xue, P.; et al. Cadmium modulates hematopoietic stem and progenitor cells and skews toward myelopoiesis in mice. Toxicol. Appl. Pharmacol. 2016, 313, 24–34. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Q.; Yang, Z.; Shao, Y.; Xue, P.; Qu, W.; Jia, X.; Cheng, L.; He, M.; He, R.; et al. Cadmium Activates Noncanonical Wnt Signaling to Impair Hematopoietic Stem Cell Function in Mice. Toxicol. Sci. 2018, 165, 254–266. [Google Scholar] [CrossRef]
- WHO (World Health Organization). Arsenic. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/arsenic (accessed on 21 September 2020).
- Hughes, M.F. Arsenic toxicity and potential mechanisms of action. Toxicol. Lett. 2002, 133, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Patterson, R.; Vega, L.; Trouba, K.; Bortner, C.; Germolec, D.R. Arsenic-induced alterations in the contact hypersensitivity response in Balb/c mice. Toxicol. Appl. Pharmacol. 2004, 198, 434–443. [Google Scholar] [CrossRef]
- Lemarié, A.; Morzadec, C.; Merino, D.; Micheau, O.; Fardel, O.; Vernhet, L. Arsenic Trioxide Induces Apoptosis of Human Monocytes during Macrophagic Differentiation through Nuclear Factor-κB-Related Survival Pathway Down-Regulation. J. Pharmacol. Exp. Ther. 2005, 316, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Bishayi, B.; Sengupta, M. Intracellular survival of Staphylococcus aureus due to alteration of cellular activity in arsenic and lead intoxicated mature Swiss albino mice. Toxicology 2003, 184, 31–39. [Google Scholar] [CrossRef]
- Harrison, M.; McCoy, K.L. Immunosuppression by arsenic: A comparison of cathepsin L inhibition and apoptosis. Int. Immunopharmacol. 2001, 1, 647–656. [Google Scholar] [CrossRef]
- Biswas, D.; Banerjee, M.; Sen, G.; Das, J.K.; Banerjee, A.; Sau, T.; Pandit, S.; Giri, A.K.; Biswas, T. Mechanism of erythrocyte death in human population exposed to arsenic through drinking water. Toxicol. Appl. Pharmacol. 2008, 230, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, D.; Gribaldo, L.; Hartung, T. Arsenic Exposure and Immunotoxicity: A Review Including the Possible Influence of Age and Sex. Curr. Environ. Health Rep. 2016, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.A.; Law, S. Microenvironmental Scenario of the Bone Marrow of Inorganic Arsenic-Exposed Experimental Mice. Biol. Trace Element Res. 2017, 181, 304–313. [Google Scholar] [CrossRef]
- Heck, J.E.; Chen, Y.; Grann, V.R.; Slavkovich, V.; Parvez, F.; Ahsan, H. Arsenic Exposure and Anemia in Bangladesh: A Population-Based Study. J. Occup. Environ. Med. 2008, 50, 80–87. [Google Scholar] [CrossRef]
- Surdu, S.; Bloom, M.S.; Neamtiu, I.A.; Pop, C.; Anastasiu, D.; Fitzgerald, E.F.; Gurzau, E.S. Consumption of arsenic-contaminated drinking water and anemia among pregnant and non-pregnant women in northwestern Romania. Environ. Res. 2015, 140, 657–660. [Google Scholar] [CrossRef] [Green Version]
- Kile, M.L.; Cardenas, A.; Rodrigues, E.; Mazumdar, M.; Dobson, C.; Golam, M.; Quamruzzaman, Q.; Rahman, M.; Christiani, D.C. Estimating effects of arsenic exposure during pregnancy on perinatal outcomes in a Bangladeshi cohort. Epidemiology 2015, 27, 173. [Google Scholar] [CrossRef] [Green Version]
- Parvez, F.; Medina, S.; Santella, R.M.; Islam, T.; Lauer, F.T.; Alam, N.; Eunus, M.; Rahman, M.; Factor-Litvak, P.; Ahsan, H.; et al. Arsenic exposures alter clinical indicators of anemia in a male population of smokers and non-smokers in Bangladesh. Toxicol. Appl. Pharmacol. 2017, 331, 62–68. [Google Scholar] [CrossRef]
- Feussner, J.R.; Shelburne, J.D.; Bredehoeft, S.; Cohen, H.J. Arsenic-induced bone marrow toxicity: Ultrastructural and electron-probe analysis. Blood 1979, 53, 820–827. [Google Scholar] [CrossRef] [Green Version]
- Saulle, E.; Riccioni, R.; Pelosi, E.; Stafness, M.; Mariani, G.; De Tuglie, G.; Peschle, C.; Testa, U. In vitro dual effect of arsenic trioxide on hemopoiesis: Inhibition of erythropoiesis and stimulation of megakaryocytic maturation. Blood Cells Mol. Dis. 2006, 36, 59–76. [Google Scholar] [CrossRef]
- Medina, S.; Xu, H.; Wang, S.C.; Lauer, F.T.; Liu, K.J.; Burchiel, S.W. Low level arsenite exposures suppress the development of bone marrow erythroid progenitors and result in anemia in adult male mice. Toxicol. Lett. 2017, 273, 106–111. [Google Scholar] [CrossRef]
- Pirrone, N.; Cinnirella, S.; Feng, X.; Finkelman, R.B.; Friedli, H.R.; Leaner, J.; Mason, R.; Mukherjee, A.B.; Stracher, G.; Streets, D.G.; et al. Global Mercury Emissions to the Atmosphere from Natural and Anthropogenic Sources. In Mercury Fate and Transport in the Global Atmosphere; Mason, R., Pirrone, N., Eds.; Springer: Boston, MA, USA, 2009; pp. 1–47. [Google Scholar]
- Agency for Toxic Substances & Disease Registry (ATSDR). Toxicological Profile for Mercury; US Department of Health and Human Services: Atlanta, GA, USA, 1999.
- Ekawanti, A.; Krisnayanti, B.D. Effect of Mercury Exposure on Renal Function and Hematological Parameters among Artisanal and Small-scale Gold Miners at Sekotong, West Lombok, Indonesia. J. Health Pollut. 2015, 5, 25–32. [Google Scholar] [CrossRef]
- Eto, K.; Takeuchi, T. Pathological changes of human sural nerves in Minamata disease (methylmercury poisoning). Virchows Archiv. B 1977, 23, 109. [Google Scholar]
- Strom, S.; Johnson, R.L.; Uyeki, E.M. Mercury toxicity to hemopoietic and tumor colony-forming cells and its reversal by selenium in vitro. Toxicol. Appl. Pharmacol. 1979, 49, 431–436. [Google Scholar] [CrossRef]
- Miszta, H. Effects of mercury on acetylcholinesterase (E.C. 3.1.1.7.) activity of erythrocytes and bone marrow in rats. Folia Haematol. Int. Mag. Klin. Morphol. Blutforsch. 1984, 111, 632–637. [Google Scholar]
- Miszta, H. Mercury and the activity of erythrocyte and bone marrow glutathione reductase (E.C. 1.6.4.2.) and glucose-6-phosphate dehydrogenase (E.C. 1.1.1.49) in rats. Folia Haematol. Int. Mag. Klin. Morphol. Blutforsch. 1984, 111, 638–644. [Google Scholar]
- Brunet, S.; Guertin, F.; Flipo, D.; Fournier, M.; Krzystyniak, K. Cytometric profiles of bone marrow and spleen lymphoid cells after mercury exposure in mice. Int. J. Immunopharmacol. 1993, 15, 811–819. [Google Scholar] [CrossRef]
- Institóris, L.; Siroki, O.; Ündeger, Ülkü; Basaran, N.; Banerjee, B.D.; Dési, I. Detection of the effects of repeated dose combined propoxur and heavy metal exposure by measurement of certain toxicological, haematological and immune function parameters in rats. Toxicology 2001, 163, 185–193. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scharf, P.; Broering, M.F.; Oliveira da Rocha, G.H.; Farsky, S.H.P. Cellular and Molecular Mechanisms of Environmental Pollutants on Hematopoiesis. Int. J. Mol. Sci. 2020, 21, 6996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21196996
Scharf P, Broering MF, Oliveira da Rocha GH, Farsky SHP. Cellular and Molecular Mechanisms of Environmental Pollutants on Hematopoiesis. International Journal of Molecular Sciences. 2020; 21(19):6996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21196996
Chicago/Turabian StyleScharf, Pablo, Milena Fronza Broering, Gustavo Henrique Oliveira da Rocha, and Sandra Helena Poliselli Farsky. 2020. "Cellular and Molecular Mechanisms of Environmental Pollutants on Hematopoiesis" International Journal of Molecular Sciences 21, no. 19: 6996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21196996