Cellular Prion Protein (PrPc): Putative Interacting Partners and Consequences of the Interaction



Abstract
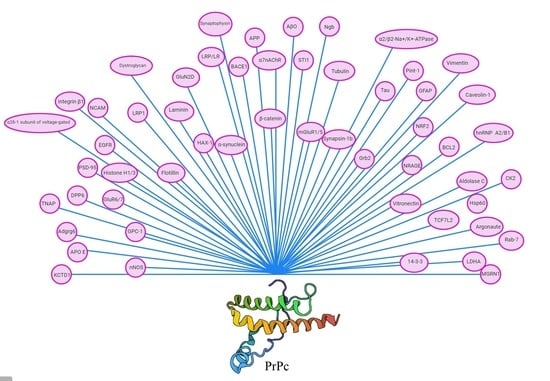
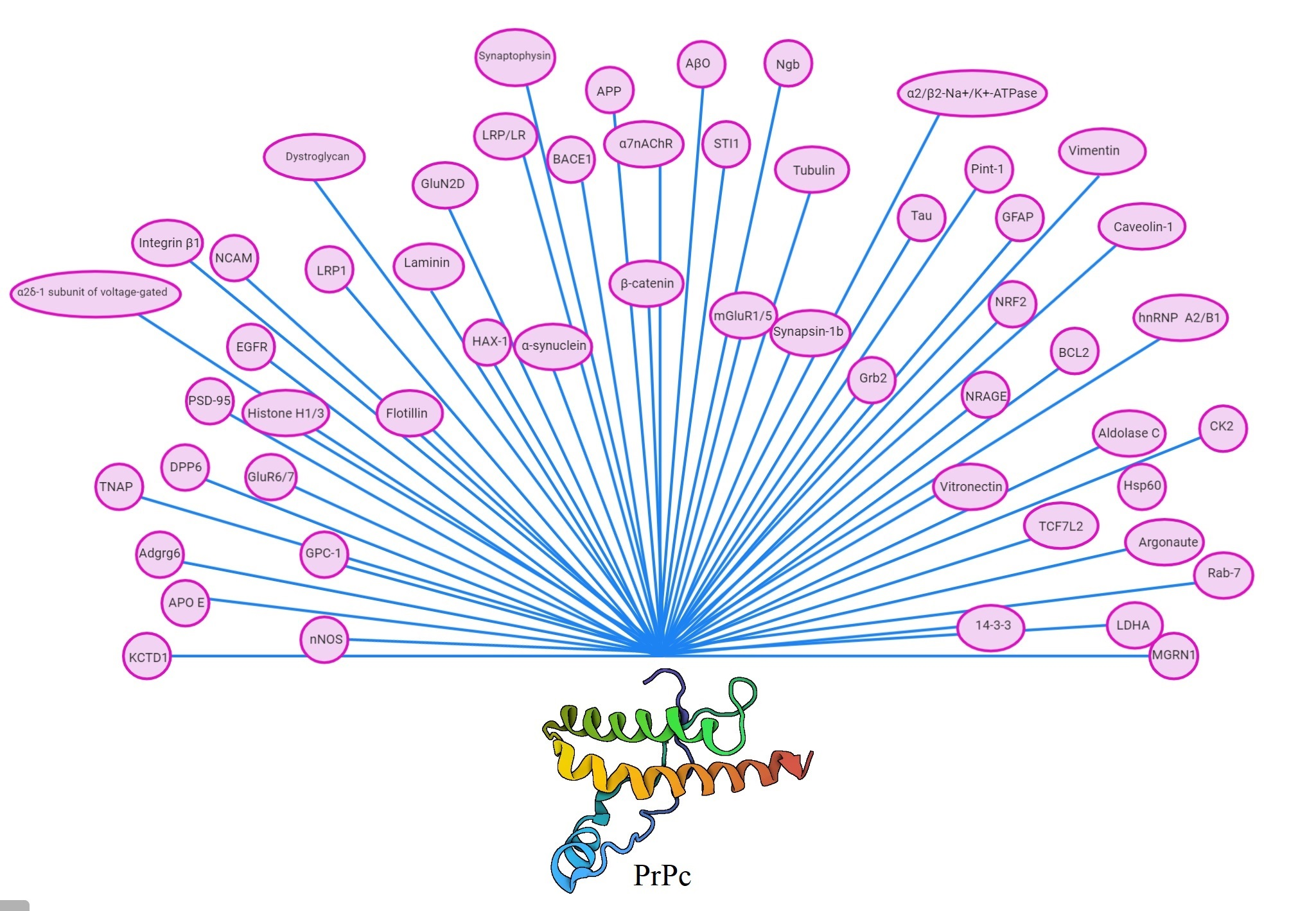
:
1. Introduction
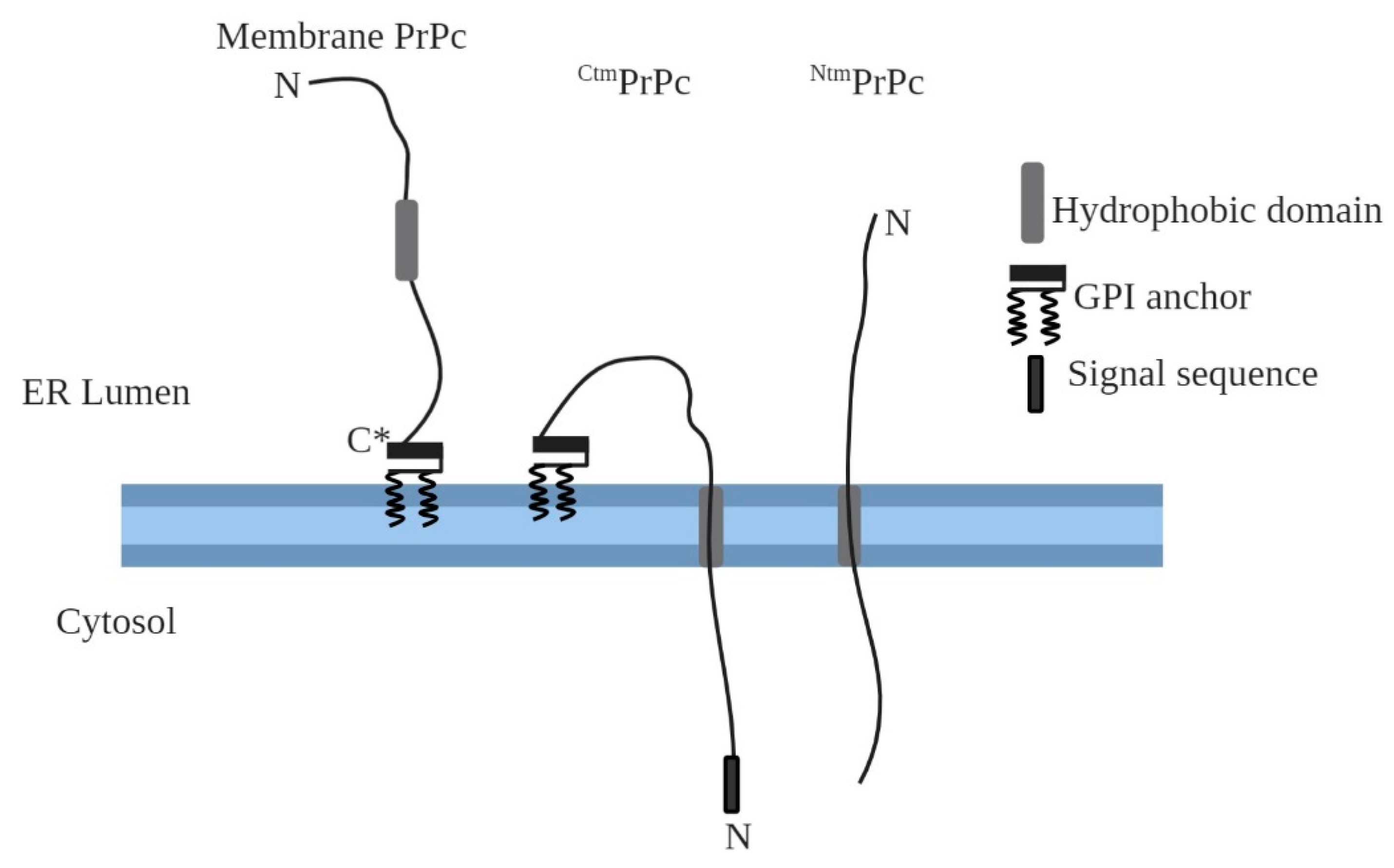
2. Transmembrane Forms of Cellular Prion Protein (PrPc)
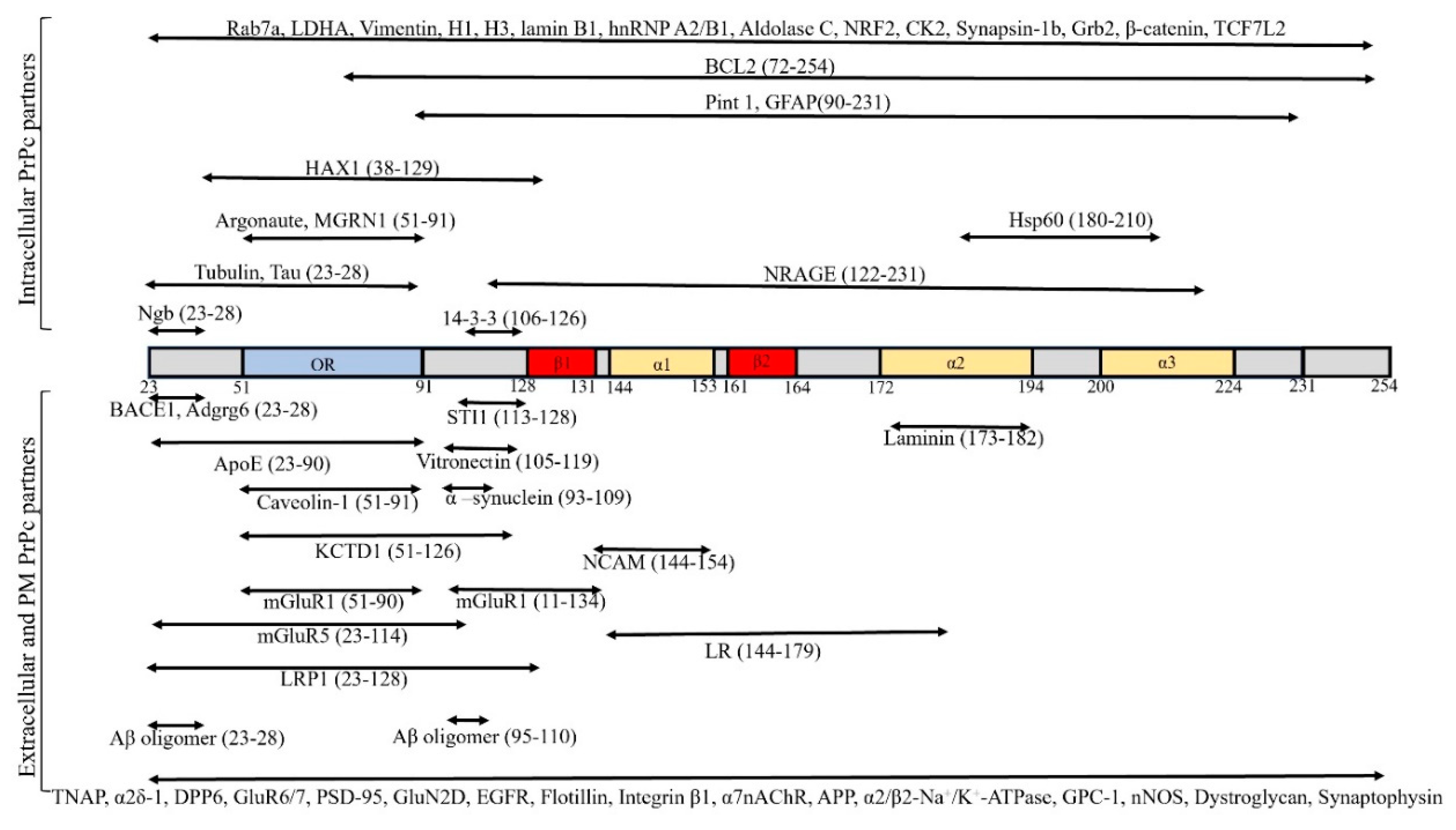
3. Itracellular Partners of PrPc
3.1. Neuroglobin
3.2. Tubulin
3.3. Tau
3.4. Synapsin-1b
3.5. Growth Factor Receptor-Bound Protein 2
3.6. Prion Interactor 1
3.7. Glial Fibrillary Acidic Protein
3.8. Heterogeneous Nuclear Ribonucleoproteins and Aldolase C
3.9. Nuclear Factor Erythroid 2-Related Factor 2
3.10. B Cell Lymphoma 2
3.11. 14-3-3 Proteins
3.12. Neurotrophin Receptor-Interacting MAGE Homolog
3.13. Casein Kinase II
3.14. Mahogunin Ring Finger 1
3.15. Chaperones
3.16. Members of the Rab Family of Small GTPases
3.17. Argonaute
3.18. Lactate Dehydrogenase A
3.19. Vimentin
3.20. Wnt Signaling Pathway Proteins
4. The Interaction Partners of PrPc in the Plasma Membrane
4.1. Stress-Inducible Phosphoprotein 1
4.2. Nicotinic Acetylcholine Receptors
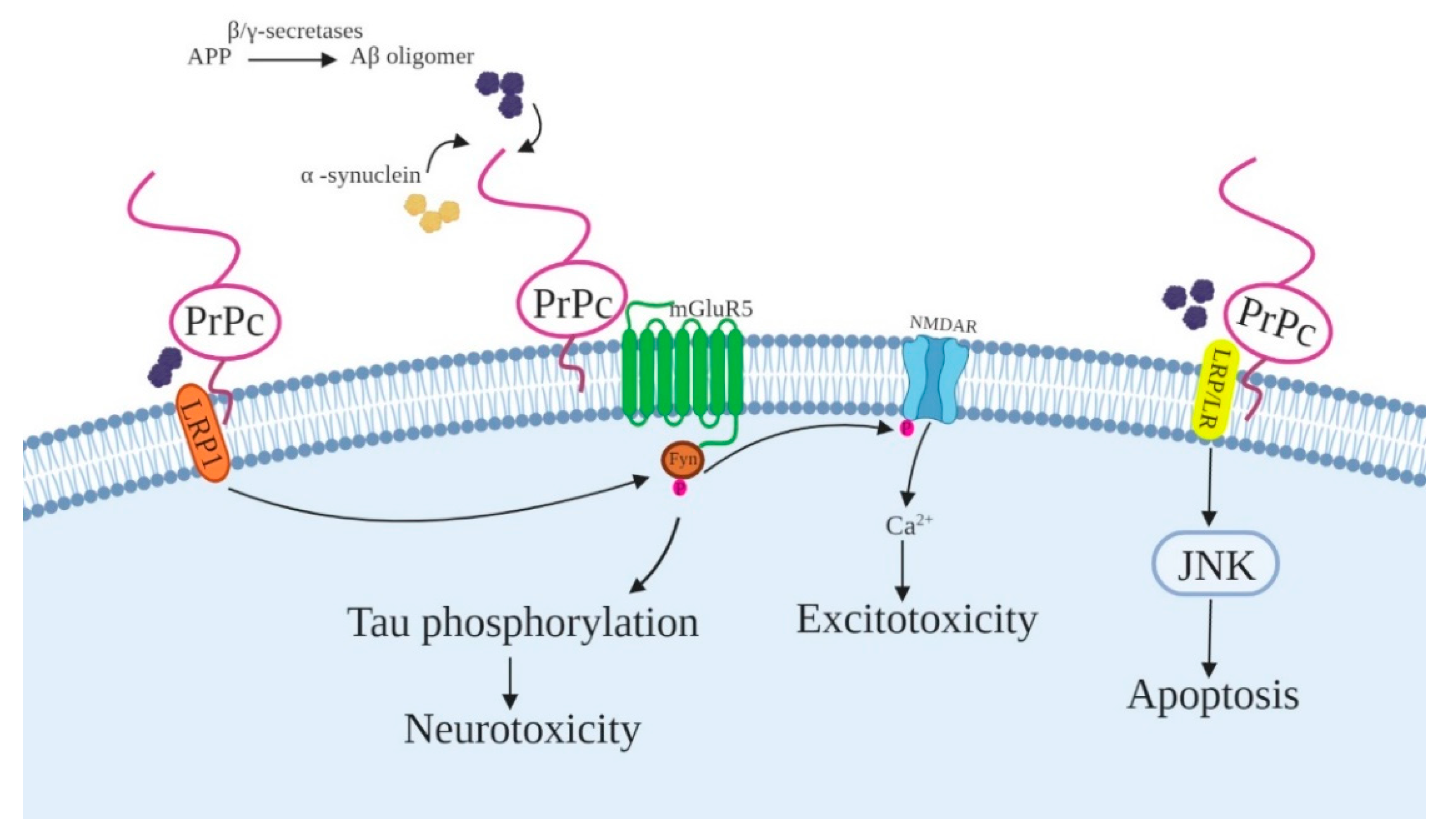
4.3. Alzheimer’s Disease-Related Proteins
4.4. Laminin
4.5. GluN2D Subunit of NMDAR
4.6. Low-Density Lipoprotein Receptor-Related Protein 1
4.7. Neural Cell Adhesion Molecule
4.8. Vitronectin
4.9. Caveolin-1
4.10. Integrin β1
4.11. Flotillin
4.12. Epidermal Growth Factor Receptor
4.13. Kainate Receptor GluR6/7 and Postsynaptic Density Protein 95
4.14. Dipeptidyl Peptidase-Like Protein 6
4.15. α2δ-1 Subunit of Voltage-Gated Calcium Channels
4.16. Tissue Nonspecific Alkaline Phosphatase
4.17. Na+/K+-ATPase
4.18. Glypican-1
4.19. Gpr126 (Adgrg6)
4.20. Neuronal Nitric Oxide Synthase (nNOS) and Dystroglycan
4.21. Potassium Channel Tetramerization Domain Containing 1
5. PrPc in the Immune System
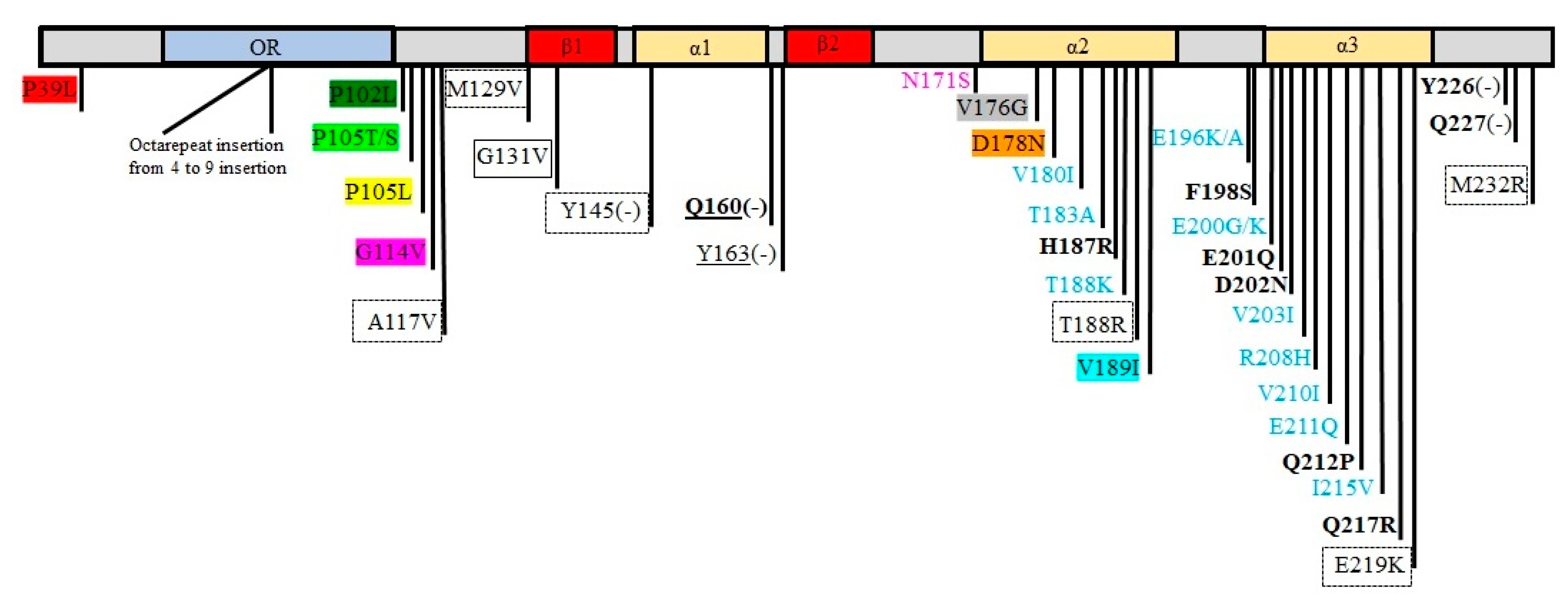
6. Polymorphisms and Mutations on the PRNP Gene
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Acevedo-Morantes, C.Y.; Wille, H. The Structure of Human Prions: From Biology to Structural Models—Considerations and Pitfalls. Viruses 2014, 6, 3875–3892. [Google Scholar] [CrossRef] [PubMed]
- Orlean, P.; Menon, A.K. Thematic review series: Lipid Posttranslational Modifications.GPI anchoring of protein in yeast and mammalian cells, or: How we learned to stop worrying and love glycophospholipids. J. Lipid Res. 2007, 48, 993–1011. [Google Scholar] [CrossRef] [Green Version]
- Béland, M.; Roucou, X. The prion protein unstructured N-terminal region is a broad spectrum molecular sensor with diverse and contrasting potential functions. J. Neurochem. 2011, 120. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, T.; Fisher, S.; Olofsson, S.; Endo, T.; Groth, D.; Tarentino, A.; Borchelt, D.R.; Teplow, D.; Hood, L.; Burlingame, A.; et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 1989, 274, 1–13. [Google Scholar] [CrossRef]
- Wulf, M.-A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Boil. 2017, 15, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, C.J.; Zhang, K.; Munn, A.L.; Wiegmans, A.; Wei, M.Q. Prion protein scrapie and the normal cellular prion protein. Prion 2015, 10, 63–82. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [Green Version]
- McKinley, M.P.; Bolton, D.C.; Prusiner, S.B. A protease-resistant protein is a structural component of the Scrapie prion. Cell 1983, 35, 57–62. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Groth, D.F.; Bolton, D.C.; Kent, S.B.; Hood, L.E. Purification and structural studies of a major scrapie prion protein. Cell 1984, 38, 127–134. [Google Scholar] [CrossRef]
- Kanaani, J.; Prusiner, S.B.; Diacovo, J.; Baekkeskov, S.; Legname, G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J. Neurochem. 2005, 95, 1373–1386. [Google Scholar] [CrossRef]
- Linden, R.; Martins, V.R.; Prado, M.A.M.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the Prion Protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef] [PubMed]
- Bounhar, Y.; Zhang, Y.; Goodyer, C.G.; Leblanc, A.C. Prion Protein Protects Human Neurons against Bax-mediated Apoptosis. J. Boil. Chem. 2001, 276, 39145–39149. [Google Scholar] [CrossRef] [Green Version]
- Roucou, X.; Giannopoulos, P.N.; Zhang, Y.; Jodoin, J.; Goodyer, C.G.; Leblanc, A.C. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005, 12, 783–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassallo, N.; Herms, J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J. Neurochem. 2003, 86, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Qin, K.; Herms, J.; Madlung, A.; Manson, J.C.; Strome, R.; Fraser, P.E.; Kruck, T.; Von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [Green Version]
- Leighton, P.L.A.; Nadolski, N.J.; Morrill, A.; Hamilton, S.; Allison, W.T. An ancient conserved role for prion protein in learning and memory. Boil. Open 2018, 7, bio025734. [Google Scholar] [CrossRef] [Green Version]
- Roguski, A.; Gill, A.C. The Role of the Mammalian Prion Protein in the Control of Sleep. Pathogens 2017, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Stahl, N.; Baldwin, M.; Hecker, R.; Pan, K.-M.; Burlingame, A.; Prusiner, S. Glycosylinositol Phospholipid Anchors of the Scrapie and Cellular Prion Proteins Contain Sialic Acid. Biochemistry 1992, 31, 5043–5053. [Google Scholar] [CrossRef]
- Vana, K.; Zuber, C.; Nikles, D.; Weiss, S. Novel Aspects of Prions, Their Receptor Molecules, and Innovative Approaches for TSE Therapy. Cell. Mol. Neurobiol. 2006, 27, 107–128. [Google Scholar] [CrossRef]
- Hajj, G.N.M.; Lopes, M.H.; Mercadante, A.F.; Veiga, S.S.; Da Silveira, R.B.; Dos Santos, T.G.; Ribeiro, K.C.B.; Juliano, M.A.; Jacchieri, S.G.; Zanata, S.M.; et al. Cellular prion protein interaction with vitronectin supports axonal growth and is compensated by integrins. J. Cell Sci. 2007, 120, 1915–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandi, P.K. Interaction of prion peptide HuPrP106–126 with nucleic acid. Arch. Virol. 1997, 142, 2537–2545. [Google Scholar] [CrossRef] [PubMed]
- Budnik, V.; Ruiz-Canada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heisler, F.F.; Pechmann, Y.; Wieser, I.; Altmeppen, H.C.; Veenendaal, L.; Muhia, M.; Schiweizer, M.; Glatzel, M.; Krasemann, S.; Kneussel, M. Muskelin Coordinates PrPC Lysosome versus Exosome Targeting and Impacts Prion Disease Progression. Neuron 2018, 99, 1155–1169.e9. [Google Scholar] [CrossRef] [Green Version]
- Godsave, S.F.; Wille, H.; Kujala, P.; Latawiec, D.; DeArmond, S.J.; Serban, A.; Prusiner, S.B.; Peters, P.J. Cryo-immunogold electron microscopy for prions: Toward identification of a conversion site. J. Neurosci. 2008, 28, 12489–12499. [Google Scholar] [CrossRef]
- Yost, C.S.; Lopez, C.D.; Prusiner, S.B.; Myers, R.M.; Lingappa, V.R. Non-hydrophobic extracytoplasmic determinant of stop transfer in the prion protein. Nature 1990, 343, 669–672. [Google Scholar] [CrossRef]
- Ivanova, L.; Barmada, S.; Kummer, T.; Harris, D.A. Mutant Prion Proteins Are Partially Retained in the Endoplasmic Reticulum. J. Boil. Chem. 2001, 276, 42409–42421. [Google Scholar] [CrossRef] [Green Version]
- Siskova, Z.; Mahad, D.J.; Pudney, C.; Campbell, G.R.; Cadogan, M.; O’Connor, V.; Asuni, A.; Perry, V.H. Morphological and Functional Abnormalities in Mitochondria Associated with Synaptic Degeneration in Prion Disease. Am. J. Pathol. 2010, 177, 1411–1421. [Google Scholar] [CrossRef]
- Faris, R.; Moore, R.A.; Ward, A.; Race, B.; Dorward, D.W.; Hollister, J.R.; Fischer, E.R.; Priola, S.A. Cellular prion protein is present in mitochondria of healthy mice. Sci. Rep. 2017, 7, 41556. [Google Scholar] [CrossRef]
- Sorice, M.; Mattei, V.; Tasciotti, V.; Manganelli, V.; Garofalo, T.; Misasi, R. Trafficking of PrPc to mitochondrial raft-like microdomains during cell apoptosis. Prion 2012, 6, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Mattei, V.; Matarrese, P.; Garofalo, T.; Tinari, A.; Gambardella, L.; Ciarlo, L.; Manganelli, V.; Tasciotti, V.; Misasi, R.; Malorni, W.; et al. Recruitment of cellular prion protein to mitochondrial raft-like microdomains contributes to apoptosis execution. Mol. Boil. Cell 2011, 22, 4842–4853. [Google Scholar] [CrossRef] [PubMed]
- Lechauve, C.; Rezaei, H.; Célier, C.; Kiger, L.; Corral-Debrinski, M.; Noinville, S.; Chauvierre, C.; Hamdane, D.; Pato, C.; Marden, M.C. Neuroglobin and Prion Cellular Localization: Investigation of a Potential Interaction. J. Mol. Boil. 2009, 388, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Palladino, P.; Scaglione, G.L.; Arcovito, A.; Vitale, R.M.; Amodeo, P.; Vallone, B.; Brunori, M.; Benedetti, E.; Rossi, F. Neuroglobin-prion protein interaction: What’s the function? J. Pept. Sci. 2011, 17, 387–391. [Google Scholar] [CrossRef]
- Dong, C.-F.; Shi, S.; Wang, X.-F.; An, R.; Li, P.; Chen, J.-M.; Wang, X.; Wang, G.-R.; Shan, B.; Zhang, B.-Y.; et al. The N-terminus of PrP is responsible for interacting with tubulin and fCJD related PrP mutants possess stronger inhibitive effect on microtubule assembly in vitro. Arch. Biochem. Biophys. 2008, 470, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Osiecka, K.M.; Nieznanska, H.; Skowronek, K.; Karolczak, J.; Schneider, G.; Nieznański, K. Prion protein region 23-32 interacts with tubulin and inhibits microtubule assembly. Proteins: Struct. Funct. Bioinform. 2009, 77, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, N.S.; Watanabe, K.; Yamada, M.; Sakasegawa, Y.; Kaneko, K. Anterograde and retrograde intracellular trafficking of fluorescent cellular prion protein. Biochem. Biophys. Res. Commun. 2004, 315, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, J.; Yao, H.; Wang, X.; Li, F.; Chen, L.; Gao, C.; Gao, J.; Nie, K.; Zhou, W.; et al. Study on interaction between microtubule associated protein tau and prion protein. Sci. China Ser. C Life Sci. 2006, 49, 473–479. [Google Scholar] [CrossRef]
- Wang, X.-F.; Dong, C.-F.; Zhang, J.; Wan, Y.-Z.; Li, F.; Huang, Y.-X.; Han, L.; Shan, B.; Gao, C.; Han, J.; et al. Human tau protein forms complex with PrP and some GSS- and fCJD-related PrP mutants possess stronger binding activities with tau in vitro. Mol. Cell. Biochem. 2007, 310, 49–55. [Google Scholar] [CrossRef]
- Osiecka, K.M.; Nieznanska, H.; Skowronek, K.; Jozwiak, J.; Nieznański, K. Tau inhibits tubulin oligomerization induced by prion protein. Biochim. Biophys. Acta Bioenerg. 2011, 1813, 1845–1853. [Google Scholar] [CrossRef] [Green Version]
- Ávila, J.; Lucas, J.J.; Pérez, M.; Hernandez, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Spielhaupter, C.; Schatzl, H.M. PrPC directly interacts with proteins involved in signaling pathways. J. Biol. Chem. 2001, 276, 44604–44612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisó, S.; Puig, B.; Varea, R.; Vidal, E.; Acín, C.; Prinz, M.; Montrasio, F.; Badiola, J.; Aguzzi, A.; Pumarola, M.; et al. Abnormal synaptic protein expression and cell death in murine scrapie. Acta Neuropathol. 2002, 103, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Clinton, J.; Forsyth, C.; Royston, M.C.; Roberts, G.W. Synaptic degeneration is the primary neuropathological feature in prion disease. NeuroReport 1993, 4, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Lysek, D.A.; Wüthrich, K. Prion Protein Interaction with the C-Terminal SH3 Domain of Grb2 Studied Using NMR and Optical Spectroscopy. Biochemistry 2004, 43, 10393–10399. [Google Scholar] [CrossRef] [PubMed]
- Oesch, B.; Teplow, D.B.; Stahl, N.; Serban, D.; Hood, L.E.; Prusiner, S.B. Identification of cellular proteins binding to the scrapie prion protein. Biochemistry 1990, 29, 5848–5855. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Scallet, A.C.; Kascsak, R.J.I.; Carp, R. Astrocytosis and amyloid deposition in scrapie-infected hamsters. Brain Res. 1998, 809, 277–287. [Google Scholar] [CrossRef]
- Dong, C.-F.; Wang, X.-F.; Wang, X.; Shi, S.; Wang, G.-R.; Shan, B.; An, R.; Li, X.-L.; Zhang, B.-Y.; Han, J.; et al. Molecular interaction between prion protein and GFAP both in native and recombinant forms in vitro. Med. Microbiol. Immunol. 2007, 197, 361–368. [Google Scholar] [CrossRef]
- Strom, A.; Diecke, S.; Hunsmann, G.; Stuke, A.W. Identification of prion protein binding proteins by combined use of far-Western immunoblotting, two dimensional gel electrophoresis and mass spectrometry. Proteomics 2006, 6, 26–34. [Google Scholar] [CrossRef]
- Yehiely, F.; Perry, B.J.; Carlson, G.A.; Bamborough, P.; Da Costa, M.; Thinakaran, G.; Cohen, F.E.; Prusiner, S.B. Identification of Candidate Proteins Binding to Prion Protein. Neurobiol. Dis. 1997, 3, 339–355. [Google Scholar] [CrossRef] [Green Version]
- Kurschner, C.; Morgan, J.I. The cellular prion protein (PrP) selectively binds to Bcl-2 in the yeast two-hybrid system. Mol. Brain Res. 1995, 30, 165–168. [Google Scholar] [CrossRef]
- Kurschner, C.; Morgan, J.I. Analysis of interaction sites in homo- and heteromeric complexes containing Bcl-2 family members and the cellular prion protein. Mol. Brain Res. 1996, 37, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Miesbauer, M.; Rapaport, D.; Bartke, T.; Baier, M.; Winklhofer, K.F.; Tatzelt, J. Association of Bcl-2 with Misfolded Prion Protein Is Linked to the Toxic Potential of Cytosolic PrP. Mol. Boil. Cell 2006, 17, 3356–3368. [Google Scholar] [CrossRef] [Green Version]
- Richard, M.; Biacabe, A.-G.; Streichenberger, N.; Ironside, J.W.; Mohr, M.; Kopp, N.; Perret-Liaudet, A. Immunohistochemical localization of 14.3.3 ζ protein in amyloid plaques in human spongiform encephalopathies. Acta Neuropathol. 2003, 105, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.-I.; Onoue, H.; Arima, K.; Yamamura, T. The 14-3-3 Protein Forms a Molecular Complex with Heat Shock Protein Hsp60 and Cellular Prion Protein. J. Neuropathol. Exp. Neurol. 2005, 64, 858–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fede, D.G.; Giaccone, G.; Limido, L.; Mangieri, M.; Suardi, S.; Puoti, G.; Morbin, M.; Mazzoleni, G.; Ghetti, B.; Tagliavini, F. The epsilon isoform of 14-3-3 protein is a component of the prion protein amyloid deposits of Gerstmann-Straussler-Scheinker disease. J. Neuropathol. Exp. Neurol. 2007, 66, 124–130. [Google Scholar]
- Mei, G.-Y.; Li, Y.; Wang, G.-R.; Zhang, B.; Tian, C.; Chen, C.; Zhou, R.-M.; Wang, X.; Li, X.-L.; Wang, K.-X.; et al. Molecular interaction between PrP protein and the signal protein 14-3-3 beta. BDXB Chin. J. Virol. 2009, 25, 208–212. [Google Scholar]
- Bragason, B.T.; Palsdottir, A. Interaction of PrP with NRAGE, a protein involved in neuronal apoptosis. Mol. Cell. Neurosci. 2005, 29, 232–244. [Google Scholar] [CrossRef]
- Meggio, F.; Negro, A.; Sarno, S.; Ruzzene, M.; Bertoli, A.; Sorgato, M.C.; Pinna, L.A. Bovine prion protein as a modulator of protein kinase CK2. Biochem. J. 2000, 352, 191–196. [Google Scholar] [CrossRef]
- Negro, A.; Meggio, F.; Bertoli, A.; Battistutta, R.; Sorgato, M.; Pinna, L.A. Susceptibility of the Prion Protein to Enzymic Phosphorylation. Biochem. Biophys. Res. Commun. 2000, 271, 337–341. [Google Scholar] [CrossRef]
- Zamponi, E.; Buratti, F.; Cataldi, G.; Caicedo, H.H.; Song, Y.; Jungbauer, L.M.; Ladu, M.J.; Bisbal, M.; Lorenzo, A.; Ma, J.; et al. Prion protein inhibits fast axonal transport through a mechanism involving casein kinase 2. PLoS ONE 2017, 12, e0188340. [Google Scholar] [CrossRef]
- Silvius, D.; Pitstick, R.; Ahn, M.; Meishery, D.; Oehler, A.; Barsh, G.S.; DeArmond, S.J.; Carlson, G.A.; Gunn, T.M. Levels of the Mahogunin Ring Finger 1 E3 Ubiquitin Ligase Do Not Influence Prion Disease. PLoS ONE 2013, 8, e55575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, A.; Amanullah, A.; Chhangani, D.; Mishra, R.; Prasad, A.; Mishra, A. Mahogunin Ring Finger-1 (MGRN1), a Multifaceted Ubiquitin Ligase: Recent Unraveling of Neurobiological Mechanisms. Mol. Neurobiol. 2015, 53, 4484–4496. [Google Scholar] [CrossRef] [PubMed]
- Edenhofer, F.; Rieger, R.; Famulok, M.; Wendler, W.; Weiss, S.; Winnacker, E.L. Prion protein PrPC interacts with molecular chaperones of the Hsp60 family. J. Virol. 1996, 70, 4724–4728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Leal, J.B.; Seabra, M.C. Evolution of the rab family of small GTP-binding proteins. J. Mol. Boil. 2001, 313, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Zerial, M.; McBride, H.M. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Boil. 2001, 2, 107–117. [Google Scholar] [CrossRef]
- Gibbings, D.; Leblanc, P.; Jay, F.; Pontier, D.; Michel, F.; Schwab, Y.; Alais, S.; Lagrange, T.; Voinnet, O. Human prion protein binds Argonaute and promotes accumulation of microRNA effector complexes. Nat. Struct. Mol. Boil. 2012, 19, 517–524. [Google Scholar] [CrossRef]
- Watts, J.C.; Hairu, H.; Yu, B.; Sepehr, E.; Amy, H.W.; Tujin, S.; Nathalie, D.; Agnes, L.; Rebecca, Y.; Lei, X.; et al. Interactome analyses identify ties of PrPC and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog. 2009, 5, e1000608. [Google Scholar] [CrossRef]
- Ramljak, S.; Schmitz, M.; Zafar, S.; Wrede, A.; Schenkel, S.; Asif, A.R.; Carimalo, J.; Doeppner, T.R.; Schulz-Schaeffer, W.J.; Weise, J.; et al. Cellular prion protein directly interacts with and enhances lactate dehydrogenase expression under hypoxic conditions. Exp. Neurol. 2015, 271, 155–167. [Google Scholar] [CrossRef]
- Zafar, S.; Von Ahsen, N.; Oellerich, M.; Zerr, I.; Schulz-Schaeffer, W.J.; Armstrong, V.W.; Asif, A.R. Proteomics Approach to Identify the Interacting Partners of Cellular Prion Protein and Characterization of Rab7a Interaction in Neuronal Cells. J. Proteome Res. 2011, 10, 3123–3135. [Google Scholar] [CrossRef]
- Gimenez, A.P.L.; Richter, L.M.L.; Atherino, M.C.; Beirão, B.C.B.; Fávaro, C.; Costa, M.D.M.; Zanata, S.M.; Malnic, B.; Mercadante, A.F. Identification of novel putative-binding proteins for cellular prion protein and a specific interaction with the STIP1 homology and U-Box-containing protein 1. Prion 2015, 9, 355–366. [Google Scholar] [CrossRef]
- Besnier, L.S.; Cardot, P.; Da Rocha, B.; Simon, A.; Loew, D.; Klein, C.; Riveau, B.; Lacasa, M.; Clair, C.; Rousset, M.; et al. The cellular prion protein PrPC is a partner of the Wnt pathway in intestinal epithelial cells. Mol. Boil. Cell 2015, 26, 3313–3328. [Google Scholar] [CrossRef]
- Strom, A.; Wang, G.-S.; Picketts, D.J.; Reimer, R.; Stuke, A.W.; Scott, F.W. Cellular prion protein localizes to the nucleus of endocrine and neuronal cells and interacts with structural chromatin components. Eur. J. Cell Boil. 2011, 90, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.-Y.; Li, X.-L.; Shi, Q.; Wang, Z.-Y.; Guo, Y.; Pan, M.-M.; Tian, C.; Zhu, S.-Y.; Chen, C.; Gong, H.-S.; et al. A Novel PrP Partner HS-1 Associated Protein X-1 (HAX-1) Protected the Cultured Cells Against the Challenge of H2O2. J. Mol. Neurosci. 2011, 45, 216–228. [Google Scholar] [CrossRef]
- Burmester, T.; Reinhardt, S.; Weich, B.; Hankeln, T. A vertebrate globin expressed in the brain. Nature 2000, 407, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-Y.; Zhou, S.; Lou, Z.-Y.; Zhu, C.; Zheng, X.-P.; Hu, X. Translocation and neuroprotective properties of transactivator-of-transcription protein-transduction domain–neuroglobin fusion protein in primary cultured cortical neurons. Biotechnol. Appl. Biochem. 2008, 49, 25. [Google Scholar] [CrossRef] [PubMed]
- Amos, L.A.; Schlieper, D. Microtubules and maps. Adv. Protein. Chem. 2005, 71, 257–298. [Google Scholar]
- Nieznański, K.; Nieznanska, H.; Skowronek, K.; Osiecka, K.M.; Stępkowski, D. Direct interaction between prion protein and tubulin. Biochem. Biophys. Res. Commun. 2005, 334, 403–411. [Google Scholar] [CrossRef]
- Hachiya, N.S.; Watanabe, K.; Sakasegawa, Y.; Kaneko, K. Microtubules-associated intracellular localization of the NH2-terminal cellular prion protein fragment. Biochem. Biophys. Res. Commun. 2004, 313, 818–823. [Google Scholar] [CrossRef]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [Green Version]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; Elidrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates With Changes in Its Function Beyond Microtubule Stability. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Xiaopeng, D. Dysfunction of microtubule-associated proteins of MAP2/tau family in Prion disease. Prion 2012, 6, 334–338. [Google Scholar]
- Navone, F.; Greengard, P.; De Camilli, P. Synapsin I in nerve terminals: Selective association with small synaptic vesicles. Science 1984, 226, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Takenawa, T.; Miki, H.; Matuoka, K. Signaling through Grb2/Ash-control of the Ras pathway and cytoskeleton. Curr. Top. Microbiol. Immunol. 1998, 228, 325–342. [Google Scholar] [PubMed]
- Soriano, J.V.; Liu, N.; Gao, Y.; Yao, Z.-J.; Ishibashi, T.; Underhill, C.; Burke, T.R.; Bottaro, D.P. Inhibition of angiogenesis by growth factor receptor bound protein 2-Src homology 2 domain bound antagonists. Mol. Cancer Ther. 2004, 3, 1289–1299. [Google Scholar]
- Eddleston, M.; Mucke, L. Molecular profile of reactive astrocytes—Implications for their role in neurologic disease. Neuroscience 1993, 54, 15–36. [Google Scholar] [CrossRef]
- Gomi, H.; Yokoyama, T.; Fujimoto, K.; Ikeda, T.; Katoh, A.; Itoh, T.; Itohara, S. Mice devoid of the glial fibrillary acidic protein develop normally and are susceptible to scrapie prions. Neuron 1995, 14, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Dormont, D.; Delpech, B.; Delpech, A.; Courcel, M.N.; Viret, J.; Markovits, P.; Court, L. Hyperproduction of glial fibrillary acidic protein (GFA) during development of experimental scrapie in mice. C. R. Acad. Sci. Ser. III 1981, 293, 53–56. [Google Scholar]
- MacKenzie, A. Immunohistochemical demonstration of glial fibrillary acidic protein in scrapie. J. Comp. Pathol. 1983, 93, 251–259. [Google Scholar] [CrossRef]
- Gomi, H.; Yokoyama, T.; Itohara, S. Role of GFAP in morphological retention and distribution of reactive astrocytes induced by scrapie encephalopathy in mice. Brain Res. 2010, 1312, 156–167. [Google Scholar] [CrossRef]
- Kozu, T.; Henrich, B.; Schäfer, K.P. Structure and expression of the gene (HNRPA2B1) encoding the human hnRNP protein A2/B1. Genomics 1995, 25, 365–371. [Google Scholar] [CrossRef]
- Gao, Y.; Tatavarty, V.; Korza, G.; Levin, M.K.; Carson, J.H. Multiplexed Dendritic Targeting of α Calcium Calmodulin-dependent Protein Kinase II, Neurogranin, and Activity-regulated Cytoskeleton-associated Protein RNAs by the A2 Pathway. Mol. Boil. Cell 2008, 19, 2311–2327. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.; Kynoch, P.A.; Willson, V.J. Cellular localization of aldolase C subunits in human brain. Brain Res. 1982, 232, 489–493. [Google Scholar] [CrossRef]
- Cañete-Soler, R.; Reddy, K.S.; Tolan, D.R.; Zhai, J. Aldolases A and C Are Ribonucleolytic Components of a Neuronal Complex That Regulates the Stability of the Light-Neurofilament mRNA. J. Neurosci. 2005, 25, 4353–4364. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Redmann, M.; Rajasekaran, N.S.; Darley-Usmar, V.; Zhang, J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 2015, 469, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Cichon, A.-C.; Brown, D.R. Nrf-2 regulation of prion protein expression is independent of oxidative stress. Mol. Cell. Neurosci. 2014, 63, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.W.-L.; Puthalakath, H. Bcl-2 family proteins: The sentinels of the mitochondrial apoptosis pathway. IUBMB Life 2008, 60, 390–397. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple Functions of BCL-2 Family Proteins. Cold Spring Harb. Perspect. Boil. 2013, 5, a008722. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Subramanian, R.R.; Masters, S.C. 14-3-3 proteins: Structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 617–647. [Google Scholar] [CrossRef]
- Berg, D.; Holzmann, C.; Riess, O. 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 2003, 4, 752–762. [Google Scholar] [CrossRef]
- Muayqil, T.; Gronseth, G.; Camicioli, R. Evidence-based guideline: Diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: Report of the guideline development subcommittee of the American Academy of Neurology. Neurology 2012, 79, 1499–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, V.K.; Sadiq, S.A. Disease Biomarkers in Multiple Sclerosis. Mol. Diagn. Ther. 2009, 13, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Song, Q.-Q.; Sun, P.; Zhang, J.; Wang, X.; Song, J.; Li, G.-Q.; Liu, Y.-H.; Mei, G.-Y.; Shi, Q.; et al. Interaction between 14-3-3β and PrPC influences the dimerization of 14-3-3 and fibrillization of PrPC106–126. Int. J. Biochem. Cell Boil. 2014, 47, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.H.; Xanthoudakis, S.; Barker, P.A. NRAGE, a p75 Neurotrophin Receptor-interacting Protein, Induces Caspase Activation and Cell Death through a JNK-dependent Mitochondrial Pathway. J. Boil. Chem. 2002, 277, 48043–48050. [Google Scholar] [CrossRef] [Green Version]
- Götz, C.; Montenarh, M. Protein kinase CK2 in development and differentiation. Biomed. Rep. 2016, 6, 127–133. [Google Scholar] [CrossRef]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef]
- Chen, J.; Gao, C.; Shi, Q.; Wang, G.; Lei, Y.; Shan, B.; Zhang, B.; Dong, C.; Shi, S.; Wang, X.; et al. Casein kinase II interacts with prion protein in vitro and forms complex with native prion protein in vivo. Acta Biochim. Biophys. Sin. 2008, 40, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-M.; Gao, C.; Shi, Q.; Shan, B.; Lei, Y.-J.; Dong, C.-F.; An, R.; Wang, G.-R.; Zhang, B.-Y.; Han, J.; et al. Different expression patterns of CK2 subunits in the brains of experimental animals and patients with transmissible spongiform encephalopathies. Arch. Virol. 2008, 153, 1013–1020. [Google Scholar] [CrossRef]
- Canton, D.A.; Litchfield, D.W. The shape of things to come: An emerging role for protein kinase CK2 in the regulation of cell morphology and the cytoskeleton. Cell. Signal. 2006, 18, 267–275. [Google Scholar] [CrossRef]
- Kim, B.Y.; Olzmann, J.A.; Barsh, G.S.; Chin, L.-S.; Li, L. Spongiform Neurodegeneration-associated E3 Ligase Mahogunin Ubiquitylates TSG101 and Regulates Endosomal Trafficking. Mol. Boil. Cell 2007, 18, 1129–1142. [Google Scholar] [CrossRef] [Green Version]
- Amit, I.; Yakir, L.; Katz, M.; Zwang, Y.; Marmor, M.D.; Citri, A.; Shtiegman, K.; Alroy, I.; Tuvia, S.; Reiss, Y.; et al. Tal, a Tsg101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 2004, 18, 1737–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Liao, J.; Ruland, J.; Mak, T.W.; Cohen, S.N. A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control. Proc. Natl. Acad. Sci. USA 2001, 98, 1619–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.H.; Cohen, S.N. Human MDM2 isoforms translated differentially on constitutive versus p53-regulated transcripts have distinct functions in the p53/MDM2 and TSG101/MDM2 feedback control loops. Mol. Cell Biol. 2007, 27, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, J. Proteins as molecular chaperones. Nature 1987, 328, 378–379. [Google Scholar] [CrossRef]
- Martins, V.R.; Graner, E.; Garcia-Abreu, J.; De Souza, S.J.; Mercadante, A.F.; Veiga, S.S.; Zanata, S.M.; Moura-Neto, V.; Brentani, R.R. Complementary hydropathy identifies a cellular prion protein receptor. Nat. Med. 1997, 3, 1376–1382. [Google Scholar] [CrossRef]
- Sun, G.; Guo, M.; Shen, A.; Mei, F.; Peng, X.; Gong, R.; Guo, D.-Y.; Wu, J.; Tien, P.; Xiao, G. Bovine PrPC directly interacts with αB-crystalline. FEBS Lett. 2005, 579, 5419–5424. [Google Scholar] [CrossRef] [Green Version]
- Dulle, J.E.; Fort, P.E. Crystallins and neuroinflammation: The glial side of the story. Biochim. Biophys. Acta Bioenerg. 2015, 1860, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Leach, M.R.; Williams, D.B. Calnexin and Calreticulin, Molecular Chaperones of the Endoplasmic Reticulum. In Wilms Tumor: Clinical and Molecular Characterization; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2003; pp. 49–62. [Google Scholar]
- Gething, M.-J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Boil. 1999, 10, 465–472. [Google Scholar] [CrossRef]
- Capellari, S.; Zaidi, S.I.A.; Urig, C.B.; Perry, G.; Smith, M.A.; Petersen, R.B. Prion Protein Glycosylation Is Sensitive to Redox Change. J. Boil. Chem. 1999, 274, 34846–34850. [Google Scholar] [CrossRef] [Green Version]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta Bioenerg. 2012, 1823, 774–787. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuka, K.; Hata, M.; Ohtsuka, M.H.K. Molecular chaperone function of mammalian Hsp70 and Hsp40—A review. Int. J. Hyperth. 2000, 16, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H.R. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Boil. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.J.; Fort, P.E. Heat Shock Proteins Regulatory Role in Neurodevelopment. Front. Mol. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Boil. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Feng, Y.; Press, B.; Chen, W.; Zimmerman, J.; Wandinger-Ness, A. Expression and properties of Rab7 in endosome function. Enzym. Eng. Evol. Gen. Methods 2001, 329, 175–187. [Google Scholar] [CrossRef]
- Feng, Y.; Press, B.; Wandinger-Ness, A. Rab 7: An important regulator of late endocytic membrane traffic. J. Cell Boil. 1995, 131, 1435–1452. [Google Scholar] [CrossRef] [Green Version]
- Edinger, A.L.; Cinalli, R.M.; Thompson, C.B. Rab7 Prevents Growth Factor-Independent Survival by Inhibiting Cell-Autonomous Nutrient Transporter Expression. Dev. Cell 2003, 5, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Rosales, K.R.; Peralta, E.R.; Guenther, G.G.; Wong, S.Y.; Edinger, A.L. Rab7 Activation by Growth Factor Withdrawal Contributes to the Induction of Apoptosis. Mol. Boil. Cell 2009, 20, 2831–2840. [Google Scholar] [CrossRef] [Green Version]
- Harrison, R.E.; Bucci, C.; Vieira, O.V.; Schroer, T.A.; Grinstein, S. Phagosomes Fuse with Late Endosomes and/or Lysosomes by Extension of Membrane Protrusions along Microtubules: Role of Rab7 and RILP. Mol. Cell. Boil. 2003, 23, 6494–6506. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.Y.; Karri, S.; Law, S.; Schatzl, H.M.; Gilch, S. Prion infection impairs lysosomal degradation capacity by interfering with rab7 membrane attachment in neuronal cells. Sci. Rep. 2016, 6, 21658. [Google Scholar] [CrossRef] [Green Version]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.; Meister, G. Argonaute Proteins: Mediators of RNA Silencing. Mol. Cell 2007, 26, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Boil. 2008, 9, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macrae, I.J.; Ma, E.; Zhou, M.; Robinson, C.V.; Doudna, J.A. In vitro reconstitution of the human RISC-loading complex. Proc. Natl. Acad. Sci. USA 2008, 105, 512–517. [Google Scholar] [CrossRef] [Green Version]
- El-Shami, M.; Pontier, D.; Lahmy, S.; Braun, L.; Picart, C.; Vega, D.; Hakimi, M.-A.; Jacobsen, S.E.; Cooke, R.; Lagrange, T. Reiterated WG/GW motifs form functionally and evolutionarily conserved ARGONAUTE-binding platforms in RNAi-related components. Genes Dev. 2007, 21, 2539–2544. [Google Scholar] [CrossRef] [Green Version]
- Gould, G.W.; Lippincott-Schwartz, J. New roles for endosomes: From vesicular carriers to multi-purpose platforms. Nat. Rev. Mol. Cell Boil. 2009, 10, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Sivaramakrishnan, M.; Sivapathasundharam, B.; Jananni, M. Evaluation of lactate dehydrogenase enzyme activity in saliva and serum of oral submucous fibrosis patients. J. Oral Pathol. Med. 2014, 44, 449–452. [Google Scholar] [CrossRef]
- Schurr, A.; Dong, W.-Q.; Reid, K.H.; West, C.A.; Rigor, B.M. Lactic acidosis and recovery of neuronal function following cerebral hypoxia in vitro. Brain Res. 1988, 438, 311–314. [Google Scholar] [CrossRef]
- Schurr, A.; Payne, R.S.; Miller, J.J.; Rigor, B.M. Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: An in vitro study. Brain Res. 1997, 744, 105–111. [Google Scholar] [CrossRef]
- Schurr, A.; Payne, R.S.; Miller, J.J.; Tseng, M.T.; Rigor, B.M. Blockade of lactate transport exacerbates delayed neuronal damage in a rat model of cerebral ischemia. Brain Res. 2001, 895, 268–272. [Google Scholar] [CrossRef]
- Valvona, C.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2015, 26, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Berthet, C.; Lei, H.; Thevenet, J.; Gruetter, R.; Magistretti, P.J.; Hirt, L. Neuroprotective Role of Lactate after Cerebral Ischemia. Br. J. Pharmacol. 2009, 29, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Puschmann, T.B.; Marasek, P.; Inagaki, M.; Pekna, M.; Wilhelmsson, U.; Pekny, M. Increased Neuronal Differentiation of Neural Progenitor Cells Derived from Phosphovimentin-Deficient Mice. Mol. Neurobiol. 2017, 55, 5478–5489. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, H.; Chen, L.; Wang, J.; Lv, Y.; Yang, X.; Zhang, B.Y.; Tian, C.; Shi, Q.; Dong, X.P.; et al. Remarkable impairment of Wnt/beta-catenin signaling in the brains of the mice infected with scrapie agents. J. Neurochem. 2016, 136, 731–740. [Google Scholar] [CrossRef]
- Khosravani, H.; Zhang, Y.; Tsutsui, S.; Hameed, S.; Altier, C.; Hamid, J.; Lina, C.; Michelle, V.; Zenobia, A.; Frank, R.; et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 2008, 181, 551–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haitao, Y.; Shigeki, T.; Shahid, H.; Thomas, J.K.; Lina, C.; Peng, X.; Jordan, D.T.; Stuart, A.L.; Peter, K.S.; Gerald, W.Z. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1737–1742. [Google Scholar]
- Chiarini, L.B.; Freitas, A.R.O.; Zanata, S.M.; Brentani, R.R.; Martins, V.R.; Linden, R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002, 21, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.M.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.O.; Cabral, A.L.B.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef]
- Lopes, M.H.; Hajj, G.N.M.; Muras, A.G.; Mancini, G.L.; Castro, R.M.P.S.; Ribeiro, K.C.B.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of Cellular Prion and Stress-Inducible Protein 1 Promotes Neuritogenesis and Neuroprotection by Distinct Signaling Pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [Green Version]
- Fabiana, A.C.; Marilene, H.L.; Glaucia, N.M.H.; Cleiton, F.M.; Camila, P.A.; Ana, C.M.; Mônica, P.B.V.; Tatiana, A.A.; Andre, R.M.; Suzette, A.P.; et al. Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci. 2008, 28, 6691–6702. [Google Scholar]
- Beraldo, F.H.; Arantes, C.P.; Dos Santos, T.G.; Queiroz, N.G.T.; Young, K.; Rylett, R.; Markus, R.; Prado, M.A.M.; Martins, V.R. Role of α7 Nicotinic Acetylcholine Receptor in Calcium Signaling Induced by Prion Protein Interaction with Stress-inducible Protein 1. J. Boil. Chem. 2010, 285, 36542–36550. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.-K.; Park, S.-Y. Neuroprotective effect of cellular prion protein (PrPC) is related with activation of alpha7 nicotinic acetylcholine receptor (α7nAchR)-mediated autophagy flux. Oncotarget 2015, 6, 24660–24674. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.M.; Acharya, M.; Leighton, P.L.A.; Wang, H.; Daude, N.; Wohlgemuth, S.; Shi, B.; Allison, W.T. Amyloid Beta Precursor Protein and Prion Protein Have a Conserved Interaction Affecting Cell Adhesion and CNS Development. PLoS ONE 2012, 7, e51305. [Google Scholar] [CrossRef] [PubMed]
- Edward, T.P.; Nicole, T.W.; Ishrut, H.; Elizabeth, A.E.; Christopher, B.E.; Jean, C.M.; Herbert, N.B.; Anthony, J.T.; Nigel, M.H. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar]
- Chen, S.; Yadav, S.P.; Surewicz, W.K. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: Role OF N-terminal residues. J. Biol. Chem. 2010, 285, 26377–26383. [Google Scholar] [CrossRef] [Green Version]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Laurén, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohler, F.; Sepulveda-Falla, D.; Krasemann, S.; Altmeppen, H.; Schlüter, H.; Hildebrand, D.; Zerr, I.; Matschke, J.; Glatzel, M. High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer’s disease. Brain 2014, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Flavio, H.B.; Camila, P.A.; Tiago, G.S.; Cleiton, F.M.; Martin, R.; Gláucia, N.H.; Kil, S.L.; Ana, C.M.; Fabiana, A.C.; Gabriel, L.M.; et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 2011, 25, 265–279. [Google Scholar]
- Kunishima, N.; Shimada, Y.; Tsuji, Y.; Sato, T.; Yamamoto, M.; Kumasaka, T.; Nakanishi, S.; Jingami, H.; Morikawa, K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 2000, 407, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Satoh, K.; Homma, T.; Nakagaki, T.; Yamaguchi, N.; Atarashi, R.; Sudo, Y.; Uezono, Y.; Ishibashi, D.; Nishida, N. Prion protein interacts with the metabotropic glutamate receptor 1 and regulates the organization of Ca2+ signaling. Biochem. Biophys. Res. Commun. 2020, 525, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Neng-Wei, H.; Andrew, J.N.; Dainan, Z.; Alexandra, J.M.; Tiernan, O.M.; Silvia, A.P.; Cassandra, T.; John, C.; Dominic, M.W.; Michael, J.R.; et al. mGlu5 receptors and cellular prion protein mediate amyloid-beta-facilitated synaptic long-term depression in vivo. Nat. Commun. 2014, 5, 3374. [Google Scholar]
- Ji, W.U.; Adam, C.K.; Mikhail, K.; Jacqueline, K.H.; Massimiliano, S.; Hideyuki, T.; Meghan, E.K.; Alexander, V.; Thomas, W.; Anthony, J.K.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar]
- Rieger, R.; Edenhofer, F.; Lasmézas, C.I.; Weiss, S.; Lasm, C.I. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat. Med. 1997, 3, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Gauczynski, S.; Peyrin, J.-M.; Haik, S.; Leucht, C.; Hundt, C.; Rieger, R.; Krasemann, S.; Deslys, J.; Dormont, M.; Lasmézas, C.I.; et al. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001, 20, 5863–5875. [Google Scholar] [CrossRef] [Green Version]
- Hundt, C.; Peyrin, J.-M.; Haik, S.; Gauczynski, S.; Leucht, C.; Rieger, R.; Riley, M.L.; Deslys, J.; Dormont, M.; Lasmézas, C.I.; et al. Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. EMBO J. 2001, 20, 5876–5886. [Google Scholar] [CrossRef]
- Urrea, L.; Ferrer, I.; Gavin, R.; Del-Rio, J.A. The cellular prion protein (PrPC) as neuronal receptor for alpha-synuclein. Prion 2017, 11, 226–233. [Google Scholar] [CrossRef] [Green Version]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Forlenza, O.V.; Cabral, A.L.; Veiga, S.S.; Juliano, M.A.; Roesler, R.; Walz, R.; Minetti, A.; et al. Cellular prion protein binds laminin and mediates neuritogenesis. Mol. Brain Res. 2000, 76, 85–92. [Google Scholar] [CrossRef]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Martins, V.R.; Jay, D.G.; Brentani, R.R. Laminin-induced PC-12 cell differentiation is inhibited following laser inactivation of cellular prion protein. FEBS Lett. 2000, 482, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Tiago, G.S.; Flavio, H.B.; Glaucia, N.M.H.; Marilene, H.L.; Martin, R.; Fernanda, C.S.L.; Valeriy, G.O.; Vania, F.P.; Marco, A.M.P.; Vilma, R.M. Laminin-gamma1 chain and stress inducible protein 1 synergistically mediate PrPC-dependent axonal growth via Ca2+ mobilization in dorsal root ganglia neurons. J. Neurochem. 2013, 124, 210–223. [Google Scholar]
- Taylor, D.R.; Hooper, N.M. The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem. J. 2007, 402, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Parkyn, C.J.; Vermeulen, E.G.M.; Mootoosamy, R.C.; Sunyach, C.; Jacobsen, C.; Oxvig, C.; Moestrup, S.K.; Liu, Q.; Bu, G.; Jen, A.; et al. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J. Cell Sci. 2008, 121, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Boil. 2001, 314, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Santuccione, A.; Sytnyk, V.; Leshchyns’Ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Boil. 2005, 169, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmey, J.; Doyle, D.; Brown, V.; Rogers, M. The Cellular Isoform of the Prion Protein, PrPC, Is Associated with Caveolae in Mouse Neuroblastoma (N2a) Cells. Biochem. Biophys. Res. Commun. 1995, 210, 753–759. [Google Scholar] [CrossRef]
- Vey, M.; Pilkuhn, S.; Wille, H.; Nixon, R.; DeArmond, S.J.; Smart, E.J.; Anderson, R.G.W.; Taraboulos, A.; Prusiner, S.B. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl. Acad. Sci. USA 1996, 93, 14945–14949. [Google Scholar] [CrossRef] [Green Version]
- Mattia, T.; Enzo, S.; Cristiana, G.; Spartaco, S.; Massimo, R.; Patrizia, L.; Vittorio, T. Cellular prion protein and caveolin-1 interaction in a neuronal cell line precedes Fyn/Erk 1/2 signal transduction. J. Biomed. Biotechnol. 2006, 5, 69469. [Google Scholar]
- Pantera, B.; Bini, C.; Cirri, P.; Paoli, P.; Camici, G.; Manao, G.; Caselli, A. PrPcactivation induces neurite outgrowth and differentiation in PC12 cells: Role for caveolin-1 in the signal transduction pathway. J. Neurochem. 2009, 110, 194–207. [Google Scholar] [CrossRef]
- Bodrikov, V.; Solis, G.P.; Stuermer, C.A. Prion Protein Promotes Growth Cone Development through Reggie/Flotillin-Dependent N-Cadherin Trafficking. J. Neurosci. 2011, 31, 18013–18025. [Google Scholar] [CrossRef]
- Solis, G.P.; Schrock, Y.; Hülsbusch, N.; Wiechers, M.; Plattner, H.; Stuermer, C.A. Reggies/flotillins regulate E-cadherin–mediated cell contact formation by affecting EGFR trafficking. Mol. Boil. Cell 2012, 23, 1812–1825. [Google Scholar] [CrossRef]
- Llorens, F.; Carulla, P.; Villa, A.; Torres, J.M.; Fortes, P.; Ferrer, I.; Del Río, J.A. PrPC regulates epidermal growth factor receptor function and cell shape dynamics in Neuro2a cells. J. Neurochem. 2013, 127. [Google Scholar] [CrossRef]
- Martellucci, S.; Manganelli, V.; Santacroce, C.; Santilli, F.; Piccoli, L.; Sorice, M.; Mattei, V. Role of Prion protein-EGFR multimolecular complex during neuronal differentiation of human dental pulp-derived stem cells. Prion 2018, 12, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Rangel, A.; Burgaya, F.; Gavín, R.; Soriano, E.; Aguzzi, A.; Del Río, J.A. Enhanced susceptibility ofPrnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: Role of AMPA/kainate receptors. J. Neurosci. Res. 2007, 85, 2741–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patricia, C.; Ana, B.; Alejandra, R.; Rosalina, G.; Isidro, F.; Carme, C.; José, A.R.; Franc, L. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell 2011, 22, 3041–3054. [Google Scholar]
- Choi, H.J.; Kang, K.S.; Fukui, M.; Zhu, B.T. Critical role of the JNK-p53-GADD45α apoptotic cascade in mediating oxidative cytotoxicity in hippocampal neurons. Br. J. Pharmacol. 2010, 162, 175–192. [Google Scholar] [CrossRef] [Green Version]
- Mercer, R.C.C.; Ma, L.; Watts, J.; Strome, R.; Wohlgemuth, S.; Yang, J.; Cashman, N.R.; Coulthart, M.B.; Schmitt-Ulms, G.; Jhamandas, J.; et al. The Prion Protein Modulates A-type K+ Currents Mediated by Kv4.2 Complexes through Dipeptidyl Aminopeptidase-like Protein 6. J. Boil. Chem. 2013, 288, 37241–37255. [Google Scholar] [CrossRef] [Green Version]
- Assunta, S.; Simona, C.; Claudia, V.; Elena, R.; Raffaella, M.; Steven, B.C.; Ilaria, B.; Susanna, M.; Mara, C.; Edoardo, M. Mutant PrPC suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC alpha(2)delta-1 Subunit. Neuron 2012, 74, 300–313. [Google Scholar]
- Bouybayoune, I.; Mantovani, S.; Del Gallo, F.; Bertani, I.; Restelli, E.; Comerio, L.; Tapella, L.; Baracchi, F.; Fernández-Borges, N.; Mangieri, M.; et al. Transgenic Fatal Familial Insomnia Mice Indicate Prion Infectivity-Independent Mechanisms of Pathogenesis and Phenotypic Expression of Disease. PLoS Pathog. 2015, 11, e1004796. [Google Scholar] [CrossRef]
- Ermonval, M.; Baudry, A.; Baychelier, F.; Pradines, E.; Pietri, M.; Oda, K.; Schneider, B.; Mouillet-Richard, S.; Launay, J.-M.; Kellermann, O. The Cellular Prion Protein Interacts with the Tissue Non-Specific Alkaline Phosphatase in Membrane Microdomains of Bioaminergic Neuronal Cells. PLoS ONE 2009, 4, e6497. [Google Scholar] [CrossRef] [Green Version]
- Kleene, R.; Loers, G.; Langer, J.; Frobert, Y.; Buck, F.; Schachner, M. Prion Protein Regulates Glutamate-Dependent Lactate Transport of Astrocytes. J. Neurosci. 2007, 27, 12331–12340. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.R.; Whitehouse, I.J.; Hooper, N.M. Glypican-1 Mediates Both Prion Protein Lipid Raft Association and Disease Isoform Formation. PLoS Pathog. 2009, 5, e1000666. [Google Scholar] [CrossRef] [Green Version]
- Radovanovic, I.; Braun, N.; Giger, O.T.; Mertz, K.; Miele, G.; Prinz, M.; Navarro, B.; Aguzzi, A. Truncated Prion Protein and Doppel Are Myelinotoxic in the Absence of Oligodendrocytic PrPC. J. Neurosci. 2005, 25, 4879–4888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küffer, A.; Lakkaraju, A.K.K.; Mogha, A.; Petersen, S.; Airich, K.; Doucerain, C.; Marpakwar, R.; Bakirci, P.; Senatore, A.; Monnard, A.; et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 2016, 536, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Keshet, G.; Bar-Peled, O.; Yaffe, D.; Nudel, U.; Gabizon, R. The Cellular Prion Protein Colocalizes with the Dystroglycan Complex in the Brain. J. Neurochem. 2002, 75, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Lei, Y.-J.; Han, J.; Shi, Q.; Chen, L.; Guo, Y.; Gao, Y.-J.; Chen, J.-M.; Jiang, H.-Y.; Zhou, W.; et al. Recombinant Neural Protein PrP Can Bind with Both Recombinant and Native Apolipoprotein E In Vitro. Acta Biochim. Biophys. Sin. 2006, 38, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Xu, J.; Xiang, J.; Lu, Y.; Chen, R.; Huang, L.; Xiao, G.; Sun, G. PrPC interacts with potassium channel tetramerization domain containing 1 (KCTD1) protein through the PrP51-136 region containing octapeptide repeats. Biochem. Biophys. Res. Commun. 2012, 417, 182–186. [Google Scholar] [CrossRef]
- Picard, D. Heat-shock protein 90, a chaperone for folding and regulation. Cell. Mol. Life Sci. 2002, 59, 1640–1648. [Google Scholar] [CrossRef]
- Chen, S. Hop as an Adaptor in the Heat Shock Protein 70 (Hsp70) and Hsp90 Chaperone Machinery. J. Boil. Chem. 1998, 273, 35194–35200. [Google Scholar] [CrossRef] [Green Version]
- Roffé, M.; Beraldo, F.H.; Bester, R.; Nunziante, M.; Bach, C.; Mancini, G.; Gilch, S.; Vorberg, I.; Castilho, B.; Martins, V.R.; et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. USA 2010, 107, 13147–13152. [Google Scholar] [CrossRef] [Green Version]
- Sakudo, A.; Lee, D.-C.; Li, S.; Nakamura, T.; Matsumoto, Y.; Saeki, K.; Itohara, S.; Ikuta, K.; Onodera, T. PrP cooperates with STI1 to regulate SOD activity in PrP-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 2005, 328, 14–19. [Google Scholar] [CrossRef]
- Sargent, P.B. The Diversity of Neuronal Nicotinic Acetylcholine Receptors. Annu. Rev. Neurosci. 1993, 16, 403–443. [Google Scholar] [CrossRef]
- Zdanowski, R.; Krzyzowska, M.; Ujazdowska, D.; Lewicka, A.; Lewicki, S. Role of α7 nicotinic receptor in the immune system and intracellular signaling pathways. Central Eur. J. Immunol. 2015, 40, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Broide, R.S.; Leslie, F.M. The α7 nicotinic acetylcholine receptor in neuronal plasticity. Mol. Neurobiol. 1999, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Santos, R.; Hung, S.-Y.; Huang, W.-P.; Liou, H.-C.; Fu, W.-M. Faculty Opinions recommendation of Autophagy protects neuron from Abeta-induced cytotoxicity. Fac. Opin. Post-Publ. Peer Rev. Biomed. Lit. 2010, 5. [Google Scholar] [CrossRef]
- Thomsen, M.; Hansen, H.; Timmerman, M.; Mikkelsen, J. Cognitive Improvement by Activation of α7 Nicotinic Acetylcholine Receptors: From Animal Models to Human Pathophysiology. Curr. Pharm. Des. 2010, 16, 323–343. [Google Scholar] [CrossRef] [PubMed]
- Srivareerat, M.; Tran, T.T.; Salim, S.; Aleisa, A.M.; Alkadhi, K.A. Chronic nicotine restores normal Aβ levels and prevents short-term memory and E-LTP impairment in Aβ rat model of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 834–844. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Koo, E.H. Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 2011, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Haas, L.T.; Kostylev, M.A.; Strittmatter, S.M. Therapeutic Molecules and Endogenous Ligands Regulate the Interaction between Brain Cellular Prion Protein (PrPC) and Metabotropic Glutamate Receptor 5 (mGluR5). J. Boil. Chem. 2014, 289, 28460–28477. [Google Scholar] [CrossRef] [Green Version]
- Ferraguti, F.; Crepaldi, L.; Nicoletti, F. Metabotropic Glutamate 1 Receptor: Current Concepts and Perspectives. Pharmacol. Rev. 2008, 60, 536–581. [Google Scholar] [CrossRef]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesne, S. The complex PrPC-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, J.V.; Jarosz-Griffiths, H.H.; Watt, N.T.; Hooper, N.M. Prion Protein-mediated Toxicity of Amyloid-β Oligomers Requires Lipid Rafts and the Transmembrane LRP1. J. Boil. Chem. 2013, 288, 8935–8951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostapchenko, V.G.; Beraldo, F.H.; Mohammad, A.H.; Xie, Y.F.; Hirata, P.H.; Magalhaes, A.C.; Guillaume, L.; Hongbin, L.; Andrzej, M.; Jillian, C.B.; et al. The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-beta oligomer toxicity. J. Neurosci. 2013, 33, 16552–16564. [Google Scholar] [CrossRef]
- Goniotaki, D.; Lakkaraju, A.K.K.; Shrivastava, A.N.; Bakirci, P.; Sorce, S.; Senatore, A.; Marpakwar, R.; Hornemann, S.; Gasparini, F.; Triller, A.; et al. Inhibition of group-I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog. 2017, 13, e1006733. [Google Scholar] [CrossRef] [PubMed]
- Leucht, C.; Simoneau, S.; Rey, C.; Vana, K.; Rieger, R.; Lasmézas, C.I.; Weiss, S. The 37 kDa/67 kDa laminin receptor is required for PrP Sc propagation in scrapie-infected neuronal cells. EMBO Rep. 2003, 4, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Damien, L.; Caroline, D.; Mathéa, P.; Elodie, P.; Sophie, B.; Jacques, C.; Hector, A.-O.; Sophie, M.-R.; Jean-Marie, L.; Odile, K.L.D.; et al. Neuritogenesis: The prion protein controls beta1 integrin signaling activity. FASEB J. 2012, 26, 678–690. [Google Scholar]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—lessons and emerging principles. Mol. Neurodegener. 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, D.G.; Temido-Ferreira, M.; Miranda, H.V.; Batalha, V.L.; Coelho, J.E.; Szegö, É.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. α-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef]
- Burgeson, R.E.; Chiquet, M.; Deutzmann, R.; Ekblom, P.; Engel, J.; Kleinman, H.; Martin, G.R.; Meneguzzi, G.; Paulsson, M.; Sanes, J.; et al. A new nomenclature for the laminins. Matrix Boil. 1994, 14, 209–211. [Google Scholar] [CrossRef]
- Grimpe, B.; Dong, S.; Doller, C.; Temple, K.; Malouf, A.T.; Silver, J. The critical role of basement membrane-independent laminin γ 1 chain during axon regeneration in the CNS. J. Neurosci. 2002, 22, 3144–3160. [Google Scholar] [CrossRef] [Green Version]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialog- Clin. Neurosci. 2000, 2, 219–232. [Google Scholar]
- Jahr, C.E.; Stevens, C.F. Calcium permeability of the N-methyl-D-aspartate receptor channel in hippocampal neurons in culture. Proc. Natl. Acad. Sci. USA 1993, 90, 11573–11577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.L.; Castillo, P.E. Synaptic plasticity of NMDA receptors: Mechanisms and functional implications. Curr. Opin. Neurobiol. 2012, 22, 496–508. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.G.M.; Moser, E.I.; Riedel, G.; Martin, S.; Sandin, J.; Day, M.; O’Carroll, C. Elements of a neurobiological theory of the hippocampus: The role of activity-dependent synaptic plasticity in memory. Philos. Trans. R. Soc. B Boil. Sci. 2003, 358, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-T.; Yang, K.-C.; Lin, W.-C. Glutamatergic Dysfunction and Glutamatergic Compounds for Major Psychiatric Disorders: Evidence from Clinical Neuroimaging Studies. Front. Psychiatry 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Gielen, M.; Retchless, B.S.; Mony, L.; Johnson, J.W.; Paoletti, P. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 2009, 459, 703–707. [Google Scholar] [CrossRef]
- Gasperini, L.; Meneghetti, E.; Pastore, B.; Benetti, F.; Legname, G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid. Redox Signal. 2015, 22, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.-B.; Tenneti, L.; Le, D.A.; Ortiz, J.; Bai, G.; Chen, H.-S.V.; Lipton, S.A. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat. Neurosci. 2000, 3, 15–21. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W. Nitric oxide synthases: Regulation and function. Eur. Hear. J. 2011, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Stys, P.K.; You, H.; Zamponi, G.W. Copper-dependent regulation of NMDA receptors by cellular prion protein: Implications for neurodegenerative disorders. J. Physiol. 2012, 590, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Biasini, E.; Unterberger, U.; Solomon, I.H.; Massignan, T.; Senatore, A.; Bian, H.; Voigtlaender, T.; Bowman, F.P.; Bonetto, V.; Chiesa, R.; et al. A mutant prion protein sensitizes neurons to glutamate-induced excitotoxicity. J. Neurosci. 2013, 33, 2408–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thellung, S.; Gatta, E.; Pellistri, F.; Corsaro, A.; Villa, V.; Vassalli, M.; Robello, M.; Florio, T. Excitotoxicity Through NMDA Receptors Mediates Cerebellar Granule Neuron Apoptosis Induced by Prion Protein 90-231 Fragment. Neurotox. Res. 2012, 23, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, A.; Willnow, T.E. The low-density lipoprotein receptor gene family: A cellular Swiss army knife? Trends Cell Boil. 2002, 12, 273–280. [Google Scholar] [CrossRef]
- Strickland, D.K.; Gonias, S.L.; Argraves, W.S. Diverse roles for the LDL receptor family. Trends Endocrinol. Metab. 2002, 13, 66–74. [Google Scholar] [CrossRef]
- Gonias, S.L.; Campana, W.M. LDL Receptor–Related Protein-1. Am. J. Pathol. 2014, 184, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Julia, H.-R.; Séverine, M.-L.; Théo, Z.H.; Elodie, P.; Aurélie, A.-B.; Benoît, S.; Anne, B.; Jean-Marie, L.; Sophie, M.-R. A PrPC-caveolin-Lyn complex negatively controls neuronal GSK3beta and serotonin 1B receptor. Sci. Rep. 2014, 4, 4881. [Google Scholar]
- Jarosz-Griffiths, H.H.; Whitehouse, I.J.; Baybutt, H.; Brown, D.; Kellett, K.A.B.; Jackson, C.D.; Turner, A.J.; Piccardo, P.; Manson, J.C.; Hooper, N.M. Prion Protein Interacts with BACE1 Protein and Differentially Regulates Its Activity toward Wild Type and Swedish Mutant Amyloid Precursor Protein. J. Boil. Chem. 2011, 286, 33489–33500. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, H.H.; Whitehouse, I.J.; Hooper, N.M. Regulation of amyloid-beta production by the prion protein. Prion 2012, 6, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Kleene, R.; Schachner, M. Glycans and neural cell interactions. Nat. Rev. Neurosci. 2004, 5, 195–208. [Google Scholar] [CrossRef]
- Walsh, F.S.; Doherty, P. Neural Cell Adhesion Molecules of the Immunoglobulin Superfamily: Role in Axon Growth and Guidance. Annu. Rev. Cell Dev. Boil. 1997, 13, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Weledji, E.P.; Assob, J.C.N. The ubiquitous neural cell adhesion molecule (N-CAM). Ann. Med. Surg. 2014, 3, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Barbas, J.A.; Chaix, J.C.; Steinmetz, M.; Goridis, C. Differential splicing and alternative polyadenylation generates distinct NCAM transcripts and proteins in the mouse. EMBO J. 1988, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Schvartz, I.; Seger, D.; Shaltiel, S. Vitronectin. Int. J. Biochem. Cell Boil. 1999, 31, 539–544. [Google Scholar] [CrossRef]
- Marti´nez-Morales, J.-R.; Martí, E.; Frade, J.M.; Rodri´guez-Te´bar, A. Developmentally regulated vitronectin influences cell differentiation, neuron survival and process outgrowth in the developing chicken retina. Neuroscience 1995, 68, 245–253. [Google Scholar] [CrossRef]
- Chidlow, J.H.; Sessa, W. Caveolae, caveolins, and cavins: Complex control of cellular signalling and inflammation. Cardiovasc. Res. 2010, 86, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Fridolfsson, H.N.; Roth, D.M.; Insel, P.A.; Patel, H.H. Regulation of intracellular signaling and function by caveolin. FASEB J. 2014, 28, 3823–3831. [Google Scholar] [CrossRef] [Green Version]
- Fu, G.; Wang, W.; Luo, B.-H. Overview: Structural Biology of Integrins. Breast Cancer 2011, 757, 81–99. [Google Scholar] [CrossRef]
- Guan, J.-L. Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB Life 2010, 62, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Stuermer, C.A. The reggie/flotillin connection to growth. Trends Cell Boil. 2010, 20, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Munderloh, C.; Solis, G.P.; Bodrikov, V.; Jaeger, F.A.; Wiechers, M.; Málaga-Trillo, E.; Stuermer, C.A. Reggies/Flotillins Regulate Retinal Axon Regeneration in the Zebrafish Optic Nerve and Differentiation of Hippocampal and N2a Neurons. J. Neurosci. 2009, 29, 6607–6615. [Google Scholar] [CrossRef] [PubMed]
- Meister, M.; Tikkanen, R. Endocytic Trafficking of Membrane-Bound Cargo: A Flotillin Point of View. Membranes 2014, 4, 356–371. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, Z.; Peng, H.; Zeng, K. Recent advances on the roles of epidermal growth factor receptor in psoriasis. Am. J. Transl. Res. 2019, 11, 520–528. [Google Scholar] [PubMed]
- Zhang, M.; Mu, X.-Y.; Jiang, S.; Liu, Q.-L.; Li, D.-W. Relationship between epidermal growth factor receptor gene expression and radiosensitivity of non-small-cell lung cancer cells. Zhonghua zhong liu za zhi Chin. J. Oncol. 2013, 35, 94–97. [Google Scholar]
- Pinheiro, P.S.; Mulle, C. Kainate receptors. Cell Tissue Res. 2006, 326, 457–482. [Google Scholar] [CrossRef] [PubMed]
- Frerking, M.; Nicoll, R.A. Synaptic kainate receptors. Curr. Opin. Neurobiol. 2000, 10, 342–351. [Google Scholar] [CrossRef]
- Niethammer, M.; Kim, E.; Sheng, M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J. Neurosci. 1996, 16, 2157–2163. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Cho, K.-O.; Rothschild, A.; Sheng, M. Heteromultimerization and NMDA Receptor-Clustering Activity of Chapsyn-110, a Member of the PSD-95 Family of Proteins. Neuron 1996, 17, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Nelson, C.D.; Li, X.; Winters, C.A.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Gainer, H.; Sheng, M.; Reese, T.S. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J. Neurosci. 2011, 31, 6329–6338. [Google Scholar] [CrossRef] [Green Version]
- Tezuka, T.; Umemori, H.; Akiyama, T.; Nakanishi, S.; Yamamoto, T. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc. Natl. Acad. Sci. USA 1999, 96, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Jerng, H.H.; Pfaffinger, P.J. Modulatory mechanisms and multiple functions of somatodendritic A-type K+ channel auxiliary subunits. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Bauer, C.S.; Van Minh, A.T.; Kadurin, I.; Dolphin, A.C. A new look at calcium channel α2δ subunits. Curr. Opin. Neurobiol. 2010, 20, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.S.; Ferron, L.; Kadurin, I.; Pratt, W.S.; Dolphin, A.C. Functional exofacially tagged N-type calcium channels elucidate the interaction with auxiliary alpha2delta-1 subunits. Proc. Natl. Acad. Sci. USA 2014, 111, 8979–8984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, M.; Bittner, T.; Mitteregger, G.; Haider, N.; Moosmang, S.; Kretzschmar, H.; Herms, J. Loss of the cellular prion protein affects the Ca2+ homeostasis in hippocampal CA1 neurons. J. Neurochem. 2006, 98, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Dossena, S.; Imeri, L.; Mangieri, M.; Garofoli, A.; Ferrari, L.; Senatore, A.; Restelli, E.; Balducci, C.; Fiordaliso, F.; Salio, M.; et al. Mutant Prion Protein Expression Causes Motor and Memory Deficits and Abnormal Sleep Patterns in a Transgenic Mouse Model. Neuron 2008, 60, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, R.; Piccardo, P.; Ghetti, B.; Harris, D.A. Neurological Illness in Transgenic Mice Expressing a Prion Protein with an Insertional Mutation. Neuron 1998, 21, 1339–1351. [Google Scholar] [CrossRef] [Green Version]
- Moss, D. Alkaline Phosphatase Isoenzymes. Enzyme 1975, 20, 20–34. [Google Scholar] [CrossRef]
- Low, M.G.; Zilversmit, D.B. Role of phosphatidylinositol in attachment of alkaline phosphatase to membranes. Biochemistry 1980, 19, 3913–3918. [Google Scholar] [CrossRef]
- Narisawa, S.; Yadav, M.C.; Millán, J.L. In Vivo Overexpression of Tissue-Nonspecific Alkaline Phosphatase Increases Skeletal Mineralization and Affects the Phosphorylation Status of Osteopontin. J. Bone Miner. Res. 2013, 28, 1587–1598. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef]
- David, G. Integral membrane heparan sulfate proteoglycans. FASEB J. 1993, 7, 1023–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karthikeyan, L.; Maurel, P.; Rauch, U.; Margolis, R.; Margolis, R. Cloning of a major heparan sulfate proteoglycan from brain and identification as the rat form of glypican. Biochem. Biophys. Res. Commun. 1992, 188, 395–401. [Google Scholar] [CrossRef]
- Hijazi, N.; Kariv-Inbal, Z.; Gasset, M.; Gabizon, R. PrPScIncorporation to Cells Requires Endogenous Glycosaminoglycan Expression. J. Boil. Chem. 2005, 280, 17057–17061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horonchik, L.; Tzaban, S.; Ben-Zaken, O.; Yedidia, Y.; Rouvinski, A.; Papy-Garcia, D.; Barritault, D.; Vlodavsky, I.; Taraboulos, A. Heparan Sulfate Is a Cellular Receptor for Purified Infectious Prions. J. Boil. Chem. 2005, 280, 17062–17067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amit, M.; Breanne, L.H.; Dan, C.; Jessica, J.; Nicholas, E.S.; Ueli, S.; Xianhua, P.; Valeria, C.; Kelly, R.M. Gpr126/Adgrg6 Has Schwann Cell Autonomous and Nonautonomous Functions in Peripheral Nerve Injury and Repair. J. Neurosci. 2016, 36, 12351–12367. [Google Scholar]
- Ravenscroft, G.; Nolent, F.; Rajagopalan, S.; Meireles, A.M.; Paavola, K.J.; Gaillard, D.; Alanio, E.; Buckland, M.; Arbuckle, S.; Krivanek, M.; et al. Mutations of GPR126 Are Responsible for Severe Arthrogryposis Multiplex Congenita. Expand. Spectr. BAF-Relat. Disord. Novo Var. SMARCC2 Cause Syndr. Intellect. Disabil. Dev. Delay 2015, 96, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Hashida-Okumura, A.; Okumura, N.; Iwamatsu, A.; Buijs, R.M.; Romijn, H.J.; Nagai, K. Interaction of Neuronal Nitric-oxide Synthase with α1-Syntrophin in Rat Brain. J. Boil. Chem. 1999, 274, 11736–11741. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, J.D.; Jackson, G.S.; Altmann, D.M. The role of the cellular prion protein in the immune system. Clin. Exp. Immunol. 2006, 146, 1–8. [Google Scholar] [CrossRef]
- Termini, C.M.; Gillette, J.M. Tetraspanins Function as Regulators of Cellular Signaling. Front. Cell Dev. Boil. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Huang, T.; Cui, Y.; Pan, B.; Shen, A.; Sun, Y.; Yi, Y.; Wang, Y.; Xiao, G.; Sun, G. PrPC interacts with tetraspanin-7 through bovine PrP154–182 containing alpha-helix 1. Biochem. Biophys. Res. Commun. 2008, 365, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Perot, B.P.; Ménager, M.M. Tetraspanin 7 and its closest paralog tetraspanin 6: Membrane organizers with key functions in brain development, viral infection, innate immunity, diabetes and cancer. Med Microbiol. Immunol. 2020, 209, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, L.; Bruni, A.C. Mutations in Prion Protein Gene: Pathogenic Mechanisms in C-Terminal vs. N-Terminal Domain, a Review. Int. J. Mol. Sci. 2019, 20, 3606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minikel, E.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.C.; Gambetti, P.; et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 8, 322ra9. [Google Scholar] [CrossRef] [Green Version]
- Scialo, C.; De Cecco, E.; Manganotti, P.; Legname, G. Prion and Prion-Like Protein Strains: Deciphering the Molecular Basis of Heterogeneity in Neurodegeneration. Viruses 2019, 11, 261. [Google Scholar] [CrossRef] [Green Version]
- Benetti, F.; Legname, G. New insights into structural determinants of prion protein folding and stability. Prion 2015, 9, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Walmsley, A.R.; Zeng, F.; Hooper, N.M. The N-terminal Region of the Prion Protein Ectodomain Contains a Lipid Raft Targeting Determinant. J. Boil. Chem. 2003, 278, 37241–37248. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, J.A.; Westergard, L.; Unterberger, U.; Biasini, E.; Harris, D. The N-Terminal, Polybasic Region Is Critical for Prion Protein Neuroprotective Activity. PLoS ONE 2011, 6, e25675. [Google Scholar] [CrossRef] [Green Version]
- Ghetti, B.; Piccardo, P.; Zanusso, G. Dominantly inherited prion protein cerebral amyloidosis—A modern view of Gerstmann–Sträussler–Scheinker. Handb. Clin. Neurol. 2018, 153, 243–269. [Google Scholar] [CrossRef]
- Nitrini, R.; Da Silva, L.S.T.; Rosemberg, S.; Caramelli, P.; Carrilho, P.E.M.; Iughetti, P.; Passos-Bueno, M.R.; Zatz, M.; Albrecht, S.; Leblanc, A. Prion disease resembling frontotemporal dementia and parkinsonism linked to chromosome 17. Arq. Neuro-Psiquiatr. 2001, 59, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.A.; Leehey, M.A.; Filley, C.M.; Steinbart, E.; Montine, T.; Schellenberg, G.D.; Bosque, P.; Nixon, R.; Bird, T. PRNP H187R mutation associated with neuropsychiatric disorders in childhood and dementia. Neurol 2005, 64, 1304–1306. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, J.; Kertesz, A.; Frohn, I.; Bauer, S.; George-Hyslop, P.S.; Bergeron, C. Gerstmann-Sträussler-Scheinker disease with the Q217R mutation mimicking frontotemporal dementia. Acta Neuropathol. 2005, 110, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Clerici, F.; Elia, A.; Girotti, F.; Contri, P.; Mariani, C.; Tagliavini, F.; Di Fede, G. Atypical presentation of Creutzfeldt–Jakob disease: The first Italian case associated with E196K mutation in the PRNP gene. J. Neurol. Sci. 2008, 275, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Giovagnoli, A.R.; Di Fede, G.; Aresi, A.; Reati, F.; Rossi, G.; Tagliavini, F. Atypical frontotemporal dementia as a new clinical phenotype of Gerstmann-Straussler-Scheinker disease with the PrP-P102L mutation. Description of a previously unreported Italian family. Neurol. Sci. 2008, 29, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Samaia, H.B.; Mari, J.D.J.; Vallada, H.; Moura, R.P.; Simpson, A.J.G.; Brentania, R.R.; Brentani, H. A prion-linked psychiatric disorder. Nature 1997, 390, 241. [Google Scholar] [CrossRef]
- Collinge, J.; Brown, J.; Hardy, J.; Mullan, M.; Rossor, M.; Baker, H.; Crow, T.J.; Lofthouse, R.; Poulter, M.; Ridley, R.; et al. Inherited prion disease with 144 base pair gene insertion: 2. Clinical and pathological features. Brain 1992, 115, 687–710. [Google Scholar] [CrossRef]
- Finckh, U.; Müller-Thomsen, T.; Mann, U.; Eggers, C.; Marksteiner, J.; Meins, W.; Binetti, G.; Alberici, A.; Hock, C.; Nitsch, R.M.; et al. High Prevalence of Pathogenic Mutations in Patients with Early-Onset Dementia Detected by Sequence Analyses of Four Different Genes. Am. J. Hum. Genet. 1999, 66, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Hayes, S.; Malacrida, B.; Kiely, M.; Kiely, P.A. Studying protein–protein interactions: Progress, pitfalls and solutions. Biochem. Soc. Trans. 2016, 44, 994–1004. [Google Scholar] [CrossRef]
- Reidenbach, A.G.; Minikel, E.; Zhao, H.; Guzman, S.G.; Leed, A.J.; Mesleh, M.F.; Kordasiewicz, H.; Schreiber, S.L.; Vallabh, S.M. Characterization of the Prion Protein Binding Properties of Antisense Oligonucleotides. Biomolecules 2019, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Schätzl, H.M.; Da Costa, M.; Taylor, L.; Cohen, F.E.; Prusiner, S.B. Prion protein gene variation among primates. J. Mol. Boil. 1997, 265, 257. [Google Scholar] [CrossRef]
- Wopfner, F.; Weidenhöfer, G.; Schneider, R.; Von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schätzl, H.M. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein 1 1Edited by A. R. Fersht. J. Mol. Boil. 1999, 289, 1163–1178. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PrPc Interacting Protein/Function | Cellular Localization | Effect of Interaction | Technique | References |
|---|---|---|---|---|
| Neuroglobin (Ngb)/hemoglobin | Cytoplasm | PrPc aggregation | Immunostaining, docking | [32,33] |
| Tubulin/cytoskeletal protein | Cytoplasm | Inhibit microtubule polymerization, Tubulin oligomerization, Retrograde and anterograde transport of PrPc | Co-IP, pull-down, co-fractionation, crosslinking | [34,35,36] |
| Tau/microtubule-associated protein | Cytoplasm | Reduction of PrPc induced oligomerization of tubulin | Co-IP, pull-down | [37,38,39,40] |
| Synapsin-1b/neuron-specific phosphoprotein | Cytoplasm | Unknown | Cofractionation, Y2H, Co-IP | [41,42,43] |
| Growth factor receptor-bound protein 2 (Grb2)/growth factor receptor | Cytoplasm, Nucleus | Unknown | Cofractionation, Y2H, Co-IP | [41,44] |
| Pint-1/unknown | Cytoplasm | Unknown | Y2H, Co-IP | [41] |
| Glial fibrillary acidic protein (GFAP)/intermediate filament | Cytoplasm | Unknown | Co-IP, pull-down, overlay | [45,46,47] |
| Heterogeneous nuclear ribonucleoproteins (hnRNP) A2/B1/RNA-binding protein | Cytoplasm, Nucleus | Unknown | Co-IP, overlay | [48] |
| Aldolase C/metabolic enzyme | Cytoplasm | Unknown | Co-IP, overlay | [48] |
| Nuclear factor erythroid 2-related factor 2 (NRF2)/transcription factor | Cytoplasm, nucleus | Unknown | Screen of bacteriophage expression library of brain cDNA | [49] |
| B cell lymphoma 2 (BCL2)/apoptosis regulator | Cytoplasm | PrPc aggregation, Inhibits BCL2 anti-apoptotic function | Co-IP, Y2H, affinity | [50,51,52] |
| 14-3-3 protein/phosphorylation-dependent scaffold protein | Cytoplasm | Unknown | Co-IP, pull-down, overlay | [53,54,55,56] |
| Neurotrophin receptor-interacting MAGE homolog (NRAGE)/cell-death inducer | Cytoplasm | Aggregation, changing mitochondrial membrane potential | Co-IP, pull-down, Y2H | [57] |
| Casein kinase II (CK2)/serine-threonine kinase | Cytoplasm, nucleus, extracellular matrix | PrPc phosphorylation, regulates CK2 enzymatic activity | Co-IP, pull-down, overlay, surface plasmon resonance | [58,59,60] |
| Mahogunin ring finger 1 (MGRN1)/ubiquitin ligase | Cytoplasm | Aggregation, disruption of mahogunin function, neurodegeneration | Pull-down, Co-IP, Y2H | [61,62] |
| Heat shock protein 60 (Hsp60)/chaperon | Mitochondria | Unknown | Y2H | [63] |
| Members of the Rab family of small GTPases/guanosine triphosphate proteins | Cytoplasm | Intracellular PrPc trafficking | Co-IP | [64,65] |
| Argonaute/small RNA binding protein | Cytoplasm | Posttranscription cytoplasmic gene-silencing mechanism | Pull-down, electron microscopy | [66] |
| Lactate dehydrogenase A (LDHA)/oxidoreductases enzyme | Cytoplasm | Activation of LDHA, neuroprotection | Co-IP | [67,68] |
| Vimentin/cytoskeletal protein | Cytoplasm | Regulate intracellular transportation | Co-IP | [69,70] |
| β-catenin/transcriptional coactivator | Cytoplasm, nucleus | Transcriptional regulation | Co-IP | [71] |
| Transcription factor 7-like 2 (TCF7L2)/transcription factor | Cytoplasm, nucleus | Transcriptional regulation | Co-IP | [71] |
| Histone H1/3, lamin B1/structural chromatin components | Nucleus | Transcriptional regulation | Far-Western blot | [72] |
| HS-1-associated protein X1 (HAX-1)/apoptosis regulator | Cytoplasm | Oxidative stress, antiapoptosis | Co-IP, microarray | [73] |
| PrPc Interacting Protein/Function | Cellular Localization | Effect of Interaction | Technique | References |
|---|---|---|---|---|
| Stress-inducible phosphoprotein 1 (STI1)/extracellular ligands | PM | Ca2+ influx induction, neuritogenesis, neuroprotection | Co-IP, pull-down, complementary hydropathy | [148,149,150,151] |
| Nicotinic acetylcholine receptor (α7nAChR)/neurotransmitter receptors | PM | Mediate autophagic flux, Ca2+ signaling | Co-IP | [152,153] |
| Amyloid beta precursor protein (APP)/cell adhesion receptors | PM | Cell adhesion, CNS development | Co-IP | [154] |
| β-site amyloid precursor protein cleaving enzyme 1 (BACE1)/membrane-bound aspartic protease | PM | APP cleavage | Co-IP | [155] |
| Amyloid beta oligomer (AβO)/ extracellular ligand | PM | Neurotoxicity, LTP suppression, NMDAR phosphorylation, Tau phosphorylation | Co-IP, immunostaining | [16,156,157,158,159] |
| Metabotropic glutamate receptor 1 (mGluR1)/neurotransmitter receptors | PM | Neurite outgrowth | Co-IP, immunostaining | [160,161,162] |
| Metabotropic glutamate receptor 5 (mGluR5)/neurotransmitter receptors | PM | AβO -mediate neurotoxicity, promoting long term depression and inhibiting long-term potentiation, neurite outgrowth | Co-IP, immunostaining | [160,163,164] |
| Laminin receptor (LRP/LR)/ cell adhesion receptors | PM | PrPc internalization, Cell survival | Co-IP, Y2H | [165,166,167] |
| α-synuclein/neurotransmitter release regulator | PM | Ca2+ influx | Co-IP | [168] |
| Laminin/Extracellular matrix protein | Extracellular matrix | Modulate neuronal plasticity and memory | Pull-down | [169,170,171] |
| N-methyl-D-aspartate (NMDA) (GluN2D)/neurotransmitter receptors | PM | Neuroprotection | Co-IP | [146,147] |
| Low-density lipoprotein receptor-related protein 1 (LRP1)/endocytic receptor | PM, Intracellular compartments | Endocytosis and trafficking of PrPc, stimulation of neurotransmitter release | Co-IP | [172,173] |
| Neural cell adhesion molecule (NCAM)/ cell adhesion receptors | PM | Neuritogenesis, neurons development | Co-IP, immunostaining, pull-down | [174,175] |
| Vitronectin/extracellular matrix protein | Extracellular matrix | Axonal growth | Immunostaining, pull-down | [21] |
| Caveolin-1/scaffold proteins | PM | Regulate exosome secretion, regulate Src kinase Fyn activation | Co-IP, immunostaining, pull-down | [176,177,178,179] |
| Integrin β1/cell adhesion receptors | PM | Neuritogenesis | Co-IP, immunostaining, pull-down, overlay | [21,179] |
| Flotillin/scaffold proteins | PM | Neurons differentiation by trafficking of N-cadherin, trafficking of EGFR | Co-IP, immunostaining, electron microscopy | [180,181] |
| Epidermal growth factor receptor (EGFR)/growth factor receptors | PM | Neuritogenesis | Co-IP | [182,183] |
| Postsynaptic density protein 95 (PSD-95)/scaffolding protein | PM | Protection against excitotoxicity | Co-IP, cDNA library | [146,182,183,184,185,186] |
| Kainate receptor GluR6/7/ ionotropic glutamate receptors | PM | Protection against excitotoxicity | Co-IP | [146,182,183,184,185,186] |
| Dipeptidyl peptidase-like protein 6 (DPP6)/potassium channel | PM | Downregulation of neuronal membrane excitability | Co_IP | [187] |
| α2δ-1 subunit of voltage-gated/calcium channel | PM | Ca2+ influx | Co-IP, immunostaining | [188,189] |
| Tissue nonspecific alkaline phosphatase (TNAP)/ectoenzyme | PM | Laminin dephosphorylation | Co-IP, immunostaining, mass spectrometry | [190] |
| α2/β2-Na+/K+-ATPase/neurotransmitter receptors | PM | Lactate transportation in astrocytic | Co-IP, immunoaffinity chromatography, pull-down | [191] |
| Glypican-1 (GPC-1)/growth factor regulator | PM | Lipid raft localization of PrPc | Co-IP, immunostaining | [192] |
| G protein-coupled receptor (Adgrg6)/adhesion G protein-coupled receptor | PM | Myelin maintenance in the peripheral nerves | Co-IP, immunostaining, electron microscopy | [193,194] |
| Neuronal nitric oxide synthase (nNOS)/flavo-hemoprotein | PM | Modulate nNOS activity | Co-IP, immunostaining | [195] |
| Dystroglycan/integral membrane glycoprotein | PM | Unknown | Co-IP, immunostaining | [195] |
| Synaptophysin/Integral membrane glycoprotein | Synaptic vesicles | Unknown | Co-IP, immunostaining | [195] |
| ApoE/cholesterol carrier | Extracellular matrix | Unknown | Co-IP, pull-down | [196] |
| Potassium channel tetramerization domain containing 1 (KCTD1)/ion channel | PM | Unknown | Co-IP, Y2H | [197] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranzadeh Mahabadi, H.; Taghibiglou, C. Cellular Prion Protein (PrPc): Putative Interacting Partners and Consequences of the Interaction. Int. J. Mol. Sci. 2020, 21, 7058. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197058
Miranzadeh Mahabadi H, Taghibiglou C. Cellular Prion Protein (PrPc): Putative Interacting Partners and Consequences of the Interaction. International Journal of Molecular Sciences. 2020; 21(19):7058. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197058
Chicago/Turabian StyleMiranzadeh Mahabadi, Hajar, and Changiz Taghibiglou. 2020. "Cellular Prion Protein (PrPc): Putative Interacting Partners and Consequences of the Interaction" International Journal of Molecular Sciences 21, no. 19: 7058. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197058