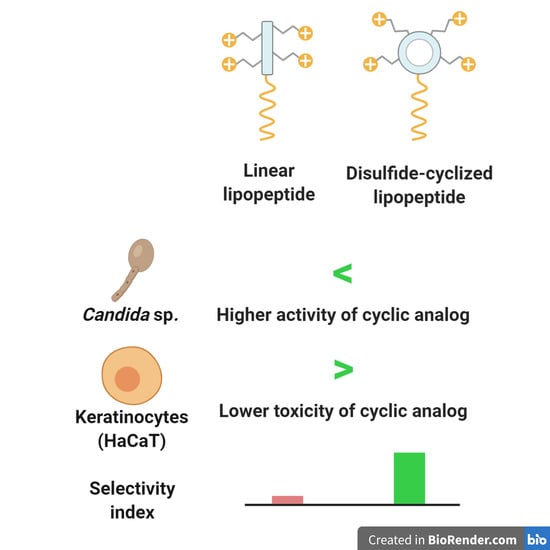
Effect of Disulfide Cyclization of Ultrashort Cationic Lipopeptides on Antimicrobial Activity and Cytotoxicity

 , , , , and
, , , , and 
Abstract
:
1. Introduction
2. Results and Discussion
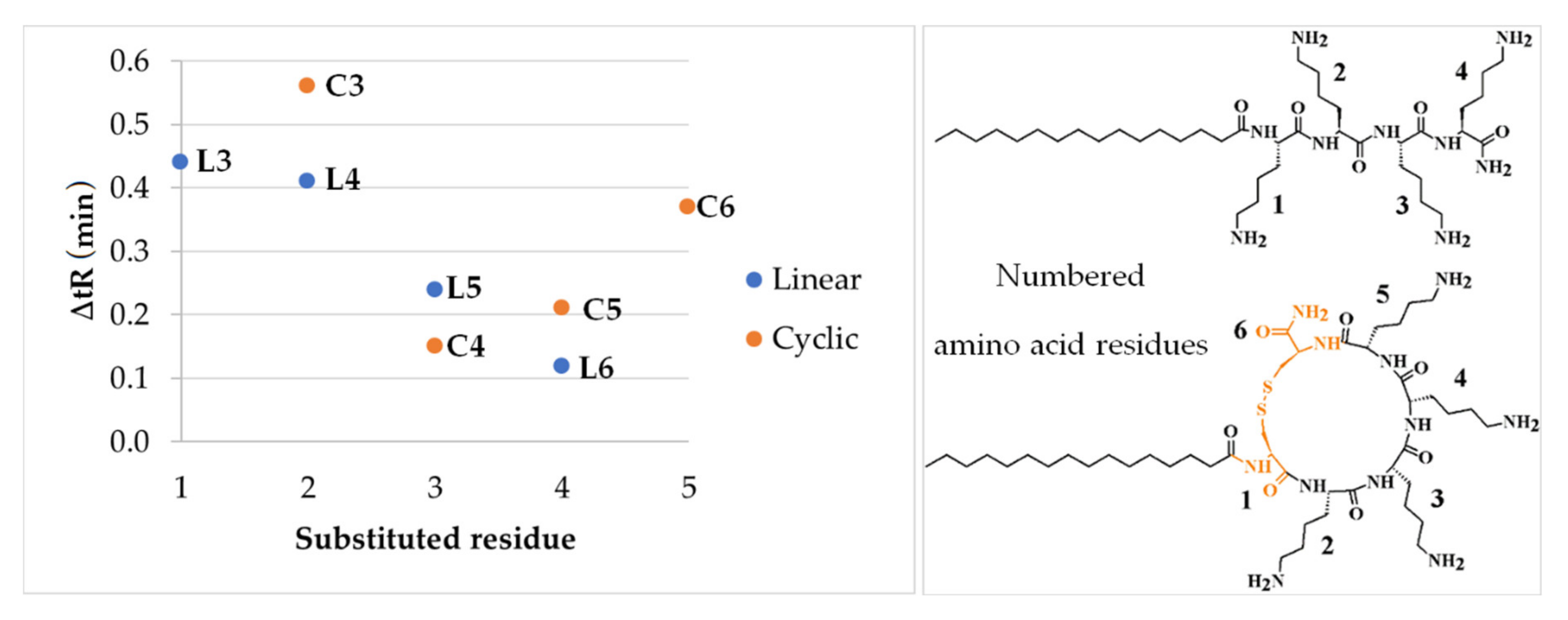
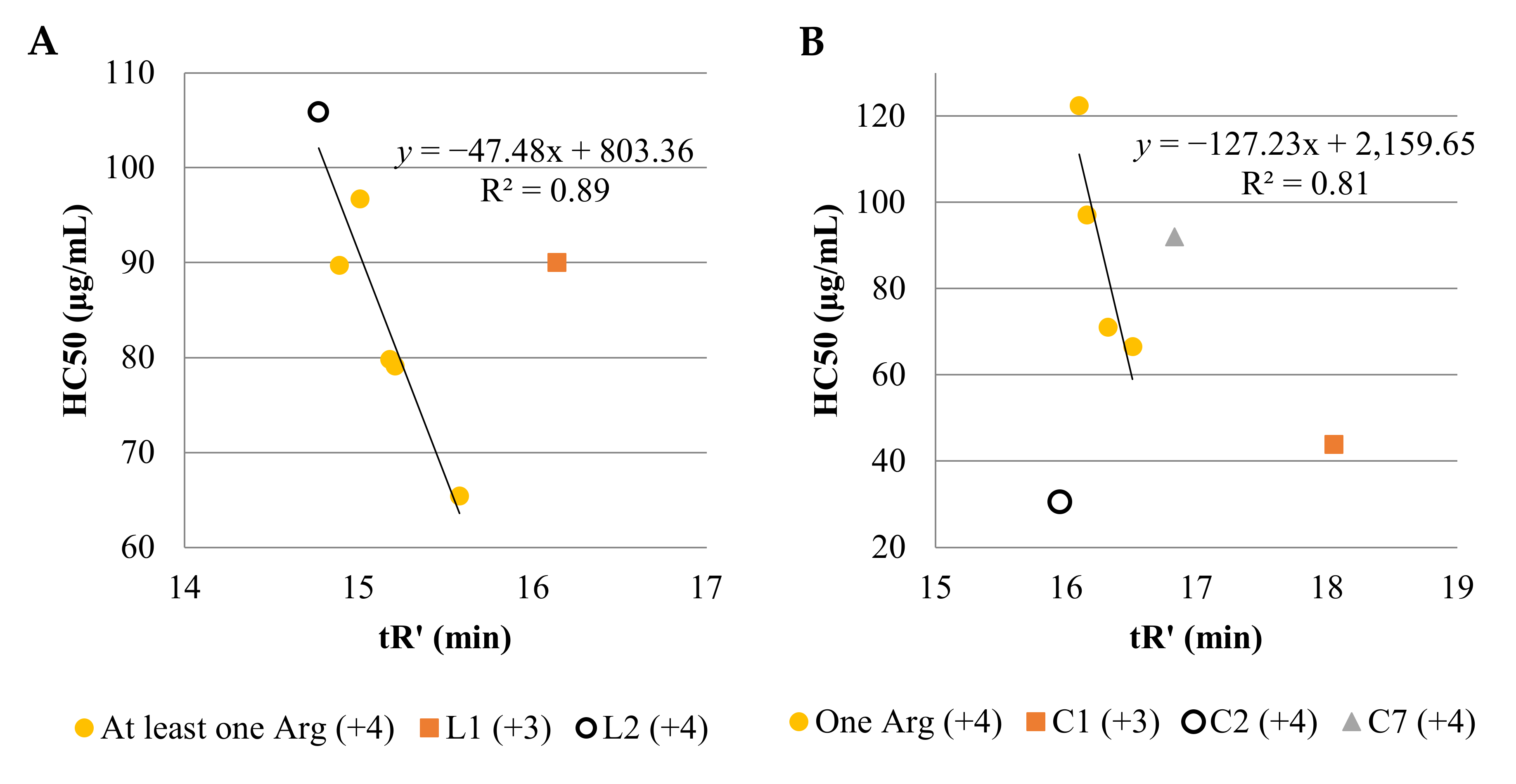
2.1. Determination of Peptide Hydrophobicity with RP-HPLC
- t’R calc. L7 = t’R L2 + t’R L3 − t’R L2 + t’R L4 − t’R L2 = t’R L3 + t’R L4 − t’R L2 = 15.21 + 15.18 − 14.77 = 15.62 min;
- t’R exp. L7 = 15.58 min;
- t’R calc. L7 − t’R exp. L7 = 0.04 min;
- t’R calc. C7 = t’R C2 + t’R C3 − t’R C2 + t’R C4 − t’R C2 = t’R C3 + t’R C4 − t’R C2 = 16.51 + 16.10 − 15.95 = 16.66 min;
- t’R exp. C7 = 16.83 min;
- t’R calc. C7 − t’R exp. C7 = −0.17 min.
2.2. Antimicrobial Activity of Lipopeptides
2.3. Antibiofilm Activity
2.4. MTT and Hemolysis Assay
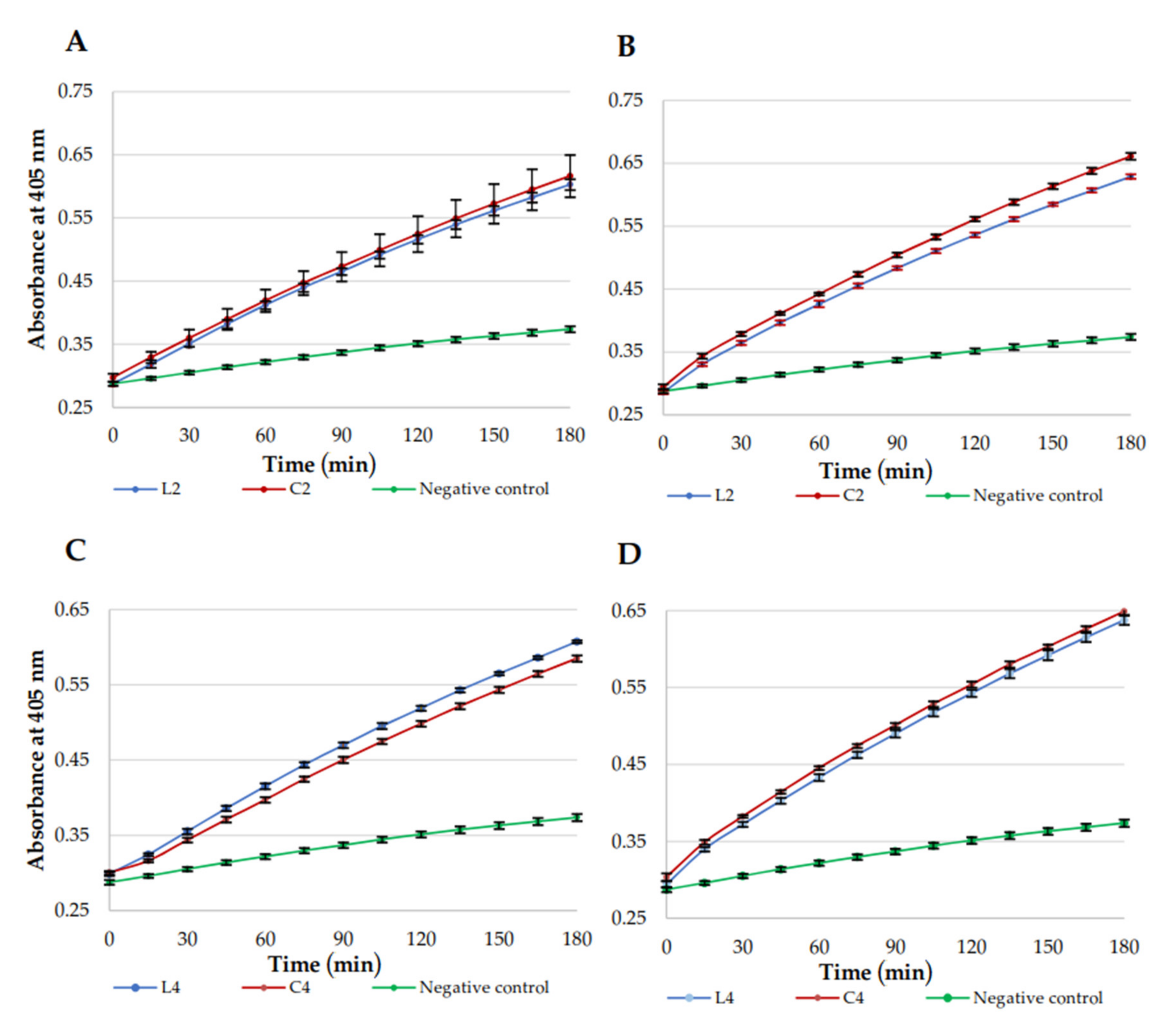
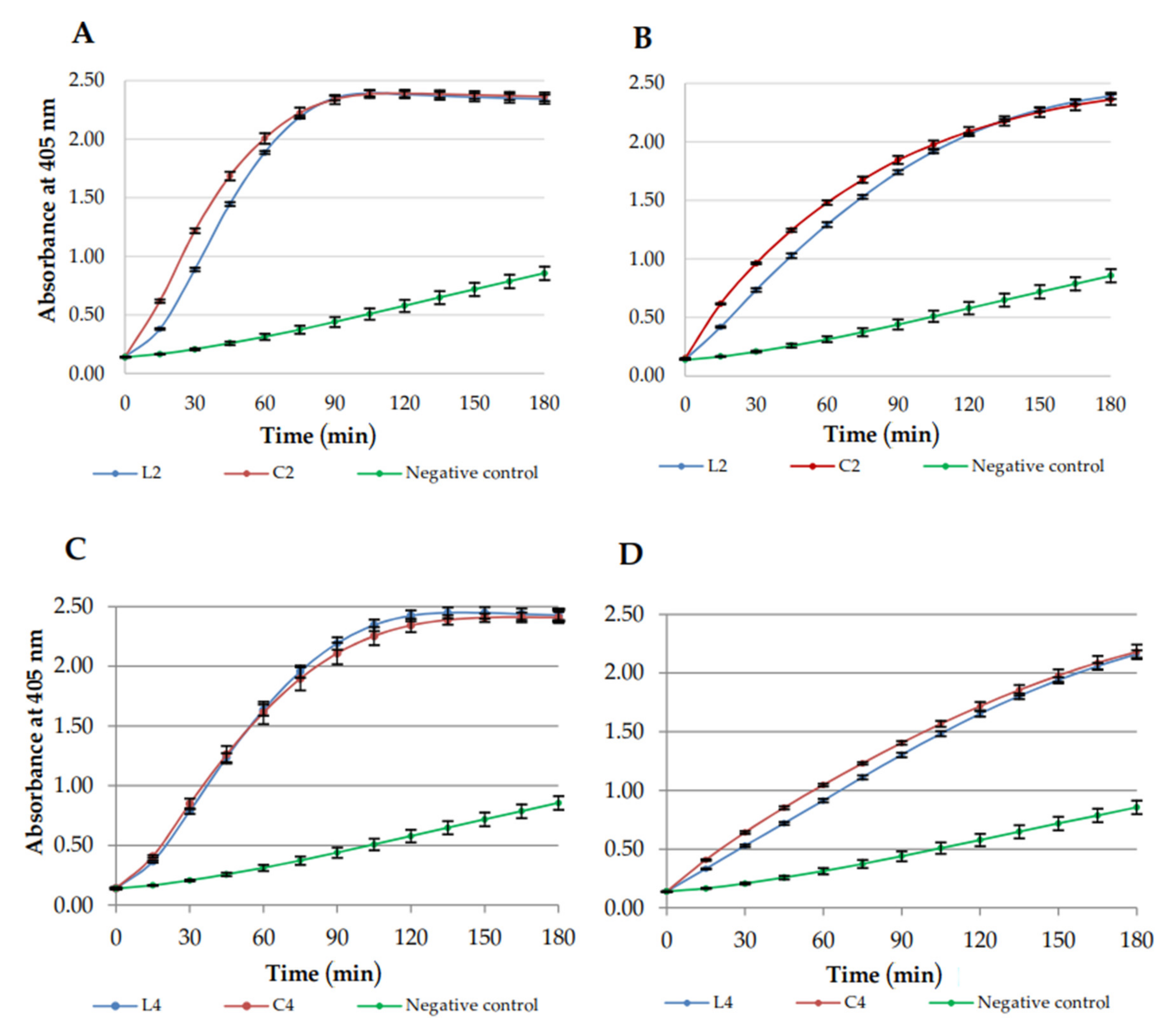
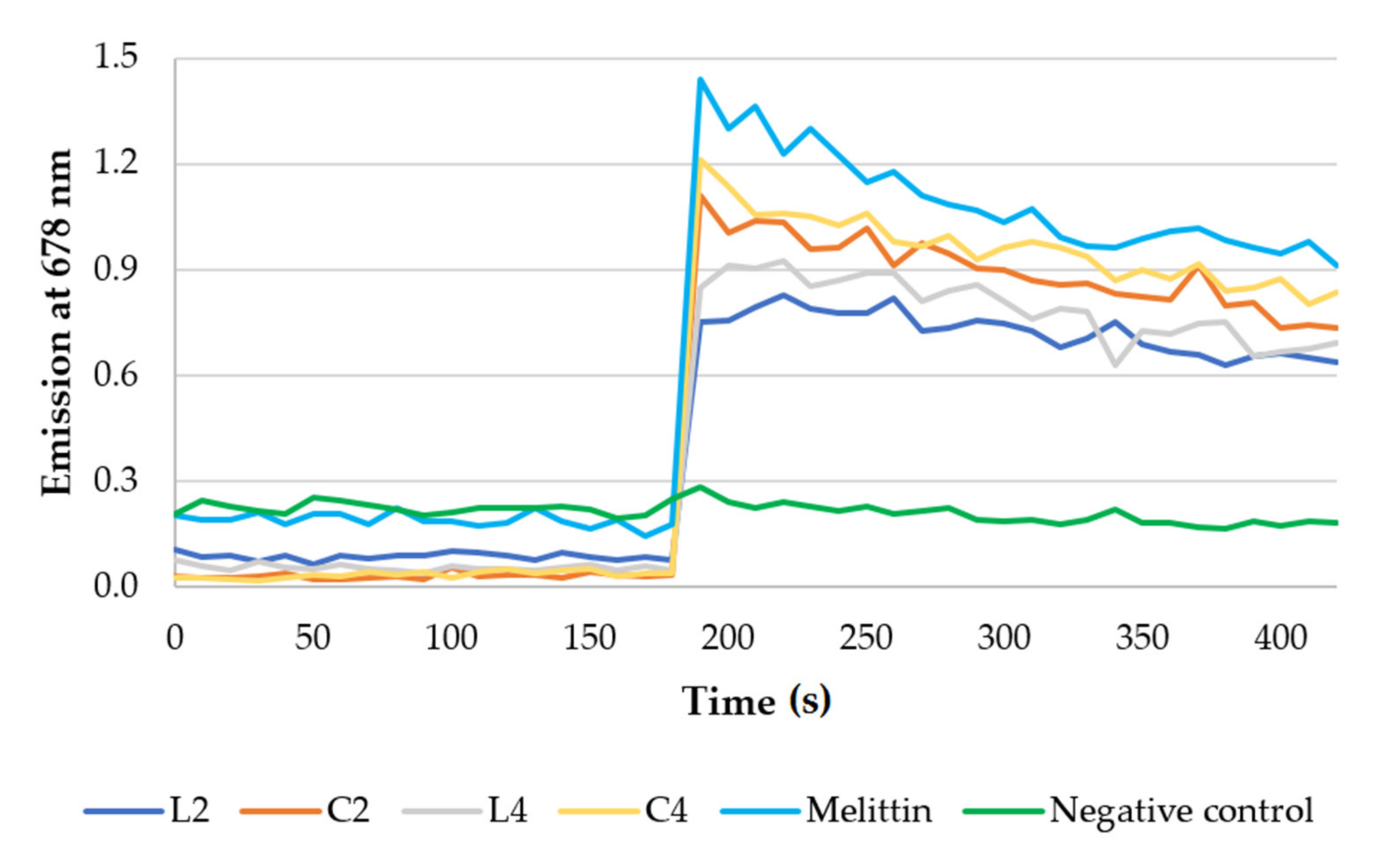
2.5. Membrane Permeabilization
2.6. Membrane Depolarization properties
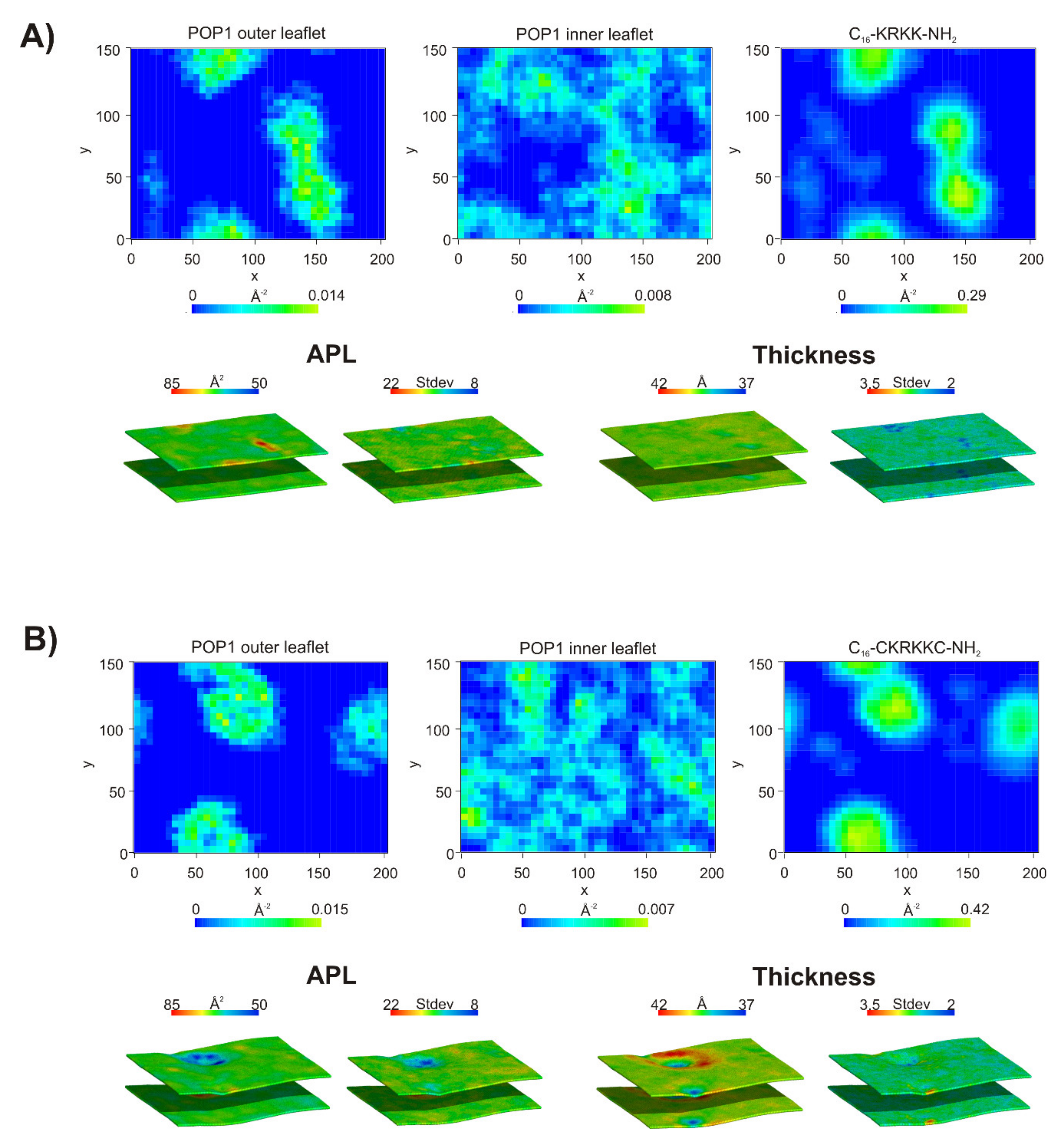
2.7. Coarse-Grained Molecular Dynamics
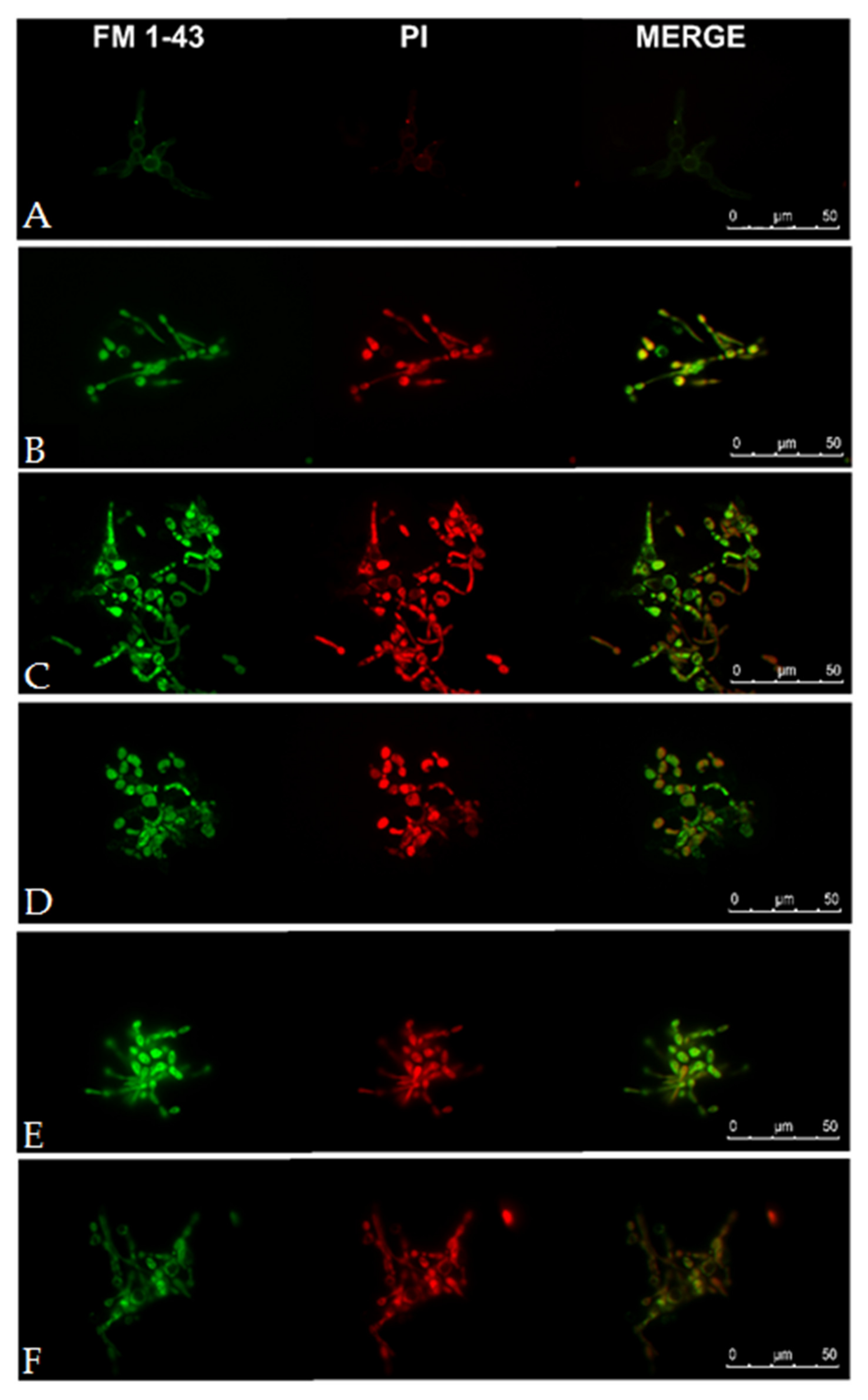
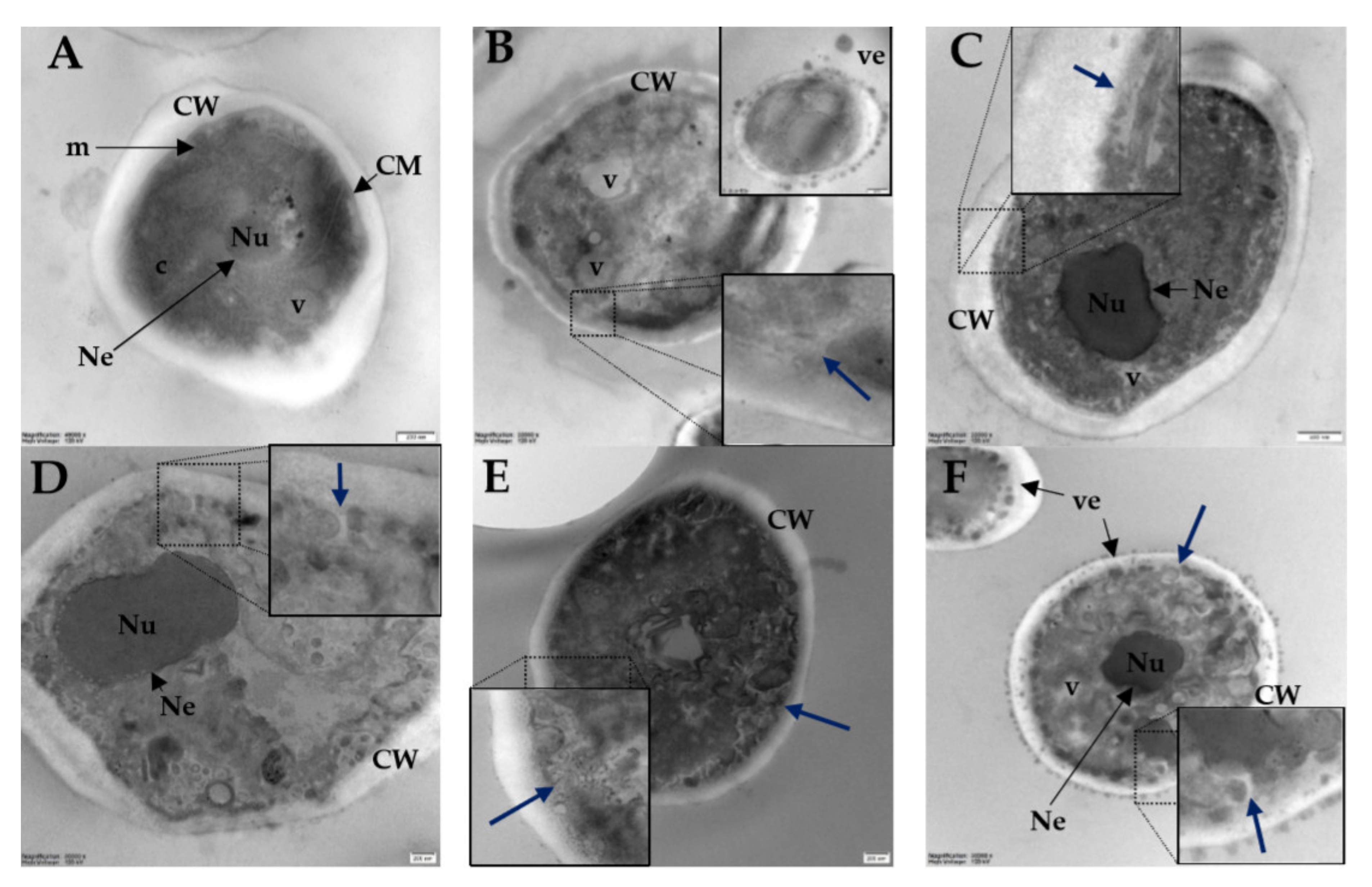
2.8. Visualization of Candida albicans Cells Treated with Selected Lipopeptides by Fluorescence Microscopy and Transmission Electron Microscopy
3. Materials and Methods
3.1. Peptide Synthesis
3.2. Determination of Peptide Hydrophobicity with RP-HPLC
3.3. Antimicrobial Activity
3.3.1. Cultivation of Microorganisms
3.3.2. Activity against Planktonic Cultures
3.3.3. Activity against Biofilm
3.3.4. Biofilm Inhibition Assay
3.4. Permeabilization of E. coli ML-35 Membranes
3.5. Membrane Depolarization Assay
3.6. Hemolysis Assay
3.7. MTT Assay
3.8. Fluorescence Microscopy
3.9. Transmission Electron Microscopy (TEM)
3.10. Molecular Dynamics Simulations
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | Bovine serum albumin |
| CENTA | 7-(thienyl-2-acetamido)-3-[(4-nitro-3-carboxyphenyl)thiomethyl]-3-cephem-4 carboxylic acid |
| CFU | Colony forming units |
| DCM | Dichloromethane |
| DIC | N,N’-diisopropylcarbodiimide |
| DiSC3(5) | 3,3′-dipropylthiacarbocyanine iodide |
| DMF | N,N-dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| EDT | 1,2-ethaneditiol |
| EDTA | Ethylenediaminetetraacetic acid |
| ESI-MS | Electrospray ionization mass spectrometry |
| FM 1-43 | N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino) styryl) pyridinium dibromide |
| Fmoc | Fluorenylmethoxycarbonyl |
| HaCaT | Human keratinocytes |
| HC50 | Peptide concentration causing 50% hemolysis |
| HeLa | Human cervical adenocarcinoma cell line |
| hRBCs | Human red blood cells |
| IC50 | Peptide concentration causing 50% inhibition of the growth |
| IM | Inner membrane |
| LB | Luria-Bertani medium |
| MBEC | Minimum biofilm eradication concentration |
| MBIC | Minimum biofilm inhibitory concentration |
| MHA | Mueller-Hinton agar |
| MHB | Mueller-Hinton broth |
| MIC | Minimum inhibitory concentration |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| ONPG | o-nitrophenyl-β-d-galactopyranoside |
| OM | Outer membrane |
| Pal | Palmitic acid (hexadecenoic acid) |
| PI | Propidium iodide |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPE | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol |
| POPI | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoinositol |
| POPS | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine |
| RP-HPLC | reversed-phase high-performance liquid chromatography |
| SDA | Sabouraud dextrose agar |
| SDB | Sabouraud dextrose broth |
| SI | Selectivity index |
| tBu | tert-butyl |
| TFA | Trifluoroacetic acid |
| TIS | Triisopropylsilane |
References
- Venter, H.; Henningsen, M.L.; Begg, S.L. Antimicrobial resistance in healthcare, agriculture and the environment: The biochemistry behind the headlines. Essays Biochem. 2017, 61, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Błażewicz, I.; Jaśkiewicz, M.; Piechowicz, L.; Neubauer, D.; Nowicki, R.; Kamysz, W.; Barańska-Rybak, W. Increasing rate of daptomycin non-susceptible strains of Staphylococcus aureus in patients with atopic dermatitis. Adv. Dermatol. Allergol. 2017, 34, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Arendrup, M.C.; Perlin, D.S. Echinocandin resistance: An emerging clinical problem? Curr. Opin. Infect. Dis. 2014, 27, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, P.; Rivard, K. Polymyxin Resistance in Gram-negative Pathogens. Curr. Infect. Dis. Rep. 2017, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Makovitzki, A.; Avrahami, D.; Shai, Y.; Sela, M. Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. USA 2006, 103, 15997–16002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerala, R. Synthetic lipopeptides: A novel class of anti-infectives. Expert Opin. Investig. Drugs 2007, 16, 1159–1169. [Google Scholar] [CrossRef]
- Meir, O.; Zaknoon, F.; Cogan, U.; Mor, A. A broad-spectrum bactericidal lipopeptide with anti-biofilm properties. Sci. Rep. 2017, 7, 2198. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, D.; Jaśkiewicz, M.; Bauer, M.; Gołacki, K.; Kamysz, W. Ultrashort Cationic Lipopeptides–Effect of N-Terminal Amino Acid and Fatty Acid Type on Antimicrobial Activity and Hemolysis. Molecules 2020, 25, 257. [Google Scholar] [CrossRef] [Green Version]
- Malanovic, N.; Lohner, K. Antimicrobial peptides targeting Gram-positive bacteria. Pharmaceuticals 2016, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Hobden, C.; Teevan, C.; Jones, L.; O’Shea, P. Hydrophobic properties of the cell surface of Candida albicans: A role in aggregation. Microbiology 1995, 141, 1875–1881. [Google Scholar] [CrossRef] [Green Version]
- Mago, N.; Khuller, G.K. Lipids of Candida albicans: Subcellular distribution and biosynthesis. J. Gen. Microbiol. 1990, 136, 993–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gährs, W.; Tigyi, Z.; Emody, L.; Makovitzky, J. Polarization optical analysis of the surface structures of various fungi. Acta Histochem. 2009, 111, 309–316. [Google Scholar] [CrossRef]
- Dawgul, M.; Greber, K.; Bartoszewska, S.; Baranska-Rybak, W.; Sawicki, W.; Kamysz, W. In Vitro Evaluation of Cytotoxicity and Permeation Study on Lysine- and Arginine-Based Lipopeptides with Proven Antimicrobial Activity. Molecules 2017, 22, 2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armas, F.; Pacor, S.; Ferrari, E.; Guida, F.; Pertinhez, T.A.; Romani, A.A.; Scocchi, M.; Benincasa, M. Design, antimicrobial activity and mechanism of action of Arg-rich ultra-short cationic lipopeptides. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawgul, M.; Maciejewska, M.; Jaskiewicz, M.; Karafova, A.; Kamysz, W. Antimicrobial peptides as potential tool to fight bacterial biofilm. Acta Pol. Pharm. 2014, 71, 39–47. [Google Scholar] [PubMed]
- Sikorska, E.; Dawgul, M.; Greber, K.; Iłowska, E.; Pogorzelska, A.; Kamysz, W. Self-assembly and interactions of short antimicrobial cationic lipopeptides with membrane lipids: ITC, FTIR and molecular dynamics studies. Biochim. Biophys. Acta 2014, 1838, 2625–2634. [Google Scholar] [CrossRef] [Green Version]
- Greber, K.E.; Dawgul, M.; Kamysz, W.; Sawicki, W. Cationic Net Charge and Counter Ion Type as Antimicrobial Activity Determinant Factors of Short Lipopeptides. Front. Microbiol. 2017, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Findlay, B.; Mookherjee, N.; Schweizer, F. Ultrashort Cationic Lipopeptides and Lipopeptoids Selectively Induce Cytokine Production in Macrophages. PLoS ONE 2013, 8, e54280. [Google Scholar] [CrossRef]
- Li, L.; Vorobyov, I.; Allen, T.W. The different interactions of lysine and arginine side chains with lipid membranes. J. Phys. Chem. B 2013, 117, 11906–11920. [Google Scholar] [CrossRef]
- Wu, Z.; Cui, Q.; Yethiraj, A. Why do arginine and lysine organize lipids differently? Insights from coarse-grained and atomistic simulations. J. Phys. Chem. B 2013, 117, 12145–12156. [Google Scholar] [CrossRef]
- Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Schöneich, C.; Borchardt, R.T. Chemical instability of protein pharmaceuticals: Mechanisms of oxidation and strategies for stabilization. Biotechnol. Bioeng. 1995, 48, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Mant, C.T.; Hodges, R.S. Context-dependent effects on the hydrophilicity/hydrophobicity of side-chains during reversed-phase high-performance liquid chromatography: Implications for prediction of peptide retention behaviour. J. Chromatogr. A 2006, 1125, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soetens, J.C.; Millot, C.; Chipot, C.; Jansen, G.; Ángyán, J.G.; Maigret, B. Effect of polarizability on the potential of mean force of two cations. The guanidimum-guanidinium ion pair in water. J. Phys. Chem. B 1997, 101, 10910–10917. [Google Scholar] [CrossRef]
- Robison, A.D.; Sun, S.; Poyton, M.F.; Johnson, G.A.; Pellois, J.P.; Jungwirth, P.; Vazdar, M.; Cremer, P.S. Polyarginine Interacts More Strongly and Cooperatively than Polylysine with Phospholipid Bilayers. J. Phys. Chem. B 2016, 120, 9287–9296. [Google Scholar] [CrossRef] [Green Version]
- Vondrášek, J.; Mason, P.E.; Heyda, J.; Collins, K.D.; Jungwirth, P. The molecular origin of like-charge arginine—Arginine pairing in water. J. Phys. Chem. B 2009, 113, 9041–9045. [Google Scholar] [CrossRef]
- Vazdar, M.; Heyda, J.; Mason, P.E.; Tesei, G.; Allolio, C.; Lund, M.; Jungwirth, P. Arginine “magic”: Guanidinium Like-Charge Ion Pairing from Aqueous Salts to Cell Penetrating Peptides. Acc. Chem. Res. 2018, 51, 1455–1464. [Google Scholar] [CrossRef] [Green Version]
- Neves, M.A.C.; Yeager, M.; Abagyan, R. Unusual arginine formations in protein function and assembly: Rings, strings, and stacks. J. Phys. Chem. B 2012, 116, 7006–7013. [Google Scholar] [CrossRef] [Green Version]
- Jaśkiewicz, M.; Neubauer, D.; Kamysz, W. Comparative Study on Antistaphylococcal Activity of Lipopeptides in Various Culture Media. Antibiotics 2017, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- McPhee, J.B.; Lewenza, S.; Hancock, R.E.W. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 2003, 50, 205–217. [Google Scholar] [CrossRef]
- Gunn, J.S.; Lim, K.B.; Krueger, J.; Kim, K.; Guo, L.; Hackett, M.; Miller, S.I. PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol. Microbiol. 1998, 27, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Thaipisuttikul, I.; Hittle, L.E.; Chandra, R.; Zangari, D.; Dixon, C.L.; Garrett, T.A.; Rasko, D.A.; Dasgupta, N.; Moskowitz, S.M.; Malmström, L.; et al. A divergent Pseudomonas aeruginosa palmitoyltransferase essential for cystic fibrosis-specific lipid A. Mol. Microbiol. 2014, 91, 158–174. [Google Scholar] [CrossRef] [Green Version]
- King, J.D.; Kocíncová, D.; Westman, E.L.; Lam, J.S. Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 2009, 15, 261–312. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.Y.; Guo, Y.; He, Q.S.; Peng, L.; Wu, S.C.; Cao, H.; Huang, S.H. Escherichia coli outer membrane protease OmpT confers resistance to urinary cationic peptides. Microbiol. Immunol. 2010, 54, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Ulvatne, H.; Haukland, H.H.; Samuelsen, Ø.; Krämer, M.; Vorland, L.H. Proteases in Escherichia coli and Staphylococcus aureus confer reduced susceptibility to lactoferricin B. J. Antimicrob. Chemother. 2002, 50, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Mangoni, M.L.; Shai, Y. Short native antimicrobial peptides and engineered ultrashort lipopeptides: Similarities and differences in cell specificities and modes of action. Cell. Mol. Life Sci. 2011, 68, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, E.; Stachurski, O.; Neubauer, D.; Małuch, L.; Wyrzykowski, D.; Bauer, M.; Brzozowski, K.; Kamysz, W. Short arginine-rich lipopeptides: From self-assembly to antimicrobial activity. Biochim. Biophys. Acta 2018, 1860, 2242–2251. [Google Scholar] [CrossRef]
- Sarig, H.; Rotem, S.; Ziserman, L.; Danino, D.; Mor, A. Impact of self-assembly properties on antibacterial activity of short acyl-lysine oligomers. Antimicrob. Agents Chemother. 2008, 52, 4308–4314. [Google Scholar] [CrossRef] [Green Version]
- Stachurski, O.; Neubauer, D.; Małuch, I.; Wyrzykowski, D.; Bauer, M.; Bartoszewska, S.; Kamysz, W.; Sikorska, E. Effect of self-assembly on antimicrobial activity of double-chain short cationic lipopeptides. Bioorg. Med. Chem. 2019, 27, 115129. [Google Scholar] [CrossRef]
- Makovitzki, A.; Baram, J.; Shai, Y. Antimicrobial lipopolypeptides composed of palmitoyl di- and tricationic peptides: In vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 2008, 47, 10630–10636. [Google Scholar] [CrossRef]
- Makovitzki, A.; Shai, Y. pH-Dependent Antifungal Lipopeptides and Their Plausible Mode of Action. Biochemistry 2005, 44, 9775–9784. [Google Scholar] [CrossRef] [PubMed]
- Vylkova, S.; Li, X.S.; Berner, J.C.; Edgerton, M. Distinct antifungal mechanisms: β-defensins require Candida albicans Ssa1 protein, while Trk1p mediates activity of cysteine-free cationic peptides. Antimicrob. Agents Chemother. 2006, 50, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schielmann, M.; Szweda, P.; Gucwa, K.; Kawczyński, M.; Milewska, M.J.; Martynow, D.; Morschhäuser, J.; Milewski, S. Transport Deficiency Is the Molecular Basis of Candida albicans Resistance to Antifungal Oligopeptides. Front. Microbiol. 2017, 8, 2154. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Chadha, S.; Saraswat, D.; Bajwa, J.S.; Li, R.A.; Conti, H.R.; Edgerton, M. Histatin 5 uptake by Candida albicans utilizes polyamine transporters Dur3 and Dur31 proteins. J. Biol. Chem. 2011, 286, 43748–43758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, J.-I.; Cho, T.; Mitarai, M.; Iohara, K.; Hayama, K.; Abe, S.; Tanaka, Y. Antifungal activity in vitro and in vivo of a salmon protamine peptide and its derived cyclic peptide against Candida albicans. FEMS Yeast Res. 2017, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Lebeaux, D.; Ghigo, J.-M.; Beloin, C. Biofilm-Related Infections: Bridging the Gap between Clinical Management and Fundamental Aspects of Recalcitrance toward Antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [Green Version]
- Maciejewska, M.; Bauer, M.; Neubauer, D.; Kamysz, W.; Dawgul, M. Influence of Amphibian Antimicrobial Peptides and Short Lipopeptides on Bacterial Biofilms Formed on Contact Lenses. Materials 2016, 9, 873. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, L.; Banat, I.M.; Teixeira, J.; Oliveira, R. Biosurfactants: Potential applications in medicine. J. Antimicrob. Chemother. 2006, 57, 609–618. [Google Scholar] [CrossRef]
- Janek, T.; Łukaszewicz, M.; Krasowska, A. Antiadhesive activity of the biosurfactant pseudofactin II secreted by the Arctic bacterium Pseudomonas fluorescens BD5. BMC Microbiol. 2012, 12, 24. [Google Scholar] [CrossRef] [Green Version]
- Alves, D.; Magalhães, A.; Grzywacz, D.; Neubauer, D.; Kamysz, W.; Pereira, M.O. Co-immobilization of Palm and DNase I for the development of an effective anti-infective coating for catheter surfaces. Acta Biomater. 2016, 44, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, M. The Effects of Proteins on Bacterial Attachment to Polystyrene. J. Gen. Microbiol. 1976, 94, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toba, F.A.; Visai, L.; Trivedi, S.; Lowy, F.D. The role of ionic interactions in the adherence of the Staphylococcus epidermidis adhesin SdrF to prosthetic material. FEMS Microbiol. Lett. 2013, 338, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Kao, W.K.; Gagnon, P.M.; Vogel, J.P.; Chole, R.A. Surface charge modification decreases Pseudomonas aeruginosa adherence in vitro and bacterial persistence in an in vivo implant model. Laryngoscope 2017, 127, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Paduszynska, M.A.; Maciejewska, M.; Neubauer, D.; Golacki, K.; Szymukowicz, M.; Bauer, M.; Kamysz, W. Influence of Short Cationic Lipopeptides with Fatty Acids of Different Chain Lengths on Bacterial Biofilms Formed on Polystyrene and Hydrogel Surfaces. Pharmaceutics 2019, 11, 506. [Google Scholar] [CrossRef] [Green Version]
- Efron, N.; Brennan, N.A.; Chalmers, R.L.; Jones, L.; Lau, C.; Morgan, P.B.; Nichols, J.J.; Szczotka-Flynn, L.B.; Willcox, M.D. Thirty years of ‘quiet eye’ with etafilcon A contact lenses. Contact Lens Anterior Eye 2020, 43, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, A.; Cirioni, O.; Barchiesi, F.; Del Prete, M.S.; Fortuna, M.; Caselli, F.; Scalise, G. In vitro susceptibility tests for cationic peptides: Comparison of broth microdilution methods for bacteria that grow aerobically. Antimicrob. Agents Chemother. 2000, 44, 1694–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laverty, G.; McLaughlin, M.; Shaw, C.; Gorman, S.P.; Gilmore, B.F. Antimicrobial Activity of Short, Synthetic Cationic Lipopeptides. Chem. Biol. Drug Des. 2010, 75, 563–569. [Google Scholar] [CrossRef]
- Lohan, S.; Cameotra, S.S.; Bisht, G.S. Systematic Study of Non-Natural Short Cationic Lipopeptides as Novel Broad-Spectrum Antimicrobial Agents. Chem. Biol. Drug Des. 2013, 82, 557–566. [Google Scholar] [CrossRef]
- Hou, J.; Liu, Z.; Cao, S.; Wang, H.; Jiang, C.; Hussain, M.A.; Pang, S. Broad-Spectrum antimicrobial activity and low cytotoxicity against human cells of a peptide derived from bovine αS1-casein. Molecules 2018, 23, 1220. [Google Scholar] [CrossRef] [Green Version]
- Avrahami, D.; Shai, Y. A New Group of Antifungal and Antibacterial Lipopeptides Derived from Non-membrane Active Peptides Conjugated to Palmitic Acid. J. Biol. Chem. 2004, 279, 12277–12285. [Google Scholar] [CrossRef] [Green Version]
- Ermakova, E.; Zuev, Y. Effect of ergosterol on the fungal membrane properties. All-atom and coarse-grained molecular dynamics study. Chem. Phys. Lipids 2017, 209, 45–53. [Google Scholar] [CrossRef]
- Lichius, A.; Zeilinger, S. Application of membrane and cell wall selective fluorescent dyes for live-cell imaging of filamentous fungi. J. Vis. Exp. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Hickey, P.C.; Swift, S.R.; Roca, M.G.; Read, N.D. Live-cell Imaging of Filamentous Fungi Using Vital Fluorescent Dyes and Confocal Microscopy. Methods Microbiol. 2004, 34, 63–87. [Google Scholar]
- Park, S.C.; Kim, J.Y.; Shin, S.O.; Jeong, C.Y.; Kim, M.H.; Shin, S.Y.; Cheong, G.W.; Park, Y.; Hahm, K.S. Investigation of toroidal pore and oligomerization by melittin using transmission electron microscopy. Biochem. Biophys. Res. Commun. 2006, 343, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Iida, K.; Matsuo, T. Electron microscopic studies on mode of action of polymyxin. J. Bacteriol. 1969, 97, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deris, Z.Z.; Swarbrick, J.D.; Roberts, K.D.; Azad, M.A.K.; Akter, J.; Horne, A.S.; Nation, R.L.; Rogers, K.L.; Thompson, P.E.; Velkov, T.; et al. Probing the penetration of antimicrobial polymyxin lipopeptides into gram-negative bacteria. Bioconjug. Chem. 2014, 25, 750–760. [Google Scholar] [CrossRef]
- Arnusch, C.J.; Ulm, H.; Josten, M.; Shadkchan, Y.; Osherov, N.; Sahl, H.G.; Shai, Y. Ultrashort peptide bioconjugates are exclusively antifungal agents and synergize with cyclodextrin and amphotericin B. Antimicrob. Agents Chemother. 2012, 56, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Neue, U.D.; Niederlaender, C.L.; Peterson, J.S. Liquid Chromatography Stationary Phases with Reduced Silanol Interactions. U.S. Patent US5374755A, 20 December 1994. [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, 9th ed.; CLSI: Wayne, PA, USA, 2012; Volume 32, ISBN 1562387839. [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI). Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Approved Standards, 2nd ed.; CLSI Document M27-2A 2002; CLSI: Wayne, PA, USA, 2002; Volume 22, ISBN 1562384694. [Google Scholar]
- Jones, R.N.; Wilson, H.W.; Novick, W.J.; Barry, A.L.; Thornsberry, C. In vitro evaluation of CENTA, a new beta-lactamase-susceptible chromogenic cephalosporin reagent. J. Clin. Microbiol. 1982, 15, 954–958. [Google Scholar] [CrossRef] [Green Version]
- Abbassi, F.; Lequin, O.; Piesse, C.; Goasdoué, N.; Foulon, T.; Nicolas, P.; Ladram, A. Temporin-SHf, a new type of phe-rich and hydrophobic ultrashort antimicrobial peptide. J. Biol. Chem. 2010, 285, 16880–16892. [Google Scholar] [CrossRef] [Green Version]
- Arcidiacono, S.; Soares, J.W.; Meehan, A.M.; Marek, P.; Kirby, R. Membrane permeability and antimicrobial kinetics of cecropin P1 against Escherichia coli. J. Pept. Sci. 2009, 15, 398–403. [Google Scholar] [CrossRef]
- Ilić, N.; Novković, M.; Guida, F.; Xhindoli, D.; Benincasa, M.; Tossi, A.; Juretić, D. Selective antimicrobial activity and mode of action of adepantins, glycine-rich peptide antibiotics based on anuran antimicrobial peptide sequences. Biochim. Biophys. Acta 2013, 1828, 1004–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romani, A.A.; Baroni, M.C.; Taddei, S.; Ghidini, F.; Sansoni, P.; Cavirani, S.; Cabassi, C.S. In vitro activity of novel in silico -developed antimicrobial peptides against a panel of bacterial pathogens. J. Pept. Sci. 2013, 19, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Periole, X.; Marrink, S.-J. The Martini Coarse-Grained Force Field. In Biomolecular Simulations; Humana Press: Totowa, NJ, USA, 2013; pp. 533–565. [Google Scholar]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; De Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chugunov, A.; Pyrkova, D.; Nolde, D.; Polyansky, A.; Pentkovsky, V.; Efremov, R. Lipid-II forms potential “landing terrain” for lantibiotics in simulated bacterial membrane. Sci. Rep. 2013, 3, 1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winger, M.; Trzesniak, D.; Baron, R.; Van Gunsteren, W.F. On using a too large integration time step in molecular dynamics simulations of coarse-grained molecular models. Phys. Chem. Chem. Phys. 2009, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gapsys, V.; de Groot, B.L.; Briones, R. Computational analysis of local membrane properties. J. Comput. Aided Mol. Des. 2013, 27, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Eckhard, L.H.; Houri-Haddad, Y.; Sol, A.; Zeharia, R.; Shai, Y.; Beyth, S.; Domb, A.J.; Bachrach, G.; Beyth, N. Sustained Release of Antibacterial Lipopeptides from Biodegradable Polymers against Oral Pathogens. PLoS ONE 2016, 11, e0162537. [Google Scholar] [CrossRef]
- Yim, V.V.; Kavianinia, I.; Cameron, A.J.; Harris, P.W.R.; Brimble, M.A. Direct synthesis of cyclic lipopeptides using intramolecular native chemical ligation and thiol-ene CLipPA chemistry. Org. Biomol. Chem. 2020, 18, 2838–2844. [Google Scholar] [CrossRef]
- Knerr, P.J.; Tzekou, A.; Ricklin, D.; Qu, H.; Chen, H.; Van Der Donk, W.A.; Lambris, J.D. Synthesis and activity of thioether-containing analogues of the complement inhibitor compstatin. ACS Chem. Biol. 2011, 6, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.S. The cyclization of peptides and depsipeptides. J. Pept. Sci. 2003, 9, 471–501. [Google Scholar] [CrossRef] [PubMed]
- Tsubery, H.; Ofek, I.; Cohen, S.; Fridkin, M. Structure—Function studies of Polymyxin B nonapeptide: Implications to sensitization of Gram-negative bacteria. J. Med. Chem. 2000, 43, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Ballantine, R.D.; Li, Y.X.; Qian, P.Y.; Cochrane, S.A. Rational design of new cyclic analogues of the antimicrobial lipopeptide tridecaptin A1. Chem. Commun. 2018, 54, 10634–10637. [Google Scholar] [CrossRef] [PubMed]
- Jelokhani-Niaraki, M.; Kondejewski, L.H.; Wheaton, L.C.; Hodges, R.S. Effect of ring size on conformation and biological activity of cyclic cationic antimicrobial peptides. J. Med. Chem. 2009, 52, 2090–2097. [Google Scholar] [CrossRef] [Green Version]
- Mulder, M.P.C.; Kruijtzer, J.A.W.; Breukink, E.J.; Kemmink, J.; Pieters, R.J.; Liskamp, R.M.J. Synthesis and evaluation of novel macrocyclic antifungal peptides. Bioorganic Med. Chem. 2011, 19, 6505–6517. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
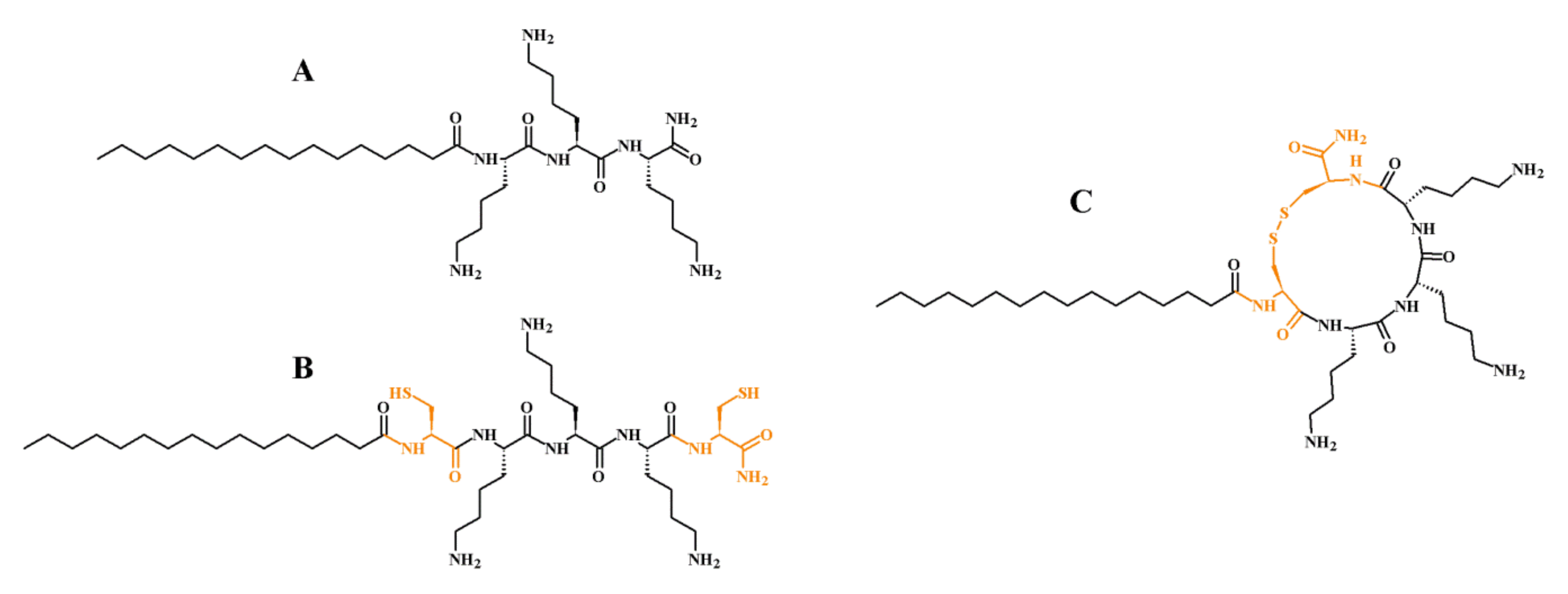
| Peptide Code | Sequence | Av. Mass (Da) | Monoisotopic Mass (Da) | MS Analysis | Net Charge | t’R (min) | ∆tR (min) * | ||
|---|---|---|---|---|---|---|---|---|---|
| z a | m/z Calc. | m/z Found | |||||||
| L1 | C16-KKK-NH2 | 639.97 | 639.54 | 1 | 640.55 | 640.69 | +3 | 16.14 | +1.91 |
| 2 | 320.78 | 321.01 | |||||||
| 3 | 214.19 | 214.39 | |||||||
| C1 | C16-CKKKC-NH2 | 844.24 | 843.54 | 1 | 844.55 | 844.70 | +3 | 18.05 | |
| 2 | 422.78 | 423.04 | |||||||
| 3 | 282.19 | 282.43 | |||||||
| L2 | C16-KKKK-NH2 | 768.14 | 767.64 | 1 | 768.64 | 768.86 | +4 | 14.77 | +1.18 |
| 2 | 384.83 | 385.18 | |||||||
| 3 | 256.89 | 257.25 | |||||||
| 4 | 192.92 | - | |||||||
| C2 | C16-CKKKKC-NH2 | 972.42 | 971.64 | 1 | 972.65 | 972.75 | +4 | 15.95 | |
| 2 | 486.83 | 487.19 | |||||||
| 3 | 324.89 | 325.20 | |||||||
| 4 | 243.92 | - | |||||||
| L3 | C16-RKKK-NH2 | 796.15 | 795.64 | 1 | 796.65 | 796.84 | +4 | 15.21 | +1.30 |
| 2 | 398.83 | 399.14 | |||||||
| 3 | 266.22 | 266.59 | |||||||
| 4 | 199.92 | - | |||||||
| C3 | C16-CRKKKC-NH2 | 1000.43 | 999.65 | 1 | 1000.65 | 1000.83 | +4 | 16.51 | |
| 2 | 500.83 | 501.19 | |||||||
| 3 | 334.22 | 334.62 | |||||||
| 4 | 250.92 | - | |||||||
| L4 | C16-KRKK-NH2 | 796.15 | 795.64 | 1 | 796.65 | 769.95 | +4 | 15.18 | +0.92 |
| 2 | 398.83 | 399.27 | |||||||
| 3 | 266.22 | 266.54 | |||||||
| 4 | 199.92 | - | |||||||
| C4 | C16-CKRKKC-NH2 | 1000.43 | 999.65 | 1 | 1000.65 | 1000.86 | +4 | 16.10 | |
| 2 | 500.83 | 501.07 | |||||||
| 3 | 334.22 | 334.68 | |||||||
| 4 | 250.92 | - | |||||||
| L5 | C16-KKRK-NH2 | 796.15 | 795.64 | 1 | 796.65 | 796.80 | +4 | 15.01 | +1.15 |
| 2 | 398.83 | 399.24 | |||||||
| 3 | 266.22 | 266.53 | |||||||
| 4 | 199.92 | - | |||||||
| C5 | C16-CKKRKC-NH2 | 1000.43 | 999.65 | 1 | 1000.65 | 1000.77 | +4 | 16.16 | |
| 2 | 500.83 | 501.11 | |||||||
| 3 | 334.22 | 334.45 | |||||||
| 4 | 250.92 | - | |||||||
| L6 | C16-KKKR-NH2 | 796.15 | 795.64 | 1 | 796.65 | 796.81 | +4 | 14.89 | +1.43 |
| 2 | 398.83 | 399.11 | |||||||
| 3 | 266.22 | 266.42 | |||||||
| 4 | 199.92 | - | |||||||
| C6 | C16-CKKKRC-NH2 | 1000.43 | 999.65 | 1 | 1000.65 | 1000.80 | +4 | 16.32 | |
| 2 | 500.83 | 501.11 | |||||||
| 3 | 334.22 | 334.51 | |||||||
| 4 | 250.92 | 251.11 | |||||||
| L7 | C16-RRKK-NH2 | 824.17 | 823.65 | 1 | 824.66 | 824.87 | +4 | 15.58 | +1.25 |
| 2 | 412.83 | 413.21 | |||||||
| 3 | 275.56 | 276.02 | |||||||
| 4 | 206.92 | - | |||||||
| C7 | C16-CRRKKC-NH2 | 1028.44 | 1027.65 | 1 | 1028.66 | 1028.79 | +4 | 16.83 | |
| 2 | 514.83 | 515.20 | |||||||
| 3 | 343.56 | 343.89 | |||||||
| 4 | 257.92 | - | |||||||
| Peptide Code | MIC (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| S. aureus | S. epidermidis | E. coli | P. aeruginosa | C. albicans | C. glabrata | C. lipolytica | C. tropicalis | |
| L1 | 32 * | 8 | 16 | 64 | 32 | 32 | 16 | 8 |
| C1 | 8 * | 4 | 16 | 256 | 8 | 4 | 4 | 4 |
| L2 | 16 | 8 | 32 | 16 | 32 | 64 | 8 | 4 |
| C2 | 32 | 8 | 64 | 32 | 4 | 4 | 2 | 2 |
| L3 | 16 * | 8 | 8 | 32 | 8 | 64 | 8 | 8 |
| C3 | 8 * | 8 | 16 | 512 | 2 | 8 | 4 | 2 |
| L4 | 8 | 8 | 8 | 32 | 16 | 32 | 1 | 2 |
| C4 | 16 | 8 | 32 | 256 | 2 | 4 | 1 | 2 |
| L5 | 16 * | 8 | 8 | 64 | 32 | 64 | 8 | 8 |
| C5 | 8 * | 4 | 16 | 256 | 16 | 8 | 4 | 4 |
| L6 | 16 * | 4 | 8 | 64 | 32 | 64 | 8 | 8 |
| C6 | 16 * | 8 | 32 | 128 | 4 | 4 | 2 | 2 |
| L7 | 4 | 4 | 8 | 64 | 32 | 16 | 8 | 8 |
| C7 | 8 | 4 | 16 | 256 | 16 | 4 | 4 | 4 |
| Lmean ** | 15.4 | 6.9 | 12.6 † | 48.0 † | 26.3 † | 48.0 † | 8.1 † | 6.6 † |
| Cmean ** | 13.7 | 6.3 | 27.4 † | 242.3 † | 7.4 † | 5.1 † | 3.0 † | 2.9 † |
| p-value | 0.633 | 0.701 | 0.030 | 0.015 | 0.009 | 0.002 | 0.025 | 0.025 |
| Peptide Code | MBEC (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| S. aureus | S. epidermidis | E. coli | P. aeruginosa | C. albicans | C. glabrata | C. lipolytica | C. tropicalis | |
| L1 | 64 * | 32 | 64 | 128 | 256 | 256 | 64 | 128 |
| C1 | 64 * | 32 | 256 | 256 | 256 | 256 | 64 | 128 |
| L2 | 64 | 32 | 64 | >256 | 256 | 256 | 64 | 128 |
| C2 | 64 | 32 | 64 | > 256 | 64 | 64 | 16 | 32 |
| L3 | 64 * | 32 | 128 | 256 | 128 | 128 | 32 | 32 |
| C3 | 128 * | 128 | 256 | 256 | 32 | 32 | 16 | 32 |
| L4 | 64 | 32 | 64 | >256 | 128 | 128 | 32 | 64 |
| C4 | 64 | 32 | 64 | >256 | 32 | 64 | 16 | 32 |
| L5 | 32 * | 32 | 64 | 256 | 256 | 256 | 64 | 128 |
| C5 | 128 * | 64 | 128 | 256 | 64 | 64 | 16 | 32 |
| L6 | 32 * | 32 | 256 | 256 | 128 | 128 | 32 | 128 |
| C6 | 64 * | 128 | >256 | >256 | 64 | 64 | 16 | 32 |
| L7 | 128 | 64 | 256 | 256 | 64 | 128 | 16 | 64 |
| C7 | 128 | 64 | 256 | 256 | 64 | 64 | 16 | 32 |
| Lmean ** | 64.0 | 36.6 | - | - | 173.7 † | 182.9 † | 43.4 † | 96.0 † |
| Cmean ** | 91.4 | 68.6 | - | - | 82.3 † | 86.9 † | 22.9 † | 45.7 † |
| p-value | 0.131 | 0.160 | - | - | 0.035 | 0.018 | 0.048 | 0.041 |
| Peptide Code | MBIC (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| S. aureus | S. epidermidis | E. coli | P. aeruginosa | C. albicans | C. glabrata | C. lipolytica | C. tropicalis | |
| L1 | 32 | 16 | 32 | 32 | 128 | 128 | 32 | 16 |
| C1 | 16 | 16 | 64 | 128 | 32 | 32 | 16 | 32 |
| L2 | 32 | 16 | 32 | 64 | 128 | 128 | 32 | 16 |
| C2 | 16 | 4 | 32 | 64 | 16 | 32 | 8 | 8 |
| L3 | 32 | 16 | 32 | 64 | 64 | 64 | 16 | 16 |
| C3 | 16 | 8 | 64 | 64 | 8 | 8 | 2 | 4 |
| L4 | 16 | 8 | 32 | 32 | 32 | 64 | 16 | 8 |
| C4 | 16 | 8 | 32 | 32 | 8 | 16 | 4 | 4 |
| L5 | 32 | 16 | 32 | 64 | 64 | 128 | 32 | 16 |
| C5 | 32 | 16 | 32 | 64 | 16 | 16 | 8 | 8 |
| L6 | 32 | 8 | 32 | 64 | 64 | 64 | 32 | 16 |
| C6 | 32 | 8 | 64 | 64 | 16 | 8 | 8 | 8 |
| L7 | 16 | 16 | 32 | 64 | 32 | 32 | 16 | 8 |
| C7 | 16 | 16 | 128 | 128 | 16 | 16 | 8 | 8 |
| Lmean * | 20.6 | 13.7 | 32.0 | 54.9 | 73.1 † | 86.9 † | 25.1 † | 13.7 † |
| Cmean * | 13.7 | 10.9 | 59.4 | 77.7 | 16.0 † | 18.3 † | 7.7 † | 10.3 † |
| p-value | 0.201 | 0.338 | 0.085 | 0.307 | 0.003 | 0.003 | 0.004 | 0.097 |
| Peptide Code | HC50 (μg/mL) | HaCaT IC50 (μg/mL) | HeLa IC50 (μg/mL) | SI (IC50 HaCaT/ IC50 HeLa) |
|---|---|---|---|---|
| L1 | 90.0 ± 6.5 | 5.2 ± 0.6 | 3.9 ± 1.1 | 1.3 |
| C1 | 43.9 ± 3.5 | 23.6 ± 6.0 | 25.5 ± 2.9 | 0.9 |
| L2 | 105.9 ± 8.0 | 23.5 ± 1.3 | 12.8 ± 1.1 | 1.8 |
| C2 | 30.6 ± 1.1 | 26.9 ± 1.9 | 17.9 ± 2.1 | 1.5 |
| L3 | 79.1 ± 6.1 | 23.6 ± 3.4 | 8.4 ± 1.7 | 2.8 |
| C3 | 66.5 ± 14.8 | 36.8 ± 2.7 | 38.8 ± 4.4 | 0.9 |
| L4 | 79.8 ± 9.0 | 4.3 ± 0.9 | 3.0 ± 0.6 | 1.4 |
| C4 | 122.4 ± 16.5 | 33.8 ± 3.1 | 27.5 ± 3.4 | 1.2 |
| L5 | 96.7 ± 1.4 | 2.4 ± 0.4 | 6.2 ± 2.7 | 0.4 |
| C5 | 97.0 ± 15.4 | 12.9 ± 4.6 | 21.9 ± 4.5 | 0.6 |
| L6 | 89.7 ± 2.4 | 7.5 ± 0.8 | 3.6 ± 0.5 | 2.1 |
| C6 | 71.0 ± 6.4 | 45.7 ± 13.5 | 30.4 ± 2.5 | 1.5 |
| L7 | 65.4 ± 1.5 | 8.1 ± 1.1 | 3.0 ± 0.4 | 2.7 |
| C7 | 92.0 ± 9.6 | 21.3 ± 8.2 | 20.9 ± 2.1 | 1.0 |
| Lmean * | 86.7 | 10.7 † | 5.8 † | 1.8 |
| Cmean * | 74.7 | 28.7 † | 26.1 † | 1.1 |
| p-value | 0.375 | 0.013 | 0.00002 | 0.064 |
| Bacteria | S. aureus | S. epidermidis | E. coli | P. aeruginosa | |||||
|---|---|---|---|---|---|---|---|---|---|
| hRBCs | Mean | 6.17 | 8.52 | 13.60 | 13.51 | 8.61 † | 3.60 † | 2.41 † | 0.43 † |
| p-value | 0.2964 | 0.9777 | 0.0039 | 0.0022 | |||||
| HaCaT | Mean | 1.37 | 2.04 | 1.61 † | 4.62 † | 0.97 | 1.26 | 0.39 | 0.23 |
| p-value | 0.3452 | 0.0003 | 0.1600 | 0.7015 | |||||
| Fungi | C. albicans | C. glabrata | C. lipolytica | C. tropicalis | |||||
| hRBCs | Mean | 4.12 † | 19.59 † | 2.17 † | 15.77 † | 20.00 | 35.44 | 17.00 | 29.07 |
| p-value | 0.0106 | 0.0022 | 0.0553 | 0.1599 | |||||
| HaCaT | Mean | 0.67 † | 8.36 † | 0.24 † | 6.29 † | 1.82 † | 13.39 † | 1.98 † | 12.29 † |
| p-value | 0.0060 | 0.0022 | 0.0033 | 0.0049 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neubauer, D.; Jaśkiewicz, M.; Sikorska, E.; Bartoszewska, S.; Bauer, M.; Kapusta, M.; Narajczyk, M.; Kamysz, W. Effect of Disulfide Cyclization of Ultrashort Cationic Lipopeptides on Antimicrobial Activity and Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 7208. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197208
Neubauer D, Jaśkiewicz M, Sikorska E, Bartoszewska S, Bauer M, Kapusta M, Narajczyk M, Kamysz W. Effect of Disulfide Cyclization of Ultrashort Cationic Lipopeptides on Antimicrobial Activity and Cytotoxicity. International Journal of Molecular Sciences. 2020; 21(19):7208. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197208
Chicago/Turabian StyleNeubauer, Damian, Maciej Jaśkiewicz, Emilia Sikorska, Sylwia Bartoszewska, Marta Bauer, Małgorzata Kapusta, Magdalena Narajczyk, and Wojciech Kamysz. 2020. "Effect of Disulfide Cyclization of Ultrashort Cationic Lipopeptides on Antimicrobial Activity and Cytotoxicity" International Journal of Molecular Sciences 21, no. 19: 7208. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197208