The Immune Microenvironment in Pancreatic Cancer

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
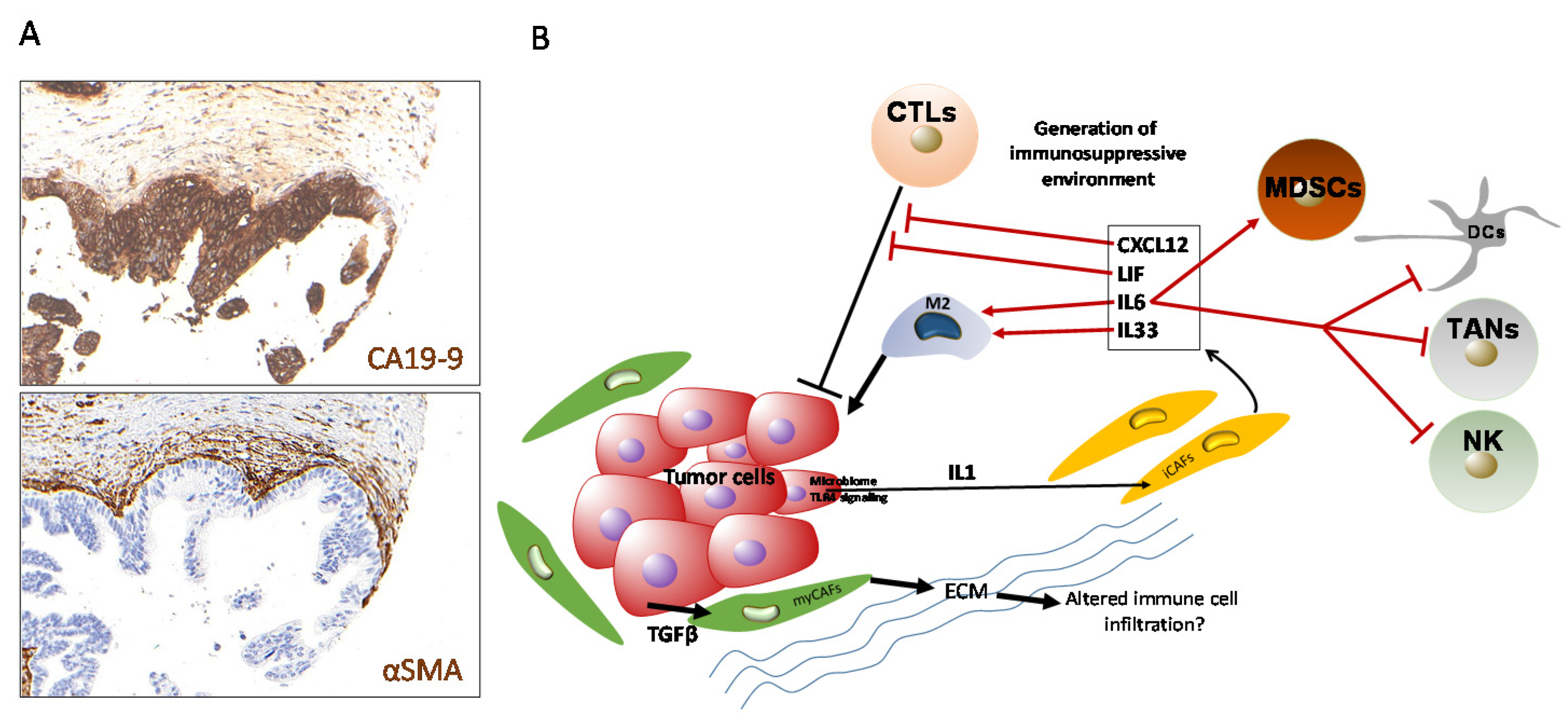{kind=link}
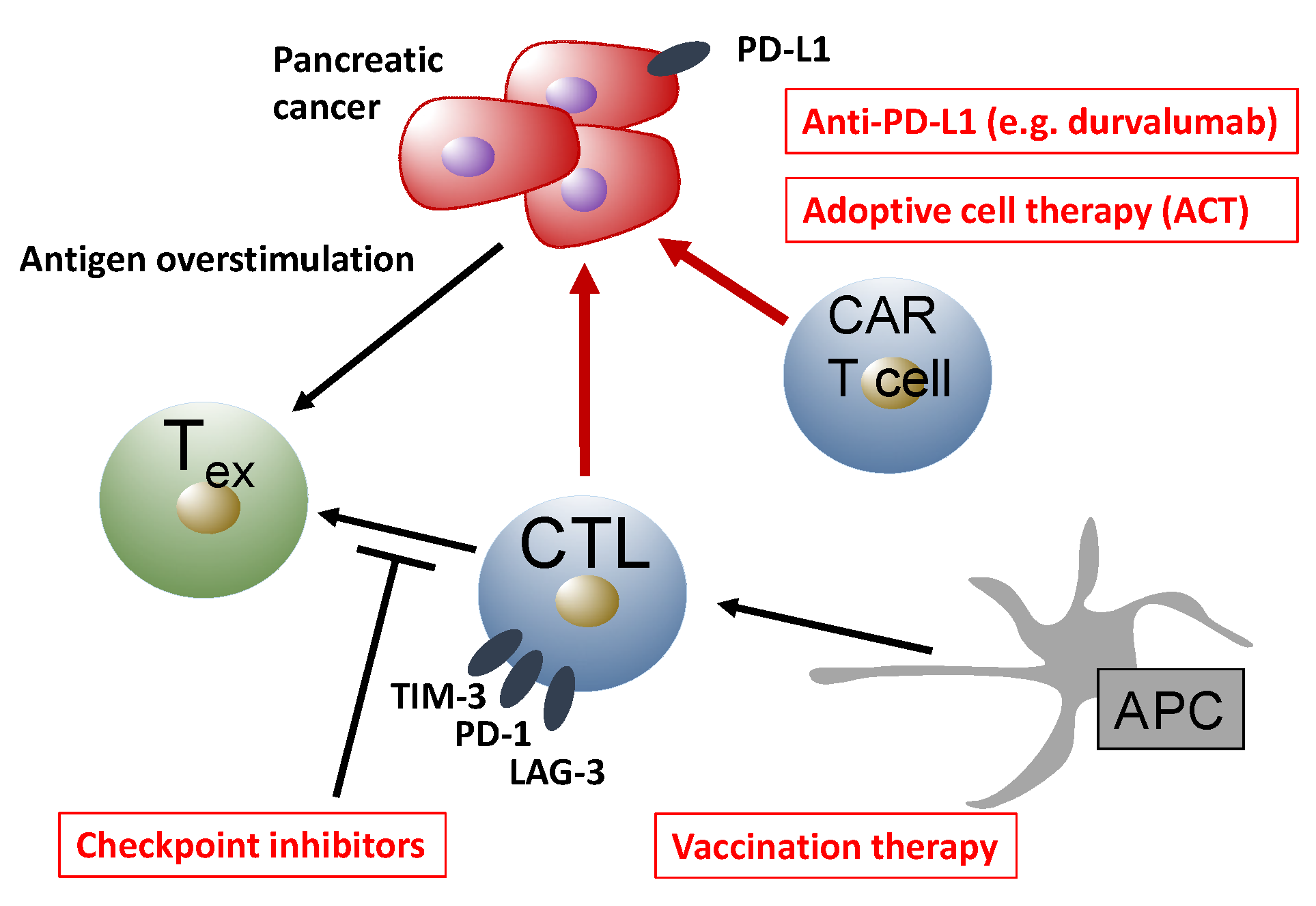{kind=link}
Abstract
:1. Pancreatic Cancer: Clinical Situation
2. Role of Innate Immune Cells in PDAC: Macrophages and Myeloid-Derived Suppressor Cells (MDSCs)
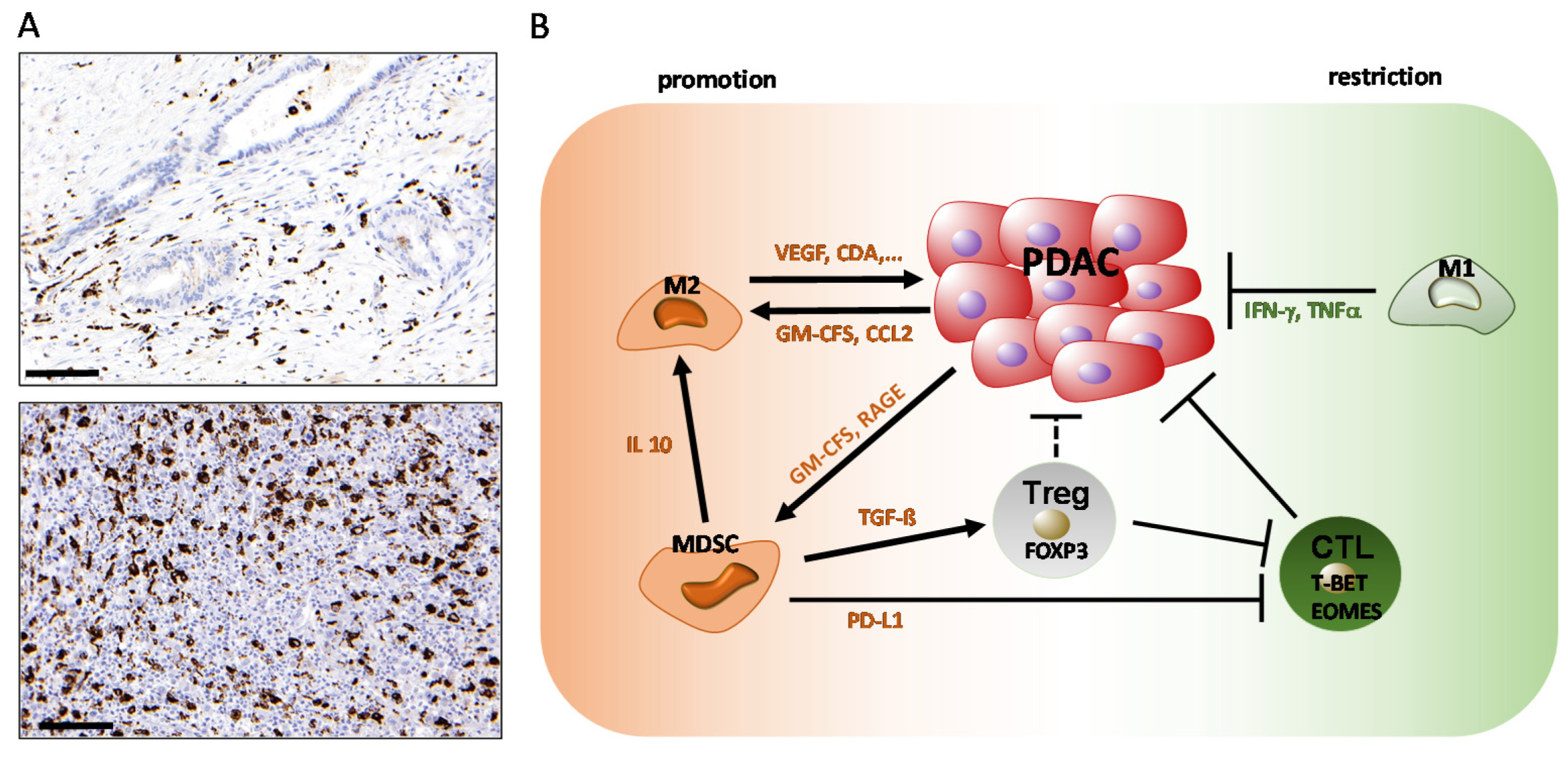
2.1. The Role of Macrophages in PDAC
2.2. The Role of Myeloid-Derived Suppressor Cells
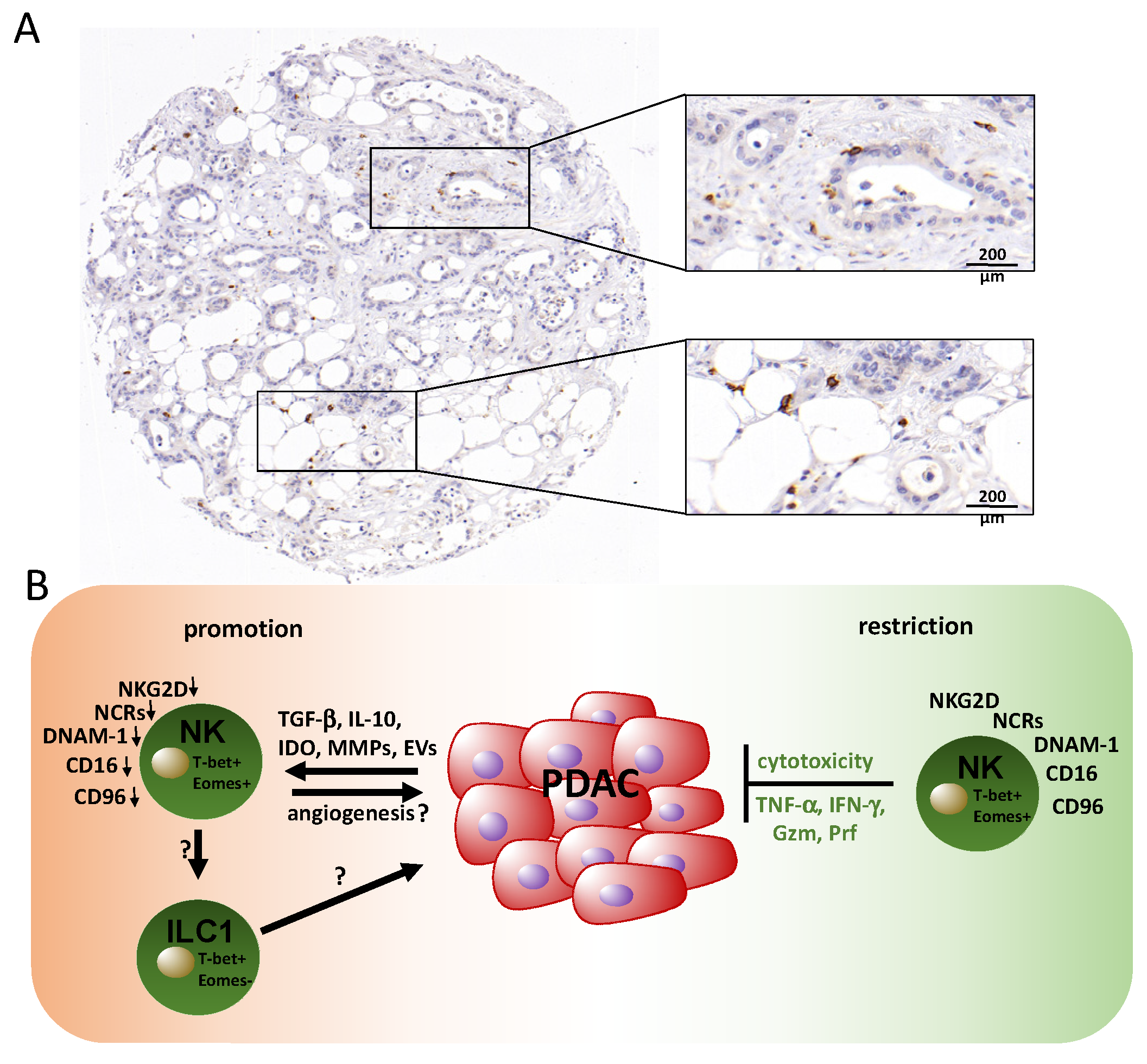
3. Contribution of Natural Killer (NK) Cells in PDAC
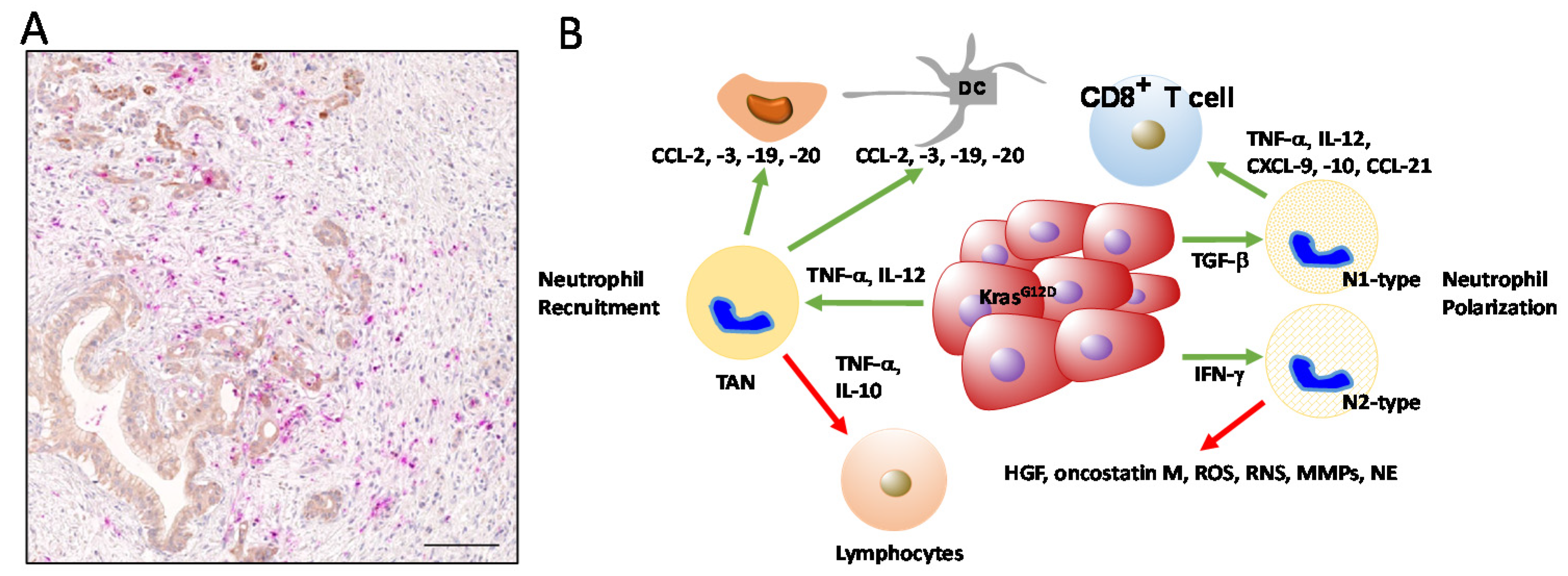
4. Contribution of Neutrophils in PDAC
5. The Role of Adaptive T Cells in PDAC
5.1. Accumulation of T Cells
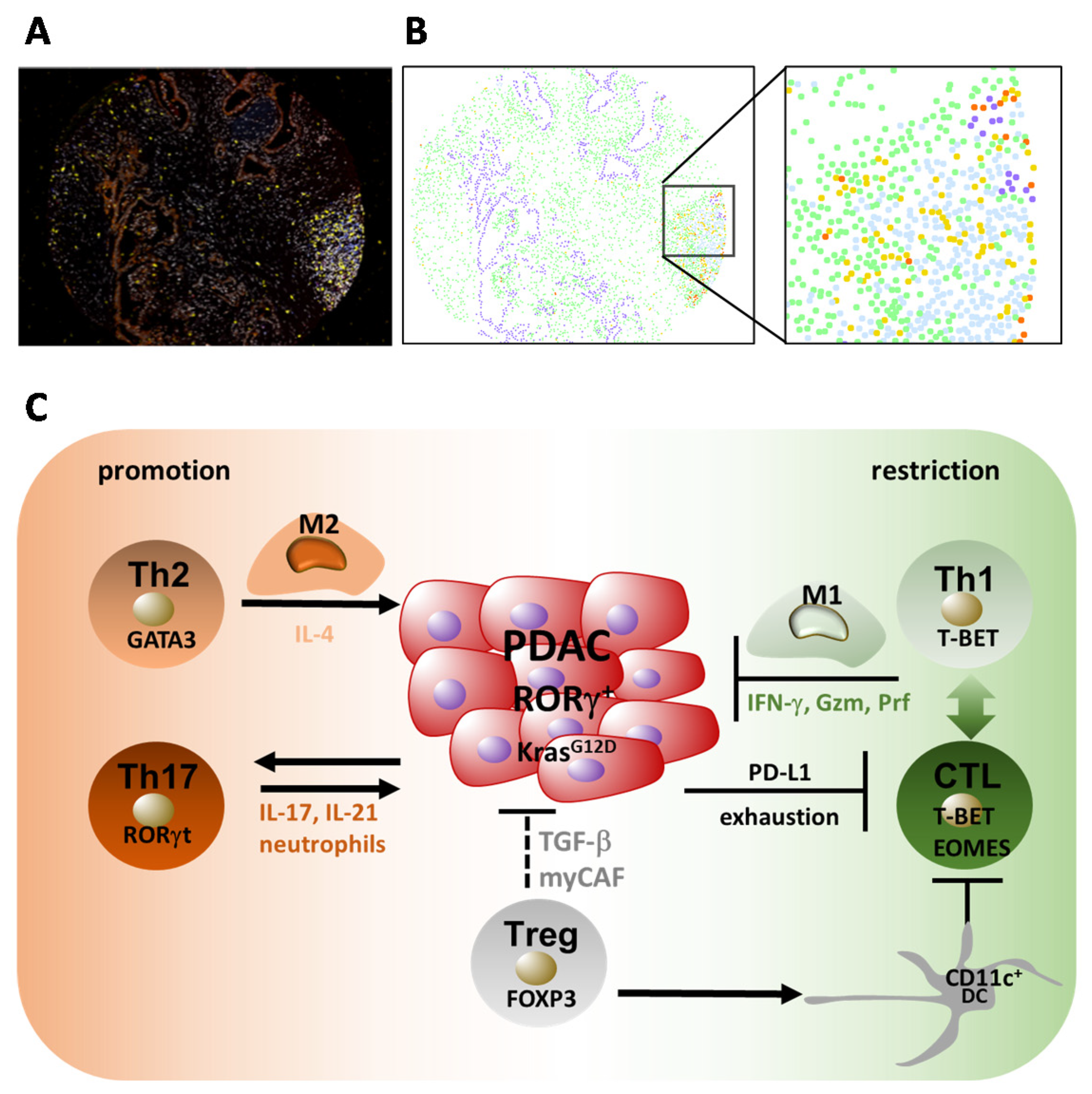
5.2. Th1 and Th2 Responses in PDAC
5.3. Th17 and Treg Responses in PDAC
5.4. Regulatory T Cells (Tregs) and PDAC
5.5. CD8+ T Cell Responses in PDAC
6. Cancer-Associated Fibroblasts (CAFs) as Immunological Modulators in PDAC
7. Immunotherapy in PDAC
8. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2018, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Versteijne, E.; Suker, M.; Groothuis, K.; Akkermans-Vogelaar, J.M.; Besselink, M.G.; Bonsing, B.A.; Buijsen, J.; Busch, O.R.; Creemers, G.-J.M.; Van Dam, R.M.; et al. Preoperative Chemoradiotherapy Versus Immediate Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Results of the Dutch Randomized Phase III PREOPANC Trial. J. Clin. Oncol. 2020, 38, 1763–1773. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Michl, P.; Gress, T.M. Current concepts and novel targets in advanced pancreatic cancer. Gut 2012, 62, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardiere, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Biankin, A.V.; Maitra, A. Subtyping Pancreatic Cancer. Cancer Cell 2015, 28, 411–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Nowak, J.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.-H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.-B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2017, 24, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.K.; Nones, K.; Quek, K. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Bender, R.J.; Halverson, D.; Rahib, L.; Hendifar, A.; Mikhail, S.; Chung, V.; Picozzi, V.J.; Sohal, D.; Blais, E.M.; et al. Molecular Profiling of Patients with Pancreatic Cancer: Initial Results from the Know Your Tumor Initiative. Clin. Cancer Res. 2018, 24, 5018–5027. [Google Scholar] [CrossRef] [Green Version]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2019, 17, 108–123. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Hechtman, J. Tumour response to TRK inhibition in a patient with pancreatic adenocarcinoma harbouring an NTRK gene fusion. Ann. Oncol. 2019, 30, viii36–viii40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.R.; Williamson, L.M.; Topham, J.T.; Lee, M.K.; Goytain, A.; Ho, J.; Denroche, R.E.; Jang, G.-H.; Pleasance, E.D.; Shen, Y.; et al. NRG1 Gene Fusions Are Recurrent, Clinically Actionable Gene Rearrangements in KRAS Wild-Type Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2019, 25, 4674–4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heining, C.; Horak, P.; Uhrig, S.; Codo, P.L.; Klink, B.; Hutter, B.; Fröhlich, M.; Bonekamp, D.; Richter, D.; Steiger, K.; et al. NRG1 Fusions in KRAS Wild-Type Pancreatic Cancer. Cancer Discov. 2018, 8, 1087–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [Green Version]
- Seppälä, T.T.; Zimmerman, J.W.; Sereni, E.; Plenker, D.; Suri, R.; Rozich, N.; Blair, A.; Thomas, D.L.; Teinor, J.; Javed, A.; et al. Patient-derived Organoid Pharmacotyping is a Clinically Tractable Strategy for Precision Medicine in Pancreatic Cancer. Ann. Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.M.; McKernin, S.E.; Khorana, A.A. Evaluating Susceptibility to Pancreatic Cancer: ASCO Clinical Practice Provisional Clinical Opinion Summary. J. Oncol. Pract. 2019, 15, 108–111. [Google Scholar] [CrossRef]
- Goggins, M.; Overbeek, K.A.; Brand, R.; Syngal, S.; Del Chiaro, M.; Bartsch, D.K.; Bassi, C.; Carrato, A.; Farrell, J.; Fishman, E.K.; et al. Management of patients with increased risk for familial pancreatic cancer: Updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2019, 69, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Corrales, L.; Gajewski, T.F. Innate Immune Recognition of Cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef]
- Stone, M.L.; Beatty, G.L. Cellular determinants and therapeutic implications of inflammation in pancreatic cancer. Pharmacol. Ther. 2019, 201, 202–213. [Google Scholar] [CrossRef]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Bracarda, S.; Nabissi, M.; Massari, F.; Conti, A.; Bria, E.; Tortora, G.; Santoni, G.; Cascinu, S. CXC and CC Chemokines as Angiogenic Modulators in Nonhaematological Tumors. BioMed. Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; LaVine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Bolli, E.; Movahedi, K.; Laoui, D.; Van Ginderachter, J.A. Novel insights in the regulation and function of macrophages in the tumor microenvironment. Curr. Opin. Oncol. 2017, 29, 55–61. [Google Scholar] [CrossRef]
- Di Caro, G.; Cortese, N.; Castino, G.F.; Grizzi, F.; Gavazzi, F.; Ridolfi, C.; Capretti, G.; Mineri, R.; Todoric, J.; Zerbi, A.; et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut 2015, 65, 1710–1720. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Q.; Peng, J.; Wang, M.; Li, T.; Liu, J.; Cui, M.; Zhang, X.; Gao, X.; Liao, Q.; et al. CXCL5 overexpression predicts a poor prognosis in pancreatic ductal adenocarcinoma and is correlated with immune cell infiltration. J. Cancer 2020, 11, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Hang, J.-J.; Han, T.; Zhuo, M.; Jiao, F.; Wang, L.-W. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumor Boil. 2016, 37, 8657–8664. [Google Scholar] [CrossRef] [PubMed]
- Griesmann, H.; Drexel, C.; Milosevic, N.; Sipos, B.; Rosendahl, J.; Gress, T.M.; Michl, P. Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer. Gut 2016, 66, 1278–1285. [Google Scholar] [CrossRef]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2017, 67, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Edenfield, B.; Zhang, L.; Dawson, D.W.; Storz, P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2014, 5, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Filippini, D.; D’Agosto, S.; Delfino, P.; Simbolo, M.; Piro, G.; Rusev, B.; Veghini, L.; Cantù, C.; Lupo, F.; Ugel, S.; et al. Immunoevolution of mouse pancreatic organoid isografts from preinvasive to metastatic disease. Sci. Rep. 2019, 9, 12286. [Google Scholar] [CrossRef] [Green Version]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.-S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nature 2016, 18, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2013, 33, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Recht, L.; Strober, S. The Promise of Targeting Macrophages in Cancer Therapy. Clin. Cancer Res. 2017, 23, 3241–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalbasi, A.; Komar, C.A.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2016, 23, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2012, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, A.; AlShehri, M.S.A.; Miller, K.L.; Sherwin, C.M.; Travers, J.B.; Sahu, R.P. Myeloid-Derived Suppressor Cells and Pancreatic Cancer: Implications in Novel Therapeutic Approaches. Cancers 2019, 11, 1627. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Trovato, R.; Fiore, A.; Sartori, S.; Canè, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; de Sanctis, F.; Poffe, O.; et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J. Immunother. Cancer 2019, 7, 255. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. CII 2009, 58, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. CII 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, J.; Brooks, T.R.; Duggan, M.C.; Paul, B.K.; Pan, X.; Wei, L.; Abrams, Z.; Luedke, E.; Lesinski, G.B.; Mundy-Bosse, B.; et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol. Immunother. CII 2015, 64, 149–159. [Google Scholar] [CrossRef]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernon, P.J.; Loux, T.J.; Schapiro, N.E.; Kang, R.; Muthuswamy, R.; Kalinski, P.; Tang, D.; Lotze, M.T.; Zeh, H.J., 3rd. The receptor for advanced glycation end products promotes pancreatic carcinogenesis and accumulation of myeloid-derived suppressor cells. J. Immunol. 2013, 190, 1372–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2016, 36, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Choueiry, F.; Torok, M.; Shakya, R.; Agrawal, K.; Deems, A.; Benner, B.; Hinton, A.; Shaffer, J.; Blaser, B.W.; Noonan, A.M.; et al. CD200 promotes immunosuppression in the pancreatic tumor microenvironment. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget 2015, 7, 1168–1184. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Siret, C.; Collignon, A.; Silvy, F.; Robert, S.; Cheyrol, T.; André, P.; Rigot, V.; Iovanna, J.; Van De Pavert, S.; Lombardo, D.; et al. Deciphering the Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells in Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef]
- Stromnes, I.M.; Brockenbrough, J.S.; Izeradjene, K.; Carlson, M.A.; Cuevas, C.; Simmons, R.M.; Greenberg, P.D.; Hingorani, S.R.; Brockenbrough, S. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014, 63, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, N.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Di Magliano, M.P. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2016, 66, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.-G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Boil. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, N.; Langers, I.; Renoux, V.M.; Thiry, M.; Delvenne, P. Natural killer cells: Role in local tumor growth and metastasis. Boil. Targets Ther. 2012, 6, 73–82. [Google Scholar] [CrossRef]
- Lanier, L.L.; Testi, R.; Bindl, J.; Phillips, J.H. Identity of Leu-19 (CD56) leukocyte differentiation antigen and neural cell adhesion molecule. J. Exp. Med. 1989, 169, 2233–2238. [Google Scholar] [CrossRef] [Green Version]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Dent, P. Prior exposure of pancreatic tumors to sorafenib + vorinostat enhances the efficacy of an anti-PD-1 antibody. Cancer Biol. Ther. 2019, 20, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Gürlevik, E.; Fleischmann-Mundt, B.; Brooks, J.; Demir, I.E.; Steiger, K.; Ribback, S.; Yevsa, T.; Woller, N.; Kloos, A.; Ostroumov, D.; et al. Administration of Gemcitabine After Pancreatic Tumor Resection in Mice Induces an Antitumor Immune Response Mediated by Natural Killer Cells. Gastroenterology 2016, 151, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Liu, S.; Dhar, P.; Wu, J.D. NK Cell Plasticity in Cancer. J. Clin. Med. 2019, 8, 1492. [Google Scholar] [CrossRef] [Green Version]
- Carrega, P.; Bonaccorsi, I.; Di Carlo, E.; Morandi, B.; Paul, P.; Rizzello, V.; Cipollone, G.; Navarra, G.; Mingari, M.C.; Moretta, L.; et al. CD56brightPerforinlow Noncytotoxic Human NK Cells Are Abundant in Both Healthy and Neoplastic Solid Tissues and Recirculate to Secondary Lymphoid Organs via Afferent Lymph. J. Immunol. 2014, 192, 3805–3815. [Google Scholar] [CrossRef] [Green Version]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.; Guo, W.; Xu, Z.; He, Y.; Liang, C.; Mo, Y.; Wang, Y.; Xiong, F.; Guo, C.; Li, Y.; et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol. Cancer 2019, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, K.; Mittrücker, H.-W.; Feger, U.; Welte, S.A.; Yokoyama, W.M.; Spies, T.; Rammensee, H.-G.; Steinle, A. Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J. Immunol. 2005, 175, 720–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmberg, K.-J.; Ljunggren, H.-G. Escape from immune- and nonimmune-mediated tumor surveillance. Semin. Cancer Biol. 2006, 16, 16–31. [Google Scholar] [CrossRef]
- Davis, M.; Conlon, K.; Bohac, G.C.; Barcenas, J.; Leslie, W.; Watkins, L.; Lamzabi, I.; Deng, Y.; Li, Y.; Plate, J.M. Effect of Pemetrexed on Innate Immune Killer Cells and Adaptive Immune T Cells in Subjects with Adenocarcinoma of the Pancreas. J. Immunother. 2012, 35, 629–640. [Google Scholar] [CrossRef]
- Funa, K.; Nilsson, B.; Jacobsson, G.; Alm, G.V. Decreased natural killer cell activity and interferon production by leucocytes in patients with adenocarcinoma of the pancreas. Br. J. Cancer 1984, 50, 231–233. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-P.; Zhang, J.-J.; Liang, W.-B.; Tu, M.; Lu, Z.; Wei, J.; Jiang, K.; Gao, W.; Wu, J.-L.; Xu, Z.; et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer 2014, 14, 738. [Google Scholar] [CrossRef] [Green Version]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Peng, Y.-P.; Xi, C.-H.; Zhu, Y.; Yin, L.-D.; Wei, J.-S.; Zhang, J.-J.; Liu, X.-C.; Guo, S.; Fu, Y.; Miao, Y. Altered expression of CD226 and CD96 on natural killer cells in patients with pancreatic cancer. Oncotarget 2016, 7, 66586–66594. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ni, R.; Chen, J.; Liu, Z.; Xiao, M.-B.; Jiang, F.; Lu, C. The Presence of IGHG1 in Human Pancreatic Carcinomas Is Associated with Immune Evasion Mechanisms. Pancreas 2011, 40, 753–761. [Google Scholar] [CrossRef]
- Huang, Q.; Huang, M.; Meng, F.; Sun, R. Activated pancreatic stellate cells inhibit NK cell function in the human pancreatic cancer microenvironment. Cell. Mol. Immunol. 2018, 16, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.A.; Kim, J.; Jeon, S.; Shin, M.H.; Kwon, J.; Kim, T.-J.; Im, K.; Han, Y.; Kwon, W.; Kim, S.-W.; et al. Defective Localization with Impaired Tumor Cytotoxicity Contributes to the Immune Escape of NK Cells in Pancreatic Cancer Patients. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, S.M.; Wen, J.; Moutsopoulos, N.M. The kiss of death: Interrupted by NK-cell close encounters of another kind. Trends Immunol. 2006, 27, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor–β–dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF—Downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro-Oncology 2009, 12, 7–13. [Google Scholar] [CrossRef]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF-β1 Secretion and Down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef]
- Von Bernstorff, W.; Voss, M.; Freichel, S.; Schmid, A.; Vogel, I.; Jöhnk, C.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Systemic and local immunosuppression in pancreatic cancer patients. Clin. Cancer Res. 2001, 7, 925–932. [Google Scholar]
- Jun, E.; Song, A.Y.; Choi, J.-W.; Lee, H.H.; Kim, M.-Y.; Ko, D.-H.; Kang, H.J.; Kim, S.W.; Bryceson, Y.; Kim, S.C.; et al. Progressive Impairment of NK Cell Cytotoxic Degranulation Is Associated with TGF-β1 Deregulation and Disease Progression in Pancreatic Cancer. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Reiners, K.S.; Topolar, D.; Henke, A.; Simhadri, V.R.; Kessler, J.; Sauer, M.; Bessler, M.; Hansen, H.P.; Tawadros, S.; Herling, M.; et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood 2013, 121, 3658–3665. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, J.-I.; Murakami, T.; Iwamoto, S.; Fujita, S. Ex vivo expansion of natural killer cells from human peripheral blood mononuclear cells co-stimulated with anti-CD3 and anti-CD52 monoclonal antibodies. Cytotherapy 2016, 18, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Stangl, S.; Wortmann, A.; Guertler, U.; Multhoff, G. Control of Metastasized Pancreatic Carcinomas in SCID/Beige Mice with Human IL-2/TKD-Activated NK Cells. J. Immunol. 2006, 176, 6270–6276. [Google Scholar] [CrossRef] [PubMed]
- van Audenaerde, J.R.; Marcq, E.; von Scheidt, B.; Davey, A.S.; Oliver, A.J.; de Waele, J.; Quatannens, D.; van Loenhout, J.; Pauwels, P.; Roeyen, G.; et al. Novel combination immunotherapy for pancreatic cancer: Potent anti-tumor effects with CD40 agonist and interleukin-15 treatment. Clin. Transl. Immunol. 2020, 9, e1165. [Google Scholar] [CrossRef]
- Lee, J.; Kang, T.H.; Yoo, W.; Choi, H.; Jo, S.; Kong, K.; Lee, S.-R.; Kim, S.-U.; Kim, J.-S.; Cho, D.; et al. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol. Res. 2018, 7, 219–229. [Google Scholar] [CrossRef]
- Lin, M.; Liang, S.; Wang, X.; Liang, Y.; Zhang, M.; Chen, J.; Niu, L.; Xu, K. Percutaneous irreversible electroporation combined with allogeneic natural killer cell immunotherapy for patients with unresectable (stage III/IV) pancreatic cancer: A promising treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 2607–2618. [Google Scholar] [CrossRef]
- Lin, M.; Liang, S.; Wang, X.; Liang, Y.; Zhang, M.; Chen, J.; Niu, L.; Xu, K. Short-term clinical efficacy of percutaneous irreversible electroporation combined with allogeneic natural killer cell for treating metastatic pancreatic cancer. Immunol. Lett. 2017, 186, 20–27. [Google Scholar] [CrossRef]
- Miyashita, T.; Miki, K.; Kamigaki, T.; Makino, I.; Nakagawara, H.; Tajima, H.; Takamura, H.; Kitagawa, H.; Fushida, S.; Ahmed, A.K.; et al. Low-dose gemcitabine induces major histocompatibility complex class I-related chain A/B expression and enhances an antitumor innate immune response in pancreatic cancer. Clin. Exp. Med. 2017, 17, 19–31. [Google Scholar] [CrossRef]
- Lin, X.; Huang, M.; Xie, F.; Zhou, H.; Yang, J.; Huang, Q. Gemcitabine inhibits immune escape of pancreatic cancer by down regulating the soluble ULBP2 protein. Oncotarget 2016, 7, 70092–70099. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, T.; Miki, K.; Kamigaki, T.; Makino, I.; Tajima, H.; Nakanuma, S.; Hayashi, H.; Takamura, H.; Fushida, S.; Ahmed, A.K.; et al. Low-dose valproic acid with low-dose gemcitabine augments MHC class I-related chain A/B expression without inducing the release of soluble MHC class I-related chain A/B. Oncol. Lett. 2017, 14, 5918–5926. [Google Scholar] [CrossRef] [PubMed]
- Ponath, V.; Frech, M.; Bittermann, M.; Neubauer, A.A.K.; Brendel, C.; Pogge von Strandmann, E. The oncoprotein SKI acts as a suppressor of NK cell-2 mediated immunosurveillance in PDAC. Cancers 2020, in press. [Google Scholar]
- Ben, Q.; An, W.; Wang, L.; Wang, W.; Yu, L.; Yuan, Y. Validation of the Pretreatment Neutrophil-Lymphocyte Ratio as a Predictor of Overall Survival in a Cohort of Patients With Pancreatic Ductal Adenocarcinoma. Pancreas 2014, 44, 1. [Google Scholar] [CrossRef] [PubMed]
- Arima, K.; Okabe, H.; Hashimoto, D.; Chikamoto, A.; Tsuji, A.; Yamamura, K.; Kitano, Y.; Inoue, R.; Kaida, T.; Higashi, T.; et al. The diagnostic role of the neutrophil-to-lymphocyte ratio in predicting pancreatic ductal adenocarcinoma in patients with pancreatic diseases. Int. J. Clin. Oncol. 2016, 21, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Takagi, T.; Hikichi, T.; Konno, N.; Sugimoto, M.; Watanabe, K.; Nakamura, J.; Waragai, Y.; Kikuchi, H.; Takasumi, M.; et al. Derived neutrophil/lymphocyte ratio predicts gemcitabine therapy outcome in unresectable pancreatic cancer. Oncol. Lett. 2016, 11, 3441–3445. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhang, L.; Peng, Y.; Tao, M.; Li, G.; Xiu, D.; Yuan, C.; Ma, C.; Jiang, B. Preoperative neutrophil-to-lymphocyte ratio and tumor-related factors to predict lymph node metastasis in patients with pancreatic ductal adenocarcinoma (PDAC). Oncotarget 2016, 7, 74314–74324. [Google Scholar] [CrossRef] [Green Version]
- Pu, N.; Yin, H.; Zhao, G.; Nuerxiati, A.; Wang, D.; Xu, X.; Kuang, T.; Jin, D.; Lou, W.; Wu, W. Independent effect of postoperative neutrophil-to-lymphocyte ratio on the survival of pancreatic ductal adenocarcinoma with open distal pancreatosplenectomy and its nomogram-based prediction. J. Cancer 2019, 10, 5935–5943. [Google Scholar] [CrossRef]
- Xiang, Z.-J.; Hu, T.; Wang, Y.; Wang, H.; Xu, L.; Cui, N. Neutrophil–lymphocyte ratio (NLR) was associated with prognosis and immunomodulatory in patients with pancreatic ductal adenocarcinoma (PDAC). Biosci. Rep. 2020, 40, 40. [Google Scholar] [CrossRef]
- Felix, K.; Gaida, M.M. Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer -Active Players in Tumor Progression. Int. J. Boil. Sci. 2016, 12, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Hirth, M.; Gandla, J.; Höper, C.; Gaida, M.M.; Agarwal, N.; Simonetti, M.; Demir, A.; Xie, Y.; Weiss, C.; Michalski, C.W.; et al. CXCL10 and CCL21 Promote Migration of Pancreatic Cancer Cells Toward Sensory Neurons and Neural Remodeling in Tumors in Mice, Associated with Pain in Patients. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Mollinedo, F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Albelda, S.M. Tumor-associated neutrophils: Friend or foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wislez, M.; Rabbe, N.; Marchal, J.; Milleron, B.; Crestani, B.; Mayaud, C.; Antoine, M.; Soler, P.; Cadranel, J. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: Role in tumor progression and death. Cancer Res. 2003, 63, 1405–1412. [Google Scholar] [PubMed]
- Brandau, S.; Dumitru, C.A.; Lang, S. Protumor and antitumor functions of neutrophil granulocytes. Semin. Immunopathol. 2012, 35, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Shaul, M.E.; Levy, L.; Sun, J.; Mishalian, I.; Singhal, S.; Kapoor, V.; Horng, W.; Fridlender, G.; Albelda, S.M.; Fridlender, Z.G. Tumor-associated neutrophils display a distinct N1 profile following TGFβ modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology 2016, 5, e1232221. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Fang, T.; Huang, L.; Wang, H.; Zhang, L.; Wang, Z.; Cui, Y. Neutrophils infiltrating pancreatic ductal adenocarcinoma indicate higher malignancy and worse prognosis. Biochem. Biophys. Res. Commun. 2018, 501, 313–319. [Google Scholar] [CrossRef]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [Green Version]
- Munder, M.; Schneider, H.; Luckner, C.; Giese, T.; Langhans, C.-D.; Fuentes, J.M.; Kropf, P.; Mueller, I.; Kolb, A.; Modolell, M.; et al. Suppression of T-cell functions by human granulocyte arginase. Blood 2006, 108, 1627–1634. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xu, X.-F.; Shi, M.; Chen, Y.; Yu, D.; Zhao, C.; Gu, Y.; Yang, B.; Guo, S.; Ding, G.; et al. CD13hi Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. OncoImmunology 2017, 6, e1258504. [Google Scholar] [CrossRef] [Green Version]
- Takesue, S.; Ohuchida, K.; Shinkawa, T.; Otsubo, Y.; Matsumoto, S.; Sagara, A.; Yonenaga, A.; Ando, Y.; Kibe, S.; Nakayama, H.; et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancer-associated fibroblasts. Int. J. Oncol. 2019, 56, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhang, L.; Peng, Y.; Tao, M.; Li, L.; Xiu, D.; Yuan, C.; Ma, Z.; Jiang, B. Neutrophils assist the metastasis of circulating tumor cells in pancreatic ductal adenocarcinoma: A new hypothesis and a new predictor for distant metastasis. Medicine (Baltimore) 2016, 95, e4932. [Google Scholar] [CrossRef] [PubMed]
- Hogendorf, P.; Durczynski, A.; Skulimowski, A.; Kumor, A.; Poznańska, G.; Strzelczyk, J. Neutrophil Gelatinase-Associated Lipocalin (NGAL) concentration in urine is superior to CA19-9 and Ca 125 in differentiation of pancreatic mass: Preliminary report. Cancer Biomark. 2016, 16, 537–543. [Google Scholar] [CrossRef]
- Leung, L.; Radulovich, N.; Zhu, C.-Q.; Organ, S.; Bandarchi, B.; Pintilie, M.; To, C.; Panchal, D.; Tsao, M.-S. Lipocalin2 Promotes Invasion, Tumorigenicity and Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. PLoS ONE 2012, 7, e46677. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e7. [Google Scholar] [CrossRef] [Green Version]
- Stromnes, I.M.; Hulbert, A.; Pierce, R.H.; Greenberg, P.D.; Hingorani, S.R. T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2017, 5, 978–991. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Forget, M.-A.; Lucas, F.A.S.; Alvarez, H.A.; Haymaker, C.; Chattopadhyay, C.; Kim, S.-H.; Ekmekcioglu, S.; Grimm, E.A.; et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci. Rep. 2016, 6, 35848. [Google Scholar] [CrossRef]
- Balli, D.; Rech, A.J.; Stanger, B.Z.; Vonderheide, R.H. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin. Cancer Res. 2016, 23, 3129–3138. [Google Scholar] [CrossRef] [Green Version]
- Carstens, J.L.; De Sampaio, P.C.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Initiative, A.P.C.G.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Ene–Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Mittrücker, H.-W.; Visekruna, A.; Huber, M. Heterogeneity in the Differentiation and Function of CD8+ T Cells. Arch. Immunol. Ther. Exp. 2014, 62, 449–458. [Google Scholar] [CrossRef]
- Huber, M.; Lohoff, M. IRF4 at the crossroads of effector T-cell fate decision. Eur. J. Immunol. 2014, 44, 1886–1895. [Google Scholar] [CrossRef]
- Dobrzanski, M.J. Expanding Roles for CD4 T Cells and Their Subpopulations in Tumor Immunity and Therapy. Front. Oncol. 2013, 3, 63. [Google Scholar] [CrossRef] [Green Version]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef]
- Tassi, E.; Gavazzi, F.; Albarello, L.; Senyukov, V.; Longhi, R.; Dellabona, P.; Doglioni, C.; Braga, M.; Di Carlo, V.; Protti, M.P. Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J. Immunol. 2008, 181, 6595–6603. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef]
- Piro, G.; Simionato, F.; Carbone, C.; Frizziero, M.; Malleo, G.; Zanini, S.; Casolino, R.; Santoro, R.; Mina, M.M.; Zecchetto, C.; et al. A circulating TH2 cytokines profile predicts survival in patients with resectable pancreatic adenocarcinoma. OncoImmunology 2017, 6, e1322242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokopchuk, O.; Liu, Y.; Henne-Bruns, D.; Kornmann, M. Interleukin-4 enhances proliferation of human pancreatic cancer cells: Evidence for autocrine and paracrine actions. Br. J. Cancer 2005, 92, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetto, E.; De Monte, L.; Balzano, G.; Camisa, B.; Laino, V.; Riba, M.; Heltai, S.; Bianchi, M.; Bordignon, C.; Falconi, M.; et al. The IL-1/IL-1 receptor axis and tumor cell released inflammasome adaptor ASC are key regulators of TSLP secretion by cancer associated fibroblasts in pancreatic cancer. J. Immunother. Cancer 2019, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- De Monte, L.; Woermann, S.; Brunetto, E.; Heltai, S.; Magliacane, G.; Reni, M.; Paganoni, A.M.; Recalde, H.; Mondino, A.; Falconi, M.; et al. Basophil Recruitment into Tumor-Draining Lymph Nodes Correlates with Th2 Inflammation and Reduced Survival in Pancreatic Cancer Patients. Cancer Res. 2016, 76, 1792–1803. [Google Scholar] [CrossRef] [Green Version]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2015, 6, 270–285. [Google Scholar] [CrossRef] [Green Version]
- Ochi, A.; Nguyen, A.H.; Bedrosian, A.S.; Mushlin, H.M.; Zarbakhsh, S.; Barilla, R.; Zambirinis, C.P.; Fallon, N.C.; Rehman, A.; Pylayeva-Gupta, Y.; et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J. Exp. Med. 2012, 209, 1671–1687. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 2013, 25, 305–312. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and Clinical Significance of Th17 Cells in the Tumor Microenvironment and Peripheral Blood of Pancreatic Cancer Patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef]
- Vizio, B.; Novarino, A.; Giacobino, A.; Cristiano, C.; Prati, A.; Ciuffreda, L.; Montrucchio, G.; Bellone, G. Potential plasticity of T regulatory cells in pancreatic carcinoma in relation to disease progression and outcome. Exp. Ther. Med. 2012, 4, 70–78. [Google Scholar] [CrossRef] [Green Version]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras Activates a Hematopoietic-to-Epithelial IL-17 Signaling Axis in Preinvasive Pancreatic Neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loncle, C.; Bonjoch, L.; Folch-Puy, E.; López, M.B.; Lac, S.; Molejon, M.I.; Chuluyan, E.; Cordelier, P.; Dubus, P.; Lomberk, G.; et al. IL17 Functions through the Novel REG3β–JAK2–STAT3 Inflammatory Pathway to Promote the Transition from Chronic Pancreatitis to Pancreatic Cancer. Cancer Res. 2015, 75, 4852–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zoltan, M.; Riquelme, E.; Xu, H.; Şahin, I.; Pando, S.C.; Montiel, M.F.; Chang, K.; Jiang, Z.; Ling, J.; et al. Immune Cell Production of Interleukin 17 Induces Stem Cell Features of Pancreatic Intraepithelial Neoplasia Cells. Gastroenterology 2018, 155, 210–223.e3. [Google Scholar] [CrossRef]
- Alam, M.S.; Gaida, M.M.; Bergmann, F.; Lasitschka, F.; Giese, T.; Giese, N.A.; Hackert, T.; Hinz, U.; Hussain, S.; Kozlov, S.V.; et al. Selective inhibition of the p38 alternative activation pathway in infiltrating T cells inhibits pancreatic cancer progression. Nat. Med. 2015, 21, 1337–1343. [Google Scholar] [CrossRef]
- Diskin, B.; Adam, S.; Cassini, M.F.; Sanchez, G.; Liria, M.; Aykut, B.; Buttar, C.; Li, E.; Sundberg, B.; Salas, R.D.; et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat. Immunol. 2020, 21, 442–454. [Google Scholar] [CrossRef]
- Linnebacher, A.; Mayer, P.; Marnet, N.; Bergmann, F.; Herpel, E.; Revia, S.; Yin, L.; Liu, L.; Hackert, T.; Giese, T.; et al. Interleukin 21 Receptor/Ligand Interaction Is Linked to Disease Progression in Pancreatic Cancer. Cells 2019, 8, 1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytle, N.K.; Ferguson, L.P.; Rajbhandari, N.; Gilroy, K.; Fox, R.G.; Deshpande, A.; Schürch, C.M.; Hamilton, M.; Robertson, N.; Lin, W.; et al. A Multiscale Map of the Stem Cell State in Pancreatic Adenocarcinoma. Cell 2019, 177, 572–586.e22. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, U.K.; Moore, T.T.; Joo, H.-G.; Tanaka, Y.; Herrmann, V.; Doherty, G.M.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ Regulatory T Cells Increases During the Progression of Pancreatic Ductal Adenocarcinoma and Its Premalignant Lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Nasca, V.; Chiaravalli, M.; Piro, G.; Esposito, A.; Salvatore, L.; Tortora, G.; Corbo, V.; Carbone, C. Intraductal Pancreatic Mucinous Neoplasms: A Tumor-Biology Based Approach for Risk Stratification. Int J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Jang, J.-E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Chellappa, S.; Hugenschmidt, H.; Hagness, M.; Line, P.D.; Labori, K.J.; Wiedswang, G.; Taskén, K.; Aandahl, E.M. Regulatory T cells that co-express RORγt and FOXP3 are pro-inflammatory and immunosuppressive and expand in human pancreatic cancer. OncoImmunology 2015, 5, e1102828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.-J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallies, A.; Zehn, D.; Utzschneider, D.T. Precursor exhausted T cells: Key to successful immunotherapy? Nat. Rev. Immunol. 2019, 20, 128–136. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, A.; Dyhl-Polk, A.; Chen, I.; Nielsen, D. Checkpoint inhibitors in pancreatic cancer. Cancer Treat. Rev. 2019, 78, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Bauer, C.A.; Öhlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2018, 68, 159–171. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Belle, J.I.; Denardo, D.G. A Single-Cell Window into Pancreas Cancer Fibroblast Heterogeneity. Cancer Discov. 2019, 9, 1001–1002. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2018, 9, 282–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, V.; Semaan, A.; Huang, J.; Lucas, F.A.S.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2018, 25, 2194–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.N.; Verma, U.; Brekken, R. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2018, 9, 64–81. [Google Scholar] [CrossRef] [Green Version]
- Goehrig, D.; Nigri, J.; Samain, R.; Wu, Z.; Cappello, P.; Gabiane, G.; Zhang, X.; Zhao, Y.; Kim, I.-S.; Chanal, M.; et al. Stromal protein βig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut 2018, 68, 693–707. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell–Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-C.; Lin, Y.-L.; Hsu, C.-C.; Chao, Y.-J.; Hou, Y.-C.; Chiu, T.-J.; Huang, P.-H.; Tang, M.-J.; Chen, L.-T.; Shan, Y.-S. Pancreatic stellate cells activated by mutant KRAS-mediated PAI-1 upregulation foster pancreatic cancer progression via IL-8. Theranostics 2019, 9, 7168–7183. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K.; Tyagi, N.; Khan, M.A.; Srivastava, S.K.; Al-Ghadhban, A.; Dugger, K.; Carter, J.E.; Singh, A.P.; Singh, A.P. Gemcitabine treatment promotes immunosuppressive microenvironment in pancreatic tumors by supporting the infiltration, growth, and polarization of macrophages. Sci. Rep. 2018, 8, 12000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, P.; Yang, L.; Hosaka, K.; Zhang, Y.; Fischer, C.; Braun, H.; Liu, S.; Yu, G.; Liu, S.; Beyaert, R.; et al. Molecular mechanisms of IL-33-mediated stromal interactions in cancer metastasis. JCI Insight 2018, 3, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moral, J.A.; Leung, J.; Rojas, L.A.; Ruan, J.; Zhao, J.; Sethna, Z.; Ramnarain, A.; Gasmi, B.; Gururajan, M.; Redmond, D.; et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 2020, 579, 130–135. [Google Scholar] [CrossRef]
- Pausch, T.M.; Aue, E.; Wirsik, N.M.; Valls, A.F.; Shen, Y.; Radhakrishnan, P.; Hackert, T.; Schneider, M.; Schmidt, T. Metastasis-associated fibroblasts promote angiogenesis in metastasized pancreatic cancer via the CXCL8 and the CCL2 axes. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Investig. New Drugs 2019, 38, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Garg, B.; Giri, B.; Modi, S.; Sethi, V.; Castro, I.; Umland, O.; Ban, Y.; Lavania, S.; Dawra, R.; Banerjee, S.; et al. NFκB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology 2018, 155, 880–891. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein-. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-G.; Shon, Y.; Kim, J.; Oh, Y.-K. Selective Activation of Anticancer Chemotherapy by Cancer-Associated Fibroblasts in the Tumor Microenvironment. J. Natl. Cancer Inst. 2016, 109, 109. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Scoazec, J.-Y.; Tartare-Deckert, S.; Féral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF Mediates Proinvasive Activation of Stromal Fibroblasts in Cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.-T.; Fer, N.; Galeas, J.; Collisson, E.A.; Kim, S.E.; Sharib, J.; McCormick, F. Blockade of leukemia inhibitory factor as a therapeutic approach to KRAS driven pancreatic cancer. Nat. Commun. 2019, 10, 3055. [Google Scholar] [CrossRef] [PubMed]
- Pascual-García, M.; Bonfill-Teixidor, E.; Planas-Rigol, E.; Rubio-Perez, C.; Iurlaro, R.; Arias, A.; Cuartas, I.; Sala-Hojman, A.; Escudero, L.; Martínez-Ricarte, F.; et al. LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8+ T cell tumor-infiltration impairing anti-PD1 therapy. Nat. Commun. 2019, 10, 2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.L.; Kaspar, P.; Brunet, L.J.; Bhatt, H.; Gadi, I.; Köntgen, F.; Abbondanzo, S.J. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 1992, 359, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Shek, F.W.-T.; Benyon, R.C.; Walker, F.M.; McCrudden, P.R.; Pender, S.L.F.; Williams, E.J.; Johnson, P.A.; Johnson, C.D.; Bateman, A.C.; Fine, D.R.; et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 2002, 160, 1787–1798. [Google Scholar] [CrossRef]
- Jesnowski, R.; Furst, D.; Ringel, J.; Chen, Y.; Schrodel, A.; Kleeff, J.; Kolb, A.; Schareck, W.D.; Lohr, M. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: Deactivation is induced by matrigel and N-acetylcysteine. Lab. Investig. 2005, 85, 1276–1291. [Google Scholar] [CrossRef]
- Cave, D.D.; Di Guida, M.; Costa, V.; Sevillano, M.; Ferrante, L.; Heeschen, C.; Corona, M.; Cucciardi, A.; Lonardo, E. TGF-β1 secreted by pancreatic stellate cells promotes stemness and tumourigenicity in pancreatic cancer cells through L1CAM downregulation. Oncogene 2020, 39, 4271–4285. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Chang, C.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinselmeyer, B.H.; Heydari, S.; Sacristan, C.; Nayak, D.; Cammer, M.; Herz, J.; Cheng, X.; Davis, S.J.; Dustin, M.L.; McGavern, D.B. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J. Exp. Med. 2013, 210, 757–774. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective Effects of PD-1 on Akt and Ras Pathways Regulate Molecular Components of the Cell Cycle and Inhibit T Cell Proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef] [Green Version]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [Green Version]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.L.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, R.; Viel, A.; Doglioni, C.; Russo, A.G.; Guidoboni, M.; Capozzi, E.; Vecchiato, N.; Macri, E.; Fornasarig, M.; Boiocchi, M. High Prevalence of Activated Intraepithelial Cytotoxic T Lymphocytes and Increased Neoplastic Cell Apoptosis in Colorectal Carcinomas with Microsatellite Instability. Am. J. Pathol. 1999, 154, 1805–1813. [Google Scholar] [CrossRef] [Green Version]
- Timmermann, B.; Kerick, M.; Roehr, C.; Fischer, A.; Isau, M.; Boerno, S.T.; Wunderlich, A.; Barmeyer, C.; Seemann, P.; Koenig, J.; et al. Somatic Mutation Profiles of MSI and MSS Colorectal Cancer Identified by Whole Exome Next Generation Sequencing and Bioinformatics Analysis. PLoS ONE 2010, 5, e15661. [Google Scholar] [CrossRef]
- Dudley, J.C.; Lin, M.-T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2014, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.S.; Sundar, R.; Lopez, J.S. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2017, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H.; Bagalà, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A.; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncology 2019, 25, e808–e815. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2017, 36, 96–102. [Google Scholar] [CrossRef]
- Azad, A.; Lim, S.Y.; D’Costa, Z.; Jones, K.; Diana, A.; Sansom, O.J.; Kruger, P.; Liu, S.K.; McKenna, W.G.; Dushek, O.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2016, 9, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Victor, C.T.-S.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Contratto, M.; Miller, G.; Leichman, L.; Wu, J. Immunotherapy in pancreatic cancer: Unleash its potential through novel combinations. World J. Clin. Oncol. 2017, 8, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Mucciolo, G.; Roux, C.; Scagliotti, A.; Brugiapaglia, S.; Novelli, F.; Cappello, P. The dark side of immunotherapy: Pancreatic cancer. Cancer Drug Resist. 2020. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 2013, 13, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Mccarty, T.M.; Liu, X.; Sun, J.Y.; Peralta, E.A.; Diamond, D.J.; Ellenhorn, J.D. Targeting p53 for adoptive T-cell immunotherapy. Cancer Res. 1998, 58, 2601–2605. [Google Scholar]
- Kawaoka, T.; Oka, M.; Takashima, M.; Ueno, T.; Yamamoto, K.; Yahara, N.; Yoshino, S.; Hazama, S. Adoptive immunotherapy for pancreatic cancer: Cytotoxic T lymphocytes stimulated by the MUC1-expressing human pancreatic cancer cell line YPK-1. Oncol. Rep. 2008, 20, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Hazama, S.; Sakamoto, K.; Shindo, Y.; Kanekiyo, S.; Nakashima, M.; Matsukuma, S.; Tokuhisa, Y.; Iida, M.; Suzuki, N.; et al. Postoperative Adjuvant Therapy for Resectable Pancreatic Cancer with Gemcitabine and Adoptive Immunotherapy. Pancreas 2017, 46, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Ruella, M.; Kenderian, S.S. Next-Generation Chimeric Antigen Receptor T-Cell Therapy: Going off the Shelf. BioDrugs 2017, 31, 473–481. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197307
Huber M, Brehm CU, Gress TM, Buchholz M, Alashkar Alhamwe B, von Strandmann EP, Slater EP, Bartsch JW, Bauer C, Lauth M. The Immune Microenvironment in Pancreatic Cancer. International Journal of Molecular Sciences. 2020; 21(19):7307. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197307
Chicago/Turabian StyleHuber, Magdalena, Corinna U. Brehm, Thomas M. Gress, Malte Buchholz, Bilal Alashkar Alhamwe, Elke Pogge von Strandmann, Emily P. Slater, Jörg W. Bartsch, Christian Bauer, and Matthias Lauth. 2020. "The Immune Microenvironment in Pancreatic Cancer" International Journal of Molecular Sciences 21, no. 19: 7307. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197307