Deferred Administration of Afobazole Induces Sigma1R-Dependent Restoration of Striatal Dopamine Content in a Mouse Model of Parkinson’s Disease

 , , , and
, , , and
Abstract
:
1. Introduction
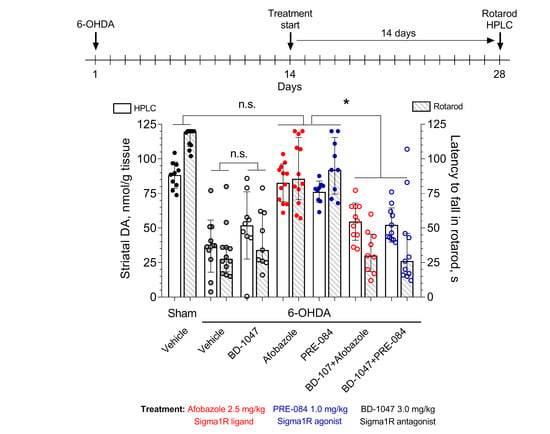
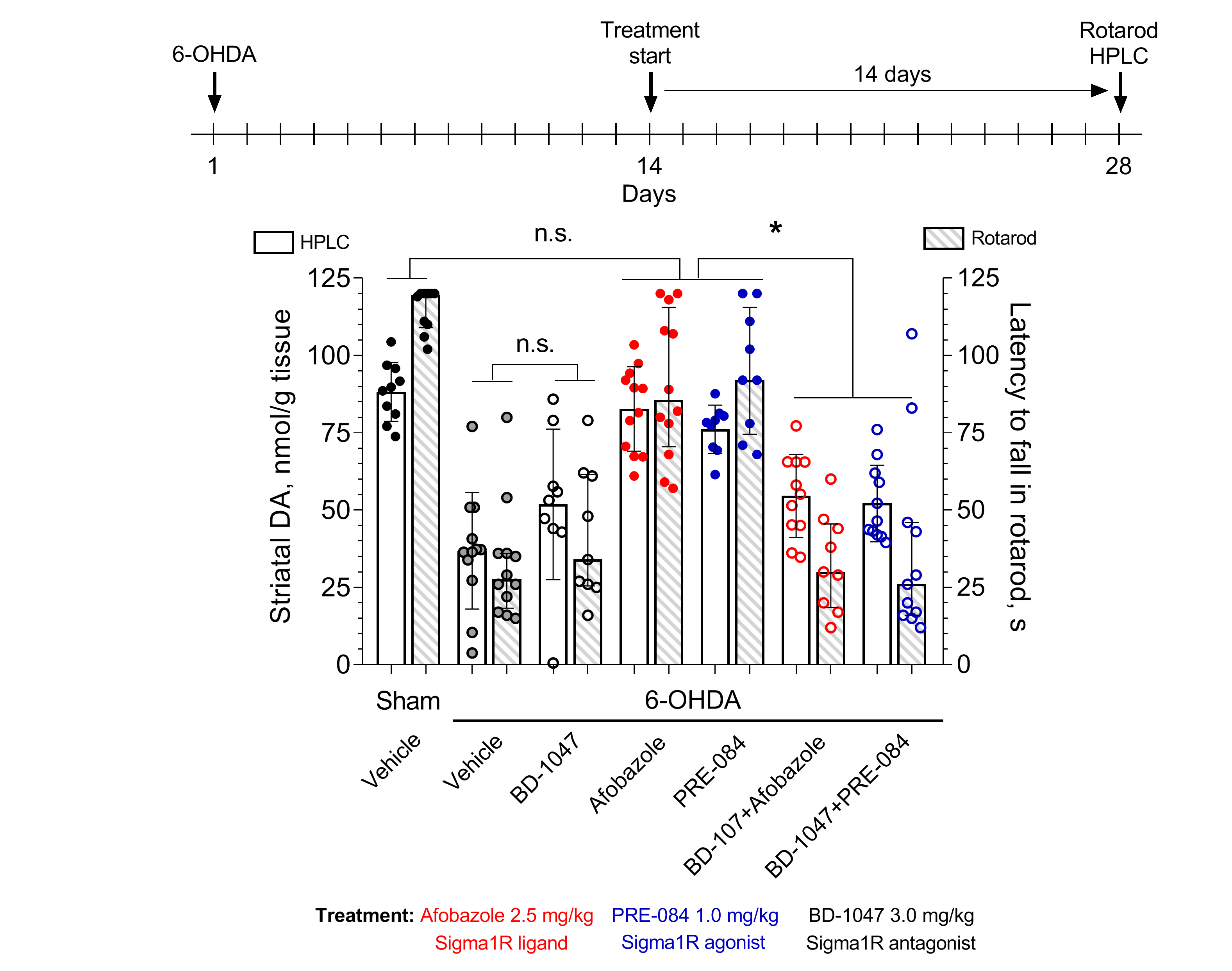
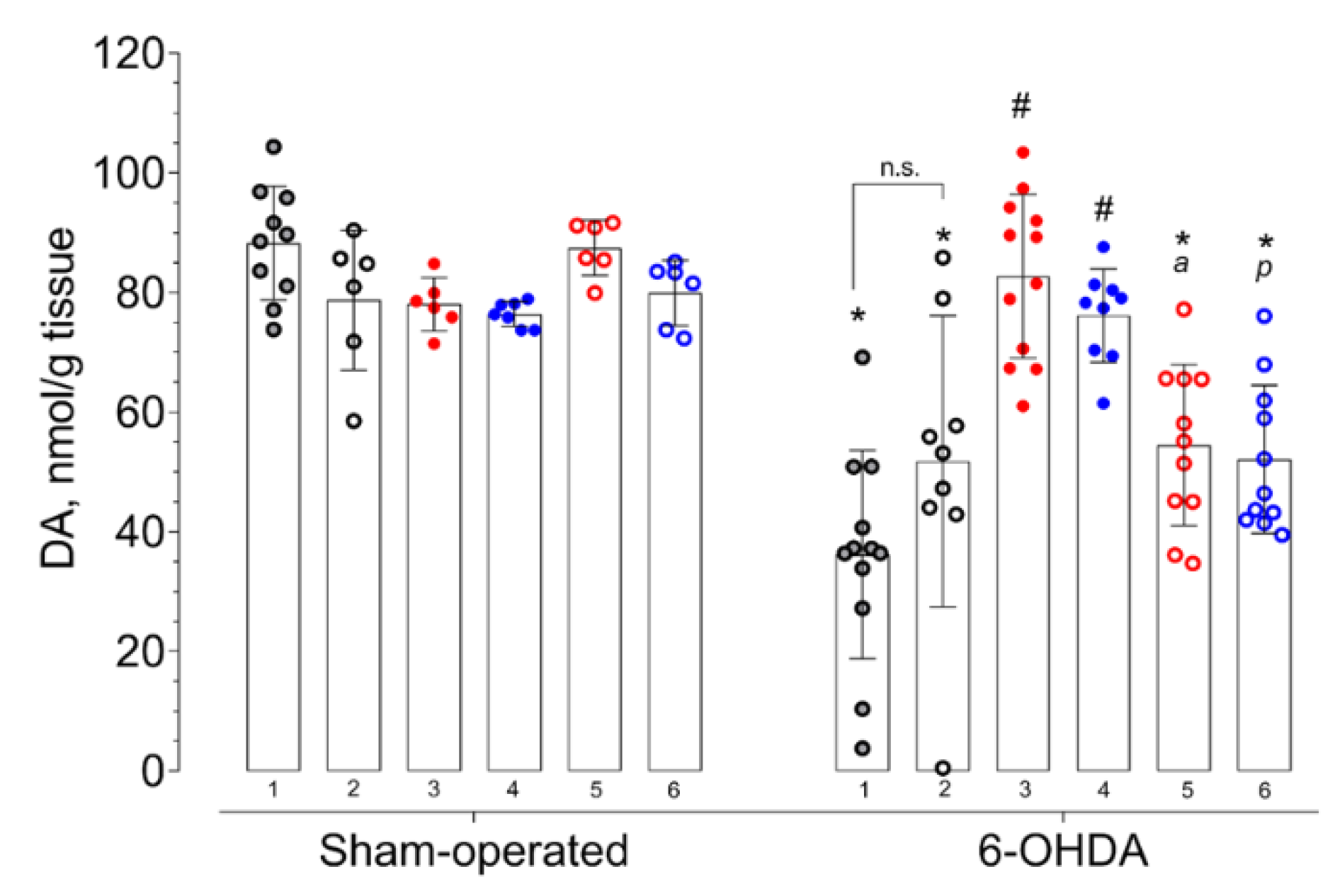
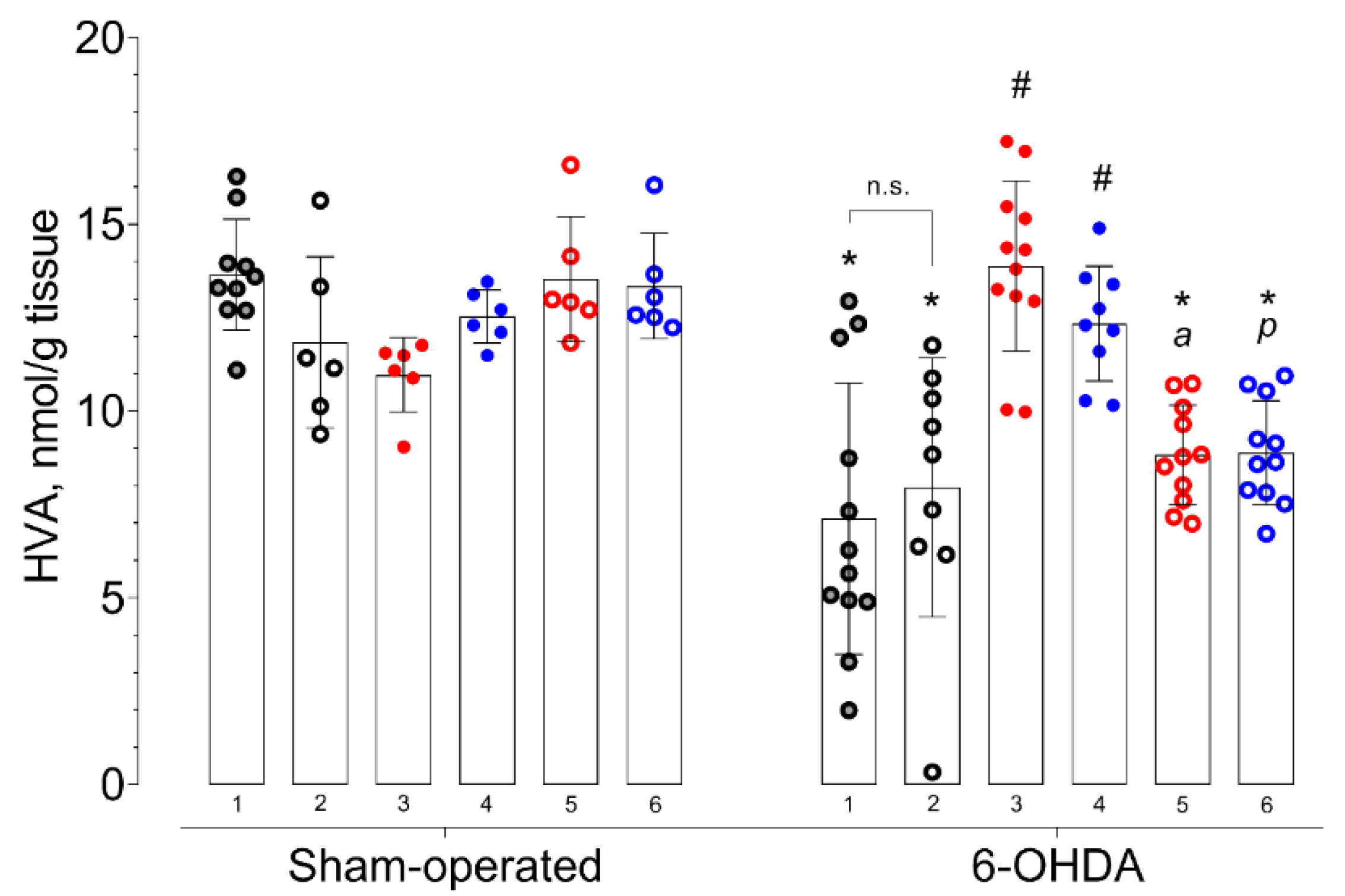
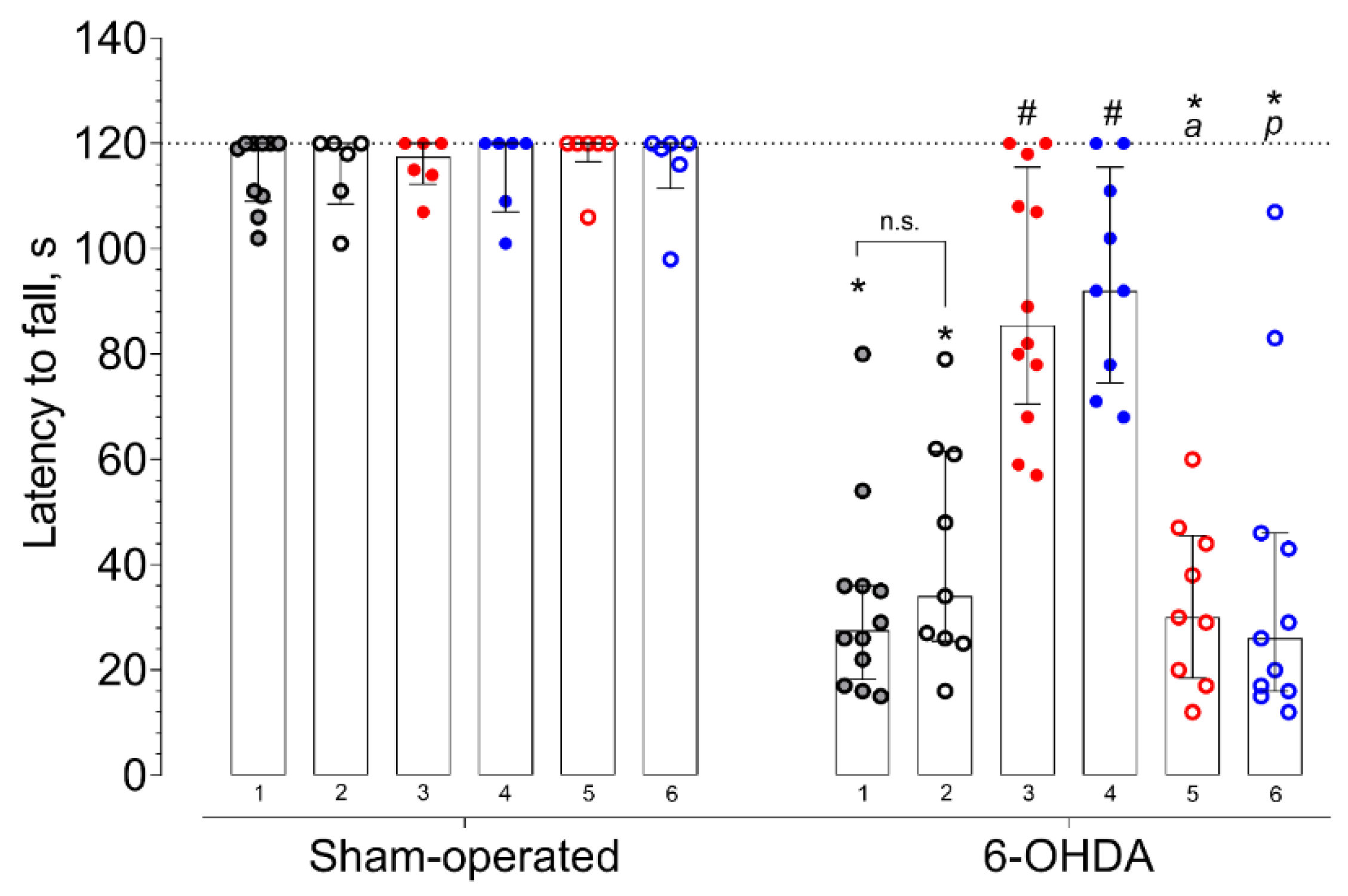
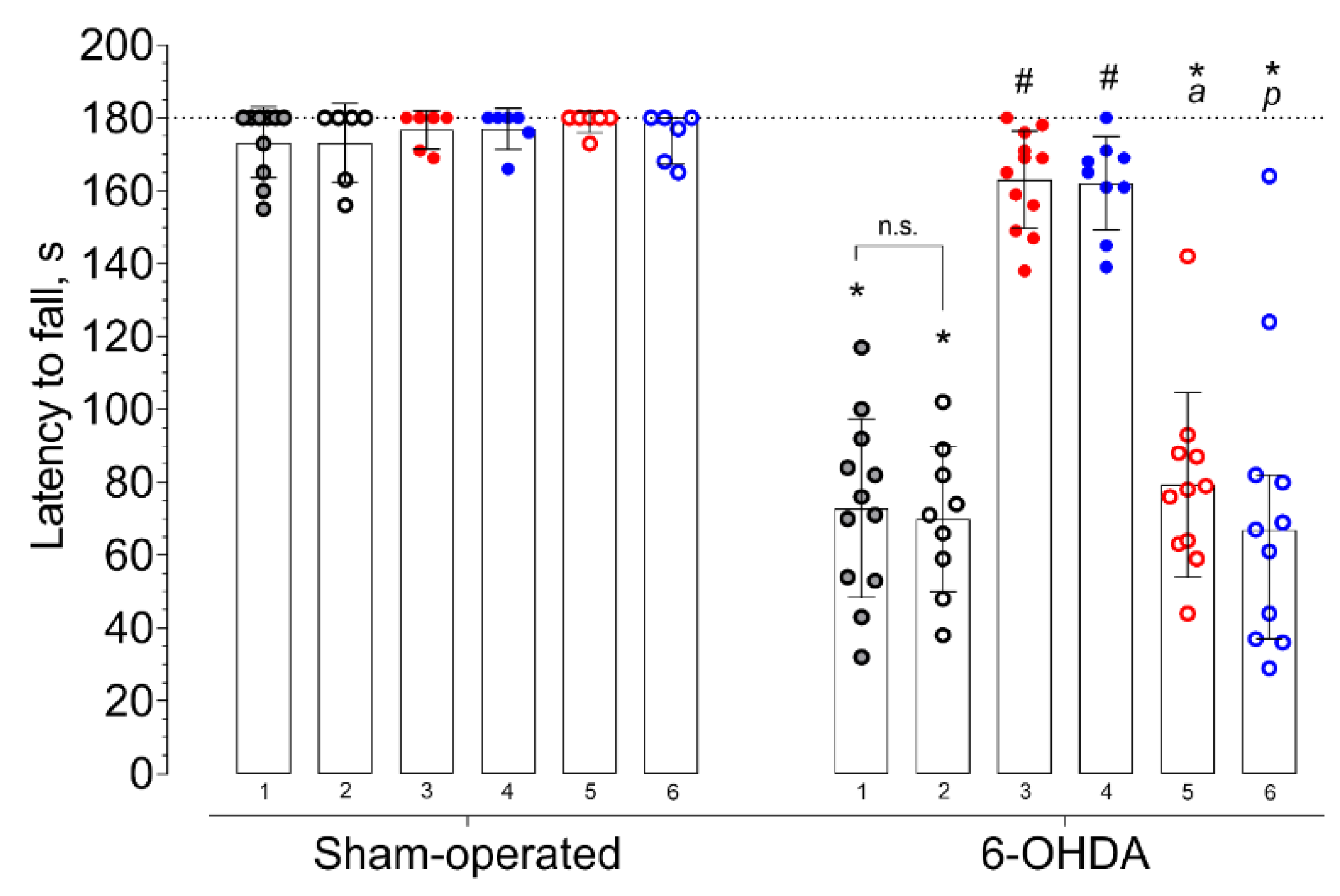
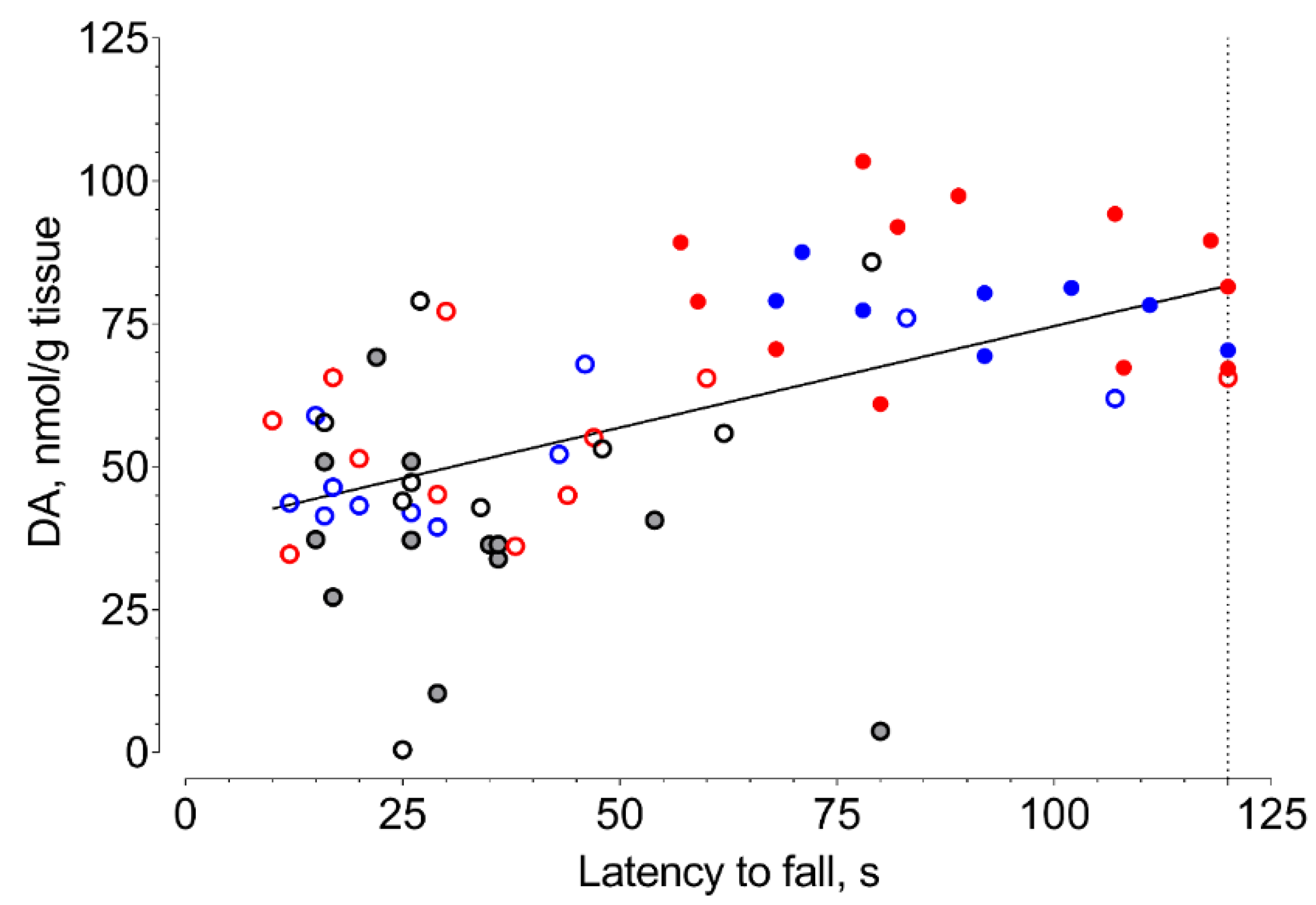
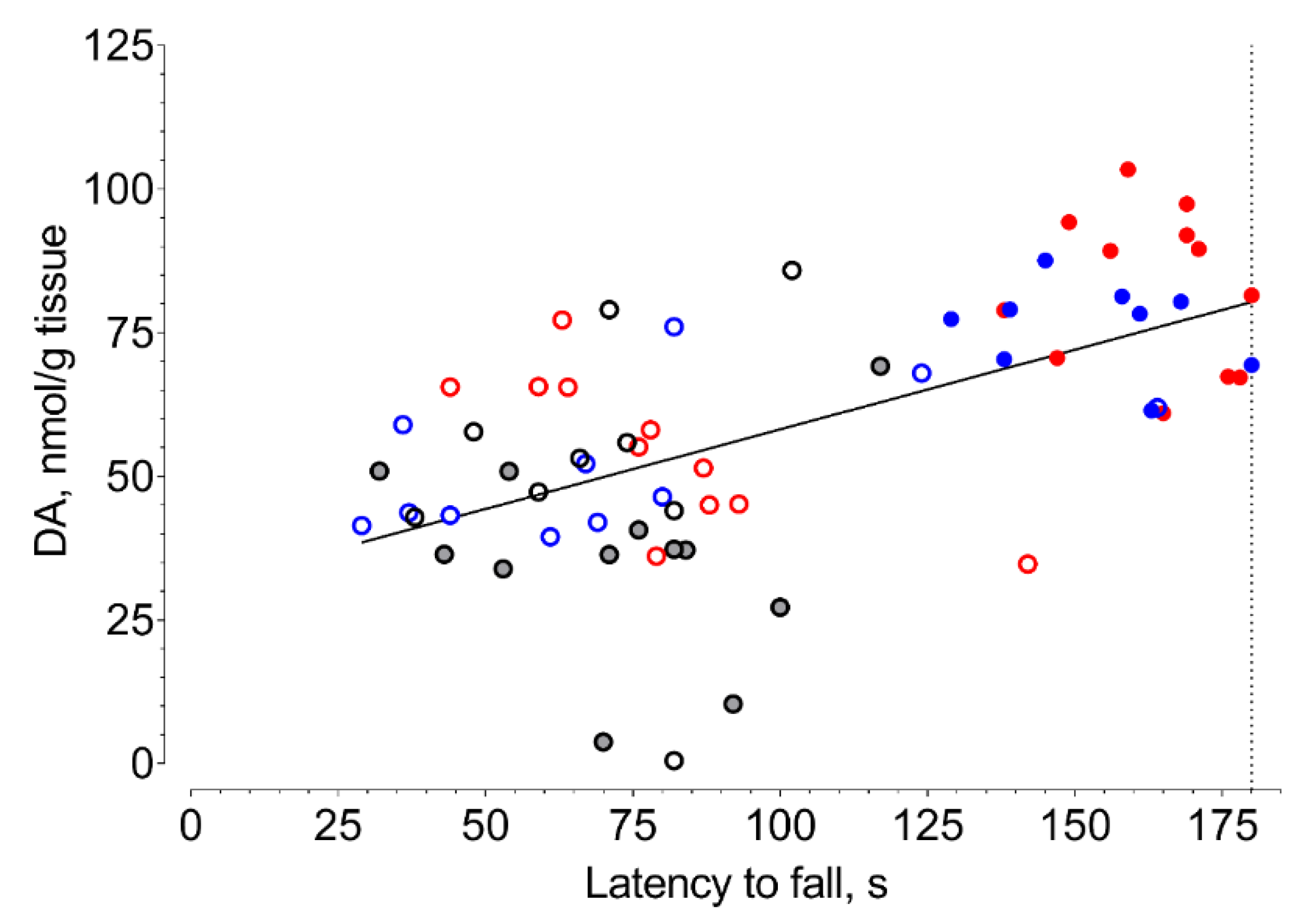
2. Results
3. Materials and Methods
3.1. Chemicals
3.2. Experimental Animals
3.3. Ethical Approval
3.4. Experimental Design
3.5. 6-OHDA Lesion
3.6. Rotarod Test
3.7. HPLC-ED Technique
3.8. Immunohistochemical Analysis
3.9. Statistical Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-Hydroxydopamine; 5-(2-aminoethyl)benzene-1,2,4-triol |
| Aβ1–42 | Amyloid-beta peptide 1–42 |
| Aβ25–35 | Amyloid-beta peptide 25–35 |
| Afobazole | 5-Ethoxy-2-[2-(morpholino)-ethylthio]benzimidazole dihydrochloride |
| ARR | Accelerated rotarod |
| BD-1047 | N′-[2-(3,4-dichlorophenyl)ethyl]-N,N,N′-trimethylethane-1,2-diamine |
| BDNF | Brain-derived neurotrophic factor |
| BiP | Endoplasmic reticulum chaperone BiP |
| BMY-14802 | 1-(4-Fluorophenyl)-4-[4-(5-fluoropyrimidin-2-yl)piperazin-1-yl]butan-1-ol |
| COMT | Catechol O-methyltransferase |
| D1 | Dopamine receptor D1 |
| D2 | Dopamine receptor D2 |
| DA | Dopamine; 4-(2-aminoethyl)benzene-1,2-diol |
| DAT | Sodium-dependent dopamine transporter |
| DOPA | 3,4-Dihydroxy-L-Phenylalanine; (2S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid |
| DOPAC | 3,4-Dihydroxyphenylacetic acid; 2-(3,4-dihydroxyphenyl)acetic acid |
| DHBA | 3,4-Dihydroxybenzylamine hydrobromide; 4-(aminomethyl)benzene-1,2-diol;hydrobromide |
| EDTA-Na2 | Ethylenediaminetetraacetic acid disodium salt dihydrate |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation system |
| ERK1 | Mitogen-activated protein kinase 3 |
| ERK2 | Mitogen-activated protein kinase 1 |
| FSRR | Fixed-speed rotarod |
| GluN | Glutamate receptor, ionotropic, N-methyl D-aspartate |
| HVA | Homovanillic acid; 2-(4-hydroxy-3-methoxyphenyl)acetic acid |
| ICR | Institute of Cancer Research outbred mice |
| INSIG1 | Insulin-induced gene 1 protein |
| IP3R3 | Inositol 1,4,5-trisphosphate receptor type 3 |
| IRE1 | Serine/threonine-protein kinase/endoribonuclease IRE1 |
| LB | Lewy body |
| MAM | Mitochondria-associated membrane of endoplasmic reticulum |
| MAO A | Amine oxidase [flavin-containing] A |
| MAO B | Amine oxidase [flavin-containing] B |
| MPTP | 1-Methyl-4-phenyl-3,6-dihydro-2H-pyridine |
| NQO2 | Ribosyldihydronicotinamide dehydrogenase [quinone]; NRH:quinone oxidoreductase 2 |
| PBS | Phosphate-buffered saline |
| PD | Parkinson’s disease |
| PRE-084 | 2-Morpholin-4-ylethyl 1-phenylcyclohexane-1-carboxylate |
| pSyn-129 | Phospho alpha-synuclein |
| ROS | Reactive oxygen species |
| SA4503 | 1-[2-(3,4-Dimethoxyphenyl)ethyl]-4-(3-phenylpropyl)piperazine |
| Sigma1R | Sigma non-opioid intracellular receptor 1, chaperone Sigma1R |
| SNc | Substantia nigra pars compacta |
| TH | Tyrosine 3-monooxygenase |
| trkB | Neurotrophic receptor tyrosine kinase 2 |
References
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Khoo, T.K.; Yarnall, A.J.; Duncan, G.W.; Coleman, S.; O’Brien, J.; Brooks, D.J.; Barker, R.A.; Burn, D.J. The Spectrum of Nonmotor Symptoms in Early Parkinson Disease. Neurology 2013, 80, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pringsheim, T.; Jetté, N.; Frolkis, A.; Steeves, T.D. The Prevalence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Ball, N.; Teo, W.-P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Aarsland, D.; Barone, P.; Burn, D.J.; Hawkes, C.H.; Oertel, W.; Ziemssen, F. Identifying Prodromal Parkinson’s Disease: Pre-Motor Disorders in Parkinson’s Disease. Mov. Disord. 2012, 27, 617–626. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J.-W. Oxidative Stress and Cellular Pathologies in Parkinson’s Disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [Green Version]
- Barrett, P.J.; Greenamyre, J.T. Post-Translational Modification of α-Synuclein in Parkinson′s Disease. Brain Res. 2015, 1628, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Evilar, M.; Campioni, S.; et al. In Vivo Demonstration That Alpha-Synuclein Oligomers Are Toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNaught, K.S.; Jenner, P. Proteasomal Function Is Impaired in Substantia Nigra in Parkinson’s Disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C. Altered Proteasomal Function in Sporadic Parkinson’s Disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s Disease: A Target for Neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological Treatment of Parkinson Disease. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.-O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R.; et al. Pathophysiology of L-Dopa-Induced Motor and Non-Motor Complications in Parkinson’s Disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef]
- Moore, T.J.; Glenmullen, J.; Mattison, D.R. Reports of Pathological Gambling, Hypersexuality, and Compulsive Shopping Associated with Dopamine Receptor Agonist Drugs. JAMA Intern. Med. 2014, 174, 1930. [Google Scholar] [CrossRef] [Green Version]
- Francardo, V.; Bez, F.; Wieloch, T.; Nissbrandt, H.; Ruscher, K.; Cenci, M.A. Pharmacological Stimulation of Sigma-1 Receptors Has Neurorestorative Effects in Experimental Parkinsonism. Brain 2014, 137, 1998–2014. [Google Scholar] [CrossRef] [Green Version]
- Francardo, V.; Geva, M.; Bez, F.; Denis, Q.; Steiner, L.; Hayden, M.R.; Cenci, M.A. Pridopidine Induces Functional Neurorestoration Via the Sigma-1 Receptor in a Mouse Model of Parkinson’s Disease. Neurotherapeutics 2019, 16, 465–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronin, M.; Kadnikov, I.A.; Voronkov, D.N.; Seredenin, S.B. Chaperone Sigma1R Mediates the Neuroprotective Action of Afobazole in the 6-OHDA Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 17020. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Wang, L.; Zhang, T.; Zhang, B.; Chen, L. Sigma-1 Receptor Knockout Increases α-Synuclein Aggregation and Phosphorylation with Loss of Dopaminergic Neurons in Substantia Nigra. Neurobiol. Aging 2017, 59, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Justinova, Z.; Hayashi, E.; Cormaci, G.; Mori, T.; Tsai, S.-Y.; Barnes, C.; Goldberg, S.R.; Su, T.-P. Regulation of Sigma-1 Receptors and Endoplasmic Reticulum Chaperones in the Brain of Methamphetamine Self-Administering Rats. J. Pharmacol. Exp. Ther. 2009, 332, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- Mishina, M.; Ishiwata, K.; Ishii, K.; Kitamura, S.; Kimura, Y.; Kawamura, K.; Oda, K.; Sasaki, T.; Sakayori, O.; Hamamoto, M.; et al. Function of Sigma-1 Receptors in Parkinson’s Disease. Acta Neurol. Scand. 2005, 112, 103–107. [Google Scholar] [CrossRef]
- Delprat, B.; Crouzier, L.; Su, T.-P.; Maurice, T. At the Crossing of ER Stress and MAMs: A Key Role of Sigma-1 Receptor? In Calcium Signaling. Advances of Experimental Medicine and Biology; Springer: Cham, Switzerland, 2020; Volume 1131, pp. 699–718. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Intracellular Dynamics of σ-1 Receptors (σ1 Binding Sites) in NG108-15 Cells. J. Pharmacol. Exp. Ther. 2003, 306, 726–733. [Google Scholar] [CrossRef]
- Su, T.-P.; Su, T.-C.; Nakamura, Y.; Tsai, S.-Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef] [Green Version]
- Navarro, G.; Moreno, E.; Bonaventura, J.; Brugarolas, M.; Farré, D.; Aguinaga, D.; Mallol, J.; Cortés, A.; Ecasado, V.; Lluis, C.; et al. Cocaine Inhibits Dopamine D2 Receptor Signaling via Sigma-1-D2 Receptor Heteromers. PLoS ONE 2013, 8, e61245. [Google Scholar] [CrossRef] [Green Version]
- Hong, W.C.; Yano, H.; Hiranita, T.; Chin, F.T.; McCurdy, C.R.; Su, T.-P.; Amara, S.G.; Katz, J.L. The Sigma-1 Receptor Modulates Dopamine Transporter Conformation and Cocaine Binding and May Thereby Potentiate Cocaine Self-Administration in Rats. J. Boil. Chem. 2017, 292, 11250–11261. [Google Scholar] [CrossRef] [Green Version]
- Balasuriya, D.; Stewart, A.P.; Edwardson, J.M. The σ-1 Receptor Interacts Directly with GluN1 but not GluN2A in the GluN1/GluN2A NMDA Receptor. J. Neurosci. 2013, 33, 18219–18224. [Google Scholar] [CrossRef] [Green Version]
- Pabba, M.; Wong, A.Y.; Ahlskog, N.; Hristova, E.; Biscaro, D.; Nassrallah, W.; Ngsee, J.K.; Snyder, M.; Beique, J.-C.; Bergeron, R. NMDA Receptors Are Upregulated and Trafficked to the Plasma Membrane after Sigma-1 Receptor Activation in the Rat Hippocampus. J. Neurosci. 2014, 34, 11325–11338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Fujita, Y.; Shibata, K.; Mori, M.; Yamashita, T. Sigma-1 Receptor Enhances Neurite Elongation of Cerebellar Granule Neurons via TrkB Signaling. PLoS ONE 2013, 8, e75760. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Roldan, J.L.L.; Ossa, F.; Schnell, J.R. Characterization of the Human Sigma-1 Receptor Chaperone Domain Structure and Binding Immunoglobulin Protein (BiP) Interactions. J. Boil. Chem. 2013, 288, 21448–21457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.-P. Sigma-1 Receptor Chaperone at the ER-Mitochondrion Interface Mediates the Mitochondrion-ER-Nucleus Signaling for Cellular Survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.-Y.A.; Chuang, J.-Y.; Tsai, M.-S.; Wang, X.-F.; Xi, Z.-X.; Hung, J.-J.; Chang, W.-C.; Bonci, A.; Su, T.-P. Sigma-1 Receptor Mediates Cocaine-Induced Transcriptional Regulation by Recruiting Chromatin-Remodeling Factors at the Nuclear Envelope. Proc. Natl. Acad. Sci. USA 2015, 112, E6562–E6570. [Google Scholar] [CrossRef] [Green Version]
- Pabba, M.; Sibille, E. Sigma-1 and N-Methyl-d-Aspartate Receptors: A Partnership with Beneficial Outcomes. Mol. Neuropsychiatry 2015, 1, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Hayashi, T.; Urfer, R.; Mita, S.; Su, T.-P. Sigma-1 Receptor Chaperones Regulate the Secretion of Brain-Derived Neurotrophic Factor. Synapse 2012, 66, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Cai, L.; Matsuhisa, K.; Ohtake, Y.; Kaneko, M.; Kanemoto, S.; Asada, R.; Imaizumi, K. Neuronal Activity-Dependent Local Activation of Dendritic Unfolded Protein Response Promotes Expression of Brain-Derived Neurotrophic Factor in Cell Soma. J. Neurochem. 2017, 144, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-C.; Alberico, S.L.; Emmons, E.; Narayanan, N.S. New Therapeutic Strategies Targeting D1-Type Dopamine Receptors for Neuropsychiatric Disease. Front. Boil. 2015, 10, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.; Singh, S.; Shukla, S. Physiological and Functional Basis of Dopamine Receptors and Their Role in Neurogenesis: Possible Implication for Parkinson’s disease. J. Exp. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutt, J.G.; Carter, J.H.; Sexton, G.J. The Dopamine Transporter: Importance in Parkinson’s Disease. Ann. Neurol. 2004, 55, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, S.; Fu, P.; Zhang, Z.; Lin, K.; Ko, J.; Yung, K. Roles of Glutamate Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 4391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson’s Disease. J. Clin. Med. 2020, 9, 257. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 Regulates the Integrity and Function of ER-Mitochondria Association Through Interaction with IP3R3-Grp75-VDAC. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Egalleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of Dopaminergic Neuron Survival by the Unfolded Protein Response Transcription Factor XBP. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Rathitharan, G.; Meyer, J.H.; Furukawa, Y.; Ang, L.-C.; Boileau, I.; Guttman, M.; Hornykiewicz, O.; Kish, S.J. Brain Monoamine Oxidase B and a in Human Parkinsonian Dopamine Deficiency Disorders. Brain 2017, 140, 2460–2474. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein Misfolding, Aggregation, and Conformational Strains in Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Fanning, S.; Selkoe, D.; Dettmer, U. Parkinson’s Disease: Proteinopathy or Lipidopathy? NPJ Park. Dis. 2020, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Alecu, I.; Bennett, S.A.L. Dysregulated Lipid Metabolism and Its Role in α-Synucleinopathy in Parkinson’s Disease. Front. Neurosci. 2019, 13, 328. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The Role of Lipids in Parkinson’s Disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.-P. σ-1 Receptors (σ1 Binding Sites) Form Raft-Like Microdomains and Target Lipid Droplets on the Endoplasmic Reticulum: Roles in Endoplasmic Reticulum Lipid Compartmentalization and Export. J. Pharmacol. Exp. Ther. 2003, 306, 718–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.-P. Cholesterol at the Endoplasmic Reticulum: Roles of the Sigma-1 Receptor Chaperone and Implications thereof in Human Diseases. Subcell. Biochem. 2010, 51, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Hayashi, E.; Fujimoto, M.; Sprong, H.; Su, T.-P. The Lifetime of UDP-galactose:Ceramide Galactosyltransferase Is Controlled by a Distinct Endoplasmic Reticulum-associated Degradation (ERAD) Regulated by Sigma-1 Receptor Chaperones. J. Boil. Chem. 2012, 287, 43156–43169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, A.; Fontanilla, D.; Gopalakrishnan, A.; Chae, Y.-K.; Markley, J.L.; Ruoho, A.E. The Sigma-1 Receptor Protects Against Cellular Oxidative Stress and Activates Antioxidant Response Elements. Eur. J. Pharmacol. 2012, 682, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Goguadze, N.; Zhuravliova, E.; Morin, D.; Mikeladze, D.G.; Maurice, T. Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox. Res. 2017, 35, 1–18. [Google Scholar] [CrossRef]
- Jia, J.; Cheng, J.; Wang, C.; Zhen, X. Sigma-1 Receptor-Modulated Neuroinflammation in Neurological Diseases. Front. Cell. Neurosci. 2018, 12, 314. [Google Scholar] [CrossRef] [Green Version]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef] [Green Version]
- Chia, S.J.; Tan, E.-K.; Chao, Y.X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [Green Version]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.-F.; Benabid, A.-L.; Sadoul, R.; Verna, J.-M. Molecular Pathways Involved in the Neurotoxicity of 6-OHDA, Dopamine and MPTP: Contribution to the Apoptotic Theory in Parkinson’s Disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Blandini, F.; Armentero, M.-T.; Martignoni, E. The 6-Hydroxydopamine Model: News from the Past. Park. Relat. Disord. 2008, 14, S124–S129. [Google Scholar] [CrossRef] [PubMed]
- Cicchetti, F.; Brownell, A.L.; Williams, K.; Chen, Y.I.; Livni, E.; Isacson, O. Neuroinflammation of the Nigrostriatal Pathway During Progressive 6-OHDA Dopamine Degeneration in Rats Monitored by Immunohistochemistry and PET Imaging. Eur. J. Neurosci. 2002, 15, 991–998. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yuan, W.; Li, Z.; Hou, Y.; Liu, F.; Feng, J. 6- Hydroxydopamine Induces Autophagic Flux Dysfunction by Impairing Transcription Factor EB Activation and Lysosomal Function in Dopaminergic Neurons and SH-SY5Y Cells. Toxicol. Lett. 2018, 283, 58–68. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; A Lashuel, H. The Process of Lewy Body Formation, Rather Than Simply α-Synuclein Fibrillization, Is One of the Major Drivers of Neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A. Do Lewy Bodies Contain Alpha-Synuclein Fibrils? and Does It Matter? a Brief History and Critical Analysis of Recent Reports. Neurobiol. Dis. 2020, 141, 104876. [Google Scholar] [CrossRef]
- Alvarez-Fischer, D.; Henze, C.; Strenzke, C.; Westrich, J.; Ferger, B.; Höglinger, G.U.; Oertel, W.H.; Hartmann, A. Characterization of the Striatal 6-OHDA Model of Parkinson’s Disease in Wild Type and α-Synuclein-Deleted Mice. Exp. Neurol. 2008, 210, 182–193. [Google Scholar] [CrossRef]
- Ganapathy, K.; Datta, I.; Sowmithra, S.; Joshi, P.; Bhonde, R. Influence of 6-Hydroxydopamine Toxicity on α-Synuclein Phosphorylation, Resting Vesicle Expression, and Vesicular Dopamine Release. J. Cell. Biochem. 2016, 117, 2719–2736. [Google Scholar] [CrossRef]
- Francardo, V. Sigma-1 Receptor: A Potential New Target for Parkinson’s Disease? Neural Regen. Res. 2014, 9, 1882–1883. [Google Scholar] [CrossRef]
- Hernandez-Baltazar, D.; Zavala-Flores, L.; Villanueva-Olivo, A. The 6-Hydroxydopamine Model and Parkinsonian Pathophysiology: Novel Findings in an Older Model. Neurología 2017, 32, 533–539. [Google Scholar] [CrossRef]
- Tieu, K. A Guide to Neurotoxic Animal Models of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef]
- Haga, H.; Matsuo, K.; Yabuki, Y.; Zhang, C.; Han, F.; Fukunaga, K. Enhancement of ATP Production Ameliorates Motor and Cognitive Impairments in a Mouse Model of MPTP-Induced Parkinson’s Disease. Neurochem. Int. 2019, 129, 104492. [Google Scholar] [CrossRef]
- Seredenin, S.B.; Voronina, A.T.; Neznamov, G.G.; Blednov, I.A.; Badyshtov, B.A.; Viglinskaia, I.V.; Kozlovskaia, M.M.; Kolotilinskaia, N.V.; Iarkova, A.M.; Savel’Ev, V.L.; et al. The pharmacogenetic concept of the anxio-selective effect. Ann. Russ. Acad. Med. Sci. 1998, 4, 3–9. [Google Scholar]
- Seredenin, S.B.; Voronin, M.V. Neuroreceptor Mechanisms of the Afobazole Effect. Eksp. Klin. Farmakol. 2009, 72, 3–11. [Google Scholar] [PubMed]
- Ryaskina, E.V.; Voronin, M.; Seredenin, S.B. Interaction of 2-Mercaptobenzimidazole Derivatives with Sigma-1 Receptors. Pharm. Chem. J. 2012, 46, 334–336. [Google Scholar] [CrossRef]
- Voronin, M.; Kadnikov, I.A. Contribution of Sigma-1 Receptor to Cytoprotective Effect of Afobazole. Pharmacol. Res. Perspect. 2016, 4, e00273. [Google Scholar] [CrossRef] [Green Version]
- Katnik, C.; Garcia, A.; Behensky, A.A.; Yasny, I.E.; Shuster, A.M.; Seredenin, S.B.; Petrov, A.V.; Cuevas, J. Activation of σ1 and σ2 Receptors by Afobazole Increases Glial Cell Survival and Prevents Glial Cell Activation and Nitrosative Stress After Ischemic Stroke. J. Neurochem. 2016, 139, 497–509. [Google Scholar] [CrossRef]
- Katnik, C.; García, A.; Behensky, A.; Yasny, I.; Shuster, A.; Seredenin, S.; Petrov, A.; Seifu, S.; McAleer, J.; Willing, A.; et al. Treatment With Afobazole at Delayed Time Points Following Ischemic Stroke Improves Long-Term Functional and Histological Outcomes. Neurobiol. Dis. 2014, 62, 354–364. [Google Scholar] [CrossRef]
- Behensky, A.A.; Yasny, I.E.; Shuster, A.M.; Seredenin, S.B.; Petrov, A.V.; Cuevas, J. Afobazole Activation of σ-1 Receptors Modulates Neuronal Responses to Amyloid-β25-35. J. Pharmacol. Exp. Ther. 2013, 347, 468–477. [Google Scholar] [CrossRef]
- Behensky, A.A.; Yasny, I.E.; Shuster, A.M.; Seredenin, S.B.; Petrov, A.V.; Cuevas, J. Stimulation of Sigma Receptors with Afobazole Blocks Activation of Microglia and Reduces Toxicity Caused by Amyloid-β25–35. J. Pharmacol. Exp. Ther. 2013, 347, 458–467. [Google Scholar] [CrossRef]
- Cuevas, J.; Rodriguez, A.; Behensky, A.; Katnik, C. Afobazole Modulates Microglial Function via Activation of Both σ-1 and σ-2 Receptors. J. Pharmacol. Exp. Ther. 2011, 339, 161–172. [Google Scholar] [CrossRef]
- Cuevas, J.; Behensky, A.; Deng, W.; Katnik, C. Afobazole Modulates Neuronal Response to Ischemia and Acidosis via Activation of σ-1 Receptors. J. Pharmacol. Exp. Ther. 2011, 339, 152–160. [Google Scholar] [CrossRef]
- Voronin, M.V.; Kadnikov, I.A.; Seredenin, S.B. Afobazole Restores the Dopamine Level in a 6-Hydroxydopamine Model of Parkinson’s Disease. Neurochem. J. 2019, 13, 49–56. [Google Scholar] [CrossRef]
- GOST 33215- Guidelines for Accommodation and Care of Animals. Environment, Housing and Management. In Interstate Council for Standartization, Metrology and Certification; Standartinform: Moscow, Russia, 2016; Available online: http://protect.gost.ru/document.aspx?control=7&id=202494 (accessed on 14 October 2020).
- National Research Council (U.S.); Committee for the Update of the Guide for the Care and Use of Laboratory Animals.; Institute for Laboratory Animal Research (U.S.); National Academies Press (U.S.). Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011; p. 220. [Google Scholar]
- Flecknell, P.A. Anaesthesia of Common Laboratory Species: Special Considerations. In Laboratory Animal Anaesthesia; Academic Press: Cambridge, MA, USA, 2009; pp. 181–241. [Google Scholar]
- Kadnikov, I.A.; Voronkov, D.N.; Voronin, M.V.; Seredenin, S.B. Analysis of Quinone Reductase 2 Implication in Mechanism of Antiparkinsonian Action of Afobazole. Neurochem. J. 2020, 14, 227–234. [Google Scholar] [CrossRef]
- Goes, A.T.; Jesse, C.R.; Antunes, M.S.; Ladd, F.V.L.; Ladd, A.A.L.; Luchese, C.; Paroul, N.; Boeira, S.P. Protective Role of Chrysin on 6-Hydroxydopamine-Induced Neurodegeneration a Mouse Model of Parkinson’s Disease: Involvement of Neuroinflammation and Neurotrophins. Chem. Interact. 2018, 279, 111–120. [Google Scholar] [CrossRef]
- Roffler-Tarlov, S.; Sharman, D.F.; Tegerdine, P. 3,4- Dihydroxyphenylacetic Acid and 4-Hydroxy-3-Methoxyphenylacetic Acid in the Mouse Striatum: A Reflection of Intra- and Extra-Neuronal Metabolism of Dopamine? Br. J. Pharmacol. 1971, 42, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Zhai, S.; Tanimura, A.; Graves, S.M.; Shen, W.; Surmeier, D.J. Striatal Synapses, Circuits, and Parkinson’s Disease. Curr. Opin. Neurobiol. 2018, 48, 9–16. [Google Scholar] [CrossRef]
- Monville, C.; Torres, E.M.; Dunnett, S.B. Comparison of Incremental and Accelerating Protocols of the Rotarod Test for the Assessment of Motor Deficits in the 6-OHDA Model. J. Neurosci. Methods 2006, 158, 219–223. [Google Scholar] [CrossRef]
- Castaneda, E.; Whishaw, I.; Robinson, T. Changes in Striatal Dopamine Neurotransmission Assessed with Microdialysis Following Recovery From a Bilateral 6-OHDA Lesion: Variation as a Function of Lesion Size. J. Neurosci. 1990, 10, 1847–1854. [Google Scholar] [CrossRef] [Green Version]
- Patrick, S.L.; Walker, J.; Perkel, J.M.; Lockwood, M.; Patrick, R.L. Increase in Rat Striatal Extracellular Dopamine and Vacous Chewing Produced by Two σ Receptor Ligands. Eur. J. Pharmacol. 1993, 231, 243–249. [Google Scholar] [CrossRef]
- Gudelsky, G.A. Effects of σ Receptor Ligands on the Extracellular Concentration of Dopamine in the Striatum and Prefrontal Cortex of the Rat. Eur. J. Pharmacol. 1995, 286, 223–228. [Google Scholar] [CrossRef]
- Iyengar, S.; Dilworth, V.M.; Mick, S.J.; Contreras, P.C.; Monahan, J.B.; Rao, T.S.; Wood, P.L. Sigma Receptors Modulate Both a9 and a10 Dopaminergic Neurons in the Rat Brain: Functional Interaction with NMDA Receptors. Brain Res. 1990, 524, 322–326. [Google Scholar] [CrossRef]
- Kobayashi, T.; Matsuno, K.; Murai, M.; Mita, S. σ1 Receptor Subtype Is Involved in the Facilitation of Cortical Dopaminergic Transmission in the Rat Brain. Neurochem. Res. 1997, 22, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Wachtel, S.R.; Abercrombie, E.D. l-3,4-Dihydroxyphenylalanine-Induced Dopamine Release in the Striatum of Intact and 6-Hydroxydopamine-Treated Rats: Differential Effects of Monoamine Oxidase A and B Inhibitors. J. Neurochem. 2002, 63, 108–117. [Google Scholar] [CrossRef]
- Johnson, M.E.; Salvatore, M.F.; Maiolo, S.; Bobrovskaya, L. Tyrosine Hydroxylase as a Sentinel for Central and Peripheral Tissue Responses in Parkinson’s Progression: Evidence From Clinical Studies and Neurotoxin Models. Prog. Neurobiol. 2018, 1–25. [Google Scholar] [CrossRef]
- Björklund, A.; Rosenblad, C.; Winkler, C.; Kirik, D. Studies on Neuroprotective and Regenerative Effects of GDNF in a Partial Lesion Model of Parkinson’s Disease. Neurobiol. Dis. 1997, 4, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, H.; Oertel, W. Progressive Degeneration of Nigrostriatal Dopamine Neurons Following Intrastriatal Terminal Lesions with 6-Hydroxydopamine: A Combined Retrograde Tracing and Immunocytochemical Study in the Rat. Neuroscience 1994, 59, 401–415. [Google Scholar] [CrossRef]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease Duration and the Integrity of the Nigrostriatal System in Parkinson’s Disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef] [Green Version]
- Booth, R.G.; Baldessarini, R.J. (+)-6,7- Benzomorphan Sigma Ligands Stimulate Dopamine Synthesis in Rat Corpus Striatum Tissue. Brain Res. 1991, 557, 349–352. [Google Scholar] [CrossRef]
- Seredin, S.B.; Melkumian, D.S.; A Val’Dman, E.; A Iarkova, M.; Seredina, T.C.; Voronin, M.V.; Lapitskaia, A.S. Effects of Afobazole on the Bdnf Content in Brain Structures of Inbred Mice with Different Phenotypes of Emotional Stress Reaction. Eksp. Klin. Farmakol. 2006, 69, 3–6. [Google Scholar]
- Pałasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [Green Version]
- Tan, F.; Guio-Aguilar, P.; Downes, C.; Zhang, M.; O’Donovan, L.; Callaway, J.; Crack, P.J. The σ1 Receptor Agonist 4-PPBP Elicits ERK1/2 Phosphorylation in Primary Neurons: A Possible Mechanism of Neuroprotective Action. Neuropharmacology 2010, 59, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Di Paolo, T.; Tremblay, M.E. Microglial Implication in Parkinson’s Disease: Loss of Beneficial Physiological Roles or Gain of Inflammatory Functions? Front. Cell. Neurosci. 2018, 12, 282. [Google Scholar] [CrossRef] [PubMed]
- Stott, S.; Barker, R.A. Time Course of Dopamine Neuron Loss and Glial Response in the 6-OHDA Striatal Mouse Model of Parkinson’s Disease. Eur. J. Neurosci. 2013, 39, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Groups | TH+ Neurons (Neurons/Slide) | Relative TH+ (%) |
|---|---|---|
| Sham + Vehicle | 93.3 (83.75–108.0) | 94.86 (89.24–104.1) |
| 6-OHDA + Vehicle | 60.2 (16.55–65.5) * p = 0.027 | 61.6 (38.65–84.96) * p = 0.022 |
| 6-OHDA + Afobazole | 61.5 (35.4–72.5) * p = 0.04 | 82.12 (57.81–87.07) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadnikov, I.A.; Verbovaya, E.R.; Voronkov, D.N.; Voronin, M.V.; Seredenin, S.B. Deferred Administration of Afobazole Induces Sigma1R-Dependent Restoration of Striatal Dopamine Content in a Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 7620. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207620
Kadnikov IA, Verbovaya ER, Voronkov DN, Voronin MV, Seredenin SB. Deferred Administration of Afobazole Induces Sigma1R-Dependent Restoration of Striatal Dopamine Content in a Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2020; 21(20):7620. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207620
Chicago/Turabian StyleKadnikov, Ilya A., Ekaterina R. Verbovaya, Dmitry N. Voronkov, Mikhail V. Voronin, and Sergei B. Seredenin. 2020. "Deferred Administration of Afobazole Induces Sigma1R-Dependent Restoration of Striatal Dopamine Content in a Mouse Model of Parkinson’s Disease" International Journal of Molecular Sciences 21, no. 20: 7620. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207620