Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems


Abstract
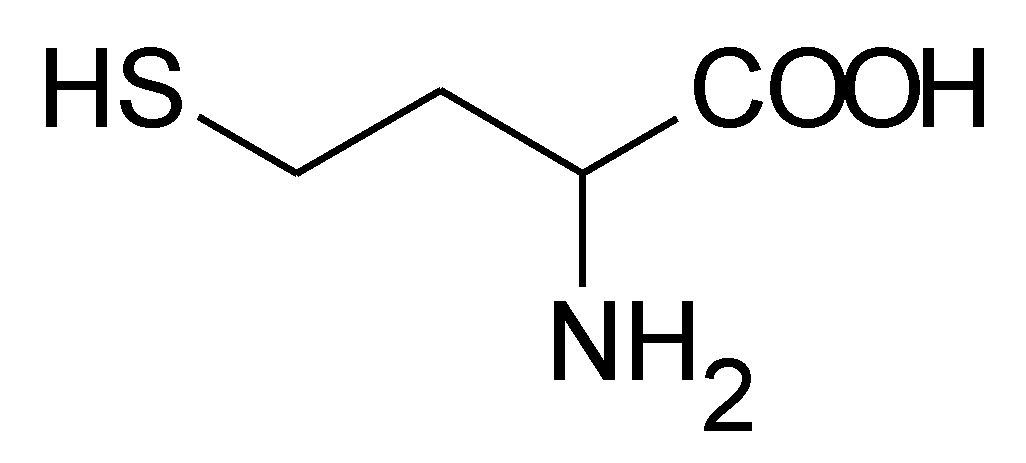
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Homocysteine Transport
3. Homocysteine and Mitochondrial Energy Metabolism
3.1. Hcy and Electron Transport Chain
3.2. Hcy and Tricarboxylic Acid Cycle
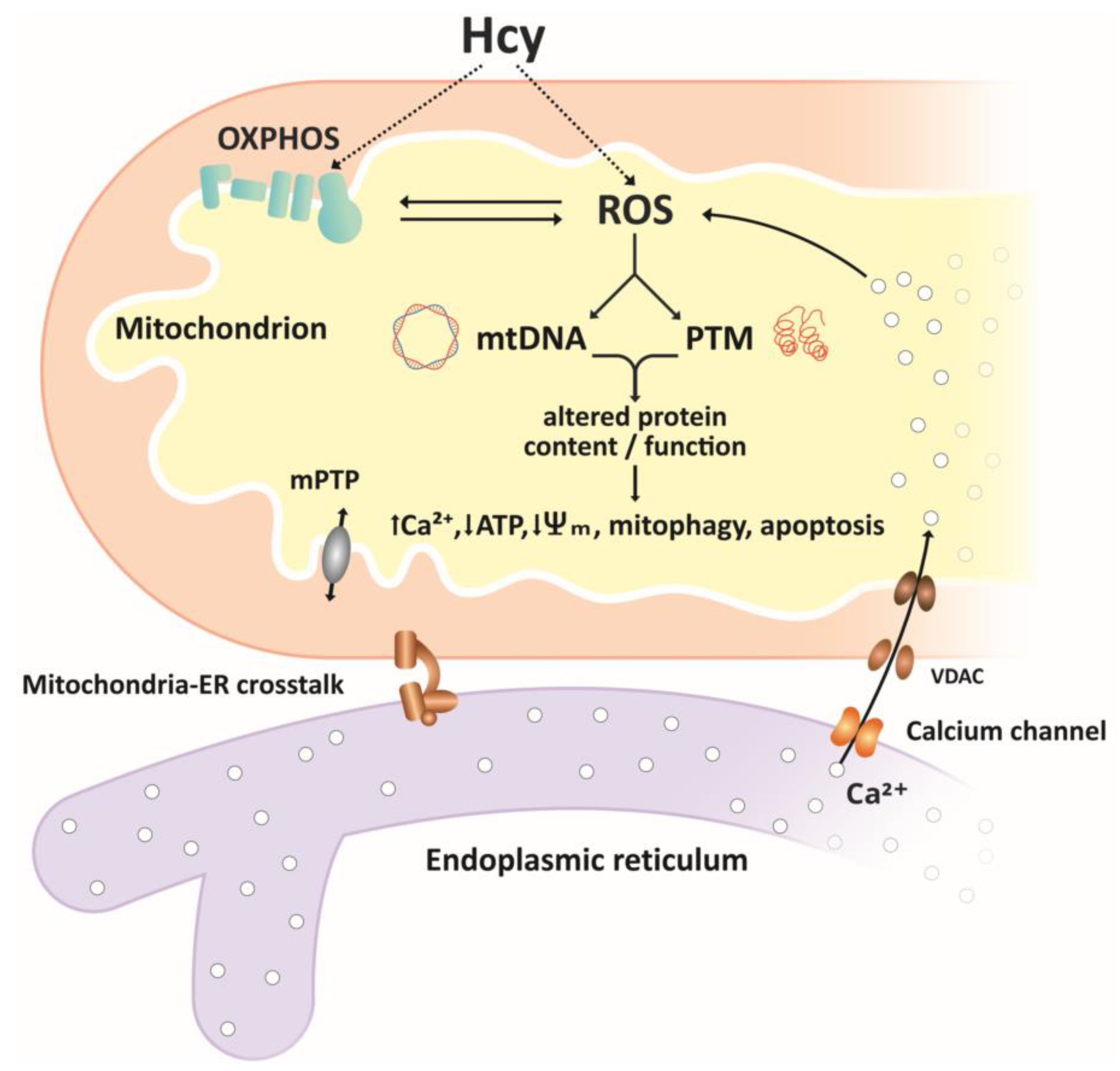
4. Homocysteine and Mitochondrial Oxidative Stress
4.1. Hcy and Mitochondrial ROS Generation
4.2. Hcy and Antioxidant Enzymes
4.3. Hcy and Hydrogen Sulfide
4.4. Hcy and ROS Producing Enzymes
4.5. Hcy and Expression of Proteins Involved in Energy Metabolism
4.6. Hcy and Mitochondrial Oxidative Damage
5. Homocysteine and Mitochondrial Apoptotic Pathway
5.1. Hcy and Pro-Apoptic and Anti-Apoptic Proteins
5.2. Hcy and Mitochondria-ER Crosstalk
5.3. Hcy, Inflammation and Apoptosis
6. Homocysteine and Mitochondrial Dynamics
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CVD | cardiovascular disease |
| DCFH | 2′,7′-dihydrodichlorofluorescein |
| DRP1 | dynamin-related protein |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| GPx | glutathione peroxidase |
| Hcy | homocysteine |
| HHcy | hyperhomocysteinemia |
| HUVECs | human umbilical vein endothelial cells |
| MAMs | mitochondria-associated membranes |
| mtDNA | mitochondrial DNA |
| mPTP | mitochondrial permeability transition pore |
| MTD | mitochondrial targeting domain |
| NMDAR | N-methyl-D-aspartate receptor |
| NOS | NO synthase |
| OXPHOS | oxidative phosphorylation |
| PTMs | post-translational modifications |
| ROS | reactive oxygen species |
| SAH | S-adenosylhomocysteine |
| SAM | S-adenosylmethionine |
| SOD | superoxide dismutase |
| TCA | tricarboxylic acid cycle |
| VDAC | voltage-dependent anion channel |
References
- Cesari, M.; Rossi, G.P.; Sticchi, D.; Pessina, A.C. Is homocysteine important as risk factor for coronary heart disease? Nutr. Metab. Cardiovasc. Dis. 2005, 15, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Loscalzo, J. The treatment of hyperhomocysteinemia. Annu. Rev. Med. 2009, 60, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halasova, E.; Lehotský, J. The molecular and cellular effect of homocysteine metabolism imbalance on human health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, X.; Kong, W. Hyperhomocysteinaemia and vascular injury: Advances in mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Gottfries, C.; Regland, B. Identification of cognitive impairment in the elderly: Homocysteine is an early marker. Dement. Geriatr. Cogn. Disord. 1998, 10, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, E.F.; Busanello, E.N.B.; Miglioranza, A.; Zanatta, Â.; Barchak, A.G.; Vargas, C.R.; Saute, J.A.M.; Rosa, C.; Carrion, M.J.; Camargo, D.; et al. Evidence that folic acid deficiency is a major determinant of hyperhomocysteinemia in Parkinson´s disease. Metab. Brain Dis. 2009, 24, 257–269. [Google Scholar] [CrossRef]
- Nygård, O.; Vollset, S.E.; Refsum, H.; Stensvold, I.; Tverdal, A.; Nordrehaug, J.E.; Ueland, P.M.; Kvåle, G. Total plasma homocysteine and cardiovascular risk profile. The Hordaland homocysteine study. JAMA 1995, 274, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Bostom, A.G.; Shemin, D.; Verhoef, P.; Nadeau, M.R.; Jacques, P.F.; Selhub, J.; Dworkin, L.; Rosenberg, I.H. Elevated fasting total plasma homocysteine levels and cardiovascular disease outcomes in maintenance dialysis patients. Arter. Thromb. Vasc. Biol. 1997, 17, 2554–2558. [Google Scholar] [CrossRef] [PubMed]
- Strauss, E.; Supinski, W.; Radziemski, A.; Oszkinis, G.; Pawlak, A.L.; Gluszek, J. Is hyperhomocysteinemia a causal factor for heart failure? The impact of the functional variants of MTHFR and PON1 on ischemic and non-ischemic etiology. Int. J. Cardiol. 2017, 228, 37–44. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidatice stress. Amino Acids 2003, 25, 409–417. [Google Scholar] [CrossRef]
- Suematsu, N.; Ojaimi, C.; Kinugawa, S.; Wang, Z.; Xu, X.; Koller, A.; Recchia, F.A.; Hintze, T.H. Hyperhomocysteinemia alters cardiac substrate metabolism by impairing nitric oxide bioavailability through oxidative stress. Circulation 2007, 115, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Kolling, J.; Scherer, E.B.; Da Cunha, A.A.; Cunha, M.J.; Wyse, A.T.S. Homocysteine induces oxidative–nitrative stress in heart of rats: Prevention by folic acid. Cardiovasc. Toxicol. 2011, 11, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Scherer, E.B.; Da Cunha, A.A.; Kolling, J.; Cunha, M.J.; Schmitz, F.; Sitta, A.; Lima, D.D.; Delwing, D.; Vargas, C.R.; Wyse, A.T.S. Development of an animal model for chronic mild hyperhomocysteinemia and its response to oxidative damage. Int. J. Dev. Neurosci. 2011, 29, 693–699. [Google Scholar] [CrossRef]
- Petras, M.; Tatarkova, Z.; Kovalska, M.; Mokra, D.; Dobrota, D.; Lehotsky, J.; Drgová, A. Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J. Physiol. Pharmacol. 2014, 65, 15–23. [Google Scholar] [PubMed]
- Perna, A.; Ingrosso, D.; Lombardi, C.; Acanfora, F.; Satta, E.; Cesare, C.M.; Violetti, E.; Romano, M.M.; De Santo, N.G. Possible mechanisms of homocysteine toxicity. Kidney Int. 2003, 63, S137–S140. [Google Scholar] [CrossRef] [Green Version]
- Esse, R.; Barroso, M.; De Almeida, I.T.; Castro, R. The contribution of homocysteine metabolism disruption to endothelial dysfunction: state-of-the-art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrakhovitch, E.A.; Tabibzadeh, S. Homocysteine and age-asscociated disorders. Aging Res. Rev. 2019, 49, 144–164. [Google Scholar] [CrossRef]
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of epigenetic mechanisms of gene expression in the pathologies of hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; Dilisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial function, biology, and role in disease. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Ghemrawi, R.; Khair, M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef] [PubMed]
- Stanga, S.; Caretto, A.; Boido, M.; Vercelli, A. Mitochondrial dysfunctions: A red thread across neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 3719. [Google Scholar] [CrossRef] [PubMed]
- Büdy, B.; O’Neill, R.; Dibello, P.M.; Sengupta, S.; Jacobsen, D.W. Homocysteine transport by human aortic endothelial cells: Identification and properties of import systems. Arch. Biochem. Biophys. 2006, 446, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Yang, F.; Brailoiu, E.; Jakubowski, H.; Dun, N.J.; Schafer, A.I.; Yang, X.; Durante, W.; Wang, H. Differential regulation of homocysteine transport in vascular endothelial and smooth muscle cells. Arter. Thromb. Vasc. Biol. 2007, 27, 1976–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, V.; Vozza, A.; Calcagnile, V.; Gorgoglione, R.; Arrigoni, R.; Fontanesi, F.; Marobbio, C.M.; Castegna, A.; Palmieri, F.; Palmieri, L. Molecular identification and functional characterization of a novel glutamate transporter in yeast and plant mitochondria. Biochim. Biophys. Acta. 2018, 1859, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Agrimi, G.; Di Noia, M.A.; Marobbio, C.M.T.; Fiermonte, G.; Lasorsa, F.M.; Palmieri, F. Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 2004, 379, 183–190. [Google Scholar] [CrossRef]
- Menga, A.; Palmieri, E.M.; Cianciulli, A.; Infantino, V.; Mazzone, M.; Scilimati, A.; Palmieri, F.; Castegna, A.; Iacobazzi, V. SLC25A26overexpression impairs cell function via mtDNAhypermethylation and rewiring of methyl metabolism. FEBS J. 2017, 284, 967–984. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Ferko, M.; Andelová, N.; Bačová, B.S.; Jašová, M. Myocardial adaptation in pseudohypoxia: signaling and regulation of mPTP via mitochondrial connexin 43 and cardiolipin. Cells 2019, 8, 1449. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, K.; Siddiqi, N.; Sing, S.; Neil, C.J.; Dawson, D.K.; Frenneaux, M.P. The breathing heart—Mitochondrial respiratory chain dysfunction in cardiac disease. Int. J. Cardiol. 2014, 171, 134–143. [Google Scholar] [CrossRef]
- Requejo, R.; Bolaños, J.P. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free. Radic. Biol. Med. 2016, 100, 123–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta. 2010, 1802, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Lehotský, J.; Tothová, B.; Kovalská, M.; Dobrota, D.; Beňová, A.; Kalenská, D.; Kaplán, P. Role of homocysteine in the ischemic stroke and development of ischemic tolerance. Front. Neurosci. 2016, 10, 538. [Google Scholar] [CrossRef]
- Lash, L.H.; Anders, M.W. Mechanism of S-(1,2-dichlorovinyl)-L-cysteine- and S-(1,2-dichlorovinyl)-L-homocysteine-induced renal mitochondrial toxicity. Mol. Pharmacol. 1987, 32, 549–556. [Google Scholar] [PubMed]
- Chang, L.; Geng, B.; Yu, F.; Zhao, J.; Jiang, H.; Du, J.; Tang, C. Hydrogen sulfide inhibits myocardial injury induced by homocysteine in rats. Amino Acids 2008, 34, 573–585. [Google Scholar] [CrossRef]
- Timkova, V.; Tatarkova, Z.; Lehotský, J.; Racay, P.; Dobrota, D.; Kaplán, P. Effects of mild hyperhomocysteinemia on electron transport chain complexes, oxidative stress, and protein expression in rat cardiac mitochondria. Mol. Cell. Biochem. 2016, 411, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Longoni, A.; Kolling, J.; Siebert, C.; Dos Santos, J.P.; Da Silva, J.S.; Pettenuzzo, L.F.; Meira-Martins, L.A.; Gonçalves, C.-A.; De Assis, A.M.; Wyse, A.T.S. 1,25-Dihydroxyvitamin D 3 prevents deleterious effects of homocysteine on mitochondrial function and redox status in heart slices. Nutr. Res. 2017, 38, 52–63. [Google Scholar] [CrossRef]
- Jakubowski, H. An overview of homocysteine metabolism. In Homocysteine in Protein Structure/Function and Human Disease, 1st ed.; Springer: Wien, Austria, 2013; pp. 7–18. [Google Scholar]
- Hasegawa, T.; Mikoda, N.; Kitazawa, M.; LaFerla, F.M. Treatment of Alzheimer’s disease with anti-homocysteic acid antibody in 3xTg-AD male mice. PLoS ONE 2010, 5, e8593. [Google Scholar] [CrossRef]
- Folbergrová, J.; Ješina, P.; Drahota, Z.; Lisý, V.; Haugvicová, R.; Pecinová, A.; Houstĕk, J. Mitochondrial complex I inhibition in cerebral cortex of immature rats following homocysteic acid-induced seizures. Exp. Neurol. 2007, 204, 597–609. [Google Scholar] [CrossRef]
- Folbergrová, J.; Ješina, P.; Haugvicová, R.; Lisý, V.; Houštěk, J. Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem. Int. 2010, 56, 394–403. [Google Scholar] [CrossRef]
- Kumar, M.; Sandhir, R. Hydrogen sulfide attenuates hyperhomocysteinemia-induced mitochondrial dysfunctions in brain. Mitochondrion 2020, 50, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Wyse, A.T.S.; Sanches, E.; Dos Santos, T.; Siebert, C.; Kolling, J.; Netto, C. Chronic mild hyperhomocysteinemia induces anxiety-like symptoms, aversive memory deficits and hippocampus atrophy in adult rats: New insights into physiopathological mechanisms. Brain Res. 2020, 1728, 146592. [Google Scholar] [CrossRef]
- Kolling, J.; Scherer, E.B.S.; Siebert, C.; Longoni, A.; Loureiro, S.; Weis, S.N.; Pettenuzzo, L.; Wyse, A.T.S. Severe hyperhomocysteinemia decreases respiratory enzyme and Na+-K+ ATPase activities, and leads to mitochondrial alterations in rat amygdala. Neurotox. Res. 2015, 29, 408–418. [Google Scholar] [CrossRef]
- Dos Santos, T.M.; Siebert, C.; De Oliveira, M.F.; Manfredini, V.; Wyse, A.T.S. Chronic mild Hyperhomocysteinemia impairs energy metabolism, promotes DNA damage and induces a Nrf2 response to oxidative stress in rats brain. Cell. Mol. Neurobiol. 2019, 39, 687–700. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Borah, A. Oxidative stress and mitochondrial dysfunction are the underlying events of dopaminergic neurodegeneration in homocysteine rat model of Parkinson’s disease. Neurochem. Int. 2016, 101, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Paul, R.; Giri, A.; Borah, A. Chronic exposure of homocysteine in mice contributes to dopamine loss by enhancing oxidative stress in nigrostriatum and produces behavioral phenotypes of Parkinson’s disease. Biochem. Biophys. Rep. 2016, 6, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Dong, Z.; Zhao, Y.; Sai, N.; Wang, X.; Liu, H.; Huang, G.; Zhang, X. Homocysteine induces mitochondrial dysfunction involving the crosstalk between oxidative stress and mitochondrial pSTAT3 in rat ischemic brain. Sci. Rep. 2017, 7, 6932. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Zhao, Y.-Q.; Yan, H.; Liu, H.; Huang, G. Inhibitory effect of homocysteine on rat neural stem cell growth in vitro is associated with reduced protein levels and enzymatic activities of aconitase and respiratory complex III. J. Bioenerg. Biomembr. 2017, 49, 131–138. [Google Scholar] [CrossRef]
- Chang, L.; Xu, J.; Yu, F.; Zhao, J.; Tang, X.; Tang, C. Taurine protected myocardial mitochondria injury induced by hyperhomocysteinemia in rats. Amino Acids 2004, 27, 37–48. [Google Scholar] [CrossRef]
- Fan, C.D.; Sun, J.Y.; Fu, X.T.; Hou, Y.J.; Li, Y.; Yang, M.F.; Sun, B.L.; Fu, X.Y. Astaxanthin attenuates homocysteine-induced cardiotoxicity in vitro and in vivo by inhibiting mitochondrial dysfunction and oxidative damage. Front. Physiol. 2017, 8, 1041. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Sandhir, R. Neuroprotective efect of hydrogen sulfide in hyperhomocysteinemia is mediated through antioxidant action involving Nrf2. NeuroMolec. Med. 2018, 20, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.; Sanchez-Roman, I.; Gómez, A.; Sanchez, C.; Suarez, H.; Lopez-Torres, M.; Barja, G. Methionine and homocysteine modulate the rate of ROS generation of isolated mitochondria in vitro. J. Bioenerg. Biomembr. 2011, 43, 377–386. [Google Scholar] [CrossRef]
- Zhang, T.; Huang, D.; Hou, J.; Li, J.; Zhang, Y.; Tian, M.; Li, Z.; Tie, T.; Cheng, Y.; Su, X.; et al. High-concentration homocysteine inhibits mitochondrial respiration function and production of reactive oxygen species in neuron cells. J. Stroke Cerebrovasc. Dis. 2020, 29, 105109. [Google Scholar] [CrossRef] [PubMed]
- Hogg, N. The effect of cist(e)ine on the auto-oxidation of homocysteine. Free Radic. Biol. Med. 1999, 27, 28–33. [Google Scholar] [CrossRef]
- Starkebaum, G.; Harlan, J.M. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J. Clin. Investig. 1986, 77, 1370–1376. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Ray, R.S.; Sandhir, R. Hydrogen sulfide attenuates homocysteine-induced neurotoxicity by preventing mitochondrial dysfunctions and oxidative damage: In vitro and in vivo studies. Neurochem. Int. 2018, 120, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Protein homocysteinylation: Possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999, 13, 2277–2283. [Google Scholar] [CrossRef]
- Sharma, G.S.; Kumar, T.; Singh, L.R. N-Homocysteinylation induces different structural and functional consequences on acidic and basic proteins. PLoS ONE 2014, 9, e116386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akchiche, N.; Bossenmeyer-Pourié, C.; Kerek, R.; Martin, N.; Pourié, G.; Koziel, V.; Helle, D.; Alberto, J.M.; Ortiou, S.; Camadro, J.; et al. Homocysteinylation of neuronal proteins contributes to folate deficiency-associated alterations of differentiation, vesicular transport, and plasticity in hippocampal neuronal cells. FASEB J. 2012, 26, 3980–3992. [Google Scholar] [CrossRef] [PubMed]
- Khodadadi, S.; Riazi, G.H.; Ahmadian, S.; Hoveizi, E.; Karima, O.S.; Aryapour, H. Effect ofN-homocysteinylation on physicochemical and cytotoxic properties of amyloid β-peptide. FEBS Lett. 2011, 586, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.S.; Kumar, T.; Dar, T.A.; Singh, L.R. Protein N-homocysteinylation: From cellular toxicity to neurodegeneration. Biochim. Biophys. Acta. 2015, 1850, 2239–2245. [Google Scholar] [CrossRef]
- Perła-Kaján, J.; Marczak, Ł.; Kaján, L.; Skowronek, P.; Twardowski, T.; Jakubowski, H. Modification by homocysteine thiolactone affects redox status of cytochromec. Biochemistry 2007, 46, 6225–6231. [Google Scholar] [CrossRef]
- Sharma, G.S.; Singh, L.R. Conformational status of cytochrome c upon N-homocysteinylation: Implications to cytochrome c release. Arch. Biochem. Biophys. 2017, 614, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Qipshidze, N.; Munjal, C.; Vacek, J.C.; Metreveli, N.; Givvimani, S.; Tyagi, S.C. Tetrahydrocurcumin ameliorates homocysteinylated cytochrome-c mediated autophagy in hyperhomocysteinemia mice after cerebral ischemia. J. Mol. Neurosci. 2012, 47, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Silla, Y.; Varshney, S.; Ray, A.; Basak, T.; Zinellu, A.; Sabareesh, V.; Carru, C.; Sengupta, S. Hydrolysis of homocysteine thiolactone results in the formation of protein-Cys-S-S-homocysteinylation. Proteins 2019, 87, 625–634. [Google Scholar] [CrossRef]
- Silla, Y.; Sundaramoorthy, E.; Talwar, P.; Sengupta, S. S-linked protein homocysteinylation: Identifying targets based on structural, physicochemical and protein–protein interactions of homocysteinylated proteins. Amino Acids 2013, 44, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P.; Rahman, H.S. Antioxidant and oxidative stress: a mutual interplay in age-related diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aminzadeh, A.; Mehrzadi, S. Cardioprotective effect of levosimendan against homocysteine-induced mitochondrial stress and apoptotic cell death in H9C2. Biochem. Biophys. Res. Commun. 2018, 507, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Gao, X.; Yang, S.; Meng, M.; Yang, X.; Ge, B. The role of endoplasmic reticulum stress in endothelial dysfunction induced by homocysteine thiolactone. Fundam. Clin. Pharmacol. 2015, 29, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, A.; Mehrzadi, S. Melatonin attenuates homocysteine-induced injury in human umbilical vein endothelial cells. Fundam. Clin. Pharmacol. 2018, 32, 261–269. [Google Scholar] [CrossRef]
- Fan, X.; Wang, E.; He, J.; Zhang, L.; Zeng, X.; Gui, Y.; Sun, Q.; Song, Y.; Yuan, H. Ligustrazine protects homocysteine-induced apoptosis in human umbilical vein endothelial cells by modulating mitochondrial dysfunction. J. Cardiovasc. Transl. Res. 2019, 12, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Yue, H.; Zhou, G.H.; Zhu, Y.Y.; Wu, T.H.; Wen, J.F.; Cho, K.W.; Jin, S.N. Protective effects of oxymatrine on homocysteine-induced endothelial injury: Involvement of mitochondria-dependent apoptosis and Akt-eNOS-NO signaling pathways. Eur. J. Pharmacol. 2019, 864, 172717. [Google Scholar] [CrossRef]
- Tyagi, N.; Qipshidze, N.; Sen, U.; Rodriguez, W.; Ovechkin, A.; Tyagi, S.C. Cystathionine beta synthase gene dose dependent vascular remodeling in murine model of hyperhomocysteinemia. Int. J. Physiol. Pathophysiol. Pharmacol. 2011, 3, 210–222. [Google Scholar]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free. Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Op. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaidery, N.A.; Ahuja, M.; Thomas, B. Crosstalk between Nrf2 signaling and mitochondrial function in Parkinson’s disease. Mol. Cell. Neurosci. 2019, 101, 103413. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Ouyang, Y.; Jin, M.; Chen, M.; Liu, P.; Chao, X.; Chen, Z.; Chen, X.; Ramassamy, C.; Gao, Y.; et al. Downregulation of Nrf2/HO-1 pathway and activation of JNK/c-Jun pathway are involved in homocysteic acid-induced cytotoxicity in HT-22 cells. Toxicol. Lett. 2013, 223, 1–8. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.-C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef]
- Navneet, S.; Cui, X.; Zhao, J.; Zhao, J.; Kaidery, N.A.; Thomas, B.; Bollinger, K.E.; Yoon, Y.; Smith, S.B. Excess homocysteine upregulates the NRF2-antioxidant pathway in retinal Müller glial cells. Exp. Eye Res. 2019, 178, 228–237. [Google Scholar] [CrossRef]
- Navneet, S.; Zhao, J.; Wang, J.; Mysona, B.; Barwick, S.; Kaidery, N.A.; Saul, A.; Kaddour-Djebbar, I.; Bollag, W.B.; Thomas, B.; et al. Hyperhomocysteinemia-induced death of retinal ganglion cells: The role of Müller glial cells and NRF2. Redox Biol. 2019, 24, 101199. [Google Scholar] [CrossRef]
- Stein, A.; Bailey, S.M. Redox biology of hydrogen sulfide: Implications for physiology, pathophysiology, and pharmacology. Redox Biol. 2013, 1, 32–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.-D.; Wang, H.; Kho, S.-H.; Rinkiko, S.; Sheng, X.; Shen, H.-M.; Zhu, Y.Z. Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PLoS ONE 2013, 8, e53147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Kan, J.-T.; Cheng, Z.-Y.; Chen, J.-F.; Shen, Y.-Q.; Xu, J.; Wu, D.; Zhu, Y.-Z. Hydrogen sulfide as an endogenous modulator in mitochondria and mitochondria dysfunction. Oxidative Med. Cell. Longev. 2012, 2012, 878052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; He, G.-W. Imbalance of homocysteine and H2S: Significance, mechanisms, and therapeutic promise in vascular injury. Oxidative Med. Cell. Longev. 2019, 2019, 7629673. [Google Scholar] [CrossRef] [Green Version]
- Kamat, P.K.; Kalani, A.; Tyagi, S.C.; Tyagi, N. Hydrogen sulfide epigenetically attenuates homocysteine-induced mitochondrial toxicity mediated through NMDA receptor in mouse brain endothelial (bEnd3) cells. J. Cell. Physiol. 2014, 230, 378–394. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wei, C.; Zhou, Y.; Yan, T.; Wang, Z.; Li, W.; Zhao, L. Homocysteine induces apoptosis of human umbilical vein endothelial cells via mitochondrial dysfunction and endoplasmic reticulum stress. Oxid. Med. Cell. Longev. 2017, 2017, 5736506. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.J.; Schiffer, T.A.; Weitzberg, I.; Lundberg, J.O. Regulation of mitochondrial function and energetics by reactive nitrogen oxides. Free. Radic. Biol. Med. 2012, 53, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Moshal, K.S.; Ovechkin, A.V.; Rodriguez, W.; Steed, M.; Henderson, B.; Roberts, A.M.; Joshua, I.G.; Tyagi, S.C. Mitochondrial mechanism of oxidative stress and systemic hypertension in hyperhomocysteinemia. J. Cell. Biochem. 2005, 96, 665–671. [Google Scholar] [CrossRef]
- Lee, S.-J.; Kim, K.-M.; Namkoong, S.; Kim, C.K.; Kang, Y.-C.; Lee, H.; Ha, K.-S.; Han, J.-A.; Chung, H.-T.; Kwon, Y.-G.; et al. Nitric oxide inhibition of homocysteine-induced human endothelial cell apoptosis by down-regulation of p53-dependent noxa expression through the formation of S-nitrosohomocysteine. J. Biol. Chem. 2004, 280, 5781–5788. [Google Scholar] [CrossRef] [Green Version]
- Lind, M.; Hayes, A.; Caprnda, M.; Petrovič, D.; Rodrigo, L.; Kruzliak, P.; Zulli, A. Inducible nitric oxide synthase: Good or bad? Biomed. Pharmacother. 2017, 93, 370–375. [Google Scholar] [CrossRef]
- Yu, X.; Ge, L.; Niu, L.; Lian, X.; Ma, H.; Pang, L. The dual role of inducible nitric oxide synthase in myocardial ischemia/reperfusion Injury: Friend or foe? Oxid. Med. Cell. Longev. 2018, 2018, 8364848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandaviya, P.R.; Stolk, L.; Heil, S.G. Homocysteine and DNA methylation: A review of animal and human literature. Mol. Genet. Metab. 2014, 113, 243–252. [Google Scholar] [CrossRef]
- Cueto, R.; Zhang, L.; Shan, H.M.; Huang, X.; Li, X.; Li, Y.-F.; Lopez, J.O.; Yang, W.Y.; Lavallee, M.; Yu, C.; et al. Identification of homocysteine-suppressive mitochondrial ETC complex genes and tissue expression profile—Novel hypothesis establishment. Redox. Biol. 2018, 17, 70–88. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; He, C.; Gou, B.; Song, M. Mitochondrial regulation of cardiac aging. Biochim. Biophys. Acta. 2019, 1865, 1853–1864. [Google Scholar] [CrossRef]
- Chou, Y.-F.; Yu, C.-C.; Huang, R.-F.S. Changes in mitochondrial DNA deletion, content, and biogenesis in folate-deficient tissues of young rats depend on mitochondrial folate and oxidative DNA injuries. J. Nutr. 2007, 137, 2036–2042. [Google Scholar] [CrossRef]
- Sanz, A.; Caro, P.; Ayala, V.; Portero-Otín, M.; Pamplona, R.; Barja, G. Methionine restriction decreases mitochondrial oxygen radical generation and leak as well as oxidative damage to mitochondrial DNA and proteins. FASEB J. 2006, 20, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Perez-De-Arce, K.; Foncea, R.; Leighton, F. Reactive oxygen species mediates homocysteine-induced mitochondrial biogenesis in human endothelial cells: Modulation by antioxidants. Biochem. Biophys. Res. Commun. 2005, 338, 1103–1109. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2013, 15, 49–63. [Google Scholar] [CrossRef]
- Sipkens, J.A.; Krijnen, P.A.J.; Meischl, C.; Cillessen, S.A.G.M.; Smulders, Y.M.; Smith, D.E.C.; Giroth, C.P.E.; Spreeuwenberg, M.D.; Musters, R.J.P.; Muller, A.; et al. Homocysteine affects cardiomyocyte viability: Concentration-dependent effects on reversible flip-flop, apoptosis and necrosis. Apoptosis 2007, 12, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruman, I.I.; Culmsee, C.; Chan, S.L.; Kruman, Y.; Guo, Z.; Penix, L.; Mattson, M.P. Homocysteine elicits a dna damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 2000, 20, 6920–6926. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Bai, X.; Chen, Y.; Zhao, Y.; Liu, X. Homocysteine induces apoptosis of rat hippocampal neurons by inhibiting 14-3-3e expression and activating calcineurin. PLoS ONE 2012, 7, e48247. [Google Scholar]
- Zieminska, E.; Matyja, E.; Kozlowska, H.; Stafiej, A.; Lazarewicz, J.W. Excitotoxic neuronal injury in acute homocysteine neurotoxicity: Role of calcium and mitochondrial alterations. Neurochem. Int. 2006, 48, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Hirashima, Y.; Seshimo, S.; Fujiki, Y.; Okabe, M.; Nishiyama, K.; Matsumoto, M.; Kanouchi, H.; Oka, T. Homocysteine and copper induce cellular apoptosis via caspase activation and nuclear translocation of apoptosis-inducing factor in neuronal cell line SH-SY5Y. Neurosci. Res. 2010, 67, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Kim, J.; Ko, J.W.; Kwon, Y.H. Homocysteine induces PUMA-mediated mitochondrial apoptosis in SH-SY5Y cells. Amino Acids 2016, 48, 2559–2569. [Google Scholar] [CrossRef]
- Vacek, J.C.; Behera, J.; George, A.K.; Kamat, P.K.; Kalani, A.; Tyagi, N. Tetrahydrocurcumin ameliorates homocysteine-mediated mitochondrial remodeling in brain endothelial cells. J. Cell. Physiol. 2017, 233, 3080–3092. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Qi, X.; Gao, Z.; Yang, X.; Zheng, X.; Duan, C.; Zheng, J. Homocysteine injures vascular endothelial cells by inhibiting mitochondrial activity. Exp. Ther. Med. 2016, 12, 2247–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Zhao, L.; Song, X.; Yan, Y.; Liu, N.; Li, T.; Yan, B.; Liu, B. HSP27 inhibits homocysteine-induced endothelial apoptosis by modulation of ROS production and mitochondrial caspase-dependent apoptotic pathway. BioMed Res. Int. 2016, 2016, 4847874. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zhang, L.; Zhang, D.; Bai, L.; Kong, W.; Huang, Y.; Tang, C.; Du, J.; Jin, H. L-Cystathionine protects against homocysteine-induced mitochondria-dependent apoptosis of vascular endothelial cells. Oxid. Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Ovechkin, A.V.; Lominadze, D.; Moshal, K.S.; Tyagi, S.C. Mitochondrial mechanism of microvascular endothelial cells apoptosis in hyperhomocysteinemia. J. Cell. Biochem. 2006, 98, 1150–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, E.S.; Brasch, N.E.; Yun, J. Vitamin B12 protects against superoxide-induced cell injury in human aortic endothelial cells. Free. Radic. Biol. Med. 2011, 51, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Baydas, G.; Reiter, R.; Akbulut, M.; Tuzcu, M.; Tamer, S. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and by regulating pro- and anti-apoptotic protein levels. Neuroscience 2005, 135, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Familtseva, A.; Chaturvedi, P.; Kalani, A.; Jeremic, N.; Metreveli, N.; Kunkel, G.H.; Tyagi, S.C. Toll-like receptor 4 mutation suppresses hyperhomocysteinemia-induced hypertension. Am. J. Physiol. Cell. Physiol. 2016, 311, C596–C606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, S.A.; Kim, W.-K.; Choi, Y.-B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [Green Version]
- Sibarov, D.A.; Eginiatullin, R.; Antonov, S.M. High sensitivity of cerebellar neurons to homocysteine is determined by expression of GluN2C and GluN2D subunits of NMDA receptors. Biochem. Biophys. Res. Commun. 2018, 506, 648–652. [Google Scholar] [CrossRef]
- Poddar, R.; Paul, S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J. Neurochem. 2009, 110, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Deep, S.N.; Mitra, S.; Rajagopal, S.; Paul, S.; Poddar, R. GluN2A-NMDA receptor–mediated sustained Ca2+ influx leads to homocysteine-induced neuronal cell death. J. Biol. Chem. 2019, 294, 11154–11165. [Google Scholar] [CrossRef]
- Wang, L.; Niu, H.; Zhang, J. Homocysteine induces mitochondrial dysfunction and oxidative stress in myocardial ischemia/reperfusion injury through stimulating ROS production and the ERK1/2 signaling pathway. Exp. Ther. Med. 2020, 20, 938–944. [Google Scholar] [CrossRef]
- Liu, S.; Sun, Z.; Chu, P.; Li, H.; Ahsan, A.; Zhou, Z.; Zhang, Z.; Sun, B.; Wu, J.; Xi, Y.; et al. EGCG protects against homocysteine-induced human umbilical vein endothelial cells apoptosis by modulating mitochondrial-dependent apoptotic signaling and PI3K/Akt/eNOS signaling pathways. Apoptosis 2017, 22, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, V.; Wolyniec, K.; Sionov, R.V.; Haupt, S.; Haupt, Y. Tumour suppression by p53: The importance of apoptosis and cellular senescence. J. Pathol. 2009, 219, 3–15. [Google Scholar] [CrossRef]
- Han, J.-H.; Park, J.; Myung, S.-H.; Lee, S.H.; Kim, H.-Y.; Kim, K.S.; Seo, Y.-W.; Kim, T.-H. Noxa mitochondrial targeting domain induces necrosis via VDAC2 and mitochondrial catastrophe. Cell Death Dis. 2019, 10, 519. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Han, J.-H.; Myung, S.-H.; Kang, H.; Cho, J.-Y.; Kim, T.-H. A peptide containing Noxa mitochondrial-targeting domain induces cell death via mitochondrial and endoplasmic reticulum disruption. Biochem. Biophys. Res. Commun. 2019, 518, 80–86. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [Green Version]
- Veeresh, P.; Kaur, H.; Sarmah, D.; Mounica, L.; Verma, G.; Kotian, V.; Kesharwani, R.; Kalia, K.; Borah, A.; Wang, X.; et al. Endoplasmic reticulum–mitochondria crosstalk: From junction to function across neurological disorders. Ann. N. Y. Acad. Sci. 2019, 1457, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gökçen, A.; Kracht, M. The crosstalk of endoplasmic reticulum (ER) stress pathways with NF-κB: Complex mechanisms relevant for cancer, inflammation and infection. Biomed. 2018, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K.; Murakami, S. Role of the unfolded protein response in cell death. Apoptosis 2006, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Casas, C. GRP78 at the centre of the stage in cancer and neuroprotection. Front. Neurosci. 2017, 11, 177. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, R.; Jin, H.; Liu, D.; Tang, X.; Tang, C.; Du, J. Hydrogen sulfide attenuates hyperhomocysteinemia-induced cardiomyocytic endoplasmic reticulum stress in rats. Antioxidants Redox Signal. 2010, 12, 1079–1091. [Google Scholar] [CrossRef]
- Tang, Y.-Y.; Wang, A.-P.; Wei, H.-J.; Li, M.-H.; Zou, W.; Li, X.; Wang, C.-Y.; Zhang, P.; Tang, X.-Q. Role of silent information regulator 1 in the protective effect of hydrogen sulfide on homocysteine-induced cognitive dysfunction: Involving reduction of hippocampal ER stress. Behav. Brain Res. 2018, 342, 35–42. [Google Scholar] [CrossRef]
- Jeremic, N.; Weber, G.J.; Tyagi, S.C. Ablation of toll-like receptor 4 mitigates cardiac mitochondrial dysfunction in hyperhomocysteinemia. Can. J. Physiol. Pharmacol. 2017, 95, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganapathy, P.S.; Perry, R.L.; Tawfik, A.; Smith, R.M.; Perry, E.; Roon, P.; Bozard, B.R.; Ha, Y.; Smith, S.B. Homocysteine-mediated modulation of mitochondrial dynamics in retinal ganglion cells. Investig. Opthalmol. Vis. Sci. 2011, 52, 5551–5558. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Vacek, J.C.; Givvimani, S.; Sen, U.; Tyagi, S.C. Cardiac specific deletion of N-methyl-d-aspartate receptor 1 ameliorates mtMMP-9 mediated autophagy/mitophagy in hyperhomocysteinemia. J. Recept. Signal. Transduct. Res. 2010, 30, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaplan, P.; Tatarkova, Z.; Sivonova, M.K.; Racay, P.; Lehotsky, J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. Int. J. Mol. Sci. 2020, 21, 7698. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207698
Kaplan P, Tatarkova Z, Sivonova MK, Racay P, Lehotsky J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. International Journal of Molecular Sciences. 2020; 21(20):7698. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207698
Chicago/Turabian StyleKaplan, Peter, Zuzana Tatarkova, Monika Kmetova Sivonova, Peter Racay, and Jan Lehotsky. 2020. "Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems" International Journal of Molecular Sciences 21, no. 20: 7698. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207698