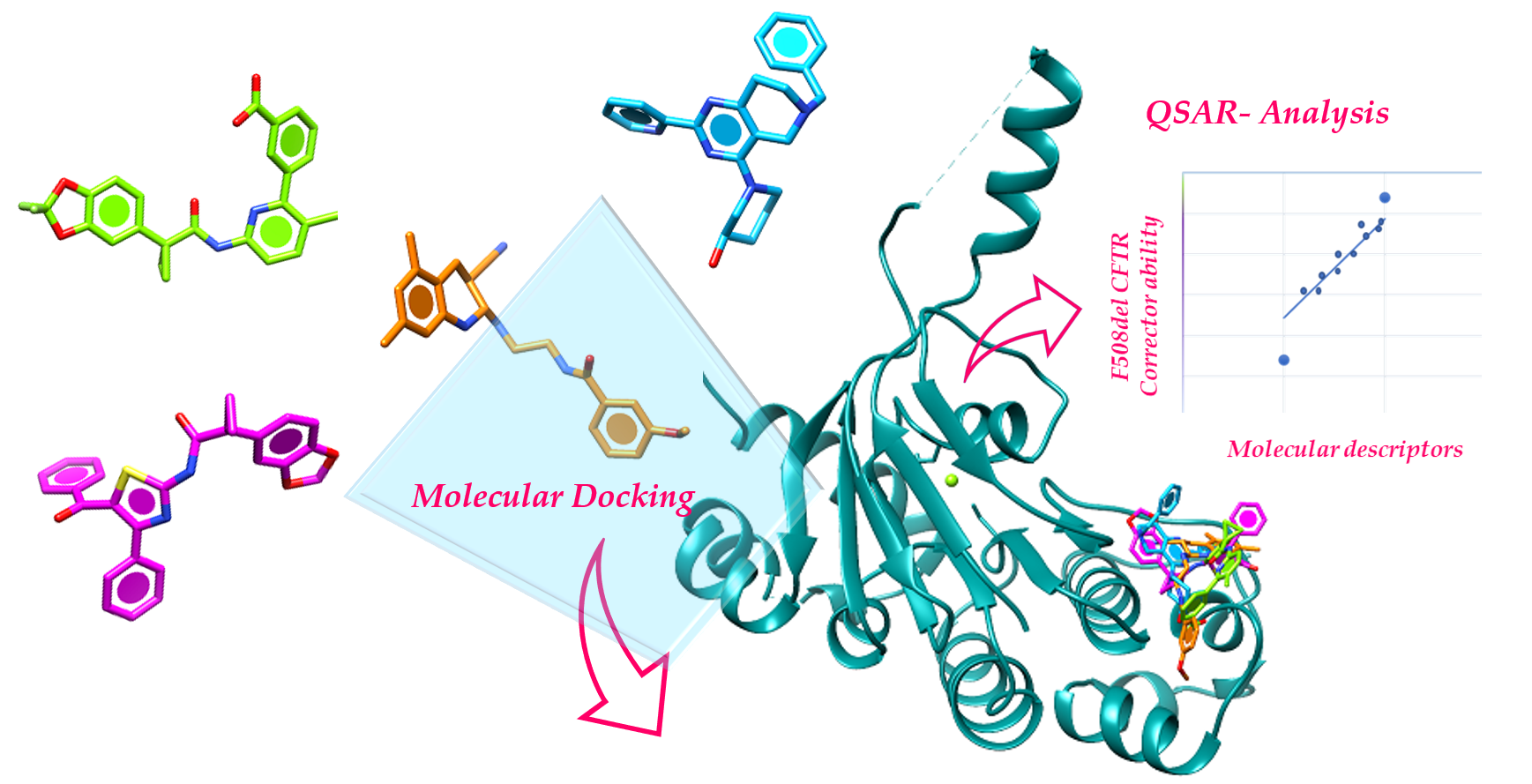
Molecular Docking and QSAR Studies as Computational Tools Exploring the Rescue Ability of F508del CFTR Correctors

 , , , , , , and
, , , , , , and 
Abstract
:
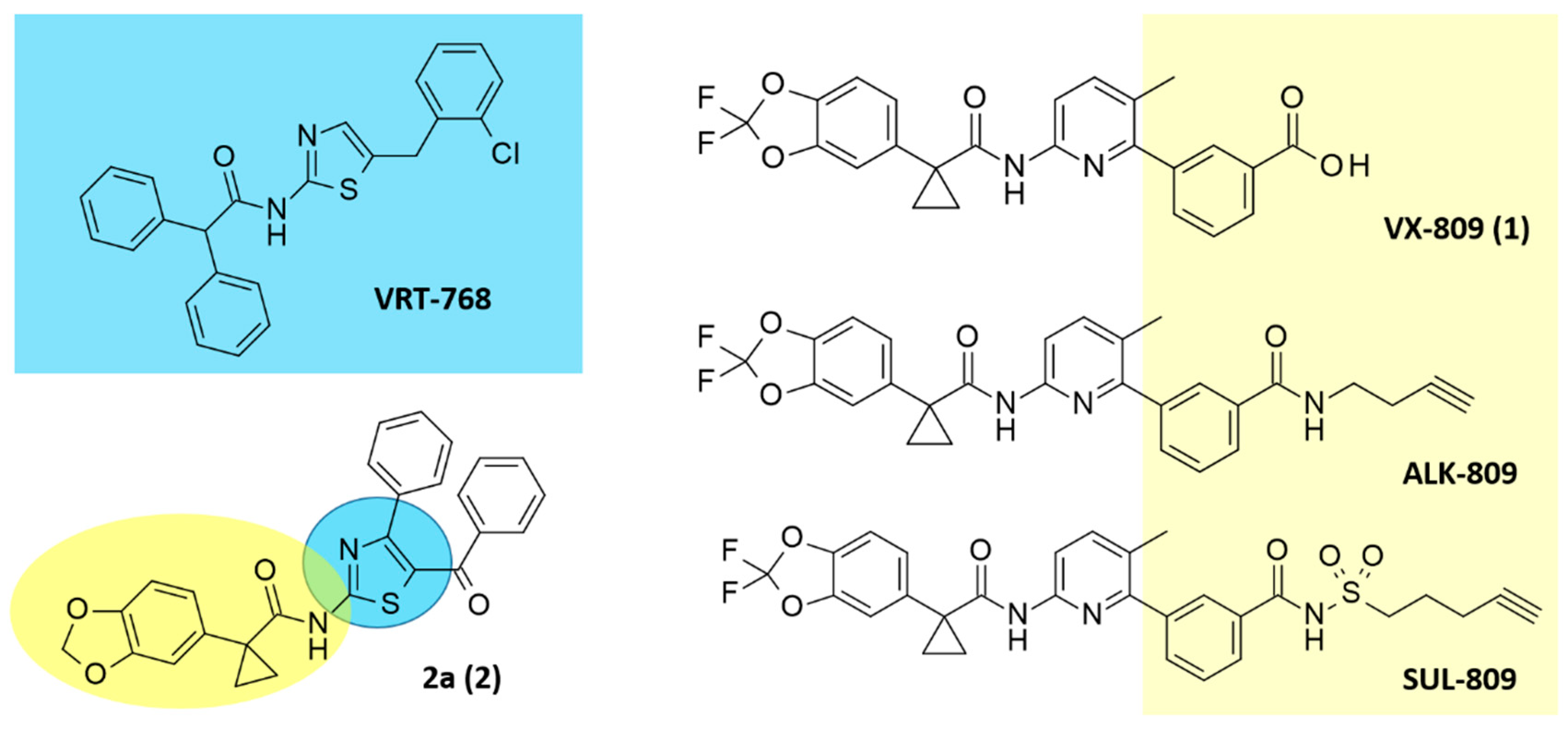
1. Introduction
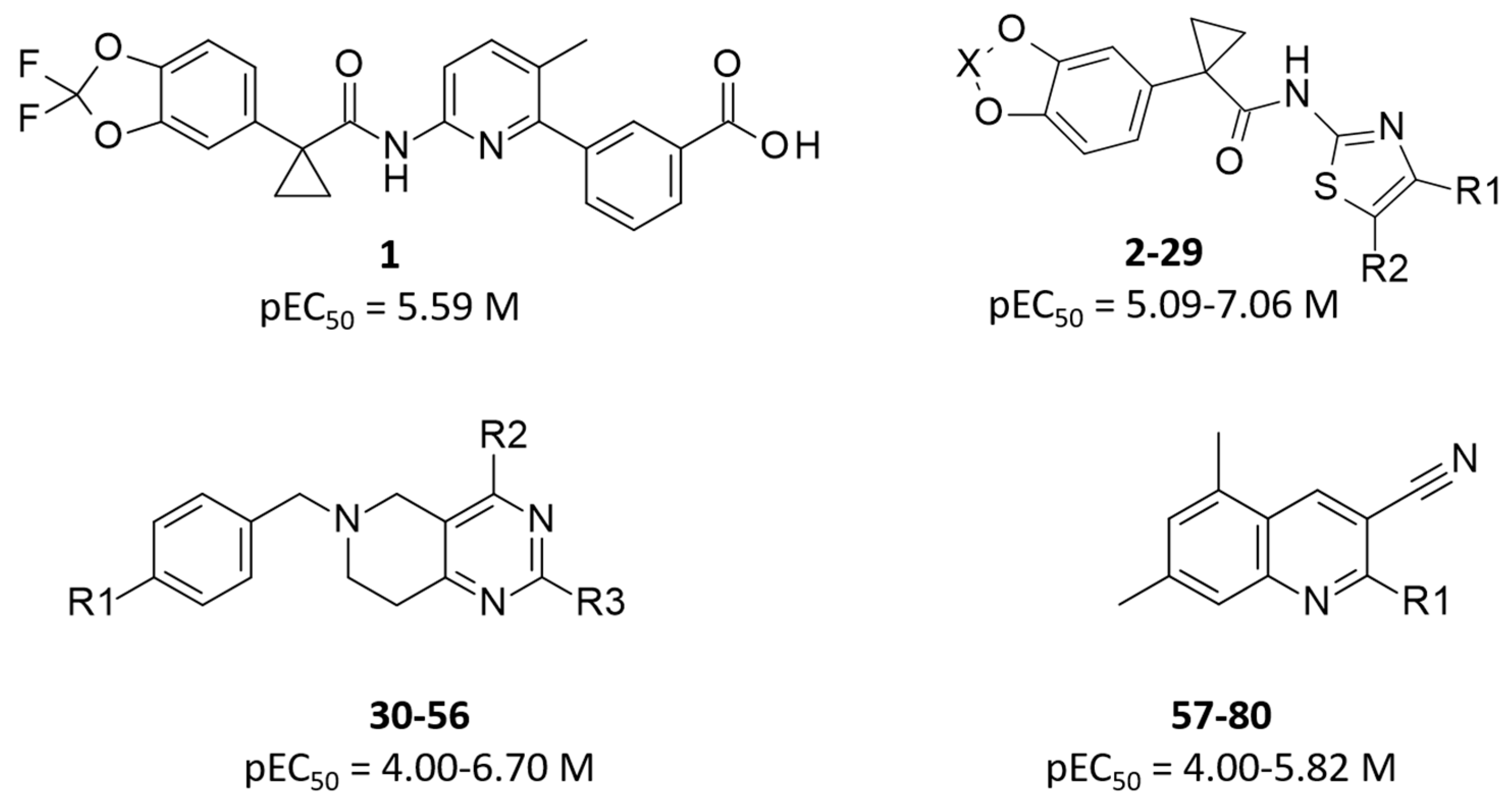
2. Results
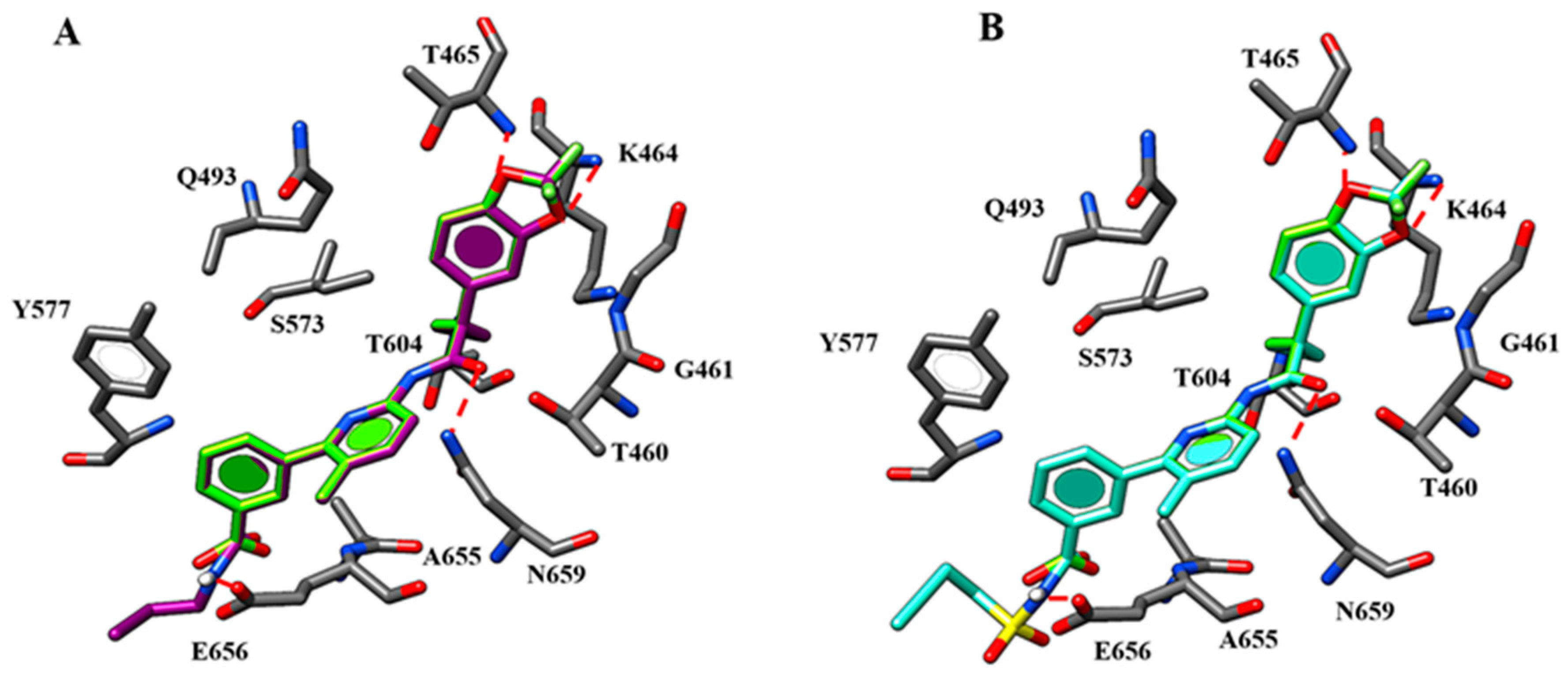
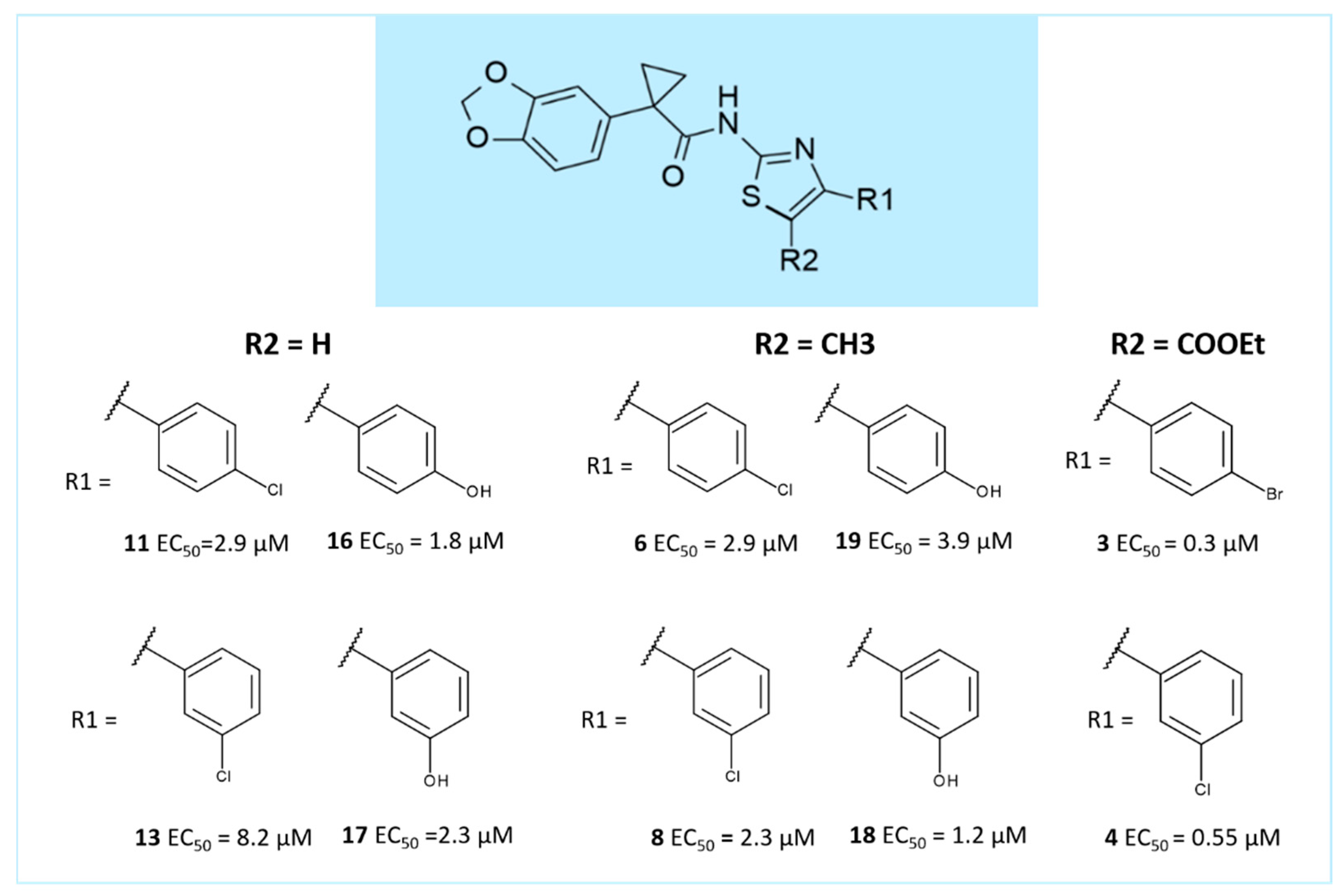
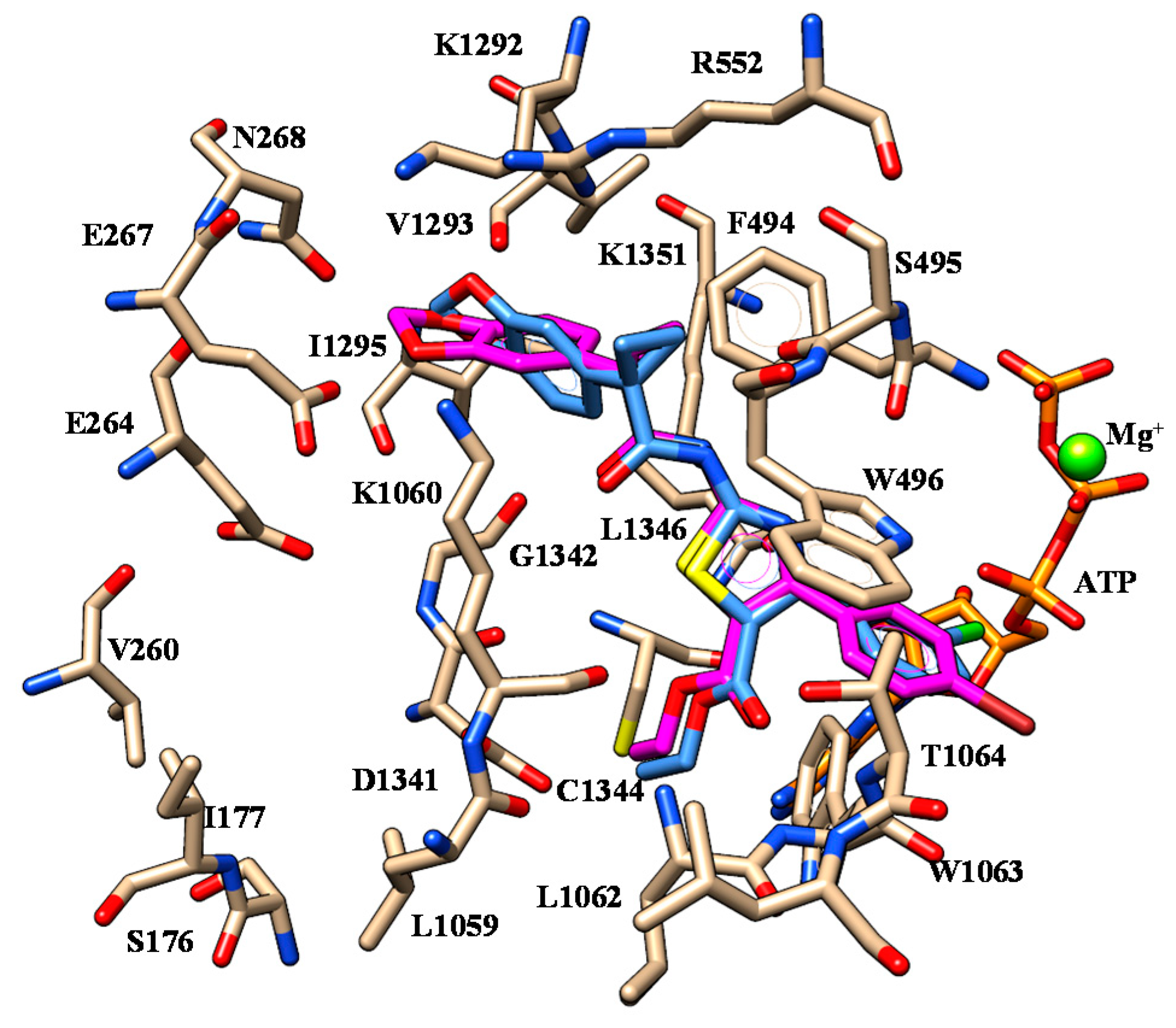
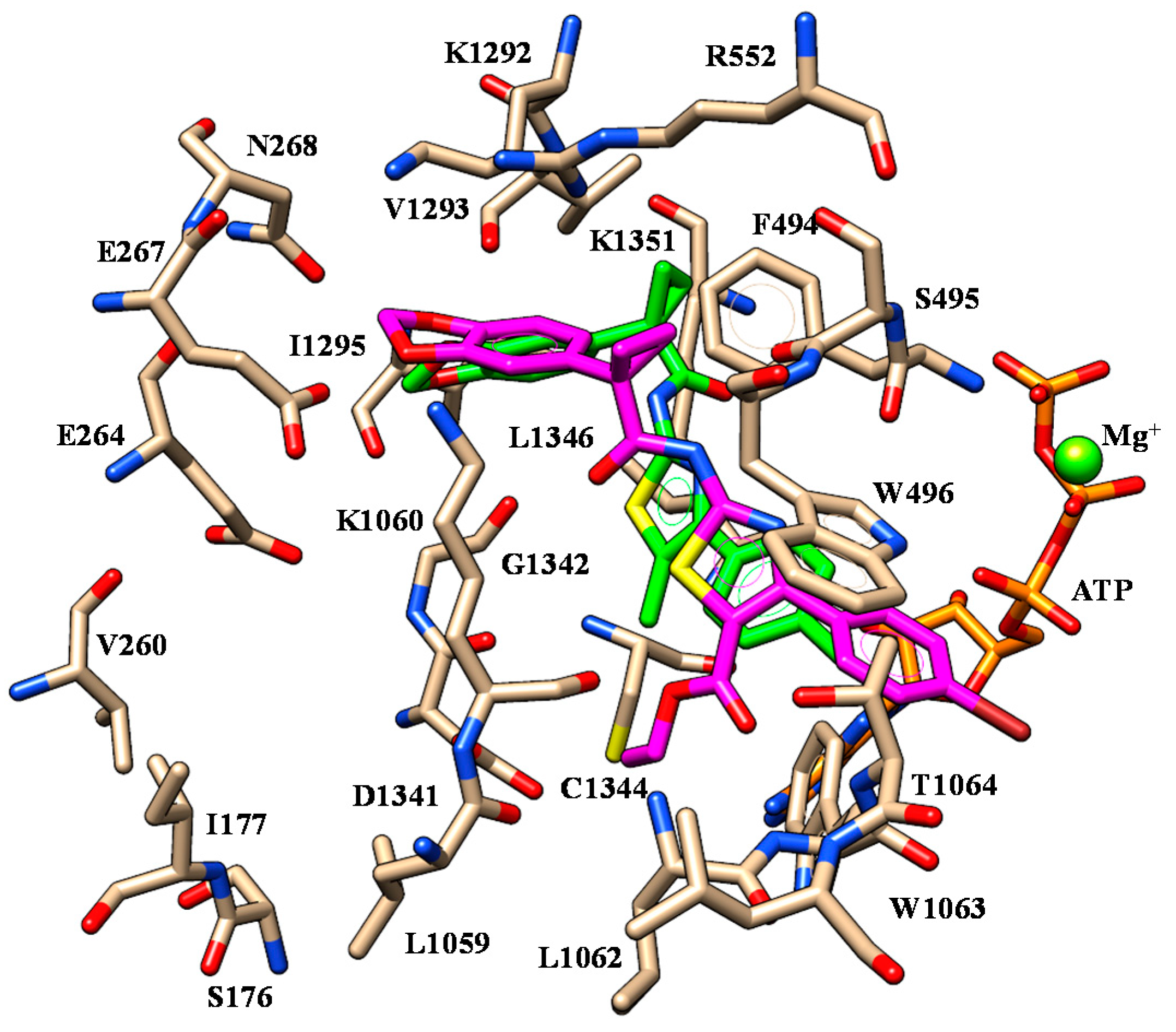
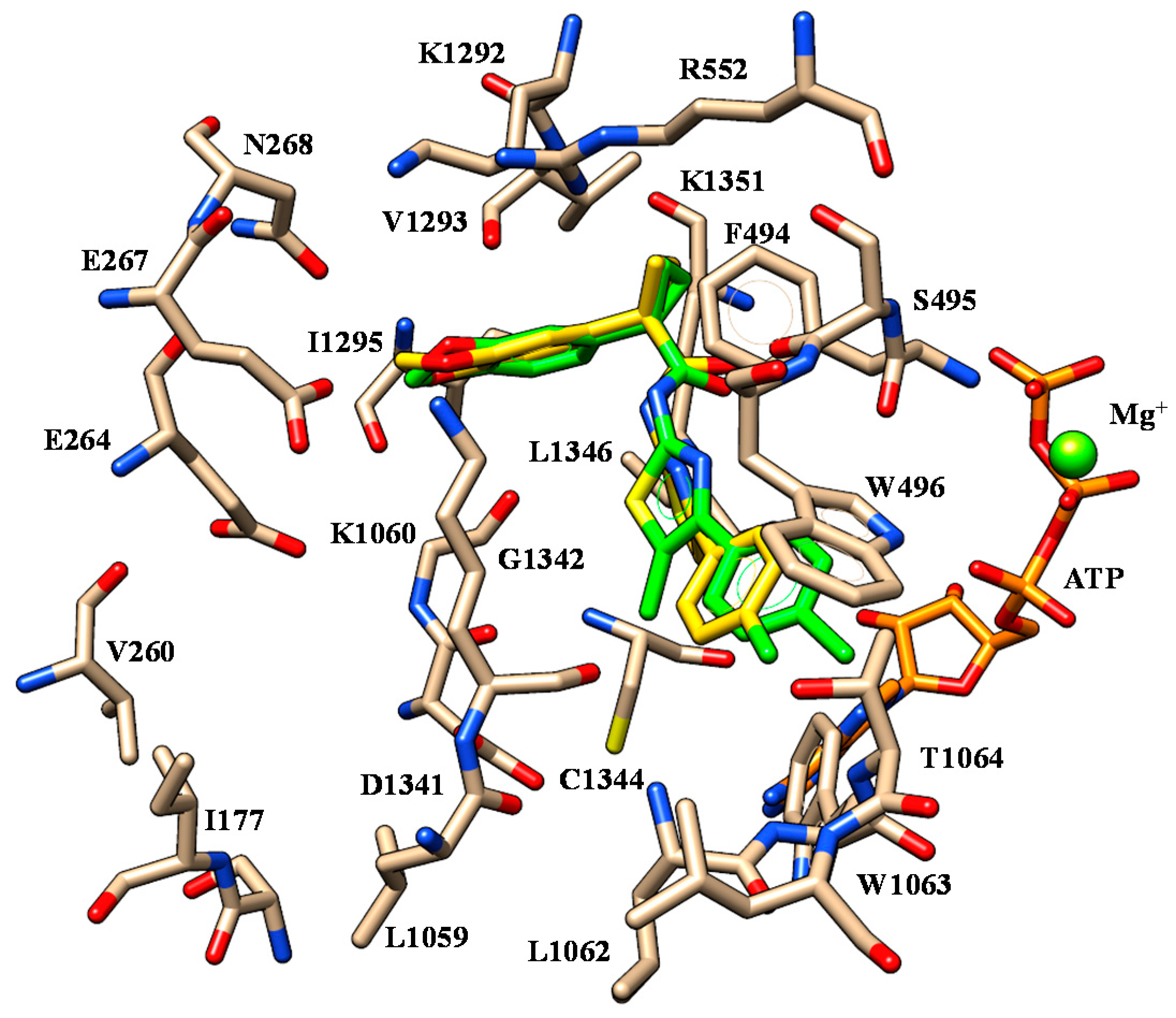
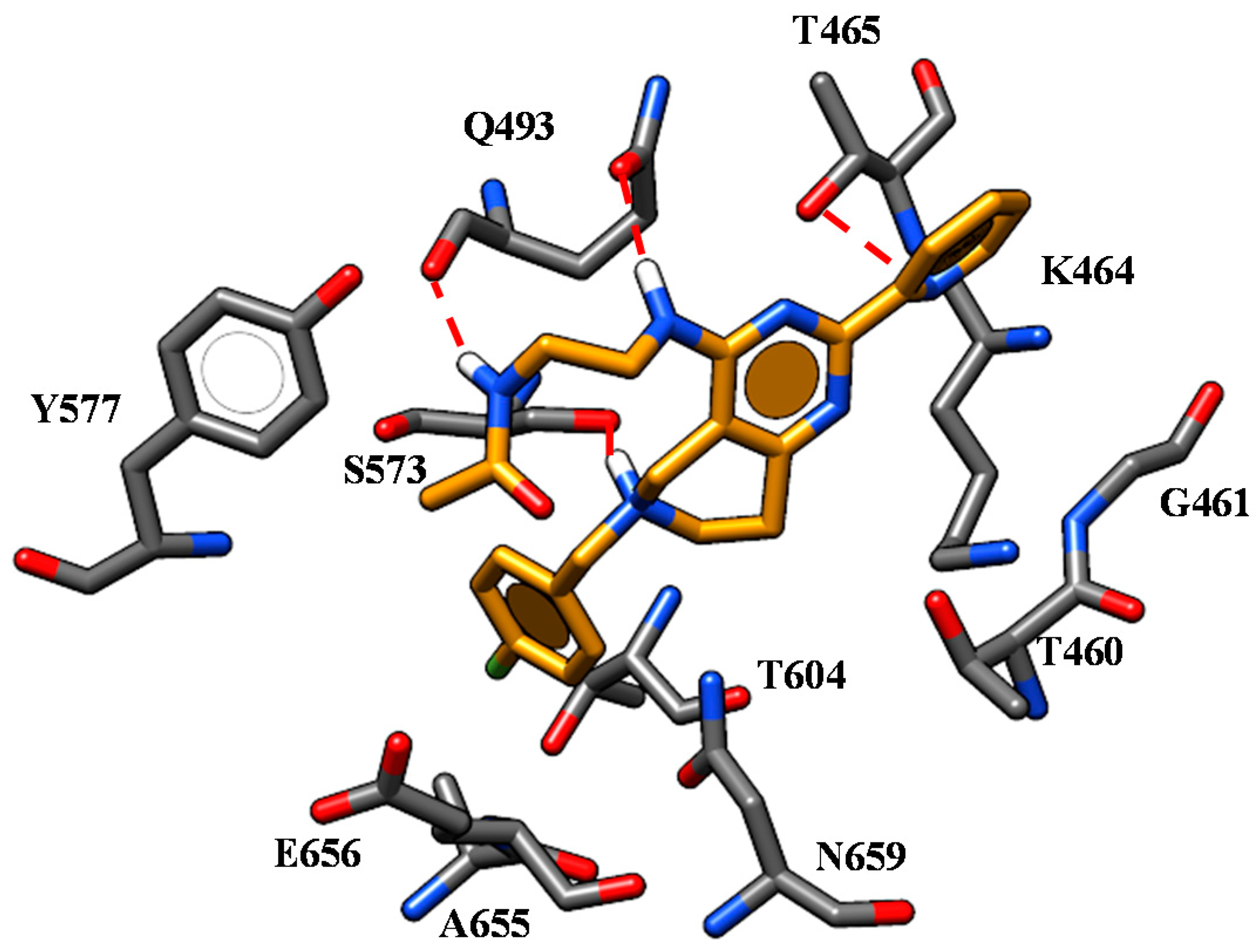
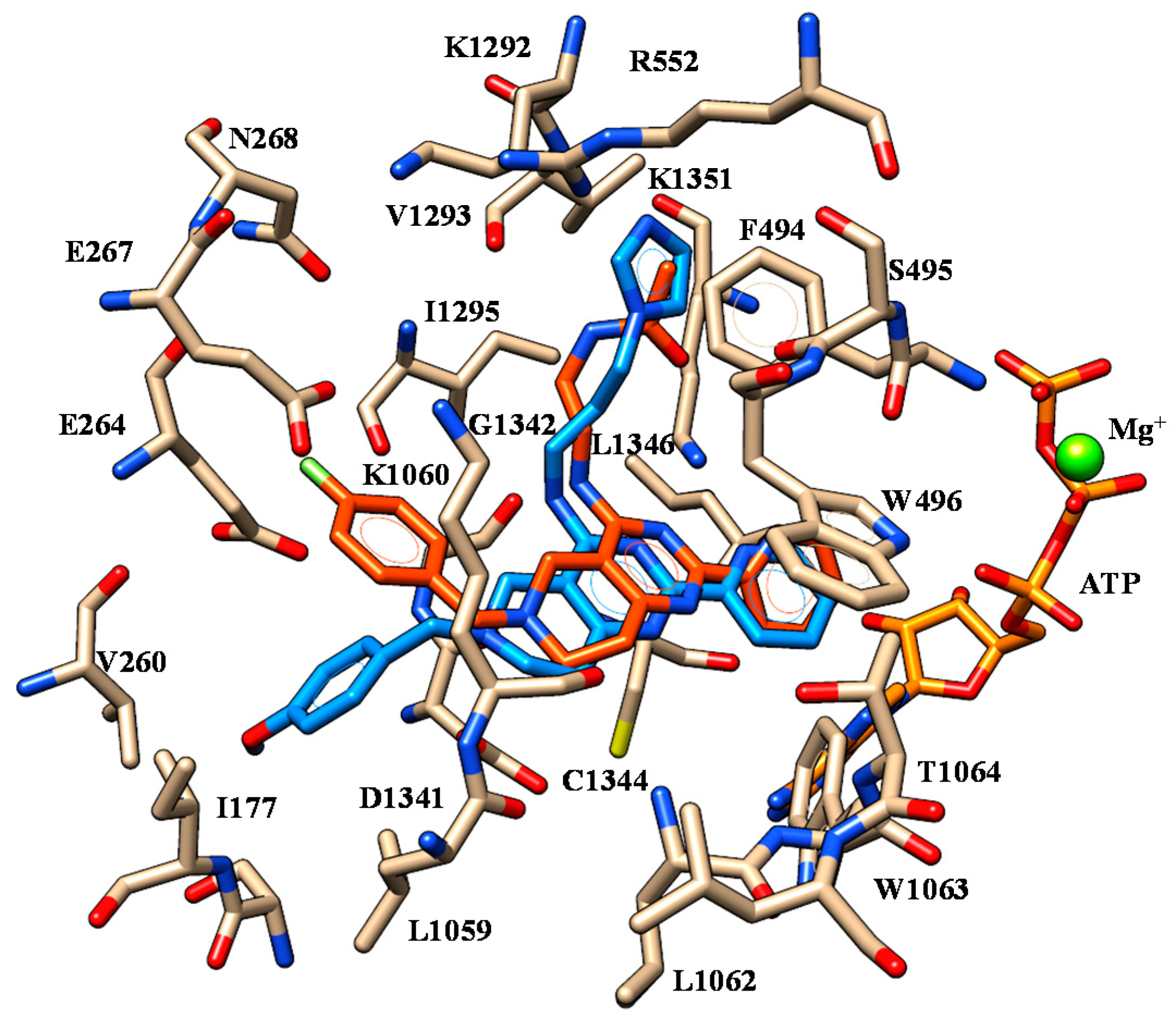
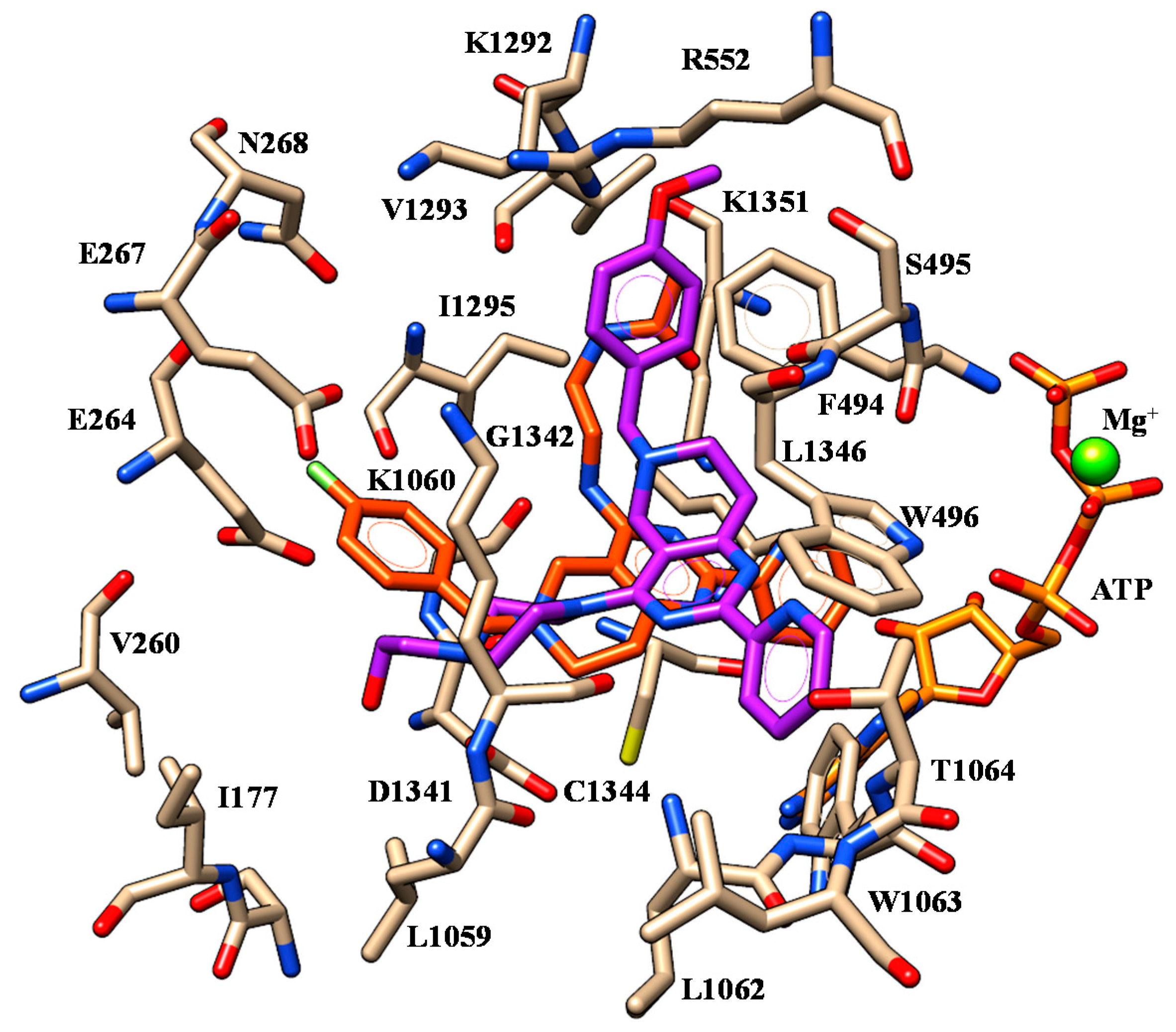
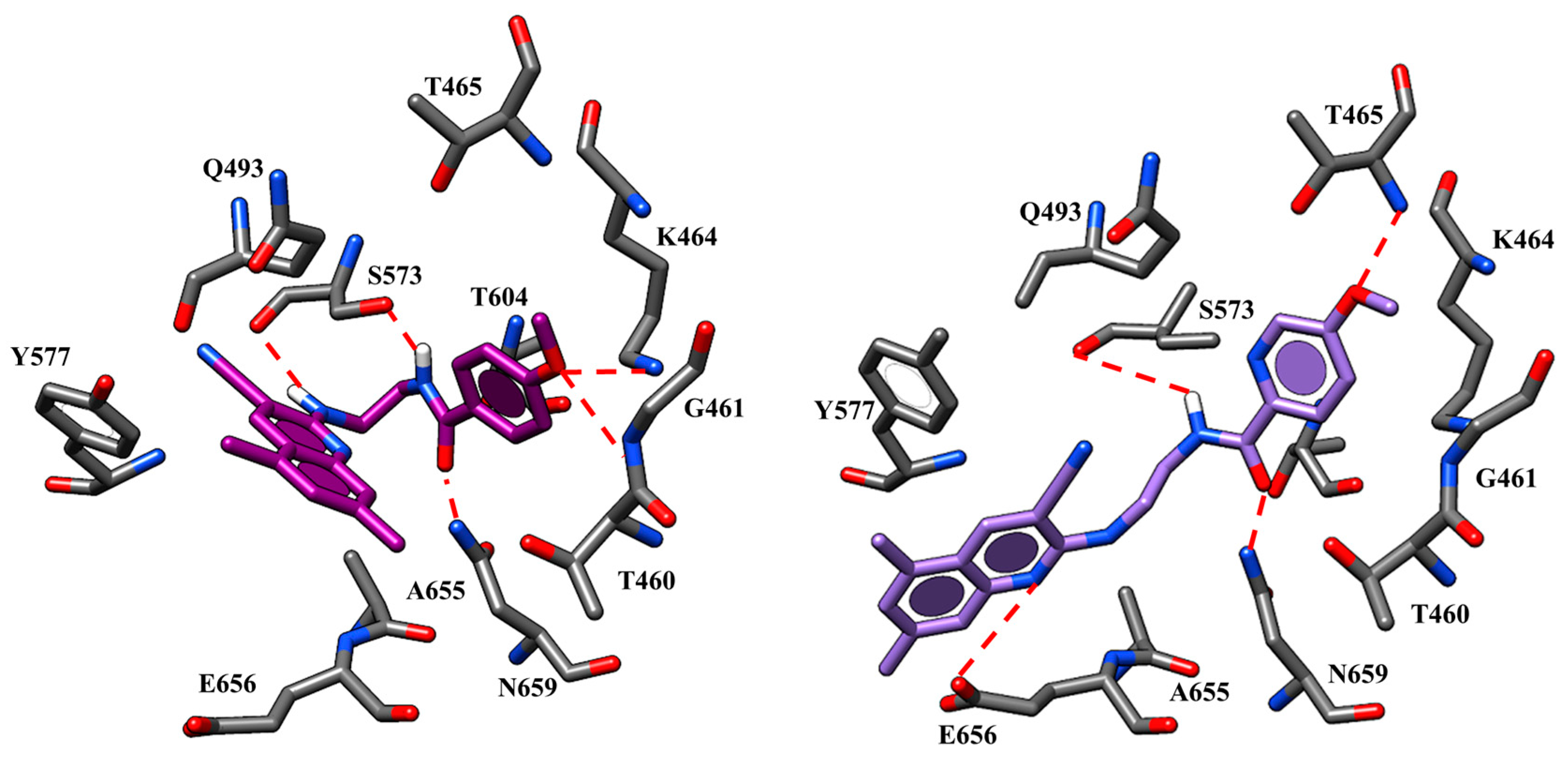
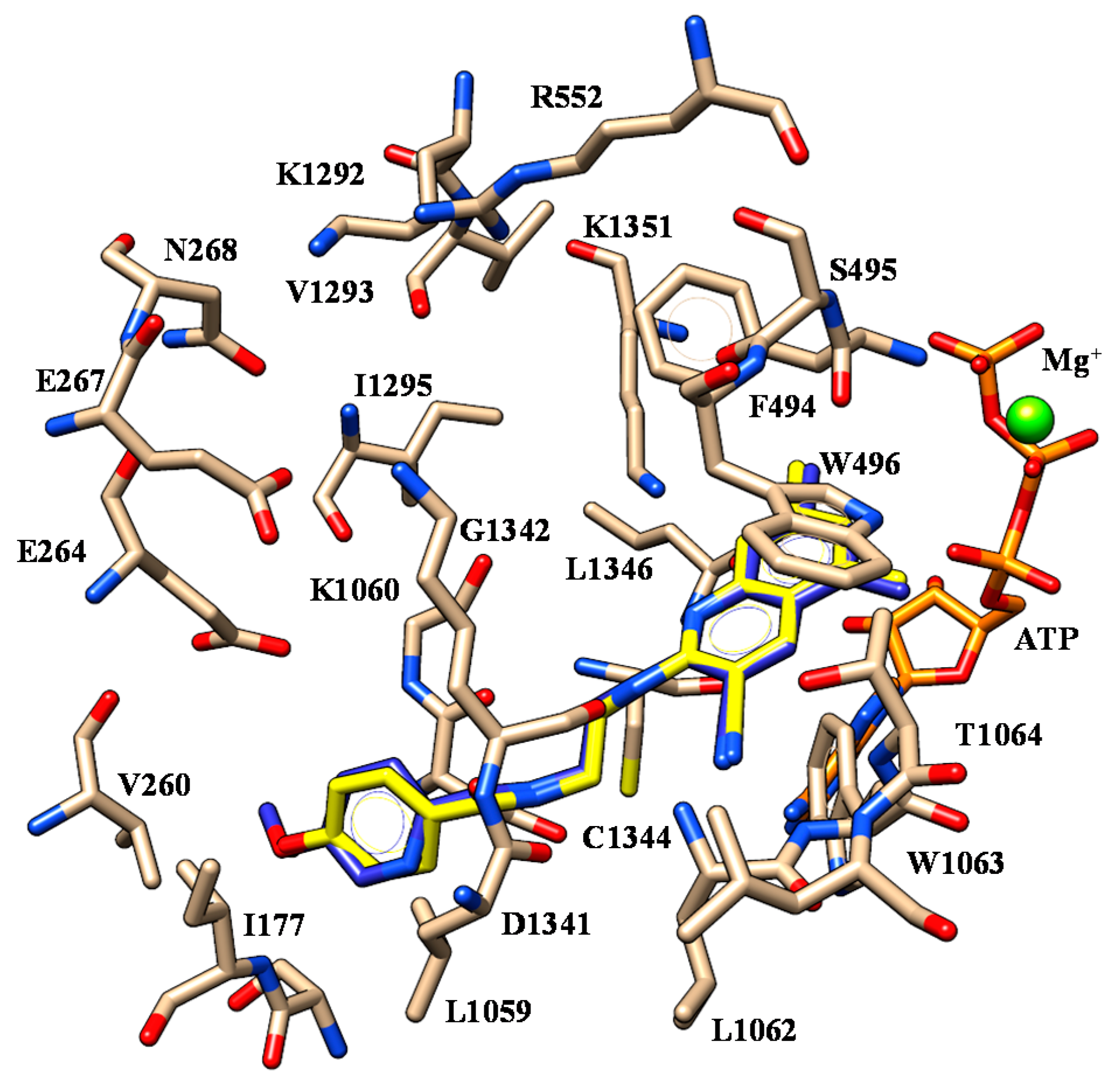
2.1. Molecular Docking
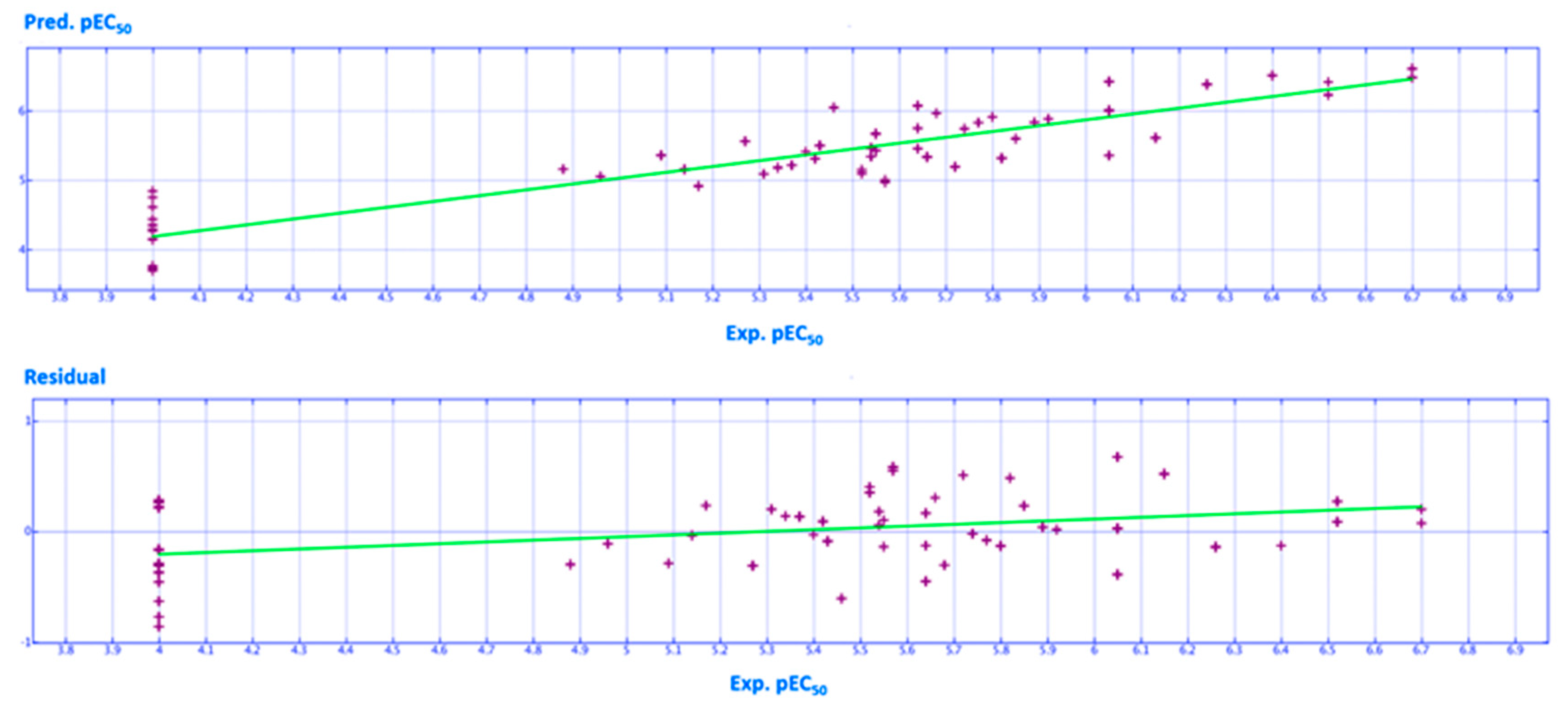
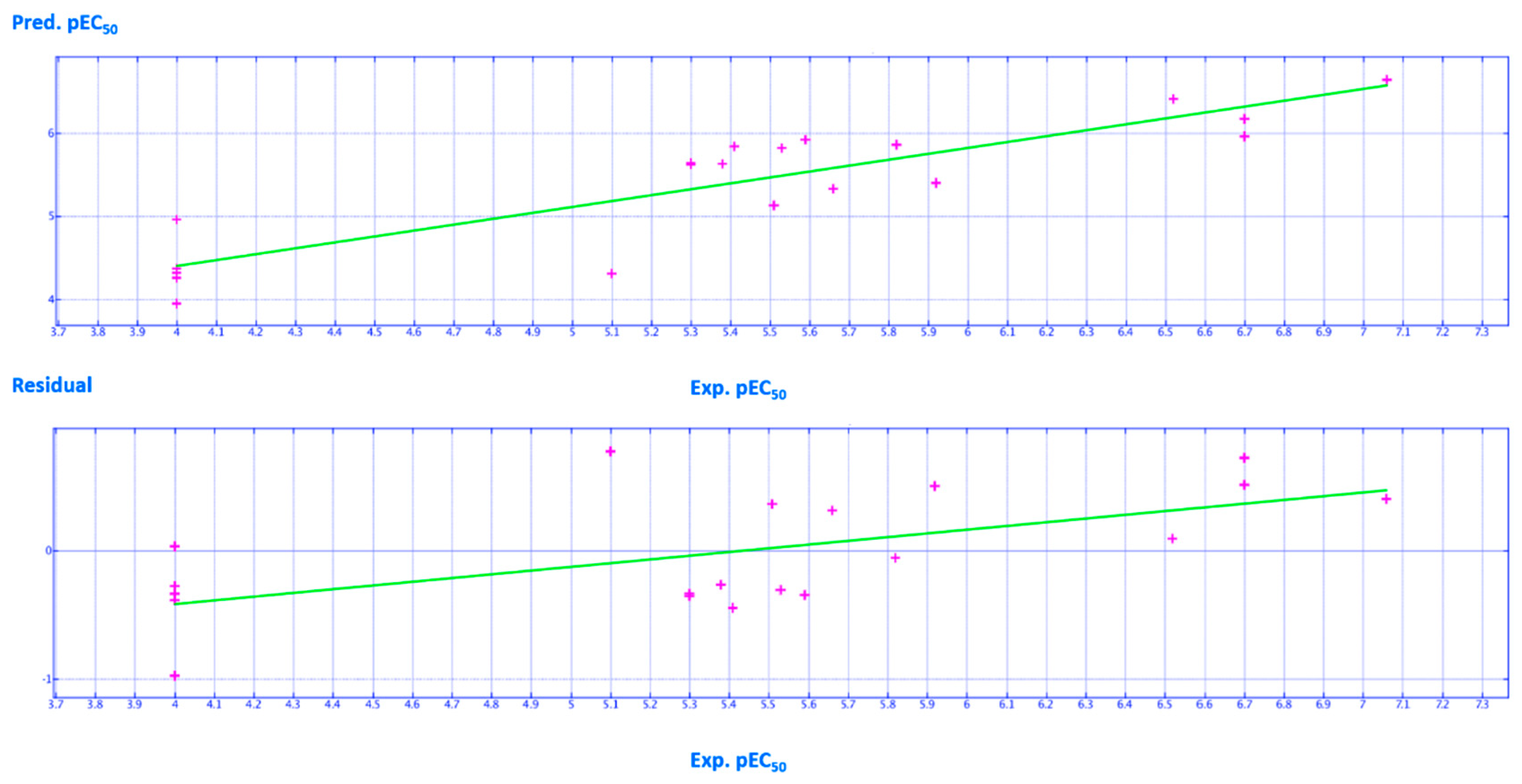
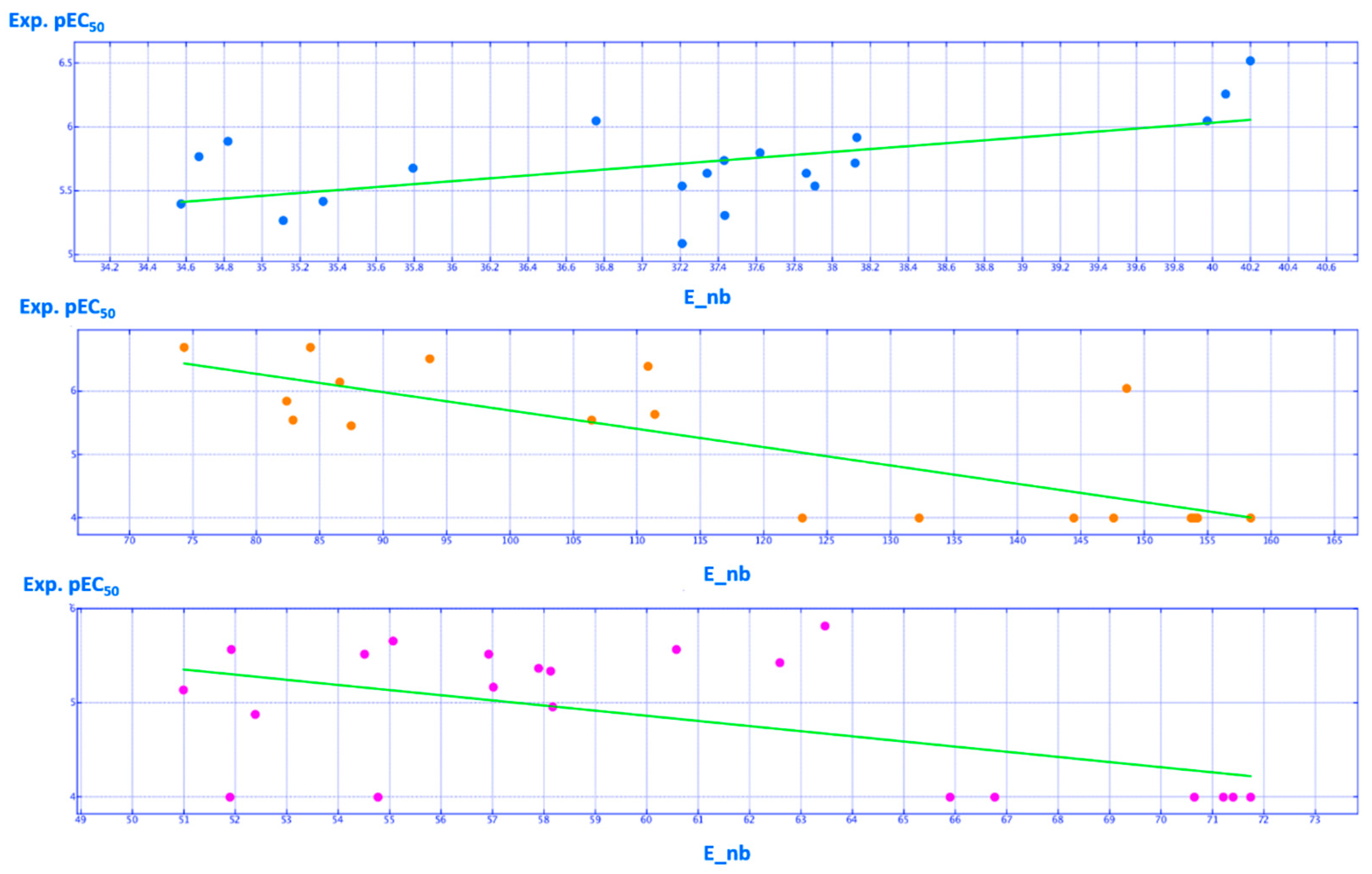
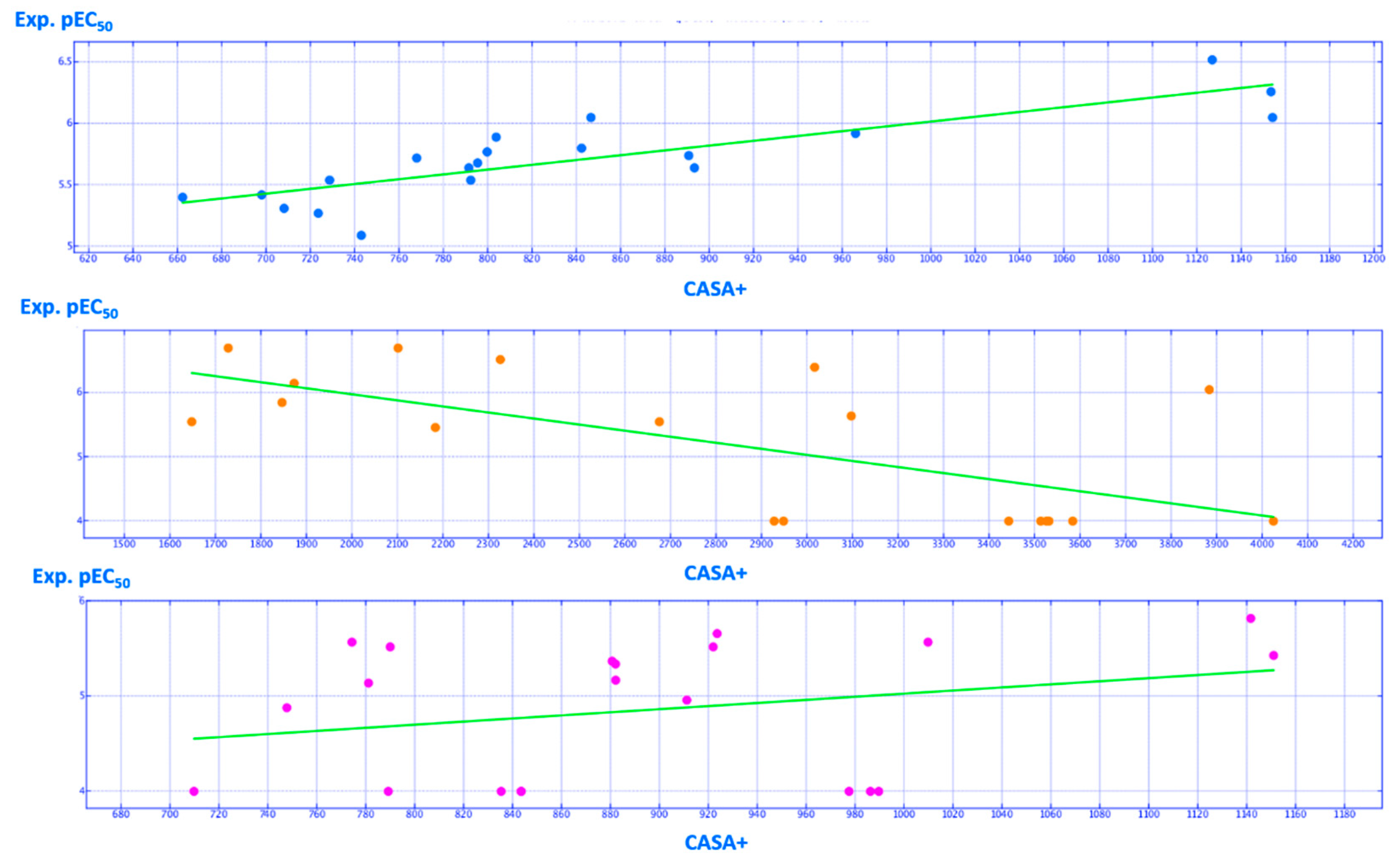
2.2. QSAR Analyses
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.2. QSAR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAT | aminoarylthiazole |
| CF | cystic fibrosis |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| CQ | cyanoquinoline |
| F508de | deletion of phenylalanine at position 508 |
| NBD1 | nucleotide binding domain 1 |
| NBD2 | nucleotide binding domain 2 |
| PDB | Protein data bank |
References
- Bobadilla, J.L.; Macek, M.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef] [PubMed]
- Gadsby, D.C.; Vergani, P.; Csanady, L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 2006, 440, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.P.; Lazdunski, M. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.N.; Welsh, M.J. Structure and function of the CFTR chloride channel. Physiol. Rev. 1999, 79, 23–45. [Google Scholar] [CrossRef] [PubMed]
- Rogan, M.P.; Stoltz, D.A.; Hornick, D.B. Cystic fibrosis transmembrane conductance regulator intracellular processing, trafficking, and opportunities for mutation-specific treatment. Chest 2011, 139, 1480–1490. [Google Scholar] [CrossRef]
- Fanen, P.; Wohlhuter-Haddad, A.; Hinzpeter, A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int. J. Biochem. Cell Biol. 2014, 52, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Amaral, M.D.; Farinha, C.M. Rescuing mutant CFTR: A multi-task approach to a better outcome in treating cystic fibrosis. Curr. Pharm. Des. 2013, 19, 3497–3508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quon, B.S.; Rowe, S.M. New and emerging targeted therapies for cystic fibrosis. BMJ 2016, 352, i859. [Google Scholar] [CrossRef] [Green Version]
- Lukacs, G.L.; Mohamed, A.; Kartner, N.; Chang, X.-B.; Riordan, J.R.; Grinstein, S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994, 13, 6076–6086. [Google Scholar] [CrossRef]
- Du, K.; Sharma, M.; Lukacs, G.L. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharmacol. 2016, 7, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedemonte, N.; Sonawane, N.D.; Taddei, A.; Hu, J.; Zegarra-Moran, O.; Suen, Y.F.; Robins, L.I.; Dicus, C.W.; Willenbring, D.; Nantz, M.H.; et al. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol. Pharmacol. 2005, 67, 1797–1807. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houch, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell. 2016, 27, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.L.; Yu, G.J.; Yang, B.; Robins, L.I.; Verkman, A.S.; Kurth, M.J. 4′-Methyl-4,5′-bithiazole-based correctors of defective delta F508-CFTR cellular processing. Bioorg. Med. Chem. Lett. 2008, 18, 2610–2614. [Google Scholar] [CrossRef] [Green Version]
- Cuthbert, A.W. New horizons in the treatment of cystic fibrosis. Br. J. Pharmacol. 2011, 163, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.J.; Yoo, C.L.; Yang, B.; Lodewyk, M.W.; Meng, L.; El-Idreesy, T.T.; Fettinger, J.C.; Tantillo, D.J.; Verkman, A.S.; Kurth, M.J. Potent s-cis-locked bithiazole correctors of Delta F508 cystic fibrosis transmembrane conductance regulator cellular processing for cystic fibrosis therapy. J. Med. Chem. 2008, 51, 6044–6054. [Google Scholar] [CrossRef] [Green Version]
- Kalid, O.; Mense, M.; Fischman, S.; Shitrit, A.; Bihler, H.; Ben-Zeev, E.; Schutz, N.; Pedemonte, N.; Thomas, P.J.; Bridges, R.J.; et al. Small molecule correctors of F508del-CFTR discovered by structure-based virtual screening. J. Comput. Aided Mol. Des. 2010, 24, 971–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ursi, P.; Uggeri, M.; Urbinati, C.; Millo, E.; Paiardi, G.; Milanesi, L.; Ford, R.C.; Clews, J.; Meng, X.; Bergese, P.; et al. Exploitation of a novel biosensor based on the full-length human F508del-CFTR with computational studies, biochemical and biological assays for the characterization of new Lumacaftor/Tezacaftor analogue. Sens. Actuators B Chem. 2019. [Google Scholar] [CrossRef]
- Pedemonte, N.; Lukacs, G.L.; Du, K.; Caci, E.; Zegarra-Moran, O.; Galietta, L.J.; Verkman, A.S. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 2005, 115, 2564–2571. [Google Scholar] [CrossRef]
- Carlile, G.W.; Robert, R.; Zhang, D.; Teske, K.A.; Luo, Y.; Hanrahan, J.W.; Thomas, D.Y. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. ChemBioChem 2007, 8, 1012–1020. [Google Scholar] [CrossRef]
- Ye, L.; Knapp, J.M.; Sangwung, P.; Fettinger, J.C.; Verkman, A.S.; Kurth, M.J. Pyrazolylthiazole as DeltaF508-cystic fibrosis transmembrane conductance regulator correctors with improved hydrophilicity compared to bithiazoles. J. Med. Chem. 2010, 53, 3772–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donald, M.B.; Rodriguez, K.X.; Shay, H.; Phuan, P.W.; Verkman, A.S.; Kurth, M.J. Click-based synthesis of triazolobithiazole ΔF508-CFTR correctors for cystic fibrosis. Bioorg. Med. Chem. 2012, 20, 5247–5253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [Green Version]
- Knapp, J.M.; Wood, A.B.; Phuan, P.W.; Lodewyk, M.W.; Tantillo, D.J.; Verkman, A.S.; Kurth, M.J. Structure-activity relationships of cyanoquinolines with corrector-potentiator activity in ΔF508 cystic fibrosis transmembrane conductance regulator protein. J. Med. Chem. 2012, 55, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Pesci, E.; Bettinetti, L.; Fanti, P.; Galietta, L.J.; La Rosa, S.; Magnoni, L.; Pedemonte, N.; Sardone, G.L.; Maccari, L. Novel Hits in the Correction of ΔF508-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Protein: Synthesis, Pharmacological, and ADME Evaluation of Tetrahydropyrido [4,3-d]pyrimidines for the Potential Treatment of Cystic Fibrosis. J. Med. Chem. 2015, 58, 9697–9711. [Google Scholar] [CrossRef] [PubMed]
- Connett, G.J. Lumacaftor-ivacaftor in the treatment of cystic fibrosis: Design, development and place in therapy. Drug Des. Devel. Ther. 2019, 13, 2405–2412. [Google Scholar] [CrossRef] [Green Version]
- Ridley, K.; Condren, M. Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy. Pediatr. Pharmacol. Ther. 2020, 25, 192–197. [Google Scholar] [CrossRef]
- Pedemonte, N.; Tomati, V.; Sondo, E.; Caci, E.; Millo, E.; Armirotti, A.; Damonte, G.; Zegarra-Moran, O.; Galietta, L.J. Dual activity of aminoarylthiazoles on the trafficking and gating defects of the cystic fibrosis transmembrane conductance regulator chloride channel caused by cystic fibrosis mutations. J. Biol. Chem. 2011, 286, 15215–15226. [Google Scholar] [CrossRef] [Green Version]
- Pesce, E.; Bellotti, M.; Liessi, N.; Guariento, S.; Damonte, G.; Cichero, E.; Galatini, A.; Salis, A.; Gianotti, A.; Pedemonte, N.; et al. Synthesis and structure-activity relationship of aminoarylthiazole derivatives as correctors of the chloride transport defect in cystic fibrosis. Eur. J. Med. Chem. 2015, 99, 14–35. [Google Scholar] [CrossRef]
- Liessi, N.; Cichero, E.; Pesce, E.; Arkel, M.; Salis, A.; Tomati, V.; Paccagnella, M.; Damonte, G.; Tasso, B.; Galietta, L.; et al. Synthesis and biological evaluation of novel thiazole- VX-809 hybrid derivatives as F508del correctors by QSAR-based filtering tools. Eur. J. Med. Chem. 2018, 144, 179–200. [Google Scholar] [CrossRef]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.J.; Brouillette, C.G.; Forman-Kay, J.D. Direct Binding of the Corrector VX-809 to Human CFTR NBD1: Evidence of an Allosteric Coupling between the Binding Site and the NBD1:CL4 Interface. Mol. Pharmacol. 2017, 92, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Rusnati, M.; D’Ursi, P.; Pedemonte, N.; Urbinati, C.; Ford, R.C.; Cichero, E.; Uggeri, M.; Orro, A.; Fossa, P. Recent Strategic Advances in CFTR Drug Discovery: An Overview. Int. J. Mol. Sci. 2020, 21, 2407. [Google Scholar] [CrossRef] [Green Version]
- Sinha, C.; Zhang, W.; Moon, C.S.; Actis, M.; Yarlagadda, S.; Arora, K.; Woodroofe, K.; Clancy, J.P.; Lin, S.; Ziady, A.G.; et al. Capturing the direct binding of CFTR correctors to CFTR using click chemistry. ChemBioChem 2015, 16, 2017–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odolczyk, N.; Fritsch, J.; Norez, C.; Servel, N.; da Cunha, M.F.; Bitam, S.; Kupniewska, A.; Wiszniewski, L.; Colas, J.; Tarnowski, K.; et al. Discovery of novel potent ΔF508-CFTR correctors that target the nucleotide binding domain. EMBO Mol. Med. 2013, 5, 1484–1501. [Google Scholar] [CrossRef]
- Rusnati, M.; Sala, D.; Orro, A.; Bugatti, A.; Trombetti, G.; Cichero, E.; Urbinati, C.; Di Somma, M.; Millo, E.; Galietta, L.; et al. Speeding Up the Identification of Cystic Fibrosis Transmembrane Conductance Regulator-Targeted Drugs: An Approach Based on Bioinformatics Strategies and Surface Plasmon Resonance. Molecules 2018, 23, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.D.; Wang, H.; Byrnes, L.J.; Shanker, S.; Wang, K.; Efremov, I.V.; Chong, P.A.; Forman-Kay, J.D.; Aulabaugh, A.E. Binding screen for cystic fibrosis transmembrane conductance regulator correctors finds new chemical matter and yields insights into cystic fibrosis therapeutic strategy. Protein Sci. 2016, 25, 360–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi, A.; Righetti, G.; Pesce, E.; Salis, A.; Tasso, B.; Urbinati, C.; Tomati, V.; Damonte, G.; Rusnati, M.; Pedemonte, N.; et al. Discovery of novel VX-809 hybrid derivatives as F508del-CFTR correctors by molecular modeling, chemical synthesis and biological assays. Eur. J. Med. Chem. 2020, 20. [Google Scholar] [CrossRef]
- Kim Chiaw, P.; Wellhauser, L.; Huan, L.J.; Ramjeesingh, M.; Bear, C.E. A chemical corrector modifies the channel function of F508del-CFTR. Mol. Pharmacol. 2010, 78, 411–418. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. MOE. Montreal. H3A2R7 Canada. Available online: http://www.chemcomp.com (accessed on 29 October 2019).
- Kennard, R.W.; Stone, L.A. Computer aided design of experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Puzyn, T.; Mostrag-Szlichtyng, A.; Gajewicz, A.; Skrzyński, M.; Worth, A.P. Investigating the influence of data splitting on the predictive ability of QSAR/QSPR models. Struct. Chem. 2011, 22, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Edelsbrunner, H.; Facello, M.; Fu, P.; Liang, J. Measuring proteins and voids in proteins. In Proceedings of the 28th Annual Hawaii International Conference on System Sciences, Kihei, HI, USA, 3–6 January 1995; pp. 256–264. [Google Scholar] [CrossRef]
- Soga, S.; Shirai, H.; Kobori, M.; Hirayama, N. Use of Amino Acid Composition to Predict Ligand-Binding Sites. J. Chem. Inf. Model. 2007, 47, 400–406. [Google Scholar] [CrossRef]
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an integrated description of hydrogen bonding and dehydration: Decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Hindle, S.; Lange, G.; Klein, R.; Albrecht, J.; Briem, H.; Beyer, K.; Claußen, H.; Gastreich, M.; Lemmen, C.; et al. Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J. Comput. Aided Mol. Des. 2012, 26, 701–723. [Google Scholar] [CrossRef]
- Guariento, S.; Tonelli, M.; Espinoza, S.; Gerasimov, A.S.; Gainetdinov, R.R.; Cichero, E. Rational design, chemical synthesis and biological evaluation of novel biguanides exploring species-specificity responsiveness of TAAR1 agonists. Eur. J. Med. Chem. 2018, 146, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Menozzi, G.; Spallarossa, A.; Mosti, L.; Fossa, P. Exploring the binding features of rimonabant analogues and acyclic CB1 antagonists: Docking studies and QSAR analysis. J. Mol. Model. 2008, 14, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Casolino, C.; Menozzi, G.; Mosti, L.; Fossa, P. Exploring the QSAR of Pyrazolo[3,4-b]Pyridine, Pyrazolo[3,4-b]Pyridone and Pyrazolo[3,4-b]Pyrimidine Derivatives as Antagonists for A1 Adenosine Receptor. QSAR Comb. Sci. 2009, 28, 426–435. [Google Scholar] [CrossRef]
- Jolliffe, I.T. Principal Component Analysis; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Type | Series | RI |
|---|---|---|---|
| E_nb | Value of the potential energy related to non-bonded terms | 3D-I | 1.000000 (1.000000) |
| CASA+ | Positive charge weighted surface area, ASA+ times max {qi > 0} [a] | 3D-V | 0.663223 (0.747889) |
| ASA− | Water accessible surface area of all atoms with negative partial charge (strictly less than 0) | 3D-V | 0.305252 (0.292524) |
| E_ang | Angle bend potential energy | 3D-I | 0.232607 (0.152808) |
| CASA− | Negative charge weighted surface area, ASA- times max {qi < 0} | 3D-V | 0.229658 (0.252628) |
| vsa_pol | Approximation to the sum of VDW surface areas (Å2) of polar atoms (atoms that are both hydrogen bond donors and acceptors) | 2D-VI | 0.136024 (0.139963) |
| Corrector | H-Bonds | π-π Stacking | ||
|---|---|---|---|---|
| Amino Acid Residues | Ligand Portion | Amino Acid Residues | Ligand Portion | |
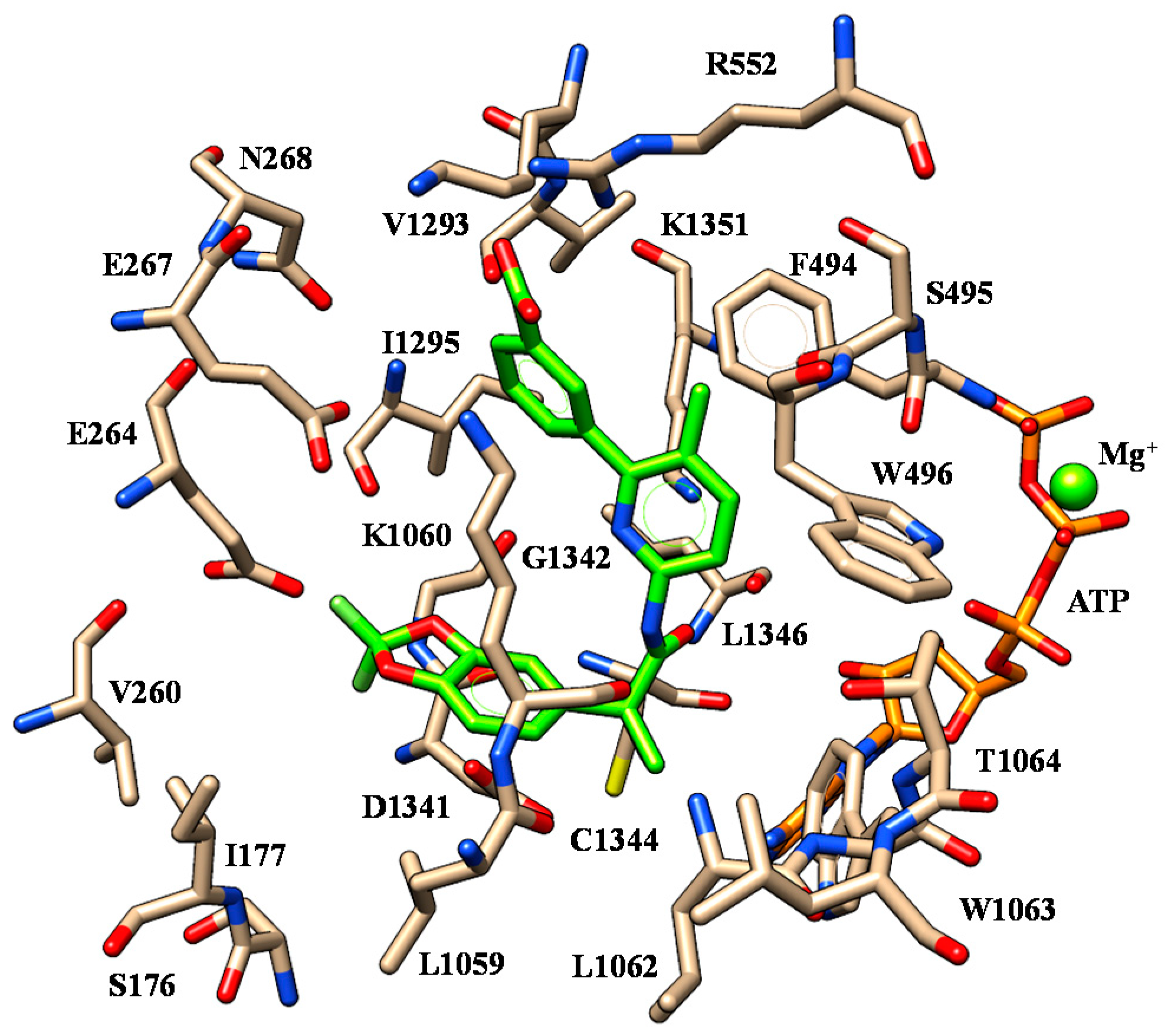
| 2 | K1060 | Oxygen atom of benzodioxole group | F494 | Benzodioxole moiety |
| S495 | Nitrogen atom of the carboxamide moiety | W496 W1063 | Phenyl ring | |
| W1063 | Ester oxygen atom | |||
| 3 | K1060 | Oxygen atom of benzodioxole group | F494 | Benzodioxole moiety |
| S495 | Nitrogen atom of the carboxamide moiety | W496 W1063 | Phenyl ring | |
| W1063 | Ester oxygen atom | |||
| 4 | K1060 | Oxygen atom of benzodioxole group | F494 | Benzodioxole moiety |
| S495 | Nitrogen atom of the carboxamide moiety | W496 W1063 | Phenyl ring | |
| W1063 | Ester oxygen atom | |||
| 6 | I1295 | Benzodioxole moiety | F494 | Benzodioxole moiety |
| K1351 | Carboxamide oxygen atom | W496 W1063 | Phenyl ring | |
| 10 | I1295 | Benzodioxole moiety | F494 | Benzodioxole moiety |
| K1351 | Carboxamide oxygen atom | W496 W1063 | Phenyl ring | |
| 11 | I1295 | Benzodioxole moiety | F494 | Benzodioxole moiety |
| K1351 | Carboxamide oxygen atom | W496 W1063 | Phenyl ring | |
| 18 | I1295 | Benzodioxole moiety | F494 | Benzodioxole moiety |
| K1351 | Carboxamide oxygen atom | W496 W1063 | Phenyl ring | |
| K1060 | Methoxy group | |||
| Corrector | H-Bonds | π-π Stacking | ||
|---|---|---|---|---|
| Amino Acid Residues | Ligand Portion | Amino Acid Residues | Ligand Portion | |
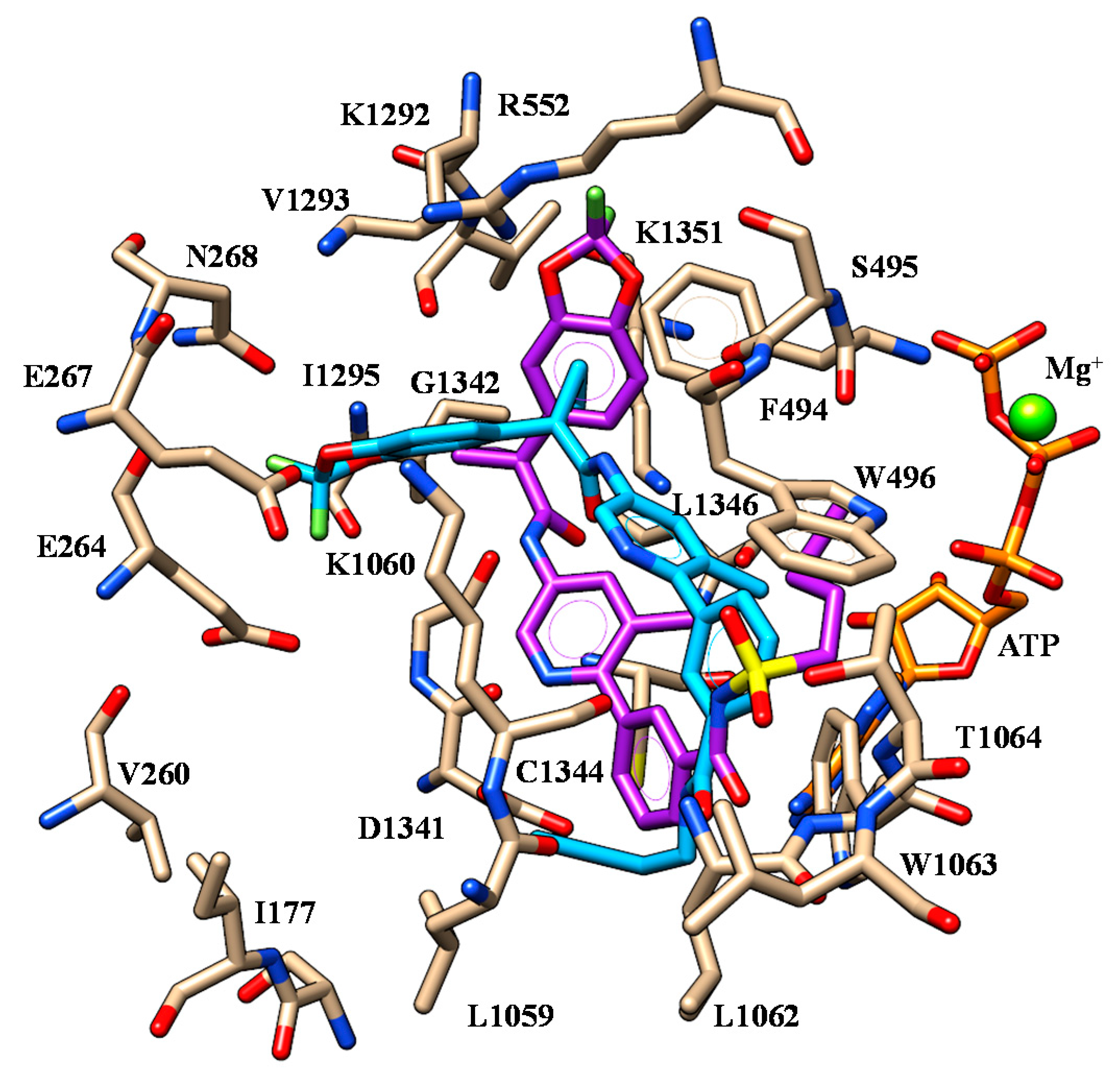
| 43 | V1293 | Methoxy group | W496 W1063 | Pyridine ring |
| D1341 | Nitrogen atom of Tetrahydropyrido-moiety | |||
| 45 | V1293 | Terminal amide Imidazolyl group | W496 W1063 | Pyridine ring |
| D1341 | Nitrogen atom of Tetrahydropyrido-moiety | |||
| G1342 | Nitrogen atom of secondary amine | |||
| K1351 | Pyridine group | |||
| 46 | V1293 | Methoxy group | W496 W1063 | Pyridine ring |
| D1341 | Nitrogen atom of Tetrahydropyrido-moiety | |||
| 50 | V1293 | Terminal amide Imidazolyl group | W496 W1063 | Pyridine ring |
| G1342 | Nitrogen atom of secondary amine | |||
| K1351 | Pyridine group | |||
| D1341 | Nitrogen atom of Tetrahydropyrido-moiety | |||
| 51 | V1293 | Terminal amide Imidazolyl goup | W496 W1063 | Pyridine ring |
| G1342 | Nitrogen atom of secondary amine | |||
| K1351 | Pyridine group | |||
| D1341 | Nitrogen atom of Tetrahydropyrido-moiety | |||
| Corrector | H-Bonds | π-π Stacking | ||
|---|---|---|---|---|
| Amino acid residues | Ligand Portion | Amino Acid Residues | Ligand Portion | |
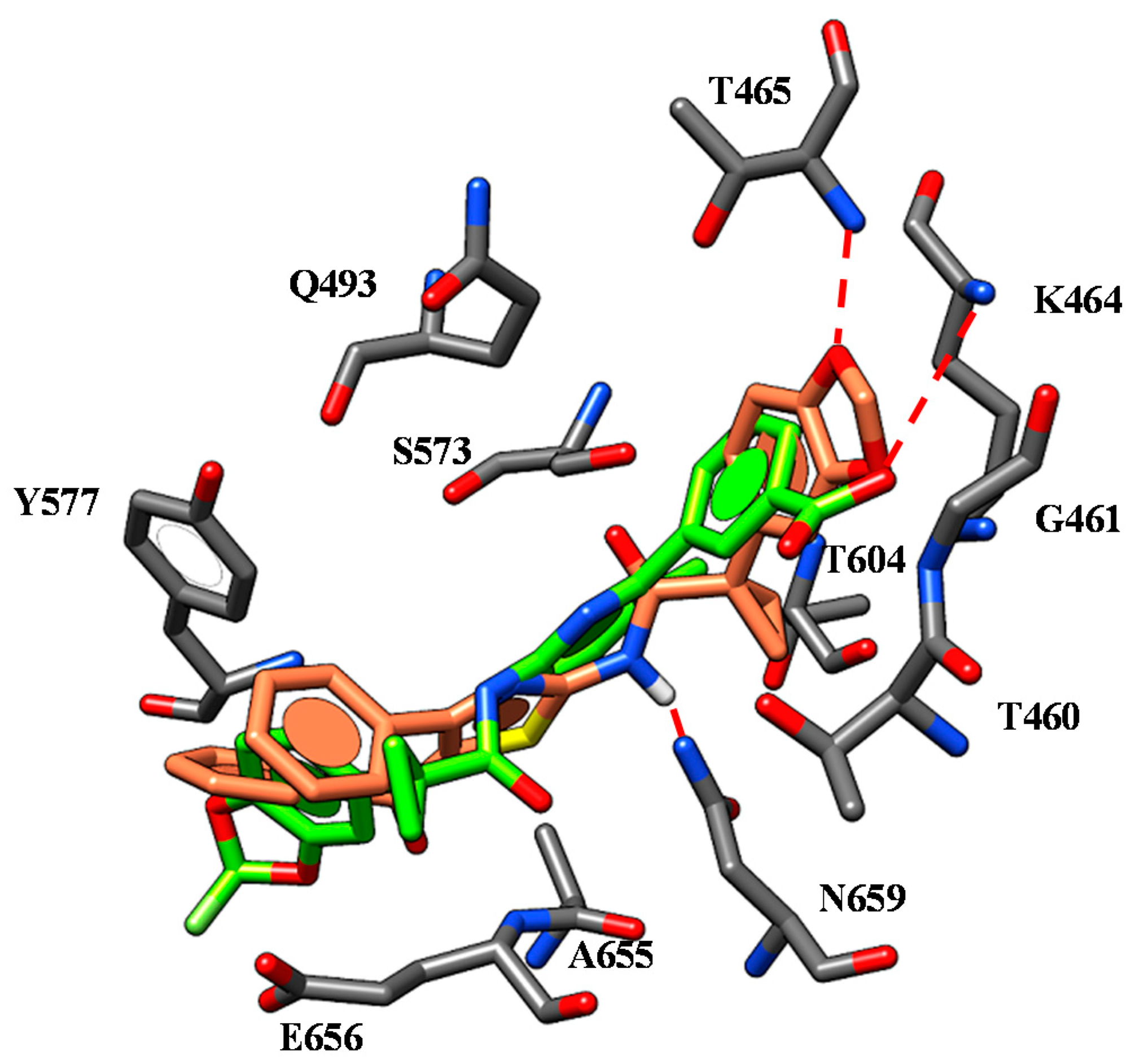
| 59 | L1062 W1063 | -CN group | W1063 F494 W496 | Quinoline core |
| K1060 D1341 | Spacer nitrogen atoms | |||
| 65 | L1062 W1063 | -CN group | W1063 F494 W496 | Quinoline core |
| K1060 D1341 | Spacer nitrogen atoms | |||
| 67 | L1062 W1063 | -CN group | W1063 F494 W496 | Quinoline core |
| K1060 D1341 | Spacer nitrogen atoms | |||
| 73 | L1062 W1063 | -CN group | W1063 F494 W496 | Quinoline core |
| K1060 D1341 | Spacer nitrogen atoms | |||
| I177 | Nicotinamide portion | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Righetti, G.; Casale, M.; Liessi, N.; Tasso, B.; Salis, A.; Tonelli, M.; Millo, E.; Pedemonte, N.; Fossa, P.; Cichero, E. Molecular Docking and QSAR Studies as Computational Tools Exploring the Rescue Ability of F508del CFTR Correctors. Int. J. Mol. Sci. 2020, 21, 8084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218084
Righetti G, Casale M, Liessi N, Tasso B, Salis A, Tonelli M, Millo E, Pedemonte N, Fossa P, Cichero E. Molecular Docking and QSAR Studies as Computational Tools Exploring the Rescue Ability of F508del CFTR Correctors. International Journal of Molecular Sciences. 2020; 21(21):8084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218084
Chicago/Turabian StyleRighetti, Giada, Monica Casale, Nara Liessi, Bruno Tasso, Annalisa Salis, Michele Tonelli, Enrico Millo, Nicoletta Pedemonte, Paola Fossa, and Elena Cichero. 2020. "Molecular Docking and QSAR Studies as Computational Tools Exploring the Rescue Ability of F508del CFTR Correctors" International Journal of Molecular Sciences 21, no. 21: 8084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218084