Peritubular Capillary Rarefaction: An Underappreciated Regulator of CKD Progression


Department of Nephrology, Takashimadaira Chūō General Hospital, 1-73-1 Takashimadaira, Itabashi City, Tokyo 175-0082, Japan
Int. J. Mol. Sci. 2020, 21(21), 8255; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218255
Submission received: 18 October 2020
/
Accepted: 29 October 2020
/
Published: 4 November 2020
(This article belongs to the Section Molecular Pathology, Diagnostics, and Therapeutics)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Peritubular capillary (PTC) rarefaction is commonly detected in chronic kidney disease (CKD) such as hypertensive nephrosclerosis and diabetic nephropathy. Moreover, PTC rarefaction prominently correlates with impaired kidney function and predicts the future development of end-stage renal disease in patients with CKD. However, it is still underappreciated that PTC rarefaction is a pivotal regulator of CKD progression, primarily because the molecular mechanisms of PTC rarefaction have not been well-elucidated. In addition to the established mechanisms (reduced proangiogenic factors and increased anti-angiogenic factors), recent studies discovered significant contribution of the following elements to PTC loss: (1) prompt susceptibility of PTC to injury, (2) impaired proliferation of PTC, (3) apoptosis/senescence of PTC, and (4) pericyte detachment from PTC. Mainly based on the recent and novel findings in basic research and clinical study, this review describes the roles of the above-mentioned elements in PTC loss and focuses on the major factors regulating PTC angiogenesis, the assessment of PTC rarefaction and its surrogate markers, and an overview of the possible therapeutic agents to mitigate PTC rarefaction during CKD progression. PTC rarefaction is not only a prominent histological characteristic of CKD but also a central driving force of CKD progression.
1. Background
Chronic kidney disease (CKD) affects one in every seven adults in the USA, suggesting that 30 million of American adults have CKD [1]. The number of patients with CKD is still growing with the increased prevalence of CKD risk factors such as aging, diabetes, hypertension, and obesity. There is no effective therapy to halt CKD progression to end-stage renal disease (ESRD, the most severe form of CKD). The mortality of patients with ESRD is approximately 15% per year [1]. Moreover, CKD is a major cause of death owing to increasing cardiovascular morbidity [2,3]. One reason for the lack of effective therapies is an incomplete understanding of pathogenesis of CKD. Regardless of initial insults, chronic injuries to the kidney frequently induce tubular atrophy, fibrosis, inflammation and peritubular capillary (PTC) rarefaction (Figure 1). PTC rarefaction is identified not only in diabetic nephropathy and hypertensive kidney diseases, two major causes of CKD [4,5,6], but also in advanced IgA nephropathy [7], congenital nephrotic syndrome [8], lupus nephritis [9], polycystic kidney disease [10,11], and allograft nephropathy [12,13], suggesting that PTC loss is a very common event in patients with CKD. Interestingly, the aging process itself accelerates PTC rarefaction [14]. Although many studies have investigated PTC rarefaction (reviewed in [15,16,17,18]), the molecular mechanisms of PTC rarefaction still remain incompletely elucidated. Recent studies have made major progress in understanding the process of PTC rarefaction. Therefore, based on those new findings, this review describes (1) the novel mechanisms of PTC rarefaction, (2) the major factors regulating PTC angiogenesis, (3) the assessment of PTC rarefaction and its surrogate markers, and (4) possible therapeutic agents to retard PTC rarefaction during CKD progression.
2. Anatomy of PTC
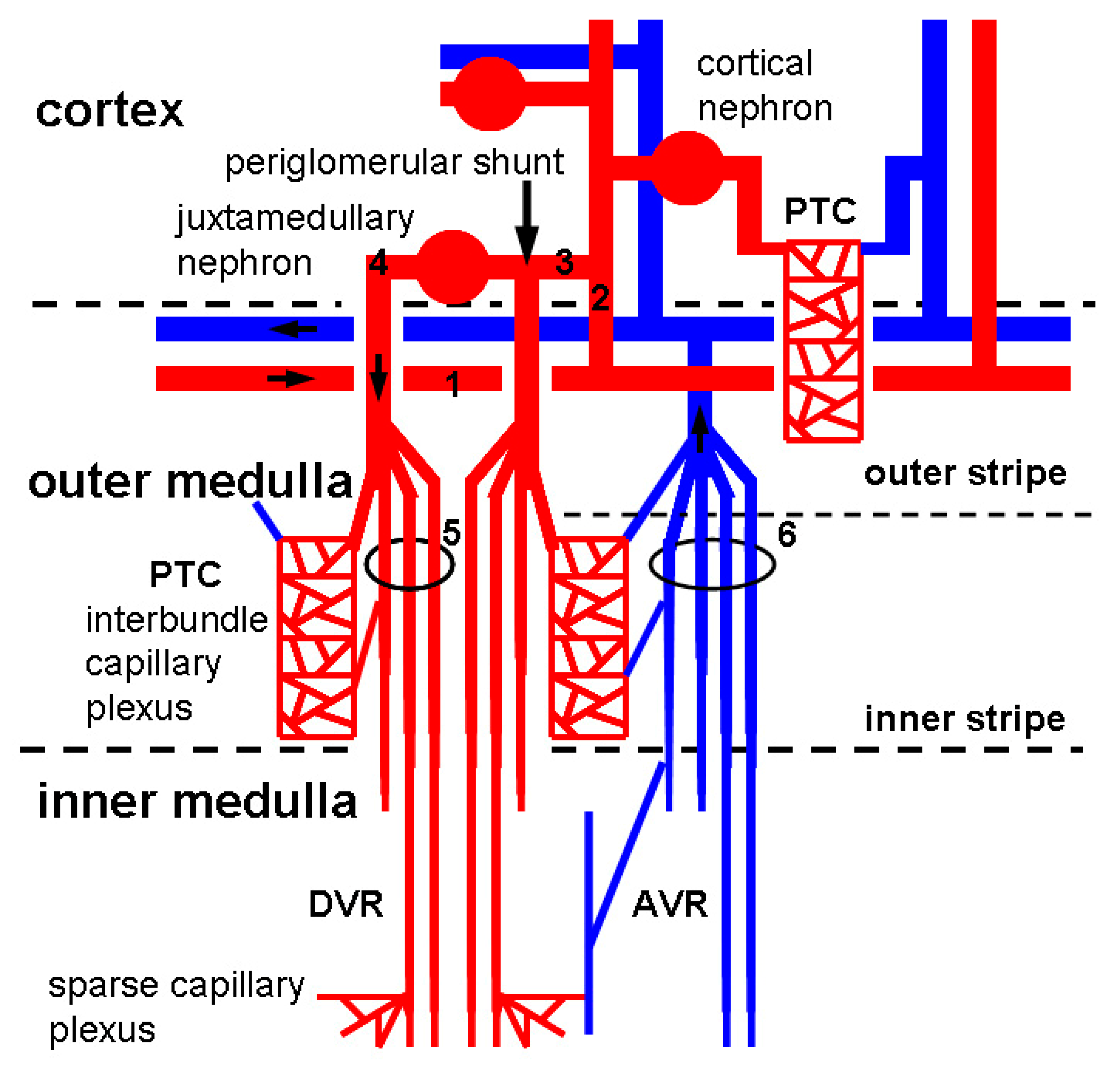
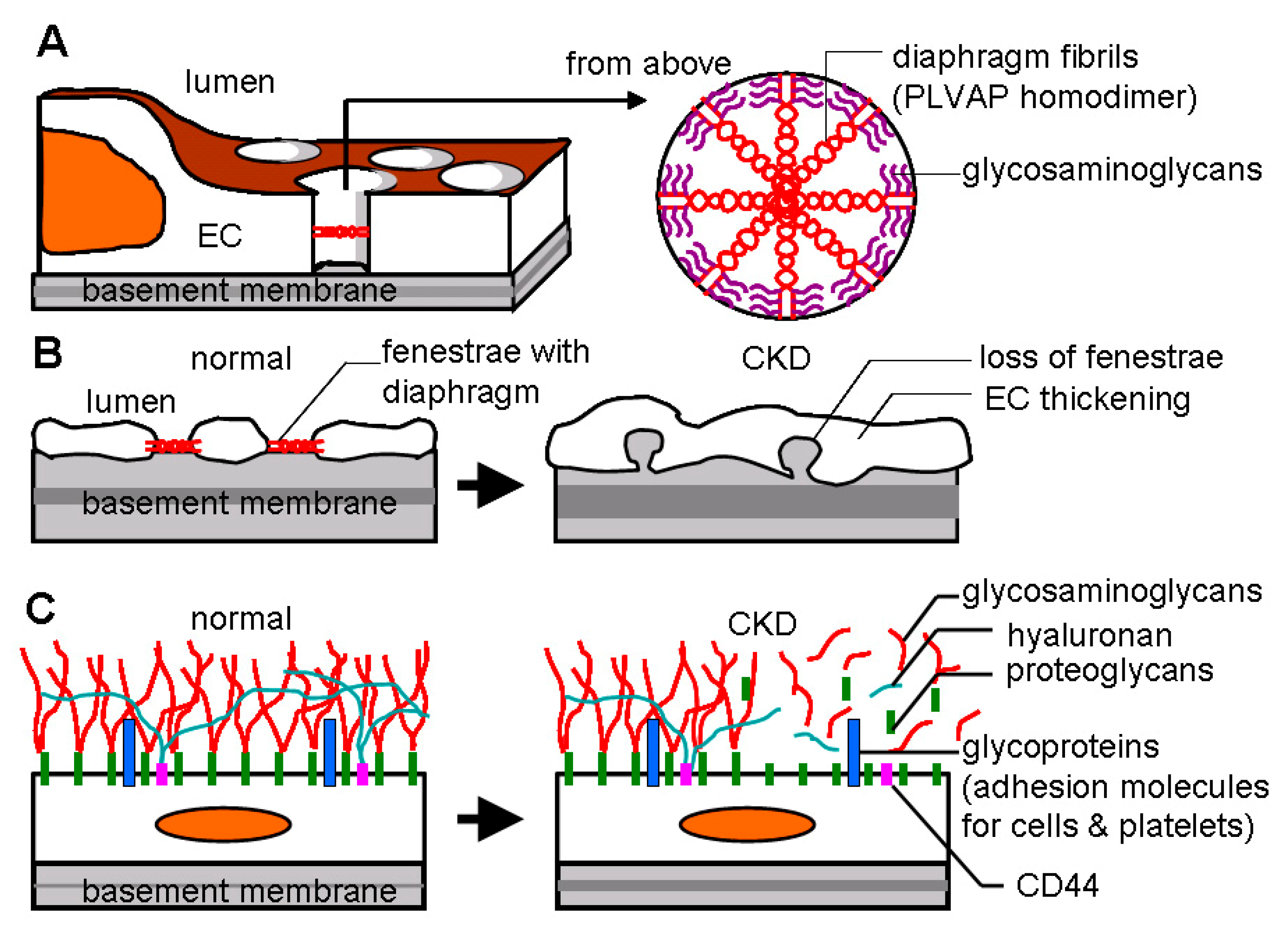
Arteriolar blood is delivered to the cortex via a series of large branches of the renal arteries such as interlobar arteries, arcuate arteries (approximate luminal diameter: 70–100 μm in rats [19]), and interlobular arteries (approximate luminal diameter: 40–50 μm in rats [19]). Cortical glomeruli (90% of total glomeruli) and juxtamedullary glomeruli (10% of total glomeruli) branch from interlobular arteries via afferent arterioles (approximate luminal diameter: 21–25 μm in rats [20]). The majority of blood flow reaches the medulla through efferent arterioles (approximate luminal diameter: 19–23 μm in rats [20]) whereas some circumvent glomeruli by a periglomerular shunt (Figure 2). Afferent arterioles are usually larger in diameter than efferent arterioles to increase blood pressure in glomeruli for ultrafiltration to take place. A bundle of glomerular capillaries (approximate luminal diameter: 6–10 μm in rats [21]) forms the efferent arteriole. Efferent arterioles follow one of the two pathways. First, efferent arterioles arise from glomeruli in the mid and outer cortex construct PTC network (approximate luminal diameter: 5–10 μm in rats [22]) running alongside the proximal and distal tubules (cortical nephron) (Figure 2). Second, efferent arterioles arise from juxtamedullary glomeruli construct vasa recta running along the loops of Henle and collecting tubules (juxtamedullary nephron) (Figure 2). Descending vasa recta (DVR, approximate luminal diameter: 13–17 μm in rats [23]) branches into several vessels, penetrating the inner medulla, which construct the sparse inner capillary network (sparse capillary plexus). Venous blood returns to the cortex via ascending vasa recta (AVR, approximate luminal diameter: 18–22 μm in rats [23]) (more details of renal vasculature anatomy are reviewed in [24,25]). Osmotically active solutes (NaCl and urea) move from AVR to medullary interstitium to DVR. This system traps solutes in the interstitium by recycling between AVR and DVR to create the countercurrent exchange for efficient urine concentration. Kidneys lacking AVR do not process the capacity to concentrate urine [26]. A trade-off of countercurrent exchange is that oxygen and nutrients are shunted from DVR to AVR, resulting in profound hypoxia in the inner medulla. Thus, DVR tightly controls perfusion to the outer and inner medulla to avoid the threat of ischemia. Indeed, DVR functions both as an exchanging vessel (capillary) and a resistance vessel (arteriole) [27]. DVR and AVR are not defined as PTCs in this review, because the proximal part of DVRs is an arteriole surrounded by smooth muscle cells [28]. Capillary endothelial cells (ECs) of the glomeruli, PTC network, AVRs, but not DVRs, possess fenestrae that are transcellular pores with 60–80 nm diameters [26,29,30] (Figure 3A,B). Fenestration pores allow water, size- and charge-restricted molecules to pass through. Fenestration pores of PTCs and AVRs display the diaphragmatic structure, but fenestrae of glomerular capillaries lack diaphragms. The diaphragm is composed of multiple radial fibrils and accessory glycosaminoglycans [31] (Figure 3A). Endothelial diaphragm deficient mice demonstrated edematous interstitium in the kidney without any proteinuria and abnormal kidney function, suggesting that PTC diaphragm is involved in transportation of water and proteins in the interstitium [29]. Another prominent structure for EC homeostasis is the glycocalyx, a gel consisting of proteoglycans, glycosaminoglycans, and glycoproteins (approximate height of glycocalyx: 50–100 nm [32]) (Figure 3C). Endothelial glycocalyx covers the luminal surface of ECs and blocks blood coagulation, platelet aggregation, and inflammatory cell adhesion [33].
3. Clinical Significance of PTC Rarefaction
While PTC rarefaction was correlated with the severity of fibrosis in patients with CKD [34,35,36], PTC rarefaction was found to be a strong predictor of future ESRD development in CKD patients [37]. Moreover, compared with tubular atrophy, interstitial fibrosis, and inflammatory cell infiltration, the extent of PTC loss reflected glomerular filtration ratio (GFR) most accurately in CKD patients [36]. By using multiple murine CKD models such as cisplatin-induced injury, rhabdomyolysis, and ischemia-reperfusion injury (IRI), another study similarly demonstrated that PTC density correlates with GFR better than fibrosis does [38]. This finding is plausible because a bundle of glomerular capillaries forms the efferent arteriole that is directly connected with PTCs or DVRs without any collateral vessels (Figure 2). As suggested before [39], once PTCs and DVRs disappear or lose their blood flow, perfusion of glomerular capillaries is also dramatically reduced, and vice versa. This idea is supported by a fact that impaired PTC/DVR perfusion causes severe loss of GFR in rat models with congested kidneys [40]. Moreover, the loss of PTC perfusion induces tissue hypoxia that triggers further PTC loss [41]. As perfused PTC number and PTC diameter were inversely correlated well with blood urea nitrogen (BUN) level both in the short term and the long term after severe IRI [42], it is strongly suggested that PTC loss is one of the major contributors to impaired GFR.
4. Mechanisms of PTC Rarefaction
4.1. Established Major Mechanisms—Loss of Endothelial Survival Factors
Healthy kidneys keep a tight balance between proangiogenic and antiangiogenic factors to prevent unnecessary angiogenesis. However, this balance is disrupted during CKD progression. In response to kidney injury, PTC ECs initially proliferate and subsequently disappear due to endothelial apoptosis [43]. Early proliferation of ECs is supported with intense expression of vascular endothelial growth factor (VEGF)-A, a major and potent proangiogenic factor, in the tubular epithelium compartment. However, expression levels of VEGF-A and its receptor vascular endothelial growth factor receptor-2 (VEGFR2) gradually decline in the later stage, resulting in enhanced EC apoptosis without compensative EC proliferation [43]. Similarly, expression level of angiopoietin (Angpt)-1, another potent proangiogenic factor, is dramatically decreased following kidney injury [44]. Concurrently, antiangiogenic factors such as thrombospondin-1 (TSP-1) and Angpt-2 are strongly induced to antagonize VEGF-A and Angpt-1 signaling [45,46]. Moreover, inflammatory macrophages infiltrate and secrete inflammatory cytokines such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α), all of which block tubular VEGF-A expression [45]. Especially macrophage-derived IL-1β and TNF-α were demonstrated to regress capillary tubes [47]. Deprivation of endothelial survival factors is believed to be a central and major mechanism for PTC rarefaction following chronic kidney injury. As described below, recent research work has discovered other important mechanisms to cause PTC loss during CKD progression.
4.2. PTC Endothelium Quickly Responds to CKD Development
A recent ultrastructural study of PTCs revealed that the number of fenestrated pores starts to decrease only 24 h after progressive kidney injury by unilateral ureteral obstruction (UUO) in mice [48]. This time point (24 h post-UUO) was a very early stage in the disease course compared with 5 days post-UUO when significant fibrosis was detectable [49]. Significant PTC rarefaction and endothelial thickening in PTC were detected 3 days and 5 days after UUO, respectively. In the same study, similar fenestration loss and endothelial thickening were observed in other animal CKD models (IRI, Alport mice) and in biopsy samples of CKD patients [48], implying that PTC ECs immediately and continuously react to kidney injury in rodents and humans regardless of the type of insults. The loss of fenestration indicates endothelial activation in glomerular and liver sinusoidal capillaries [50,51,52]. Activated ECs induce heparanase and hyaluronidase that degrade endothelial glycocalyx [53]. Without glycocalyx coverage, ECs become pro-coagulant, pro-thrombotic and pro-adhesive (Figure 3C). For example, the glycoprotein molecule of CD44 on PTC was increased and exposed to the blood stream by glycocalyx stripping after kidney injury, resulting in enhanced neutrophil recruitment/influx, exaggerated inflammation, and PTC loss [54,55] (Figure 3C). Consequently, EC activation leads to impaired blood flow and lowered laminar shear stress on ECs. As shear stress is one of the primary regulators of glycocalyx formation [56], low perfusion of PTCs leads to destabilization and further loss of endothelial glycocalyx. Actually, GFR was inversely correlated with glycocalyx shedding in patients with CKD [57]. Finally, this whole process triggers endothelial apoptosis [58]. These findings suggest that ECs in PTC are promptly activated in response to kidney injury, culminating in PTC loss due to EC apoptosis during CKD progression.
4.3. PTC Endothelium Is a Unique Population with Low Proliferative Potential
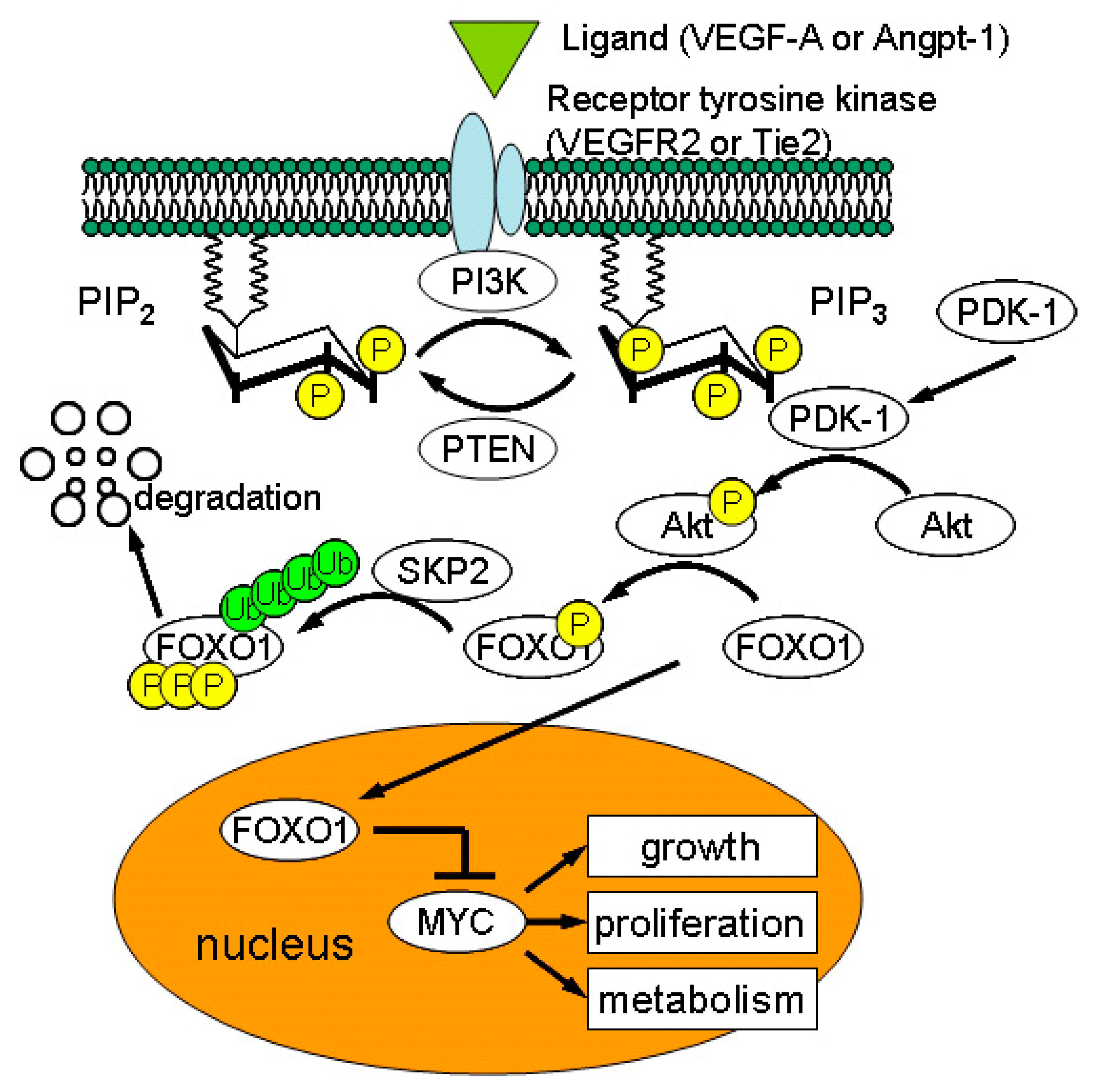
In response to IRI, PTC ECs progressively disappeared with marginal endothelial proliferation [59]. While 0.5–1.0% of PTC ECs were proliferative in normal kidneys, subtle increase in EC proliferation was detected in the early phase of UUO injury [60]. However, the mechanisms underlying low proliferation of PTC ECs have remained unclear. Recent studies identified impaired proliferation of PTC ECs isolated from mouse, rat, and human compared with that of ECs from other organs such as lung and aorta [61,62]. Dang et al. demonstrated that constitutively active phosphatase and tensin homolog (PTEN) suppresses proliferation/growth of kidney microvascular ECs by counteracting phosphoinositide-3-kinase (PI3K)/Akt signaling whereas VEGF-A potentiates PI3K/Akt pathway for capillary growth [62] (Figure 4). In ECs, activated PI3K/Akt signaling prevents nuclear translocation of forkhead box O-1 (FOXO-1) transcription factor, promoting cell cycle progression by enhanced MYC activity [63] (Figure 4). Chemical inhibitor of PTEN not only restored angiogenesis of PTC ECs in vitro but also antagonized PTC rarefaction in IRI in vivo [62]. This finding explains why PTCs are easy to regress after injury.
4.4. EC Apoptosis Is a Pivotal Cause of PTC Dropout
CKD progression enhances endothelial expression of active caspase-3 [64] and endothelial apoptosis [43]. In global caspase-3 deficient mice, endothelial apoptosis was significantly reduced post-IRI, resulting in reduced PTC rarefaction [65]. Moreover, loss of caspase-3 lowered hypoxia-inducible factor (HIF)-1α expression following IRI, indicating that preserved PTC network counterbalances tissue hypoxia. Global caspase-3 defect also rescued tubular cells from IRI, probably preserving tubular VEGF-A expression. This would inhibit PTC dropout in mutant mice although VEGF-A level was not measured in this study [65]. Kidney injury increased expression of anti-angiogenic factors such as TSP-1 and endostatin [45,66], both of which induce EC apoptosis by caspase-3 activation [67,68]. Another molecule, CD44, one of endothelial glycocalyx components and induced by kidney injury, strongly enhanced EC apoptosis by caspase-3 activation [69].
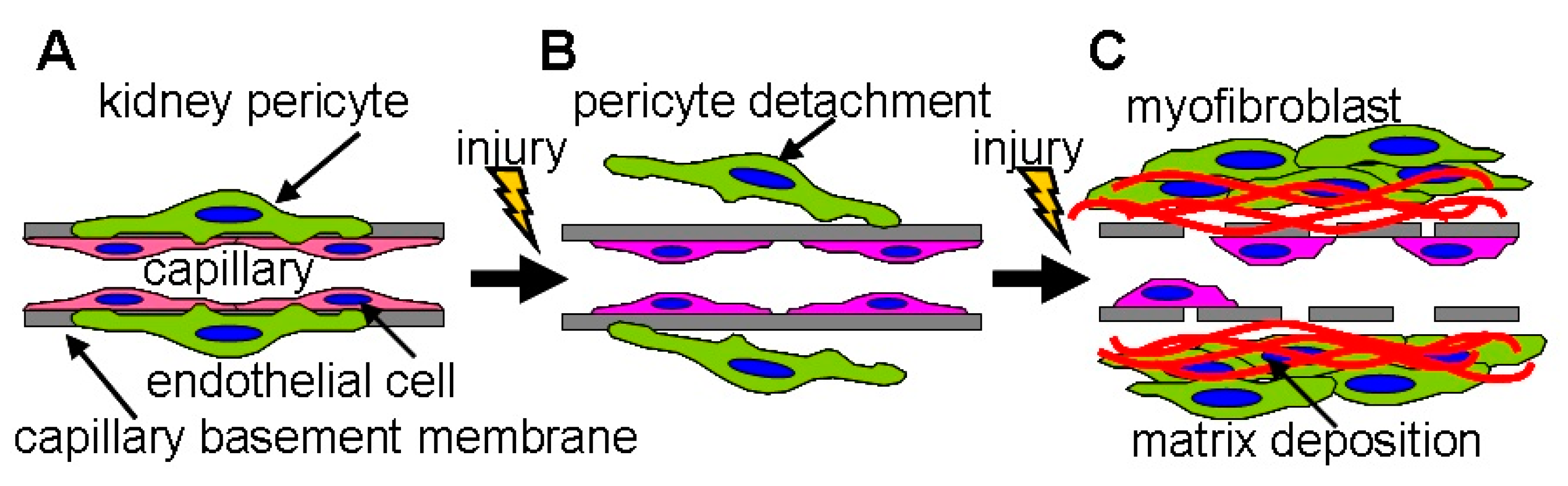
4.5. Pericyte Detachment Worsens PTC Loss
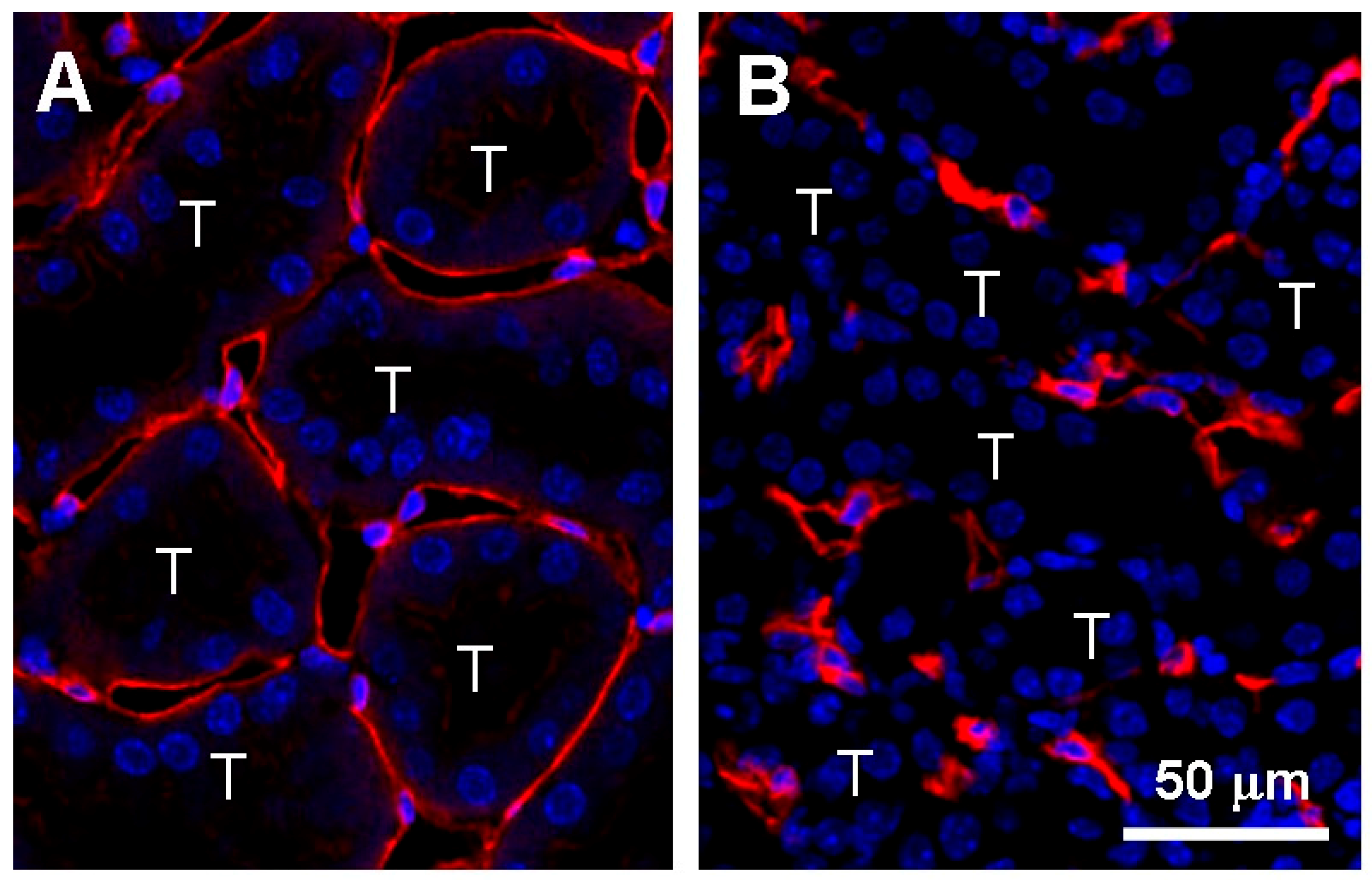
Kidney pericytes are closely attached to PTC ECs and maintain the structure and function of PTCs [70,71]. For example, kidney pericytes contact ECs and intensify synthesis of capillary basement membrane to maintain capillary integrity [60]. While kidney pericytes stabilize endothelial tube formation of PTCs in healthy kidneys, pericytes promptly migrate away from PTC following kidney injury, leading to PTC disintegration and rarefaction [60,72] (Figure 5). Images by two photon microscopy revealed that many processes from the cell body of pericytes are attached to PTC in normal kidneys whereas those processes are detached from PTCs and reattached to walls of tubular epithelial cells 3 days after UUO [71]. After injury, pericytes differentiated into two scar-forming populations (α-smooth muscle actin positive myofibroblasts and α-smooth muscle actin negative activated fibroblasts) [73]. Neither fibrotic population could stabilize PTCs after injury [74]. These facts offer the mechanisms by which PTC rarefaction accompanies tissue fibrosis (Figure 5). Kramann et al. identified that Gli1+ cells are a perivascular population of mesenchymal stem cell-like cells [75]. In the kidney, Gli1+ cells functioned as pericytes in normal kidneys, and Gli1+ pericytes were significantly detached from PTCs following IRI [76]. Genetic ablation of Gli1+ pericytes resulted in PTC rarefaction (predominant rarefaction of smaller capillaries whose diameter was less than 7 μm), tissue hypoxia, and transient hypoxic tubular epithelial injury 10 days post-ablation. Interestingly, Gli1+ pericyte ablation induced an inflammatory response with upregulation of renal TNF-α and IL-6 expression. Lemos et al. genetically ablated a forkhead box D-1 (FoxD1) positive population, which induced PTC loss, tubular injury, and albuminuria without acute inflammatory response 3 days after ablation [77]. Loss of FoxD1 was severe enough to sacrifice all mice within 3 days post-treatment. While Gli1+ cells represented a small fraction of kidney pericytes, FoxD1+ population contained pericytes, perivascular fibroblasts, glomerular mesangial cells, podocytes, and vascular smooth muscle cells in the kidney [78]. Both studies reported that pericytes loss causes tubular damage. Injured tubules increased transforming growth factor (TGF)-β expression, promoting pericyte-myofibroblast differentiation and further tissue damage [79]. Studies of pericyte ablation indicate that kidney pericytes are essential for PTC integrity as well as tubular integrity and the loss of pericytes accelerates PTC rarefaction.
5. Major Factors Affecting PTC Loss
5.1. VEGF-A
VEGF-A is the potent regulator to maintain PTC network. In the adult kidney, VEGF-A is mainly expressed in podocytes and the thick ascending limbs of Henle’s loop and, to a lesser extent, in proximal and distal tubules [80]. Loss of tubular epithelial VEGF-A induced PTC rarefaction even without kidney injury [80]. Moreover, tubular VEGF-A deletion resulted in pronounced polycythemia due to elevated erythropoietin (EPO) production, suggesting that PTC loss induces tissue hypoxia and thereby stimulates renal EPO producing cells [80]. Consistently, expression of VEGF-A was remarkably decreased in rodent CKD models and in CKD patients mainly due to tubular cell atrophy [37,43,45,81]. Conversely, overexpression of VEGF-A in tubular epithelial cells caused overgrowth of PTC ECs that synthesize excess amounts of platelet-derived growth factor (PDGF)-B and TGF-β [82]. This sequence of events resulted in tissue fibrosis because PDGF-B and TGF-β simulates pericyte proliferation and pericyte-myofibroblast transition, respectively [79]. As both loss and overexpression of tubular VEGF-A disorganize PTC architecture, a proper range of tubular VEGF-A expression is indispensable for healthy PTC integrity. Separately, soluble fms-like tyrosine kinase-1 (sFlt-1, soluble VEGFR1) was found to be elevated in patients during CKD progression [83]. sFlt-1 is a truncated form of VEGFR1 and a potent circulating antagonist for VEGF-A. Renal biopsy samples demonstrated that sFlt-1 is expressed in CD68+ histiocytes (a part of macrophage population) and PTC network around sFlt-1+ cells are diminished [84]. In patients receiving cardiac surgery, perioperative low VEGF-A and high sFlt-1 levels in the plasma significantly predicted future development of acute kidney injury (AKI) [85]. In addition, other VEGF-A antagonists such as endostatin [86,87,88] and TSP-1 [89,90,91,92] were increased in patients with CKD, which worsened PTC rarefaction [88,92]. Finally, VEGF-A rich angiogenic macrophages were identified in damaged kidneys from mice and patients [93]. These macrophages were kidney resident, not derived from circulating monocytes, and supported proliferation of PTC ECs. Taken together, reduced tubular VEGF-A expression and increased VEGF-A antagonists cooperatively promote PTC rarefaction during CKD progression.
5.2. Angiopoietin/Tie
Angpt-1 and Angpt-2 are another critical angiogenic factors that act on Tie (tyrosine kinase with Ig and EGF homology domains) receptors, Tie1 and Tie2. In the adult kidney, Angpt-1 is expressed in tubular epithelial cells, podocytes, and pericytes, whereas Angpt-2 is detected in ECs, with lower levels in tubular epithelial cells. Tie2 is expressed in glomerular and peritubular ECs in addition to hematopoietic cells. Generally, Angpt-1 binds to Tie2, enhancing EC survival and vascular stabilization, while Angpt-2 competitively inhibits the action of Angpt-1 on Tie2 [94]. In patients with CKD, circulating levels of VEGF-A and Angpt-1 were decreased and those of Angpt-2 were elevated, generating anti-angiogenic environment [46]. Furthermore, elevated Angpt-2 level was a strong predictor of mortality in CKD patients [95]. Inducible and global loss of Angpt-1 accelerated PTC loss and fibrosis only after kidney injury in mice [44], indicating a protective role of Angpt-1 in kidney injury. While Tie2 activation (Tie2 phosphorylation) was reduced by vascular endothelial protein tyrosine phosphatase (VE-PTP) during hypertensive or diabetic kidney injury, inhibition of VE-PTP activated endothelial Tie2 signaling and protected kidneys from such damage [96]. VE-PTP was highly expressed in glomerular and peritubular capillaries and repressed Tie2 activity by its dephosphorylation [96]. Although the role of Tie1 has been ambiguous, recent study showed that Tie1 enhances Angpt-1/Tie2 signaling in ECs [97,98]. However, acute inflammation promoted endothelial Angpt-2 expression and Tie1 ectodomain cleavage, counteracting angiogenesis by impairment of Tie2 signaling [97,98]. Genetic loss of endothelial Tie1 caused capillary regression via markedly enhanced EC apoptosis even if ECs expressed Tie2 [99]. Endothelial overexpression of Angpt-2 induced pericyte detachment from capillary EC, promoting capillary destabilization [100]. Collectively, Angpt-1/Tie2 signaling protects against PTC rarefaction.
5.3. HIF
Hypoxia-inducible factor (HIF) is a master regulator of cellular adaptation to hypoxia. HIF is a heterodimeric transcription factor that is composed of HIF-α and HIF-β subunits. HIF-β is constitutively expressed whereas HIF-α expression is tightly controlled by oxygen-dependent degradation. In normoxia, two conserved proline residues of HIF-α are hydroxylated by prolyl hydroxylase domain-containing proteins (PHDs). Hydroxylated HIF-α is detected by the von Hippel-Lindau protein and is subjected to polyubiquitination and following proteasomal degradation. In hypoxia, however, HIF-α escapes from hydroxylation by PHDs and binds to HIF-β. This functional heterodimeric HIF translocates to the nucleus and upregulates the transcription of HIF responsive genes such as EPO and VEGF-A. HIF-α has two major isoforms, HIF-1α and HIF-2α. In the hypoxic kidney, HIF-1α is expressed in tubular and glomerular epithelial cells, whereas HIF-2α is detected in glomerular and peritubular ECs and fibroblasts. Deletion of tubular HIF-1α attenuated tissue fibrosis and macrophage infiltration post-UUO injury [101]. While endothelial-specific HIF-1α inactivation did not show any influence on kidney injury, HIF-2α deletion in the endothelium worsened PTC rarefaction, glomerular capillary loss, albuminuria, and tissue fibrosis following kidney damage [102,103]. Loss of endothelial HIF-2α prolonged inflammatory response [104] and impaired protection against oxidative stresses [105]. Moreover, HIF-2α+ pericytes/fibroblasts were the unique source of renal EPO production [106]. EPO production in kidney pericytes/fibroblasts was exclusively dependent on HIF-2α [107], implying that HIF-2α in pericytes/fibroblasts is protective against anemia and tissue hypoxia. Taken together, renal HIF-1α exaggerates kidney injury, and renal HIF-2α antagonizes PTC loss.
5.4. Sirtuin
Silent information regulator two protein (Sirtuin)-1 is a protein with nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase activity and is highly expressed in ECs. Sirtuin-1 antagonizes endothelial cellular senescence (aging process accompanying EC dysfunction) through multiple pathways [108]. ECs stop proliferation after a limited number of doublings. Cessation of cell division induces cell growth arrest, which is termed replicative senescence. Some stresses such as oxidative stress and DNA damage elicit quite similar cell growth arrest in the short term, referred to as stress-induced premature senescence (SIPS). Kidney injury antagonizes PTC angiogenesis by inducing endothelial SIPS. Endothelial Sirtuin-1 prevented capillary loss in the hindlimb model by repressing FOXO1 activity [109]. Furthermore, Sirtuin-1 inactivated p53 by its deacetylation and induced cellular growth in ECs [110]. As FOXO1 and p53 strongly induce endothelial SIPS by cell cycle arrest, Sirtuin-1 maintains the angiogenic properties of PTC ECs by counteracting SIPS [108]. Deletion of endothelial Sirtuin-1 enhanced PTC loss via down-regulation of matrix metalloproteinase (MMP)-14 and activation of Notch1 signaling [111,112]. ECs needed MMP-14 to degrade the extracellular matrix for new microvessel formation [111]. In ECs, Notch1 induced SIPS and cell cycle arrest by elevated PTEN expression [112,113]. Separately, loss of endothelial Sirtuin-1 extensively reduced endothelial glycocalyx [114]. Reduced glycocalyx was shown to lower microvascular perfusion in rodents and humans [115,116,117], suggesting that Sirtuin-1 deficient PTCs lose their perfusion and induce tissue hypoxia following kidney injury. Taken together, endothelial Sirtuin-1 is protective against PTC loss.
5.5. Vasohibin
Vasohibin-1 (VASH-1) was initially identified as a novel antiangiogenic factor derived from ECs [118], while Vasohibin-2 (VASH-2) was identified as a VASH-1 homolog and a novel proangiogenic factor [119]. VASH-1 is expressed in glomerular and capillary ECs of the normal kidneys, and kidney injury induces VASH-1 expression in glomerular mesangial cells and inflammatory cells in addition to ECs [120]. Later study found that endothelial VASH-1 increases the expression of superoxide dismutase-2 and the synthesis of Sirtuin-1 in ECs, indicating that endothelial VASH-1 promotes stress tolerance by quenching reactive oxygen species and SIPS [121]. In response to cisplatin-induced kidney injury, VASH-1 deficient mice enhanced loss of renal function, tubular injury, macrophage infiltration, and PTC rarefaction compared with control mice [122]. Kidney injury decreased VASH-1 expression in whole kidneys and accelerated PTC loss [122]. VASH-2 expression is observed in ECs of PTC/vasa recta and cortical/medullary tubules in the normal kidney and is strongly induced in tubular epithelial cells after kidney injury [123]. Following IRI, VASH-2 knockout mice exaggerated loss of renal function, tubular injury, neutrophil infiltration, and PTC rarefaction compared with control mice [124]. VASH-2 expression was intensely increased in damaged kidneys, antagonizing PTC rarefaction. Collectively, VASH protects against PTC loss.
5.6. Pericyte-Endothelial Cell Interaction
As mentioned above, endothelial-pericyte crosstalk is critical for angiogenesis and vascular stabilization. This crosstalk involves multiple ligand-receptor interactions including PDGF-B/PDGF receptor-β (PDGFRβ) and Angpt-1/Tie2 [125]. When either PDGFRβ signaling in pericytes or VEGFR2 signaling in ECs was blocked by circulating soluble receptor ectodomains, both PTC rarefaction and fibrosis were remarkably alleviated during CKD progression [126]. This result indicates that (1) bidirectional signaling between pericytes and PTC ECs is necessary to prevent pericyte detachment from PTCs, and (2) kidney injury excessively enhances this bidirectional signaling, resulting in pericyte loss and unstable vasculatures. Once pericytes were detached from PTCs, angiogenesis of functional PTC was disrupted and inefficient, which could be corrected by PDGFRβ or VEGFR-2 blockade [126]. While pericytes synthesized EPO in normal kidneys, myofibroblasts lost the capability of producing EPO, which induced renal anemia and thereby enhanced tissue hypoxia [71,127,128]. Impaired EPO/EPO receptor signaling was shown to cause PTC rarefaction post-IRI [129]. Moreover, tissue hypoxia played a critical role in acceleration of PTC loss after kidney injury [41].
5.7. Endothelial Progenitor Cells
Endothelial progenitor cells (EPCs) are defined as non-ECs that are capable of differentiating into ECs. Asahara et al. provided the first evidence that bone marrow-derived circulating EPCs differentiate into mature ECs to form new vessels in vivo [130]. Following this primary publication, numerous studies have shown that EPCs contribute to vascular regeneration in patients and multiple models of tissue injury including AKI and CKD. However, later studies detected only marginal EPC incorporation into the vasculatures in animal models of AKI and CKD [131,132,133]. Furthermore, all new blood vessels in the damaged heart were derived from pre-existing ECs, not from EPC [134]. These results suggest an idea that EPCs stimulate PTC angiogenesis via paracrine mechanisms such as secretion of proangiogenic factors (VEGF-A and Angpt-1) to repair damaged vasculatures [131]. Alternatively, EPCs may have the potential to prevent pericyte detachment from PTCs [135].
5.8. Endothelial to Mesenchymal Transition (EndMT)
The concept of EndMT is that endothelial cells are capable of differentiating into mesenchymal cells (fibroblasts and myofibroblasts) under certain conditions. The initial study demonstrated that 30–50% of myofibroblasts are derived from ECs during kidney injury [136]. However, the later study revealed that only 10% of myofibroblasts emerged via EndMT following kidney injury [137]. Thus, renal ECs mainly disappear without becoming fibroblasts in damaged kidneys. Many studies exploring the role of EndMT in renal fibrosis have used vascular endothelial (VE)-cadherin-Cre (Cdh5-Cre) mice and Tie2-Cre mice for linage tracing or deletion of specific factors in ECs. However, VE-cadherin and Tie2 have been shown to be non-specific for ECs and were expressed in hematopoietic cells during development, because of endothelial origin of hematopoietic stem cells [138]. The lack of a specific marker for ECs for lineage tracing makes it difficult to interpret these studies properly. As no evidence of EndMT was detected in cardiac fibrosis following injury [139], further study is necessary to determine the minor contribution of EndMT to PTC rarefaction.
6. Assessment of PTC Loss or Its Surrogate Marker, Tissue Hypoxia
6.1. Histological Assessment
Histological evaluation is the popular method to assess PTC density. Many researchers have immunostained kidney sections with EC marker(s) and measured PTC density by a grid method (% of EC marker-positive grids among total grids) or by an area fraction method (% of EC marker-positive area among total area). However, these methods may overestimate PTC density, because ECs of clogged or collapsed capillaries with micro-embolisms are still stained and positive for EC marker(s), even if PTCs totally lose their perfusion. Kramann et al. injected the fluorescence microbead (0.02-μm diameter)-agarose mixture into the beating heart to visualize actual capillary lumen in mouse kidneys post-IRI [42]. They found that total perfused PTC cross-sectional area, PTC number, individual capillary cross-sectional area, and individual capillary perimeter are significantly decreased in damaged kidneys compared with sham-operated control kidneys 8 weeks post-severe IRI. Moreover, these factors of PTC density were inversely correlated with renal function assessed by BUN. For more accurate assessment of PTC density, Babickova and colleagues proposed a normalization of PTC counts to the number of adjacent tubular segments, because the number of tubules differed between rodent CKD models [48]. For example, the number of tubules per area remained constant (with the appearance of dilated and atrophic tubules), but the number of PTCs per field decreased during CKD progression in UUO model, whereas the number of PTCs was not significantly lower, but the number of tubules was higher (given the many atrophic tubules and only very few dilated tubules) in IRI model. In Alport mice, both the number of PTC and tubules reduced, but loss of PTC was more significant [48]. Further study is necessary to determine which method is most optimized for the assessment of PTC density in the course of CKD progression. Although a histological assessment of PTC density by immunostaining is the most popular method for animal models of CKD, it could not be used for follow-up of CKD patients because tissue sampling is invasive.
6.2. Micro-Computed Tomography (Micro-CT)
Micro-computed tomography (Micro-CT) provided the ability to create high resolution images of renal microvasculature such as arterioles (their diameter is bigger than 20 μm) [49]. As the diameter of PTC is typically 5–10 μm [22], Micro-CT does not have sufficient power to visualize the smallest size of capillaries. However, the recently developed microangio-CT method was utilized to visualize capillaries with high resolution [140]. This new method used a new polymerizing contrast agent and successfully visualized PTCs in the mouse, showing that created PTC images are comparable to immunostaining images [140]. Although microangio-CT could assess PTC density much faster than histology-based methods, further study is necessary for its general use.
6.3. Renal Resistive Index
Renal resistive index (RRI) is noninvasively measured by renal Doppler ultrasonography and is calculated with the following formula: (peak systolic velocity—end diastolic velocity)/peak systolic velocity. RRI not only reflects changes in intrarenal perfusion but is also related to systemic hemodynamics and the presence of subclinical microvascular atherosclerosis in the kidney. In 30 patients with CKD, RRI positively correlated with systolic blood pressure, interstitial fibrosis, and arteriosclerosis, and negatively correlated with PTC density and creatinine clearance (renal function) [141]. A multivariate analysis demonstrated that PTC loss emerged as one of independent variables associated with RRI elevation. As RRI could be measured repeatedly, RRI may be a feasible method to periodically assess PTC rarefaction in CKD patients.
6.4. Endothelial Micro-Particles
Endothelial micro-particles (EMPs) are extracellular vesicles that are shed by damaged (activated) ECs. In urine samples collected from patients with essential and renovascular hypertension, the levels of EMPs, which were positive for plasmalemmal-vesicle-associated protein (PL-VAP) and negative for CD31 as well as CD144 (VE-cadherin), significantly increased compared with those in urine samples from healthy subjects [142]. In the kidney, PL-VAP (PV-1) was abundantly expressed in diaphragms of endothelial fenestrae of PTCs and AVRs (Figure 3A), while it was undetectable in arterial and glomerular ECs [29,143]. Urine PL-VAP+ EMP levels, but not circulating EMPs, were inversely correlated with histologically assessed PTC density, GFR, and renal blood flow [142], suggesting that increased levels of urine EMPs reflect PTC rarefaction. However, glomerular injury promoted PL-VAP expression in glomerular ECs [144,145], and PL-VAP was abundantly expressed in ECs of other organs [29]. These issues must be cleared before its clinical use for PTC assessment.
6.5. BOLD-MRI
One of the major outcomes of PTC rarefaction is tissue hypoxia. Blood oxygen level-dependent (BOLD) magnetic resonance imaging (MRI) could be an indirect and non-invasive assessment of PTC rarefaction by evaluating renal hypoxia. BOLD-MRI used the principle that magnetic properties of hemoglobin depend on its oxygenated status. Increased local deoxyhemoglobin levels caused decreased T2* (tissue parameter and expressed in sec) or increased R2* (decay rate, defined as 1/T2* and expressed in 1/sec) [146]. CKD patients showed significantly decreased T2* value in their kidneys compared with healthy controls [147]. Decline in GFR correlated with increased R2* value of the cortical layers [148], suggesting that cortical hypoxia deteriorates renal function. BOLD-MRI does not require the administration of contrast agents (possibly nephrotoxic) and could be repeated multiple times without side effects.
7. Therapy to Mitigate PTC Rarefaction
7.1. Anti-Hypertensive Drugs
Hypertensive kidney diseases are one of the major causes of CKD. Several anti-hypertensive drugs were demonstrated to counteract PTC loss (or microvascular loss) in experimental animal models. In the rat model with age-dependent progressive kidney diseases, angiotensin-converting enzyme (ACE) inhibitor or angiotensin II receptor blocker (ARB) antagonized PTC rarefaction [149]. These drugs inhibited EC apoptosis and augmented EC proliferation, though the control of blood pressure was not reported [149]. In the rat hypertensive model with angiotensin II infusion, ACE inhibitor and ARB antagonized PTC rarefaction independent of blood pressure lowering effect [150]. Moreover, ARB preserved PTC perfusion and ameliorated tissue hypoxia in damaged kidneys [151]. In the rat uninephrectomy model with saline and mineralocorticoid infusion, spironolactone (SPL) counteracted hypertension, loss of creatinine clearance (renal function), and PTC loss [6]. SPL inhibited endothelial apoptosis and TSP-1 expression although it did not affect endothelial proliferation. In the pig model with renovascular diseases (renal artery stenosis), endothelin-A receptor (ET-A) blocker, but not ET-B blocker, ameliorated hypertension, reduction in GFR, and loss of microvasculature [152]. As the diameter of microvasculature was defined as less than 200 μm in that study, it mainly consisted of arterioles and venules instead of PTCs [152]. Consistent with this finding, long term use of ET-A blocker retarded CKD progression in patients with type 2 diabetes in the international trial, though water retention was its major and serious side effect [153]. These anti-hypertensive drugs could be effective to block PTC rarefaction during CKD progression.
7.2. Sodium Glucose Cotransporter Inhibitor for Diabetic Nephropathy
PTC rarefaction was identified in diabetic nephropathy, a leading cause of CKD [4]. In large clinical trials in patients with type 2 diabetes, inhibitors of sodium-glucose cotransporter 2 (SGLT2) significantly prevented the progression of diabetic nephropathy to ESRD [154,155]. SGLT2 inhibitor protected tubular epithelial cells from the toxicity of chronic hyperglycemia, resulting in preservation of tubular VEGF-A synthesis [156]. In addition to blood glucose lowering effect, SGLT2 inhibitor maintained tubular VEGF-A expression following IRI, antagonizing PTC rarefaction and fibrosis [156]. Interestingly SGLT2 inhibitor retarded CKD progression in patients without diabetes [157].
7.3. Tie2 Activator
While kidney injury compromised Tie2 activity by increased VE-PTP expression, chemical or genetic Tie2 activation sustained PTC integrity following injury, preserving GFR [96,158]. As mentioned above, VE-PTP inhibition maintained Tie2 activation (Tie2 phosphorylation) independent of Angpt-1 or Angpt-2. In clinical trials of patients with diabetic macular edema, subcutaneous injections of VE-PTP phosphatase inhibitor (AKB-9778) were well-tolerated for 12 weeks [159]. In some of such diabetic patients, AKB-9778 improved albuminuria, suggesting that AKB-9779 is a renal protective agent.
7.4. Nicotinamide (NAM)
Although Sirtuin-1 activators such as resveratrol have been demonstrated to successfully treat kidney injury in animal models [160], there have been no clinical trials to assess Sirtuin-1 activators for patients with kidney diseases [108]. NAD+ is required for Sirtuin-1 function. Supplementation with nicotinamide mononucleotide (NMN), an NAD+ precursor, protected mice from cisplatin-induced AKI via restored renal Sirtuin-1 activity [161]. Since AKI lowered renal NAD+ level, patients were orally administered with nicotinamide (NAM) for 3 days before and after cardiac surgery to assess whether NAM could block AKI development [162]. NAM is converted to NAD+ through the intermediate NMN. NAM treatment significantly increased circulating NAD+ level and prevented perioperative AKI compared with placebo, suggesting that increasing NAD+ level is protective against PTC loss and kidney injury by enhancing Sirtuin-1 activity.
7.5. PHD Inhibitor
PHD inhibitor robustly stabilizes HIF-1α and HIF-2α, which dimerize with HIF-1β and increase transcription of HIF target genes including EPO and VEGF-A. Since tissue hypoxia worsens PTC loss, HIF stabilizers are expected to improve PTC rarefaction by correcting renal anemia. In two clinical trials with non-dialysis and dialysis patients, PHD inhibitors (roxadustat) were tolerated well for 26 weeks and effectively improved renal anemia [163,164]. Moreover, PHD inhibitors effectively promoted erythropoiesis by lowering circulating hepcidin level in treated patients. In the study of knockout mice, HIF-2α promoted hepatic and renal EPO production, which inhibited hepatic hepcidin expression [165]. Reduced hepcidin level increases dietary iron absorption and iron release from macrophages, facilitating erythropoiesis. PHD inhibitors also increased VEGF-A level in CKD animal models, which may favor PTC preservation [166].
7.6. Nintedanib
Targeting endothelial-pericyte cross-talk may provide a novel therapeutic opportunity to prevent PTC loss and fibrosis. In the clinical setting, Richeldi et al. treated patients with idiopathic pulmonary fibrosis (IPF) for 1 year with nintedanib (formerly known as BIBF 1120), an intracellular inhibitor that can block multiple tyrosine kinase receptors such as PDGFRβ and VEGFR-2 [167]. Nintedanib significantly retarded IPF progression, suggesting a beneficial effect of the drug on tissue fibrosis. In an animal model of systemic sclerosis, nintedanib was shown to reduce capillary rarefaction and tissue fibrosis [168]. Although the most frequent adverse effect of nintedanib was diarrhea, more than 95% of patients tolerated the drug intake during the study period [167]. Nintedanib may have a potential to normalize endothelial-pericyte crosstalk and prevent pericyte detachment as well as PTC rarefaction during CKD progression.
8. Capillary Rarefaction in Other Organs
This review focused on the roles of PTC rarefaction in the progress of CKD. However, capillary rarefaction has been found to be correlated with organ dysfunction in heart [169], lung [170,171], skin [172], muscle [173], retina [174], and brain [175], suggesting that capillary rarefaction could be an important and universal component to determine future declines in the function of various organs.
9. Conclusions
AKI and CKD universally cause PTC rarefaction in the kidney. As glomerular capillaries and PTCs/DVRs are interconnected without collateral vessels, PTC loss causes the loss of GFR (the loss of kidney function) and vice versa. Recent studies have identified novel mechanisms of PTC rarefaction and indicated that PTC rarefaction is not only a prominent histological characteristic of CKD but also a central driving force of CKD progression. As we currently obtain new tools to assess PTC density and multiple drugs to mitigate PTC rarefaction in patients, PTC rarefaction would become a practical therapeutic target to halt the progression of CKD.
Author Contributions
Y.K. wrote and finalized the manuscript, created all figures. The author has read and agreed to the published version of the manuscript.
Funding
This study did not have any external funding support.
Acknowledgments
The author thanks Joseph Zullo (University of Utah) for putting valuable comments on the manuscript. The author apologizes for any omission of relevant literature in this review.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
| CKD | chronic kidney disease |
| PTC | peritubular capillary |
| ESRD | end-stage renal disease |
| DVR | descending vasa recta |
| AVR | ascending vasa recta |
| EC | endothelial cell |
| GFR | glomerular filtration ratio |
| VEGF-A | vascular endothelial growth factor-A |
| VEGFR2 | vascular endothelial growth factor receptor-2 |
| BUN | blood urea nitrogen |
| Angpt-1 | angiopoietin-1 |
| TSP-1 | thrombospondin-1 |
| TNF-α | tumor necrosis factor-α |
| IL-1β | interleukin-1β |
| UUO | unilateral ureteral obstruction |
| IRI | ischemia reperfusion injury |
| PI3K | phosphoinositide-3-kinase |
| FOXO1 | forkhead box O-1 |
| PTEN | phosphatase and tensin homolog |
| HIF | hypoxia-inducible factor |
| FoxD1 | forkhead box D-1 |
| TGF-β | transforming growth factor-β |
| EPO | erythropoietin |
| PDGF-B | platelet-derived growth factor-B |
| AKI | acute kidney injury |
| VE-PTP | vascular endothelial-protein tyrosine phosphatase |
| PHD | prolyl hydroxylase domain-containing protein |
| NAD | nicotinamide adenine dinucleotide |
| SIPS | stress-induced premature senescence |
| MMP | matrix metalloproteinase |
| VASH | vasohibin |
| PDGFRβ | platelet-derived growth factor receptor-β |
| EPC | endothelial progenitor cell |
| EndMT | endothelial to mesenchymal transition |
| RRI | renal resistive index |
| EMP | endothelial microparticles |
| PL-VAP | plasmalemmal-vesicle-associated protein |
| BOLD-MRI | blood oxygen level-dependent-magnetic resonance imaging |
| ACE | angiotensin-converting enzyme |
| ARB | angiotensin II receptor blocker |
| SPL | spironolactone |
| ET-A | endothelin-A receptor |
| SGLT2 | sodium glucose cotransporter 2 |
| NMN | nicotinamide mononucleotide |
| NAM | nicotinamide |
| IPF | idiopathic pulmonary fibrosis |
References
- Saran, R.; Robinson, B.; Abbott, K.C.; Agodoa, L.Y.C.; Bragg-Gresham, J.; Balkrishnan, R.; Bhave, N.; Dietrich, X.; Ding, Z.; Eggers, P.W.; et al. US Renal Data System 2018 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2019, 73, A7–A8. [Google Scholar] [CrossRef] [Green Version]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension 2003, 42, 1050–1065. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Lindenmeyer, M.T.; Kretzler, M.; Boucherot, A.; Berra, S.; Yasuda, Y.; Henger, A.; Eichinger, F.; Gaiser, S.; Schmid, H.; Rastaldi, M.P.; et al. Cohen, Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J. Am. Soc. Nephrol. 2007, 18, 1765–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, D.; Gordon, K.L.; Polinsky, P.; Suga, S.; Schwartz, S.M.; Johnson, R.J. Salt-sensitive hypertension develops after short-term exposure to Angiotensin II. Hypertension 1999, 33, 1013–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwazu, Y.; Muto, S.; Fujisawa, G.; Nakazawa, E.; Okada, K.; Ishibashi, S.; Kusano, E. Spironolactone suppresses peritubular capillary loss and prevents deoxycorticosterone acetate/salt-induced tubulointerstitial fibrosis. Hypertension 2008, 51, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Namikoshi, T.; Satoh, M.; Horike, H.; Fujimoto, S.; Arakawa, S.; Sasaki, T.; Kashihara, N. Implication of peritubular capillary loss and altered expression of vascular endothelial growth factor in IgA nephropathy. Nephron. Physiol. 2006, 102, 9–16. [Google Scholar] [CrossRef]
- Kaukinen, A.; Lautenschlager, I.; Helin, H.; Karikoski, R.; Jalanko, H. Peritubular capillaries are rarefied in congenital nephrotic syndrome of the Finnish type. Kidney Int. 2009, 75, 1099–1108. [Google Scholar] [CrossRef] [Green Version]
- Anutrakulchai, S.; Titipungul, T.; Pattay, T.; Mesung, P.; Puapairoj, A.; Sirivongs, D.; Pongsakul, C.; Futrakul, P.; Thinkhamrop, B.; Johnson, R.J. Relation of peritubular capillary features to class of lupus nephritis. BMC Nephrol. 2016, 17, 169. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Popov, V.; Walocha, J.A.; Wen, J.; Bello-Reuss, E. Evidence of angiogenesis and microvascular regression in autosomal-dominant polycystic kidney disease kidneys: A corrosion cast study. Kidney Int. 2006, 70, 1261–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, K.; Saravanabavan, S.; Zhang, J.Q.J.; Wong, A.T.Y.; Munt, A.; Burgess, J.S.; Rangan, G.K. Regression of Peritubular Capillaries Coincides with Angiogenesis and Renal Cyst Growth in Experimental Polycystic Kidney Disease. Int. J. Nephrol. Renovasc. Dis. 2020, 13, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Ishii, Y.; Sawada, T.; Kubota, K.; Fuchinoue, S.; Teraoka, S.; Shimizu, A. Injury and progressive loss of peritubular capillaries in the development of chronic allograft nephropathy. Kidney Int. 2005, 67, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Steegh, F.M.; Gelens, M.A.; Nieman, F.H.; van Hooff, J.P.; Cleutjens, J.P.; van Suylen, R.J.; Daemen, M.J.; van Heurn, E.L.; Christiaans, M.H.; Peutz-Kootstra, C.J. Early loss of peritubular capillaries after kidney transplantation. J. Am. Soc. Nephrol. 2011, 22, 1024–1029. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, R.; Melk, A. Molecular mechanisms of renal aging. Kidney Int. 2017, 92, 569–579. [Google Scholar] [CrossRef]
- Kida, Y.; Tchao, B.N.; Yamaguchi, I. Peritubular capillary rarefaction: A new therapeutic target in chronic kidney disease. Pediatr. Nephrol. 2014, 29, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Mayer, G. Capillary rarefaction, hypoxia, VEGF and angiogenesis in chronic renal disease: Official publication of the European Dialysis and Transplant Association—European Renal Association. Nephrol. Dial. Transplant. 2011, 26, 1132–1137. [Google Scholar] [CrossRef] [Green Version]
- Long, D.A.; Norman, J.T.; Fine, L.G. Restoring the renal microvasculature to treat chronic kidney disease, Nature reviews. Nephrology 2012, 8, 244–250. [Google Scholar]
- Chade, A.R. Renal vascular structure and rarefaction. Compr. Physiol. 2013, 3, 817–831. [Google Scholar]
- Inscho, E.W.; Ohishi, K.; Navar, L.G. Effects of ATP on pre- and postglomerular juxtamedullary microvasculature. Am. J. Physiol. 1992, 263, F886–F893. [Google Scholar] [CrossRef]
- Carmines, P.K.; Navar, L.G. Disparate effects of Ca channel blockade on afferent and efferent arteriolar responses to ANG II. Am. J. Physiol. 1989, 256, F1015–F1020. [Google Scholar] [CrossRef]
- Nyengaard, J.R. Number and dimensions of rat glomerular capillaries in normal development and after nephrectomy. Kidney Int. 1993, 43, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.K.; Steven, K. Influence of intratubular pressure on proximal tubular compliance and capillary diameter in the rat kidney. Pflug. Arch. Eur. J. Physiol. 1979, 382, 179–187. [Google Scholar] [CrossRef]
- Holliger, C.; Lemley, K.V.; Schmitt, S.L.; Thomas, F.C.; Robertson, C.R.; Jamison, R.L. Direct determination of vasa recta blood flow in the rat renal papilla. Circ. Res. 1983, 53, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.K.; Molitoris, B.A. Renal endothelial injury and microvascular dysfunction in acute kidney injury. Semin. Nephrol. 2015, 35, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Molema, G.; Aird, W.C. Vascular heterogeneity in the kidney. Semin. Nephrol. 2012, 32, 145–155. [Google Scholar] [CrossRef]
- Kenig-Kozlovsky, Y.; Scott, R.P.; Onay, T.; Carota, I.A.; Thomson, B.R.; Gil, H.J.; Ramirez, V.; Yamaguchi, S.; Tanna, C.E.; Heinen, S.; et al. Ascending Vasa Recta Are Angiopoietin/Tie2-Dependent Lymphatic-Like Vessels. J. Am. Soc. Nephrol. 2018, 29, 1097–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef] [Green Version]
- Moffat, D.B. The fine structure of the blood vessels of the renal medulla with particular reference to the control of the medullary circulation. J. Ultrastruct. Res. 1967, 19, 532–545. [Google Scholar] [CrossRef]
- Stan, R.V.; Tse, D.; Deharvengt, S.J.; Smits, N.C.; Xu, Y.; Luciano, M.R.; McGarry, C.L.; Buitendijk, M.; Nemani, K.V.; Elgueta, R.; et al. The diaphragms of fenestrated endothelia: Gatekeepers of vascular permeability and blood composition. Dev. Cell 2012, 23, 1203–1218. [Google Scholar] [CrossRef] [Green Version]
- Stolz, D.B.; Sims-Lucas, S. Unwrapping the origins and roles of the renal endothelium. Pediatr. Nephrol. 2015, 30, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Bearer, E.L.; Orci, L. Endothelial fenestral diaphragms: A quick-freeze, deep-etch study. J. Cell Biol. 1985, 100, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Arkill, K.P.; Knupp, C.; Michel, C.C.; Neal, C.R.; Qvortrup, K.; Rostgaard, J.; Squire, J.M. Similar endothelial glycocalyx structures in microvessels from a range of mammalian tissues: Evidence for a common filtering mechanism? Biophys. J. 2011, 101, 1046–1056. [Google Scholar] [CrossRef] [Green Version]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; Egbrink, M.G.O. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef] [Green Version]
- Bohle, A.; Mackensen-Haen, S.; Wehrmann, M. Significance of postglomerular capillaries in the pathogenesis of chronic renal failure. Kidney Blood Press. Res. 1996, 19, 191–195. [Google Scholar] [CrossRef]
- Choi, Y.J.; Chakraborty, S.; Nguyen, V.; Nguyen, C.; Kim, B.K.; Shim, S.I.; Suki, W.N.; Truong, L.D. Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: Altered expression of vascular endothelial growth factor. Hum. Pathol. 2000, 31, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Seron, D.; Alexopoulos, E.; Raftery, M.J.; Hartley, B.; Cameron, J.S. Number of interstitial capillary cross-sections assessed by monoclonal antibodies: Relation to interstitial damage: Official publication of the European Dialysis and Transplant Association, European Renal Association. Nephrol. Dial. Transplant. 1990, 5, 889–893. [Google Scholar] [CrossRef]
- Eardley, K.S.; Kubal, C.; Zehnder, D.; Quinkler, M.; Lepenies, J.; Savage, C.O.; Howie, A.J.; Kaur, K.; Cooper, M.S.; Adu, D.; et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008, 74, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Menshikh, A.; Scarfe, L.; Delgado, R.; Finney, C.; Zhu, Y.; Yang, H.; de Caestecker, M.P. Capillary rarefaction is more closely associated with CKD progression after cisplatin, rhabdomyolysis, and ischemia-reperfusion-induced AKI than renal fibrosis. Am. J. Physiol. Renal Physiol. 2019, 317, F1383–F1397. [Google Scholar] [CrossRef] [PubMed]
- Schlondorff, D.O. Overview of factors contributing to the pathophysiology of progressive renal disease. Kidney Int. 2008, 74, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Shimada, S.; Hirose, T.; Takahashi, C.; Sato, E.; Kinugasa, S.; Ohsaki, Y.; Kisu, K.; Sato, H.; Ito, S.; Mori, T. Pathophysiological and molecular mechanisms involved in renal congestion in a novel rat model. Sci. Rep. 2018, 8, 16808. [Google Scholar] [CrossRef]
- Matsumoto, M.; Tanaka, T.; Yamamoto, T.; Noiri, E.; Miyata, T.; Inagi, R.; Fujita, T.; Nangaku, M. Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J. Am. Soc. Nephrol. 2004, 15, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Kramann, R.; Tanaka, M.; Humphreys, B.D. Fluorescence microangiography for quantitative assessment of peritubular capillary changes after AKI in mice. J. Am. Soc. Nephrol. 2014, 25, 1924–1931. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, R.; Shimizu, A.; Masuda, Y.; Kitamura, H.; Ishizaki, M.; Sugisaki, Y.; Yamanaka, N. Peritubular capillary regression during the progression of experimental obstructive nephropathy. J. Am. Soc. Nephrol. 2002, 13, 1795–1805. [Google Scholar] [CrossRef] [Green Version]
- Loganathan, K.; Said, E.S.; Winterrowd, E.; Orebrand, M.; He, L.; Vanlandewijck, M.; Betsholtz, C.; Quaggin, S.E.; Jeansson, M. Angiopoietin-1 deficiency increases renal capillary rarefaction and tubulointerstitial fibrosis in mice. PLoS ONE 2018, 13, e0189433. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.H.; Joly, A.H.; Oh, S.W.; Hugo, C.; Kerjaschki, D.; Gordon, K.L.; Mazzali, M.; Jefferson, J.A.; Hughes, J.; Madsen, K.M.; et al. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J. Am. Soc. Nephrol. 2001, 12, 1434–1447. [Google Scholar]
- Futrakul, N.; Butthep, P.; Futrakul, P. Altered vascular homeostasis in chronic kidney disease. Clin. Hemorheol. Microcirc. 2008, 38, 201–207. [Google Scholar]
- Koller, G.M.; Schafer, C.; Kemp, S.S.; Aguera, K.N.; Lin, P.K.; Forgy, J.C.; Griffin, C.T.; Davis, G.E. Proinflammatory Mediators, IL (Interleukin)-1β, TNF (Tumor Necrosis Factor) α, and Thrombin Directly Induce Capillary Tube Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 365–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babickova, J.; Klinkhammer, B.M.; Buhl, E.M.; Djudjaj, S.; Hoss, M.; Heymann, F.; Tacke, F.; Floege, J.; Becker, J.U.; Boor, P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017, 91, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Ehling, J.; Babickova, J.; Gremse, F.; Klinkhammer, B.M.; Baetke, S.; Knuechel, R.; Kiessling, F.; Floege, J.; Lammers, T.; Boor, P. Quantitative Micro-Computed Tomography Imaging of Vascular Dysfunction in Progressive Kidney Diseases. J. Am. Soc. Nephrol. 2016, 27, 520–532. [Google Scholar] [CrossRef]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Chang, A.; Hack, B.K.; Eadon, M.T.; Alper, S.L.; Cunningham, P.N. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 2014, 85, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Wang, X.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012, 142, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Rabelink, T.J.; de Zeeuw, D. The glycocalyx—Linking albuminuria with renal and cardiovascular disease, Nature reviews. Nephrology 2015, 11, 667–676. [Google Scholar]
- Rouschop, K.M.; Roelofs, J.J.; Claessen, N.; Martins, P.d.; Zwaginga, J.J.; Pals, S.T.; Weening, J.J.; Florquin, S. Protection against renal ischemia reperfusion injury by CD44 disruption. J. Am. Soc. Nephrol. 2005, 16, 2034–2043. [Google Scholar] [CrossRef] [Green Version]
- Rouschop, K.M.; Claessen, N.; Pals, S.T.; Weening, J.J.; Florquin, S. CD44 disruption prevents degeneration of the capillary network in obstructive nephropathy via reduction of TGF-beta1-induced apoptosis. J. Am. Soc. Nephrol. 2006, 17, 746–753. [Google Scholar] [CrossRef] [Green Version]
- Weinbaum, S.; Zhang, X.; Han, Y.; Vink, H.; Cowin, S.C. Mechanotransduction and flow across the endothelial glycocalyx. Proc. Natl. Acad. Sci. USA 2003, 100, 7988–7995. [Google Scholar] [CrossRef] [Green Version]
- Padberg, J.S.; Wiesinger, A.; di Marco, G.S.; Reuter, S.; Grabner, A.; Kentrup, D.; Lukasz, A.; Oberleithner, H.; Pavenstadt, H.; Brand, M.; et al. Damage of the endothelial glycocalyx in chronic kidney disease. Atherosclerosis 2014, 234, 335–343. [Google Scholar] [CrossRef]
- Traub, O.; Berk, B.C. Laminar shear stress: Mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Friedrich, J.L.; Spahic, J.; Knipe, N.; Mang, H.; Leonard, E.C.; Changizi-Ashtiyani, S.; Bacallao, R.L.; Molitoris, B.A.; Sutton, T.A. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am. J. Physiol. Renal Physiol. 2011, 300, F721–F733. [Google Scholar] [CrossRef] [Green Version]
- Kida, Y.; Ieronimakis, N.; Schrimpf, C.; Reyes, M.; Duffield, J.S. EphrinB2 reverse signaling protects against capillary rarefaction and fibrosis after kidney injury. J. Am. Soc. Nephrol. 2013, 24, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Zeng, P.; Friedrich, J.L.; Leonard, E.C.; Yoder, M.C. Low proliferative potential and impaired angiogenesis of cultured rat kidney endothelial cells. Microcirculation 2012, 19, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, L.T.H.; Aburatani, T.; Marsh, G.A.; Johnson, B.G.; Alimperti, S.; Yoon, C.J.; Huang, A.; Szak, S.; Nakagawa, N.; Gomez, I.; et al. Hyperactive FOXO1 results in lack of tip stalk identity and deficient microvascular regeneration during kidney injury. Biomaterials 2017, 141, 314–329. [Google Scholar] [CrossRef]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Horbelt, M.; Lee, S.Y.; Mang, H.E.; Knipe, N.L.; Sado, Y.; Kribben, A.; Sutton, T.A. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am. J. Physiol. Renal Physiol. 2007, 293, F688–F695. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Lan, S.; Dieude, M.; Sabo-Vatasescu, J.P.; Karakeussian-Rimbaud, A.; Turgeon, J.; Qi, S.; Gunaratnam, L.; Patey, N.; Hebert, M.J. Caspase-3 Is a Pivotal Regulator of Microvascular Rarefaction and Renal Fibrosis after Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2018, 29, 1900–1916. [Google Scholar] [CrossRef] [Green Version]
- Thakar, C.V.; Zahedi, K.; Revelo, M.P.; Wang, Z.; Burnham, C.E.; Barone, S.; Bevans, S.; Lentsch, A.B.; Rabb, H.; Soleimani, M. Identification of thrombospondin 1 (TSP-1) as a novel mediator of cell injury in kidney ischemia. J. Clin. Investig. 2005, 115, 3451–3459. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef]
- Dhanabal, M.; Ramchandran, R.; Waterman, M.J.; Lu, H.; Knebelmann, B.; Segal, M.; Sukhatme, V.P. Endostatin induces endothelial cell apoptosis. J. Biol. Chem. 1999, 274, 11721–11726. [Google Scholar] [CrossRef] [Green Version]
- Tsuneki, M.; Madri, J.A. CD44 regulation of endothelial cell proliferation and apoptosis via modulation of CD31 and VE-cadherin expression. J. Biol. Chem. 2014, 289, 5357–5370. [Google Scholar] [CrossRef] [Green Version]
- Kida, Y.; Duffield, J.S. Pivotal role of pericytes in kidney fibrosis. Clin. Exp. Pharmacol. Physiol. 2011, 38, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souma, T.; Nezu, M.; Nakano, D.; Yamazaki, S.; Hirano, I.; Sekine, H.; Dan, T.; Takeda, K.; Fong, G.H.; Nishiyama, A.; et al. Erythropoietin Synthesis in Renal Myofibroblasts Is Restored by Activation of Hypoxia Signaling. J. Am. Soc. Nephrol. 2016, 27, 428–438. [Google Scholar] [CrossRef]
- Lin, S.L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008, 173, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.H.; Chang, Y.; Reed, N.I.; Sheppard, D. α-Smooth muscle actin is an inconsistent marker of fibroblasts responsible for force-dependent TGFβ activation or collagen production across multiple models of organ fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L824–L836. [Google Scholar] [CrossRef] [Green Version]
- Schrimpf, C.; Xin, C.; Campanholle, G.; Gill, S.E.; Stallcup, W.; Lin, S.L.; Davis, G.E.; Gharib, S.A.; Humphreys, B.D.; Duffield, J.S. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J. Am. Soc. Nephrol. 2012, 23, 868–883. [Google Scholar] [CrossRef] [Green Version]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Kramann, R.; Wongboonsin, J.; Chang-Panesso, M.; Machado, F.G.; Humphreys, B.D. Gli1(+) Pericyte Loss Induces Capillary Rarefaction and Proximal Tubular Injury. J. Am. Soc. Nephrol. 2017, 28, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Lemos, D.R.; Marsh, G.; Huang, A.; Campanholle, G.; Aburatani, T.; Dang, L.; Gomez, I.; Fisher, K.; Ligresti, G.; Peti-Peterdi, J.; et al. Maintenance of vascular integrity by pericytes is essential for normal kidney function. Am. J. Physiol. Renal Physiol. 2016, 311, F1230–F1242. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Mugford, J.W.; Krautzberger, A.M.; Naiman, N.; Liao, J.; McMahon, A.P. Identification of a multipotent self-renewing stromal progenitor population during mammalian kidney organogenesis. Stem Cell Rep. 2014, 3, 650–662. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.F.; Chiang, W.C.; Lai, C.F.; Chang, F.C.; Chen, Y.T.; Chou, Y.H.; Wu, T.H.; Linn, G.R.; Ling, H.; Wu, K.D.; et al. Transforming growth factor beta-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am. J. Pathol. 2013, 182, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Dimke, H.; Sparks, M.A.; Thomson, B.R.; Frische, S.; Coffman, T.M.; Quaggin, S.E. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J. Am. Soc. Nephrol. 2015, 26, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.T.; Li, X.Z.; Pitera, J.E.; Long, D.A.; Woolf, A.S. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am. J. Pathol. 2003, 163, 2289–2301. [Google Scholar] [CrossRef]
- Hakroush, S.; Moeller, M.J.; Theilig, F.; Kaissling, B.; Sijmonsma, T.P.; Jugold, M.; Akeson, A.L.; Traykova-Brauch, M.; Hosser, H.; Hahnel, B.; et al. Effects of increased renal tubular vascular endothelial growth factor (VEGF) on fibrosis, cyst formation, and glomerular disease. Am. J. Pathol. 2009, 175, 1883–1895. [Google Scholar] [CrossRef] [Green Version]
- Di Marco, G.S.; Reuter, S.; Hillebrand, U.; Amler, S.; Konig, M.; Larger, E.; Oberleithner, H.; Brand, E.; Pavenstadt, H.; Brand, M. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2009, 20, 2235–2245. [Google Scholar] [CrossRef] [Green Version]
- Zsengeller, Z.K.; Lo, A.; Tavasoli, M.; Pernicone, E.; Karumanchi, S.A.; Rosen, S. Soluble fms-Like Tyrosine Kinase 1 Localization in Renal Biopsies of CKD. Kidney Int. Rep. 2019, 4, 1735–1741. [Google Scholar] [CrossRef] [Green Version]
- Mansour, S.G.; Zhang, W.R.; Moledina, D.G.; Coca, S.G.; Jia, Y.; Thiessen-Philbrook, H.; McArthur, E.; Inoue, K.; Koyner, J.L.; Shlipak, M.G.; et al. The Association of Angiogenesis Markers with Acute Kidney Injury and Mortality After Cardiac Surgery. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2019, 74, 36–46. [Google Scholar] [CrossRef]
- Chen, J.; Hamm, L.L.; Kleinpeter, M.A.; Husserl, F.; Khan, I.E.; Chen, C.S.; Liu, Y.; Mills, K.T.; He, C.; Rifai, N.; et al. Elevated plasma levels of endostatin are associated with chronic kidney disease. Am. J. Nephrol. 2012, 35, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Hwang, S.; Pyun, B.J.; Kim, T.Y.; Lee, S.T.; Gho, Y.S.; Kwon, Y.G. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J. Biol. Chem. 2002, 277, 27872–27879. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Chen, J.; Zhang, Z.; Johnson, G.V.; Cooper, A.J.; Feola, J.; Bank, A.; Shein, J.; Ruotsalainen, H.J.; Pihlajaniemi, T.A.; et al. Endostatin and transglutaminase 2 are involved in fibrosis of the aging kidney. Kidney Int. 2016, 89, 1281–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenstein, B.; Daniel, C.; Hausknecht, B.; Boehmer, K.; Riess, R.; Amann, K.U.; Hugo, C.P. Correlation of enhanced thrombospondin-1 expression, TGF-beta signaling and proteinuria in human type-2 diabetic nephropathy: Official publication of the European Dialysis and Transplant Association—European Renal Association. Nephrol. Dial. Transplant. 2008, 23, 3880–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.; Gupta, P.; Wild, R.; Ramakrishnan, S.; Hebbel, R.P. Binding and displacement of vascular endothelial growth factor (VEGF) by thrombospondin: Effect on human microvascular endothelial cell proliferation and angiogenesis. Angiogenesis 1999, 3, 147–158. [Google Scholar] [CrossRef]
- Kaur, S.; Martin-Manso, G.; Pendrak, M.L.; Garfield, S.H.; Isenberg, J.S.; Roberts, D.D. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J. Biol. Chem. 2010, 285, 38923–38932. [Google Scholar] [CrossRef] [Green Version]
- Bige, N.; Shweke, N.; Benhassine, S.; Jouanneau, C.; Vandermeersch, S.; Dussaule, J.C.; Chatziantoniou, C.; Ronco, P.; Boffa, J.J. Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction. Kidney Int. 2012, 81, 1226–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puranik, A.S.; Leaf, I.A.; Jensen, M.A.; Hedayat, A.F.; Saad, A.; Kim, K.W.; Saadalla, A.M.; Woollard, J.R.; Kashyap, S.; Textor, S.C.; et al. Kidney-resident macrophages promote a proangiogenic environment in the normal and chronically ischemic mouse kidney. Sci. Rep. 2018, 8, 13948. [Google Scholar] [CrossRef] [Green Version]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- David, S.; John, S.G.; Jefferies, H.J.; Sigrist, M.K.; Kumpers, P.; Kielstein, J.T.; Haller, H.; McIntyre, C.W. Angiopoietin-2 levels predict mortality in CKD patients: Official publication of the European Dialysis and Transplant Association—European Renal Association. Nephrol. Dial. Transplant. 2012, 27, 1867–1872. [Google Scholar] [CrossRef]
- Carota, I.A.; Kenig-Kozlovsky, Y.; Onay, T.; Scott, R.; Thomson, B.R.; Souma, T.; Bartlett, C.S.; Li, Y.; Procissi, D.; Ramirez, V.; et al. Targeting VE-PTP phosphatase protects the kidney from diabetic injury. J. Exp. Med. 2019, 216, 936–949. [Google Scholar] [CrossRef] [Green Version]
- Korhonen, E.A.; Lampinen, A.; Giri, H.; Anisimov, A.; Kim, M.; Allen, B.; Fang, S.; D’Amico, G.; Sipilä, T.J.; Lohela, M.; et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J. Clin. Investig. 2016, 126, 3495–3510. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Allen, B.; Korhonen, E.A.; Nitschké, M.; Yang, H.W.; Baluk, P.; Saharinen, P.; Alitalo, K.; Daly, C.; Thurston, G.; et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J. Clin. Investig. 2016, 126, 3511–3525. [Google Scholar] [CrossRef]
- Savant, S.; La Porta, S.; Budnik, A.; Busch, K.; Hu, J.; Tisch, N.; Korn, C.; Valls, A.F.; Benest, A.V.; Terhardt, D.; et al. The Orphan Receptor Tie1 Controls Angiogenesis and Vascular Remodeling by Differentially Regulating Tie2 in Tip and Stalk Cells. Cell Rep. 2015, 12, 1761–1773. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, T.; Horstkotte, J.; Schwab, C.; Pfetsch, V.; Weinmann, K.; Dietzel, S.; Rohwedder, I.; Hinkel, R.; Gross, L.; Lee, S.; et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J. Clin. Investig. 2013, 123, 3436–3445. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Luque, Y.; Lenoir, O.; Bonnin, P.; Hardy, L.; Chipont, A.; Placier, S.; Vandermeersch, S.; Xu-Dubois, Y.-C.; Robin, B.; Lazareth, H.; et al. Endothelial Epas1 Deficiency Is Sufficient To Promote Parietal Epithelial Cell Activation and FSGS in Experimental Hypertension. J. Am. Soc. Nephrol. 2017, 28, 3563–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalucka, J.; Schley, G.; Georgescu, A.; Klanke, B.; Rössler, S.; Baumgärtl, J.; Velden, J.; Amann, K.; Willam, C.; Johnson, R.S.; et al. Kidney injury is independent of endothelial HIF-1alpha. J. Mol. Med. 2015, 93, 891–904. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.-Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Investig. 2014, 124, 2396–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, I.; Tanaka, T.; Inagi, R.; Kato, H.; Yamashita, T.; Sakiyama, A.; Ohneda, O.; Takeda, N.; Sata, M.; Miyata, T.; et al. Protective Role of Hypoxia-Inducible Factor-2α against Ischemic Damage and Oxidative Stress in the Kidney. J. Am. Soc. Nephrol. 2007, 18, 1218–1226. [Google Scholar] [CrossRef]
- Paliege, A.; Rosenberger, C.; Bondke, A.; Sciesielski, L.; Shina, A.; Heyman, S.N.; Flippin, L.A.; Arend, M.P.; Klaus, S.J.; Bachmann, S. Hypoxia-inducible factor-2α-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int. 2010, 77, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Liu, Q.; Binns, T.C.; Urrutia, A.A.; Davidoff, O.; Kapitsinou, P.P.; Pfaff, A.S.; Olauson, H.; Wernerson, A.; Fogo, A.B.; et al. Distinct subpopulations of FOXD1 stroma-derived cells regulate renal erythropoietin. J. Clin. Investig. 2016, 126, 1926–1938. [Google Scholar] [CrossRef] [Green Version]
- Kida, Y.; Goligorsky, M.S. Sirtuins, Cell Senescence, and Vascular Aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Potente, M.; Ghaeni, L.; Baldessari, D.; Mostoslavsky, R.; Rossig, L.; Dequiedt, F.; Haendeler, J.; Mione, M.; Dejana, E.; Alt, F.W.; et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007, 21, 2644–2658. [Google Scholar] [CrossRef] [Green Version]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell. Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef]
- Vasko, R.; Xavier, S.; Chen, J.; Lin, C.H.S.; Ratliff, B.; Rabadi, M.; Maizel, J.; Tanokuchi, R.; Zhang, F.; Cao, J.; et al. Endothelial Sirtuin 1 Deficiency Perpetrates Nephrosclerosis through Downregulation of Matrix Metalloproteinase-14: Relevance to Fibrosis of Vascular Senescence. J. Am. Soc. Nephrol. 2013, 25, 276–291. [Google Scholar] [CrossRef] [Green Version]
- Kida, Y.; Zullo, J.A.; Goligorsky, M.S. Endothelial sirtuin 1 inactivation enhances capillary rarefaction and fibrosis following kidney injury through Notch activation. Biochem. Biophys. Res. Commun. 2016, 478, 1074–1079. [Google Scholar] [CrossRef] [Green Version]
- Serra, H.; Chivite, I.; Angulo-Urarte, A.; Soler, A.; Sutherland, J.D.; Arruabarrena-Aristorena, A.; Ragab, A.; Lim, R.; Malumbres, M.; Fruttiger, M.; et al. PTEN mediates Notch-dependent stalk cell arrest in angiogenesis. Nat. Commun. 2015, 6, 7935. [Google Scholar] [CrossRef] [Green Version]
- Lipphardt, M.; Song, J.W.; Ratliff, B.B.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Endothelial dysfunction is a superinducer of syndecan-4: Fibrogenic role of its ectodomain. Am. J. Physiol. Circ. Physiol. 2018, 314, H484–H496. [Google Scholar] [CrossRef]
- Cabrales, P.; Vázquez, B.Y.S.; Tsai, A.G.; Intaglietta, M. Microvascular and capillary perfusion following glycocalyx degradation. J. Appl. Physiol. 2007, 102, 2251–2259. [Google Scholar] [CrossRef] [Green Version]
- Machin, D.R.; Bloom, S.I.; Campbell, R.A.; Phuong, T.T.T.; Gates, P.E.; Lesniewski, L.A.; Rondina, M.T.; Donato, A.J. Advanced age results in a diminished endothelial glycocalyx. Am. J. Physiol. Circ. Physiol. 2018, 315, H531–H539. [Google Scholar] [CrossRef]
- Weissgerber, T.L.; Garcia-Valencia, O.; Milic, N.M.; Codsi, E.; Cubro, H.; Nath, M.C.; White, W.M.; Nath, K.A.; Garovic, V.D. Early Onset Preeclampsia Is Associated With Glycocalyx Degradation and Reduced Microvascular Perfusion. J. Am. Hear. Assoc. 2019, 8, e010647. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Hasegawa, Y.; Yamashita, H.; Shimizu, K.; Ding, Y.; Abe, M.; Ohta, H.; Imagawa, K.; Hojo, K.; Maki, H.; et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J. Clin. Investig. 2004, 114, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, T. Isolation and Characterization of Vasohibin-2 as a Homologue of VEGF-Inducible Endothelium-Derived Angiogenesis Inhibitor Vasohibin. Arter. Thromb. Vasc. Biol. 2006, 26, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- Hinamoto, N.; Maeshima, Y.; Saito, D.; Yamasaki, H.; Tanabe, K.; Nasu, T.; Watatani, H.; Ujike, H.; Kinomura, M.; Sugiyama, H.; et al. Renal distribution of Vasohibin-1 in patients with chronic kidney disease. Acta Med. Okayama 2014, 68, 219–233. [Google Scholar]
- Miyashita, H.; Watanabe, T.; Hayashi, H.; Suzuki, Y.; Nakamura, T.; Ito, S.; Ono, M.; Hoshikawa, Y.; Okada, Y.; Kondo, T.; et al. Angiogenesis Inhibitor Vasohibin-1 Enhances Stress Resistance of Endothelial Cells via Induction of SOD2 and SIRT1. PLoS ONE 2012, 7, e46459. [Google Scholar] [CrossRef] [Green Version]
- Tanimura, S.; Tanabe, K.; Miyake, H.; Masuda, K.; Tsushida, K.; Morioka, T.; Sugiyama, H.; Sato, Y.; Wada, J. Renal tubular injury exacerbated by vasohibin-1 deficiency in a murine cisplatin-induced acute kidney injury model. Am. J. Physiol. Physiol. 2019, 317, F264–F274. [Google Scholar] [CrossRef] [PubMed]
- Arata, Y.; Tanabe, K.; Hinamoto, N.; Yamasaki, H.; Sugiyama, H.; Maeshima, Y.; Kanomata, N.; Sato, Y.; Wada, J. Immunohistochemistry of Vasohibin-2 in Human Kidney Disease: Implications in Impaired Glucose Tolerance and Reduced Renal Function. Acta Med. Okayama 2017, 71, 369–380. [Google Scholar]
- Miyake, H.; Tanabe, K.; Tanimura, S.; Nakashima, Y.; Morioka, T.; Masuda, K.; Sugiyama, H.; Sato, Y.; Wada, J. Genetic Deletion of Vasohibin-2 Exacerbates Ischemia-Reperfusion-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2020, 21, 4545. [Google Scholar] [CrossRef]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/Pericyte Interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-L.; Chang, F.-C.; Schrimpf, C.; Chen, Y.-T.; Wu, C.-F.; Wu, V.-C.; Chiang, W.-C.; Kuhnert, F.; Kuo, C.J.; Chen, Y.-M.; et al. Targeting Endothelium-Pericyte Cross Talk by Inhibiting VEGF Receptor Signaling Attenuates Kidney Microvascular Rarefaction and Fibrosis. Am. J. Pathol. 2011, 178, 911–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-T.; Yang, C.-C.; Pan, S.-Y.; Chou, Y.-H.; Chang, F.-C.; Lai, C.-F.; Tsai, M.-H.; Hsu, H.-L.; Lin, C.-H.; Chiang, W.-C.; et al. DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. J. Clin. Investig. 2016, 126, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, N.; Takase, M.; Nakamura, J.; Oguchi, A.; Asada, M.; Suzuki, N.; Yamamura, K.-I.; Nagoshi, N.; Shibata, S.; Rao, T.N.; et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Investig. 2011, 121, 3981–3990. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Flores, B.; Li, P.; Gillings, N.; McMillan, K.L.; Ye, J.; Huang, L.J.-S.; Sidhu, S.S.; Zhong, Y.-P.; Grompe, M.T.; et al. Effects of erythropoietin receptor activity on angiogenesis, tubular injury, and fibrosis in acute kidney injury: A “U-shaped” relationship. Am. J. Physiol. Physiol. 2018, 314, F501–F516. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; Van Der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of Putative Progenitor Endothelial Cells for Angiogenesis. Science 1997, 275, 964–966. [Google Scholar] [CrossRef]
- Li, B.; Cohen, A.; Hudson, T.E.; Motlagh, D.; Amrani, D.L.; Duffield, J.S. Mobilized human hematopoietic stem/progenitor cells promote kidney repair after ischemia/reperfusion injury. Circulation 2010, 121, 2211–2220. [Google Scholar] [CrossRef] [Green Version]
- Schirutschke, H.; Vogelbacher, R.; Stief, A.; Parmentier, S.; Daniel, C.; Hugo, C. Injured kidney endothelium is only marginally repopulated by cells of extrarenal origin. Am. J. Physiol. Physiol. 2013, 305, F1042–F1052. [Google Scholar] [CrossRef] [Green Version]
- Sradnick, J.; Rong, S.; Luedemann, A.; Parmentier, S.P.; Bartaun, C.; Todorov, V.T.; Gueler, F.; Hugo, C.P.; Hohenstein, B. Extrarenal Progenitor Cells Do Not Contribute to Renal Endothelial Repair. J. Am. Soc. Nephrol. 2015, 27, 1714–1726. [Google Scholar] [CrossRef] [Green Version]
- Xiuzhen, H.; Huang, X.; Kanisicak, O.; Li, Y.; Wang, Y.; Li, Y.; Pu, W.; Liu, Q.; Zhang, H.; Tian, X.; et al. Preexisting endothelial cells mediate cardiac neovascularization after injury. J. Clin. Investig. 2017, 127, 2968–2981. [Google Scholar]
- Yang, J.; Wang, M.; Zhu, F.; Sun, J.; Xu, H.; Shin, O.L.-S.C.L.; Zhao, Z.; Pei, G.; Zhu, H.; Cao, C.; et al. Putative endothelial progenitor cells do not promote vascular repair but attenuate pericyte-myofibroblast transition in UUO-induced renal fibrosis. Stem Cell Res. Ther. 2019, 10, 104. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in Kidney Fibrosis Emerge via Endothelial-to-Mesenchymal Transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Taduri, G.; Oconnell, J.T.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Zovein, A.C.; Hofmann, J.J.; Lynch, M.; French, W.J.; Turlo, K.A.; Yang, Y.; Becker, M.S.; Zanetta, L.; Dejana, E.; Gasson, J.C.; et al. Fate Tracing Reveals the Endothelial Origin of Hematopoietic Stem Cells. Cell Stem Cell 2008, 3, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- Hlushchuk, R.; Zubler, C.; Barré, S.; Shokiche, C.C.; Schaad, L.; Röthlisberger, R.; Wnuk, M.L.; Daniel, C.; Khoma, O.; Tschanz, S.A.; et al. Cutting-edge microangio-CT: New dimensions in vascular imaging and kidney morphometry. Am. J. Physiol. Physiol. 2018, 314, F493–F499. [Google Scholar] [CrossRef]
- Kimura, N.; Kimura, H.; Takahashi, N.; Hamada, T.; Maegawa, H.; Mori, M.; Imamura, Y.; Kusaka, Y.; Yoshida, H.; Iwano, M. Renal resistive index correlates with peritubular capillary loss and arteriosclerosis in biopsy tissues from patients with chronic kidney disease. Clin. Exp. Nephrol. 2015, 19, 1114–1119. [Google Scholar] [CrossRef]
- Sun, I.O.; Santelli, A.; Abumoawad, A.; Eirin, A.; Ferguson, C.; Woollard, J.R.; Lerman, L.O.; Textor, S.C.; Puranik, A.S.; Lerman, L.O. Loss of Renal Peritubular Capillaries in Hypertensive Patients is Detectable by Urinary Endothelial Microparticle Levels. Hypertension 2018, 72, 1180–1188. [Google Scholar] [CrossRef]
- Herrnberger, L.; Seitz, R.; Kuespert, S.; Bösl, M.R.; Fuchshofer, R.; Tamm, E.R. Lack of endothelial diaphragms in fenestrae and caveolae of mutant Plvap-deficient mice. Histochem. Cell Biol. 2012, 138, 709–724. [Google Scholar] [CrossRef]
- Kuppe, C.; Rohlfs, W.; Grepl, M.; Schulte, K.; Verón, D.; Elger, M.; Sanden, S.K.; Saritas, T.; Andrae, J.; Betsholtz, C.; et al. Inverse correlation between vascular endothelial growth factor back-filtration and capillary filtration pressures: Official publication of the European Dialysis and Transplant Association—European Renal Association. Nephrol. Dial. Transpl. 2018, 33, 1514–1525. [Google Scholar]
- Ichimura, K.; Stan, R.V.; Kurihara, H.; Sakai, T. Glomerular endothelial cells form diaphragms during development and pathologic conditions. J. Am. Soc. Nephrol. 2008, 19, 1463–1471. [Google Scholar] [CrossRef] [Green Version]
- Pruijm, M.; Milani, B.; Burnier, M. Blood Oxygenation Level-Dependent MRI to Assess Renal Oxygenation in Renal Diseases: Progresses and Challenges. Front. Physiol. 2017, 7, 539. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Kozawa, E.; Okada, H.; Inukai, K.; Watanabe, S.; Kikuta, T.; Watanabe, Y.; Takenaka, T.; Katayama, S.; Tanaka, J.; et al. Noninvasive evaluation of kidney hypoxia and fibrosis using magnetic resonance imaging. J. Am. Soc. Nephrol. 2011, 22, 1429–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruijm, M.; Milani, B.; Pivin, E.; Podhajska, A.; Vogt, B.; Stuber, M.; Burnier, M. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int. 2018, 93, 932–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remuzzi, A.; Sangalli, F.; Macconi, D.; Tomasoni, S.; Cattaneo, I.; Rizzo, P.; Bonandrini, B.; Bresciani, E.; Longaretti, L.; Gagliardini, E.; et al. Regression of Renal Disease by Angiotensin II Antagonism Is Caused by Regeneration of Kidney Vasculature. J. Am. Soc. Nephrol. 2015, 27, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Kitayama, H.; Maeshima, Y.; Takazawa, Y.; Yamamoto, Y.; Wu, Y.; Ichinose, K.; Hirokoshi, K.; Sugiyama, H.; Yamasaki, Y.; Makino, H. Regulation of Angiogenic Factors in Angiotensin II Infusion Model in Association With Tubulointerstitial Injuries. Am. J. Hypertens. 2006, 19, 718–727. [Google Scholar] [CrossRef] [Green Version]
- Manotham, K.; Tanaka, T.; Matsumoto, M.; Ohse, T.; Miyata, T.; Inagi, R.; Kurokawa, K.; Fujita, T.; Nangaku, M. Evidence of Tubular Hypoxia in the Early Phase in the Remnant Kidney Model. J. Am. Soc. Nephrol. 2004, 15, 1277–1288. [Google Scholar] [CrossRef]
- Chade, A.R.; Stewart, N.J.; Peavy, P.R. Disparate effects of single endothelin-A and -B receptor blocker therapy on the progression of renal injury in advanced renovascular disease. Kidney Int. 2014, 85, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; Von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nakano, D.; Guan, Y.; Hitomi, H.; Uemura, A.; Masaki, T.; Kobara, H.; Sugaya, T.; Nishiyama, A. A sodium-glucose cotransporter 2 inhibitor attenuates renal capillary injury and fibrosis by a vascular endothelial growth factor–dependent pathway after renal injury in mice. Kidney Int. 2018, 94, 524–535. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Rübig, E.; Stypmann, J.; Van Slyke, P.; Dumont, D.J.; Spieker, T.; Buscher, K.; Reuter, S.; Goerge, T.; Pavenstädt, H.; Kümpers, P. The Synthetic Tie2 Agonist Peptide Vasculotide Protects Renal Vascular Barrier Function in Experimental Acute Kidney Injury. Sci. Rep. 2016, 6, 22111. [Google Scholar] [CrossRef] [Green Version]
- Campochiaro, P.A.; Khanani, A.; Singer, M.; Patel, S.; Boyer, D.; Dugel, P.; Kherani, S.; Withers, B.; Gambino, L.; Peters, K.; et al. Enhanced Benefit in Diabetic Macular Edema from AKB-9778 Tie2 Activation Combined with Vascular Endothelial Growth Factor Suppression. Ophthalmology 2016, 123, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wang, Y.; Zhang, M.-Z.; You, L.; Davis, L.S.; Fan, H.; Yang, H.-C.; Fogo, A.B.; Zent, R.; Harris, R.C.; et al. Sirt1 activation protects the mouse renal medulla from oxidative injury. J. Clin. Investig. 2010, 120, 1056–1068. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehr, A.P.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Liu, B.-C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef]
- Liu, Q.; Davidoff, O.; Niss, K.; Haase, V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Investig. 2012, 122, 4635–4644. [Google Scholar] [CrossRef] [Green Version]
- Schellinger, I.N.; Cordasic, N.; Panesar, J.; Buchholz, B.; Jacobi, J.; Hartner, A.; Klanke, B.; Jakubiczka-Smorag, J.; Burzlaff, N.; Heinze, E.; et al. Hypoxia inducible factor stabilization improves defective ischemia-induced angiogenesis in a rodent model of chronic kidney disease. Kidney Int. 2017, 91, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Maier, C.; Zhang, Y.; Soare, A.; Dees, C.; Beyer, C.; Harre, U.; Chen, C.-W.; Distler, O.; Schett, G.; et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary Microvascular Rarefaction and Myocardial Fibrosis in Heart Failure with Preserved Ejection Fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Ebina, M.; Shimizukawa, M.; Shibata, N.; Kimura, Y.; Suzuki, T.; Endo, M.; Sasano, H.; Kondo, T.; Nukiwa, T. Heterogeneous Increase in CD34-positive Alveolar Capillaries in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2004, 169, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Farkas, L.; Farkas, D.; Ask, K.; Möller, A.; Gauldie, J.; Margetts, P.; Inman, M.; Kolb, M. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J. Clin. Investig. 2009, 119, 1298–1311. [Google Scholar] [CrossRef] [Green Version]
- Koenig, M.; Joyal, F.; Fritzler, M.J.; Roussin, A.; Abrahamowicz, M.; Boire, G.; Goulet, J.-R.; Rich, É.; Grodzicky, T.; Raymond, Y.; et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: A twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum. 2008, 58, 3902–3912. [Google Scholar] [CrossRef] [PubMed]
- Borisov, A.B.; Huang, S.-K.; Carlson, B.M. Remodeling of the vascular bed and progressive loss of capillaries in denervated skeletal muscle. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2000, 258, 292–304. [Google Scholar] [CrossRef] [Green Version]
- Grant, Z.L.; Whitehead, L.; Wong, V.H.; He, Z.; Yan, R.Y.; Miles, A.R.; Benest, A.V.; Bates, D.O.; Prahst, C.; Bentley, K.; et al. Blocking endothelial apoptosis revascularizes the retina in a model of ischemic retinopathy. J. Clin. Invest. 2020, 130, 4235–4251. [Google Scholar] [CrossRef]
- Warrington, J.P.; Csiszar, A.; Johnson, D.A.; Herman, T.S.; Ahmad, S.; Lee, Y.W.; Sonntag, W.E. Cerebral microvascular rarefaction induced by whole brain radiation is reversible by systemic hypoxia in mice. Am. J. Physiol. Circ. Physiol. 2010, 300, H736–H744. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Peritubular capillary rarefaction in the kidney cortex. (A) Peritubular capillaries surround tubules in the normal kidneys. (B) Peritubular capillaries rarefy after kidney injury, leading to capillary regression. Peritubular capillary endothelial cells are visualized with immunofluorescence staining for CD31 (red). Note that many of peritubular capillaries lose their lumina in the injured kidney whereas the capillary lumen is clearly maintained in the normal kidney. Kidney injury is induced by unilateral ureteral obstruction (UUO) in the mouse. T indicates a tubular epithelial compartment. Blue represents nuclei. Scale bar, 50 μm.
Figure 1.
Peritubular capillary rarefaction in the kidney cortex. (A) Peritubular capillaries surround tubules in the normal kidneys. (B) Peritubular capillaries rarefy after kidney injury, leading to capillary regression. Peritubular capillary endothelial cells are visualized with immunofluorescence staining for CD31 (red). Note that many of peritubular capillaries lose their lumina in the injured kidney whereas the capillary lumen is clearly maintained in the normal kidney. Kidney injury is induced by unilateral ureteral obstruction (UUO) in the mouse. T indicates a tubular epithelial compartment. Blue represents nuclei. Scale bar, 50 μm.

Figure 2.
Peritubular microvasculature in the kidney. Arterial blood is delivered to glomerular capillaries in cortical nephrons (90% of total nephrons) and juxtamedullary nephrons (10% of total nephrons) via arcuate artery (labeled as 1), interlobular artery (labeled as 2), and afferent arteriole (labeled as 3). Afferent arterioles occasionally create a periglomerular shunt (indicated with an arrow) but are mostly connected with capillaries in glomeruli without shunting. Efferent arterioles (labeled as 4) in juxtamedullary nephrons construct vascular bundle (labeled as 5) to supply arterial blood for interbundle dense capillary plexus (PTC) and DVR. Venous blood returns to AVR and vascular bundle (labeled as 6) via interbundle dense capillary plexus or sparse capillary plexus. In cortical nephrons, efferent arterioles construct PTC without branches of vasa recta.
Figure 2.
Peritubular microvasculature in the kidney. Arterial blood is delivered to glomerular capillaries in cortical nephrons (90% of total nephrons) and juxtamedullary nephrons (10% of total nephrons) via arcuate artery (labeled as 1), interlobular artery (labeled as 2), and afferent arteriole (labeled as 3). Afferent arterioles occasionally create a periglomerular shunt (indicated with an arrow) but are mostly connected with capillaries in glomeruli without shunting. Efferent arterioles (labeled as 4) in juxtamedullary nephrons construct vascular bundle (labeled as 5) to supply arterial blood for interbundle dense capillary plexus (PTC) and DVR. Venous blood returns to AVR and vascular bundle (labeled as 6) via interbundle dense capillary plexus or sparse capillary plexus. In cortical nephrons, efferent arterioles construct PTC without branches of vasa recta.

Figure 3.
Endothelial fenestrae and glycocalyx. (A) Endothelium of PTC including AVR owns fenestrae with 60–80 nm diameters. Each fenestration pore is spanned with multiple units of diaphragm fibrils composed of PLVAP (plasmalemmal-vesicle-associated protein) dimers and glycosaminoglycans. (B) In early response to injury, endothelial fenestrae disappear. Endothelial cells (ECs) are subsequently thickening. (C) The luminal surface of ECs is coated with glycocalyx consisting of proteoglycans (membrane-bound proteins such as syndecans and glypicans); glycosaminoglycans (long, linear polysaccharides that impart a strong negative charge, including heparan sulfate, chondroitin sulfate, and hyaluronan or hyaluronic acid); and glycoproteins (cell surface receptors such as selectins, integrins, intercellular adhesion molecule [ICAM], and vascular cell adhesion molecule [VCAM]). Injury promotes glycocalyx shedding, which exposes glycoproteins to blood stream, resulting in inflammatory cell adhesion, platelet aggregation, and following coagulation. CD44, one of principle glycoproteins in glycocalyx, anchors hyaluronan chains and stripping of glycocalyx facilitates CD44-leukocyte interaction and leukocyte extravasation. CKD: chronic kidney disease.
Figure 3.
Endothelial fenestrae and glycocalyx. (A) Endothelium of PTC including AVR owns fenestrae with 60–80 nm diameters. Each fenestration pore is spanned with multiple units of diaphragm fibrils composed of PLVAP (plasmalemmal-vesicle-associated protein) dimers and glycosaminoglycans. (B) In early response to injury, endothelial fenestrae disappear. Endothelial cells (ECs) are subsequently thickening. (C) The luminal surface of ECs is coated with glycocalyx consisting of proteoglycans (membrane-bound proteins such as syndecans and glypicans); glycosaminoglycans (long, linear polysaccharides that impart a strong negative charge, including heparan sulfate, chondroitin sulfate, and hyaluronan or hyaluronic acid); and glycoproteins (cell surface receptors such as selectins, integrins, intercellular adhesion molecule [ICAM], and vascular cell adhesion molecule [VCAM]). Injury promotes glycocalyx shedding, which exposes glycoproteins to blood stream, resulting in inflammatory cell adhesion, platelet aggregation, and following coagulation. CD44, one of principle glycoproteins in glycocalyx, anchors hyaluronan chains and stripping of glycocalyx facilitates CD44-leukocyte interaction and leukocyte extravasation. CKD: chronic kidney disease.

Figure 4.
Major signaling pathways for endothelial growth/proliferation. When ligands (vascular endothelial growth factor-A (VEGF-A) or angiopietin-1 (Angpt-1)) bind to receptor tyrosine kinase, activated PI3K (phosphoinositide-3-kinase) converts PIP2 (phosphatidylinositol-4,5-biphosphate) to PIP3 (phosphatidylinositol-3,4,5-triphosphate) by phosphorylation. Conversely, PTEN (phosphatase and tensin homolog deleted from chromosome ten) turns PIP3 into PIP2 by dephosphorylation. PIP3 is a membrane bound and intracellular messenger that recruits PDK-1 (phosphatidylinositol-dependent kinase 1) and Akt to the plasma membrane. PDK-1 phosphorylates Akt, which, in turn, inhibits nuclear translocation of forkhead box O-1 (FOXO1). As cytoplasmic FOXO1 cannot suppress transcription factor, MYC, enhanced MYC activity leads to increased cellular metabolisms, growth, and proliferation. Phosphorylated FOXO1 is ubiquitinated by SKP2 (S-phase kinase associated protein2 E3 ligase) and is subjected to degradation. When VEGF-A or Angpt-1 is not available, nuclear FOXO1 inhibits MYC activity, limiting endothelial growth/proliferation.
Figure 4.
Major signaling pathways for endothelial growth/proliferation. When ligands (vascular endothelial growth factor-A (VEGF-A) or angiopietin-1 (Angpt-1)) bind to receptor tyrosine kinase, activated PI3K (phosphoinositide-3-kinase) converts PIP2 (phosphatidylinositol-4,5-biphosphate) to PIP3 (phosphatidylinositol-3,4,5-triphosphate) by phosphorylation. Conversely, PTEN (phosphatase and tensin homolog deleted from chromosome ten) turns PIP3 into PIP2 by dephosphorylation. PIP3 is a membrane bound and intracellular messenger that recruits PDK-1 (phosphatidylinositol-dependent kinase 1) and Akt to the plasma membrane. PDK-1 phosphorylates Akt, which, in turn, inhibits nuclear translocation of forkhead box O-1 (FOXO1). As cytoplasmic FOXO1 cannot suppress transcription factor, MYC, enhanced MYC activity leads to increased cellular metabolisms, growth, and proliferation. Phosphorylated FOXO1 is ubiquitinated by SKP2 (S-phase kinase associated protein2 E3 ligase) and is subjected to degradation. When VEGF-A or Angpt-1 is not available, nuclear FOXO1 inhibits MYC activity, limiting endothelial growth/proliferation.

Figure 5.
Kidney pericytes are essential to the integrity of peritubular capillaries. (A) In peritubular capillaries of the normal kidney, kidney pericytes (green) are attached to capillary endothelial cells (red) to stabilize capillary tube formation. Pericytes share the capillary basement membrane with endothelial cells. (B) In response to injury, kidney pericytes promptly migrate away from the capillary basement membrane, resulting in activation of endothelial cells. (C) Following further injury, kidney pericytes differentiate into scar-forming myofibroblasts or activated fibroblasts. These populations synthesize extracellular matrices such as collagens, promoting tissue fibrosis. Capillary endothelial cells are not able to maintain the capillary basement membrane without pericytes. Finally, peritubular capillaries start to disappear due to apoptosis, as myofibroblasts can no longer stabilize capillary tube formation.
Figure 5.
Kidney pericytes are essential to the integrity of peritubular capillaries. (A) In peritubular capillaries of the normal kidney, kidney pericytes (green) are attached to capillary endothelial cells (red) to stabilize capillary tube formation. Pericytes share the capillary basement membrane with endothelial cells. (B) In response to injury, kidney pericytes promptly migrate away from the capillary basement membrane, resulting in activation of endothelial cells. (C) Following further injury, kidney pericytes differentiate into scar-forming myofibroblasts or activated fibroblasts. These populations synthesize extracellular matrices such as collagens, promoting tissue fibrosis. Capillary endothelial cells are not able to maintain the capillary basement membrane without pericytes. Finally, peritubular capillaries start to disappear due to apoptosis, as myofibroblasts can no longer stabilize capillary tube formation.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kida, Y. Peritubular Capillary Rarefaction: An Underappreciated Regulator of CKD Progression. Int. J. Mol. Sci. 2020, 21, 8255. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218255
AMA Style
Kida Y. Peritubular Capillary Rarefaction: An Underappreciated Regulator of CKD Progression. International Journal of Molecular Sciences. 2020; 21(21):8255. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218255
Chicago/Turabian StyleKida, Yujiro. 2020. "Peritubular Capillary Rarefaction: An Underappreciated Regulator of CKD Progression" International Journal of Molecular Sciences 21, no. 21: 8255. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218255
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.