Cell Death Mechanisms Induced by CLytA-DAAO Chimeric Enzyme in Human Tumor Cell Lines

 , ,
, ,  ,
,  , and
, and 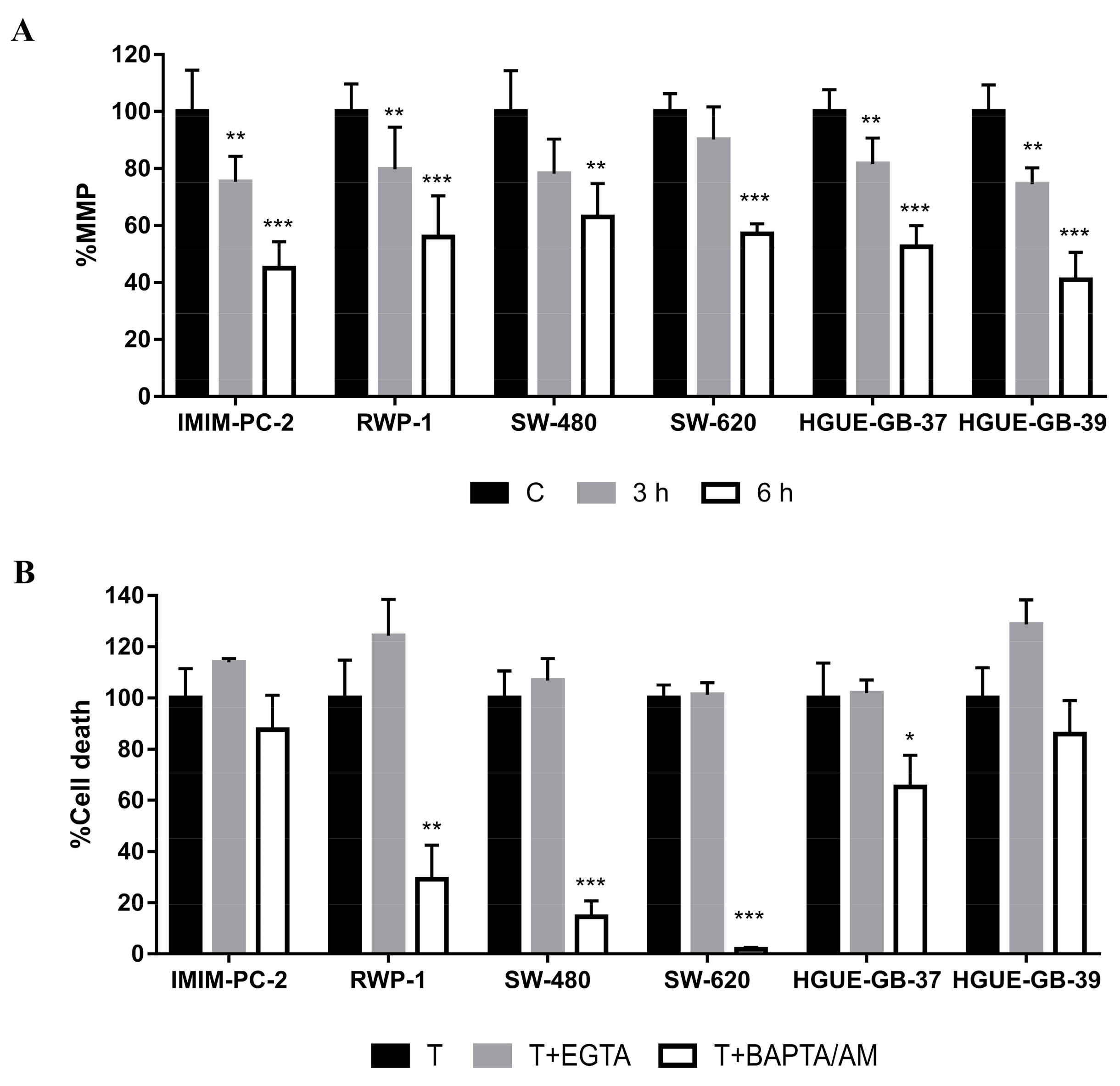{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
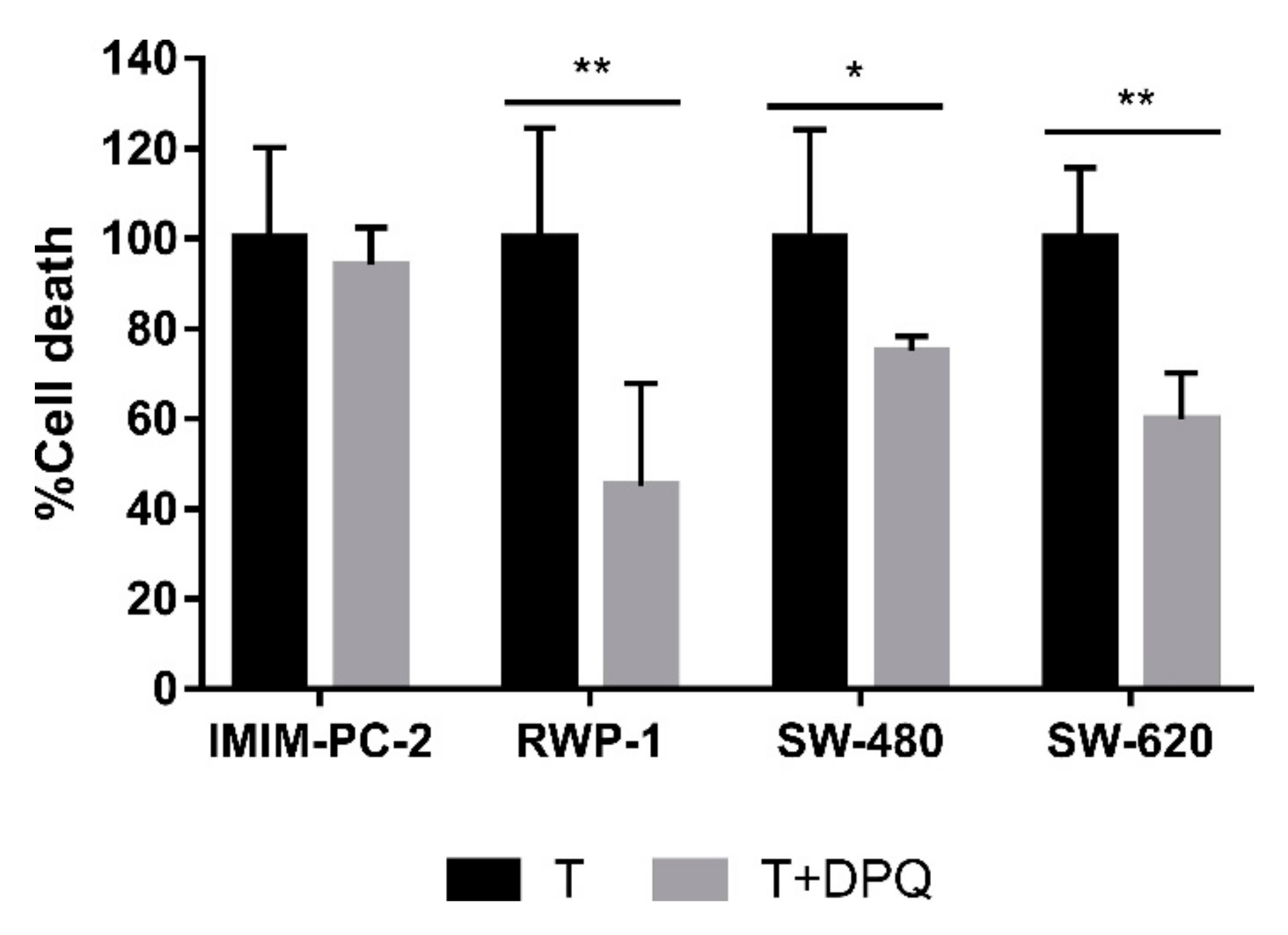
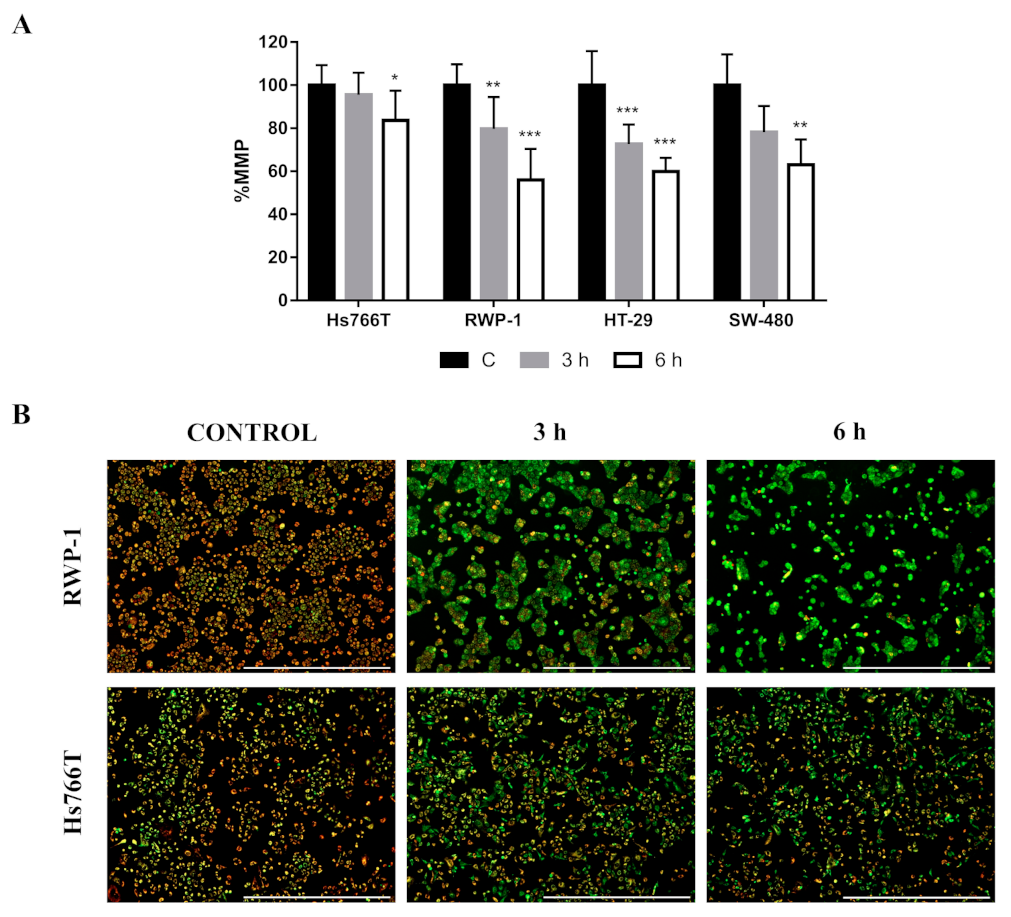
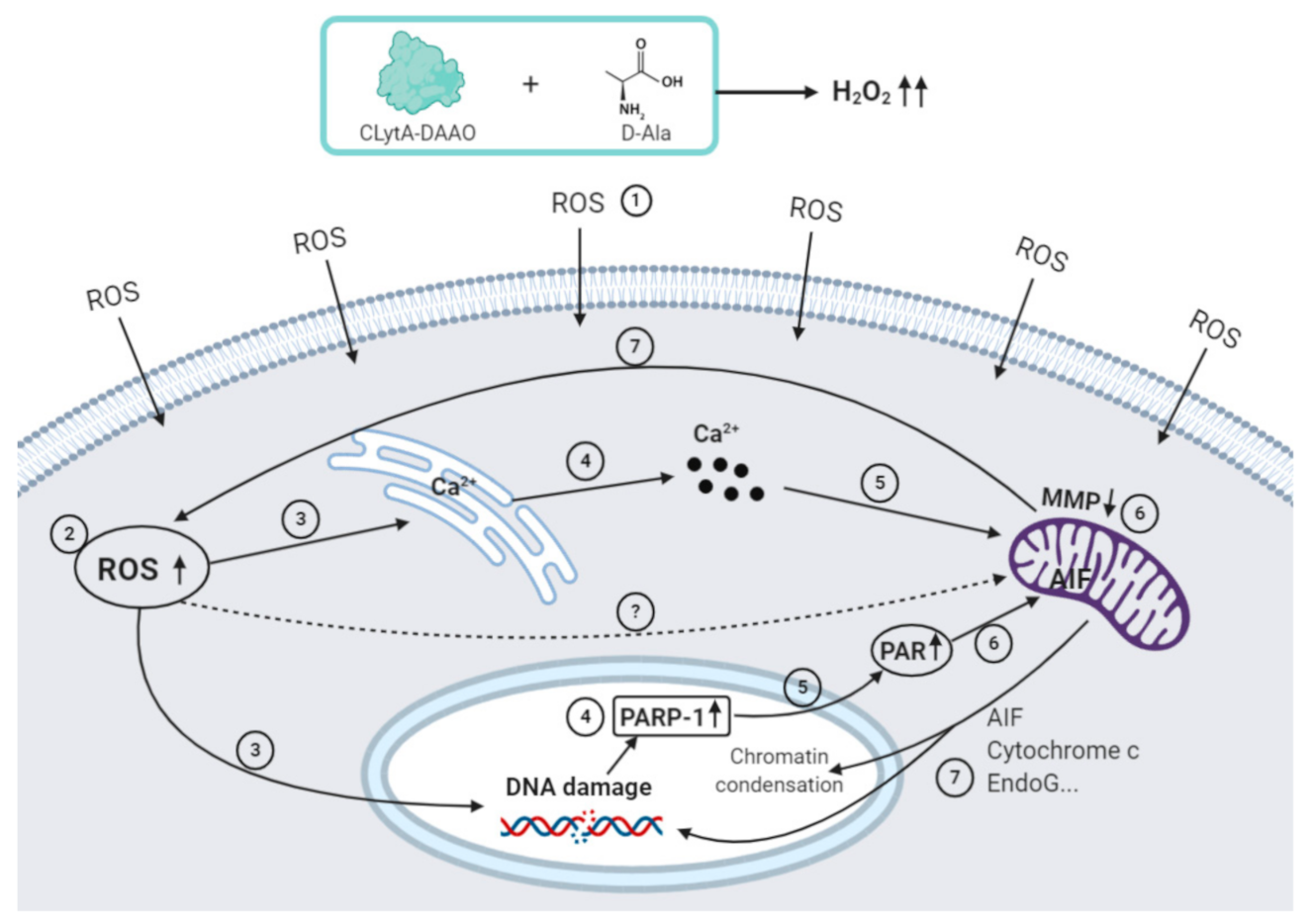
2.1. Calcium Release from Cellular Reservoirs Is Responsible for the MMP Decrease That Leads to Cell Death in Most of the Cell Lines after CLytA-DAAO Treatment
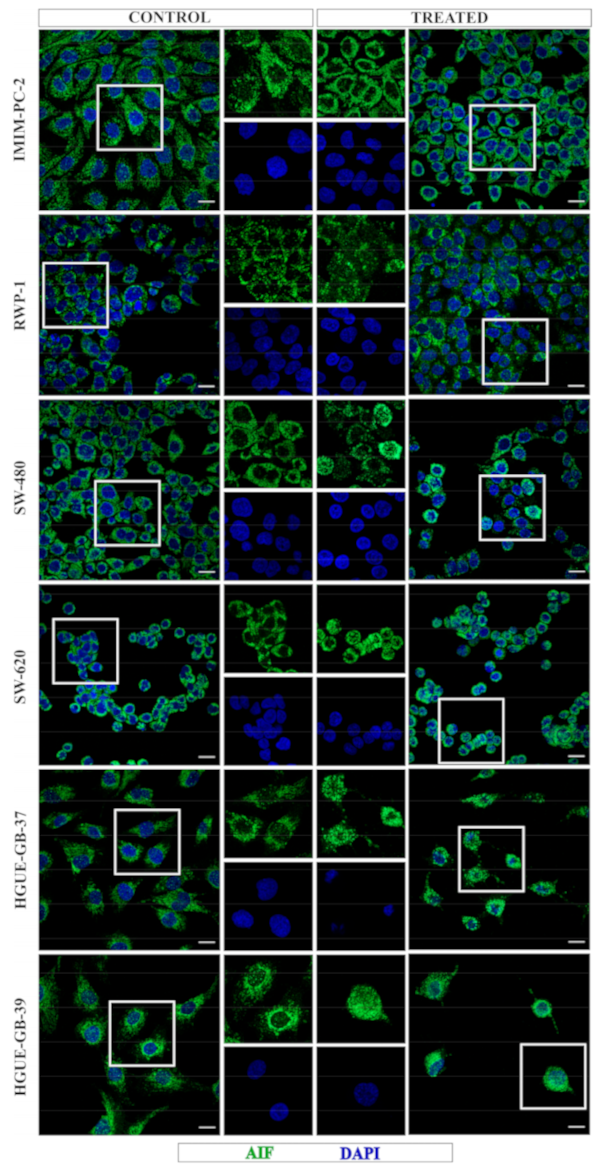
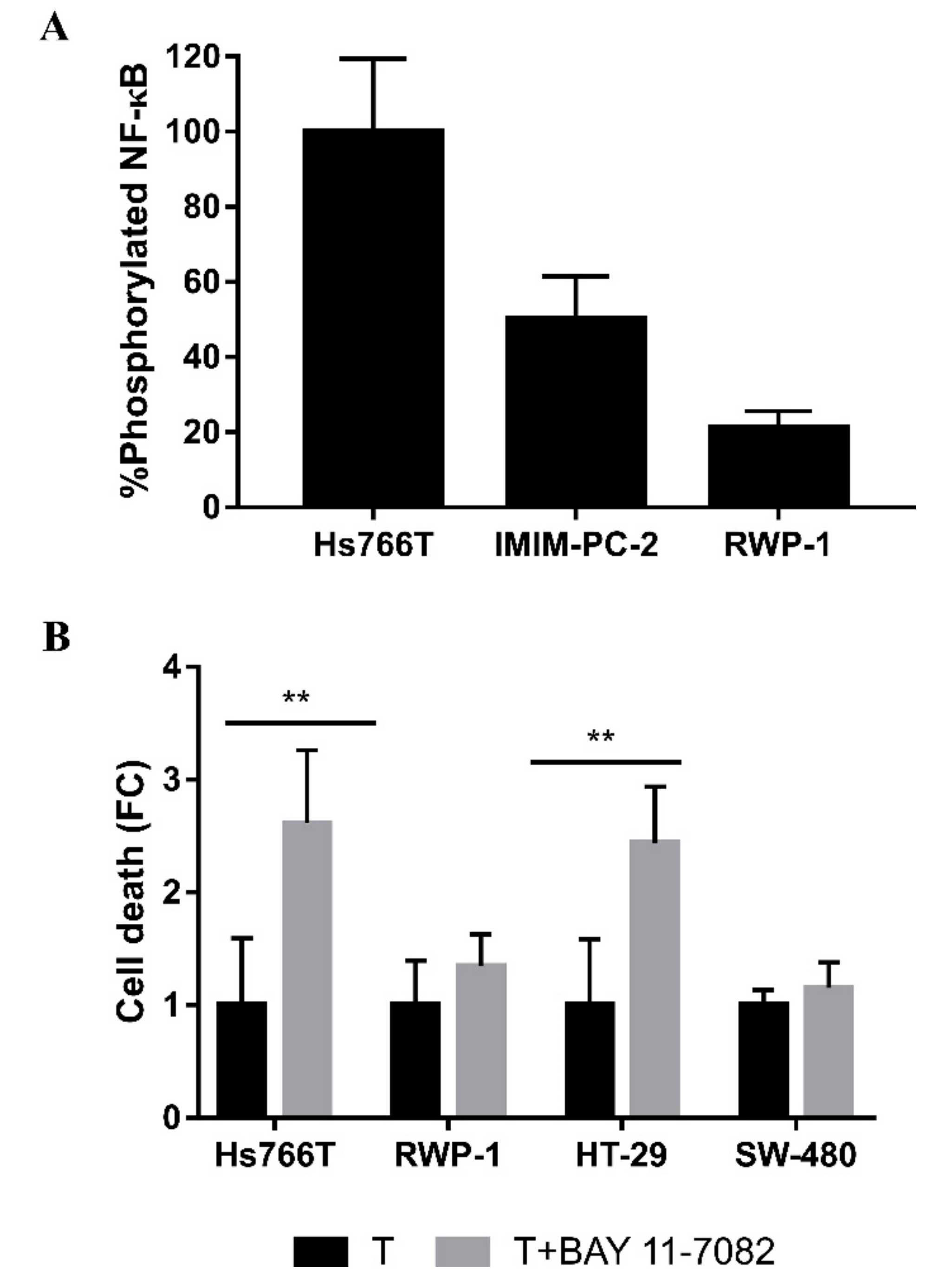
2.2. CLytA-DAAO Induces Different Cell Death Mechanisms Depending on the Carcinoma Cell Lines
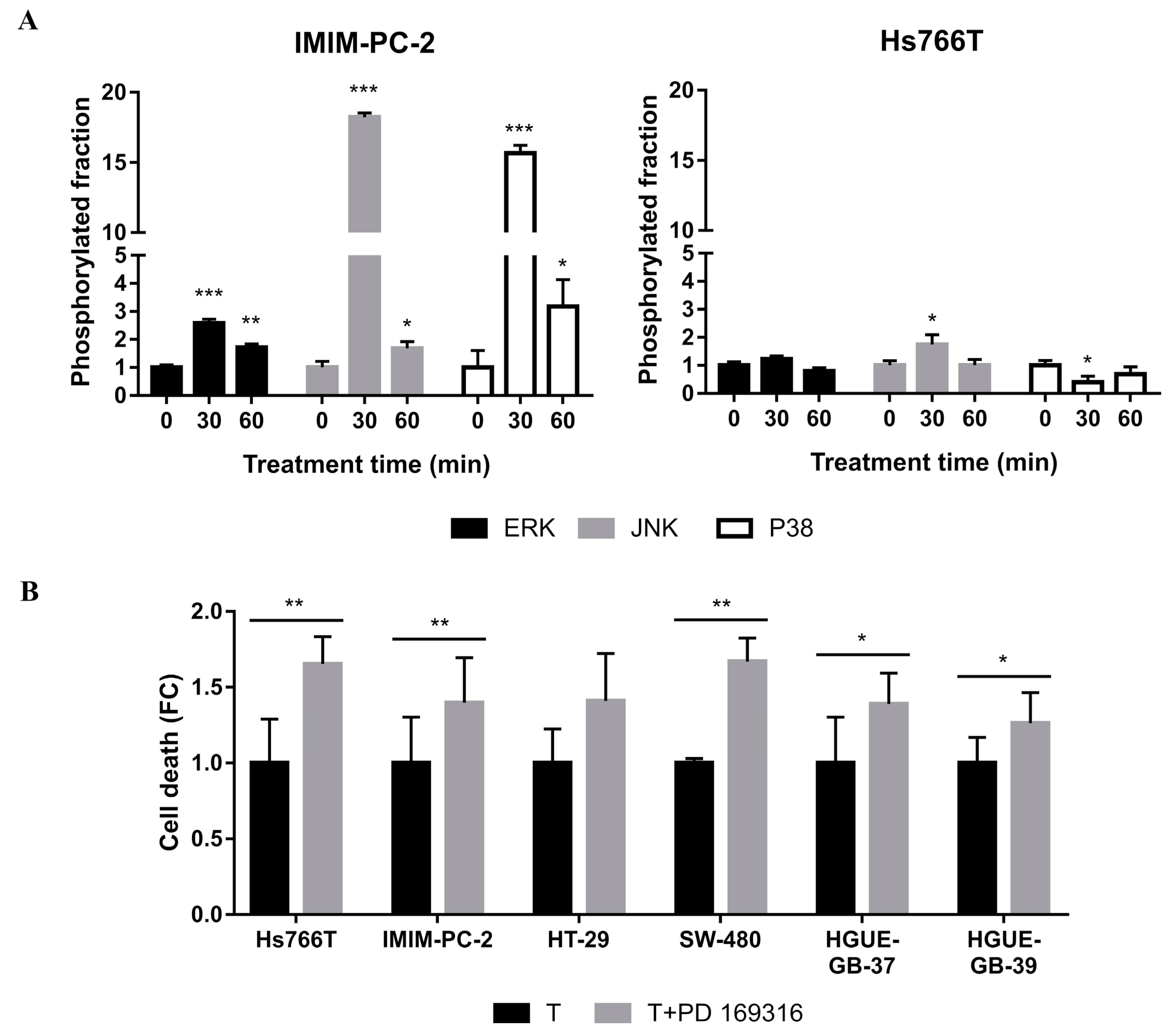
2.3. MAPKs Pathways Are Involved in CLytA-DAAO-Induced Cell Death
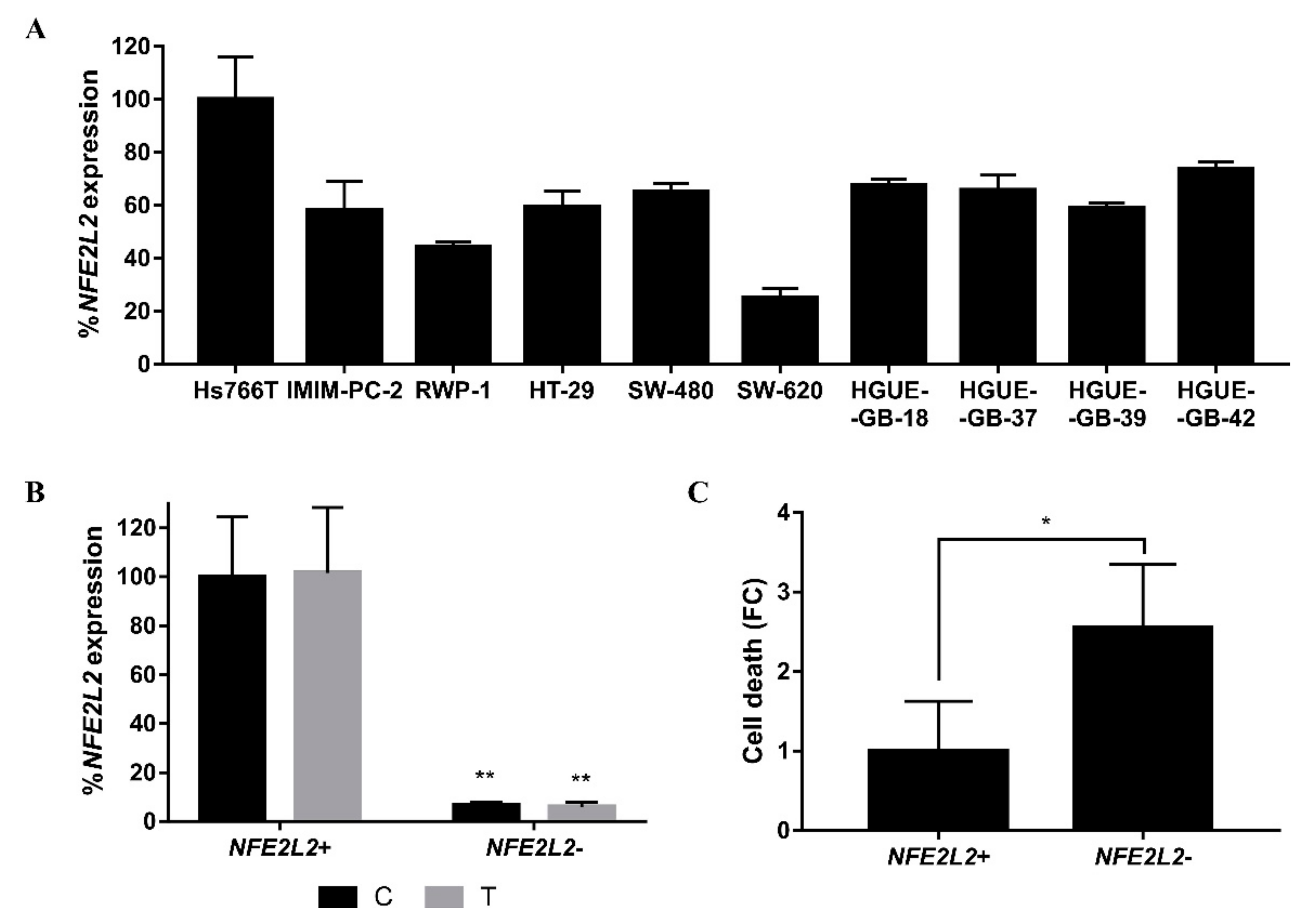
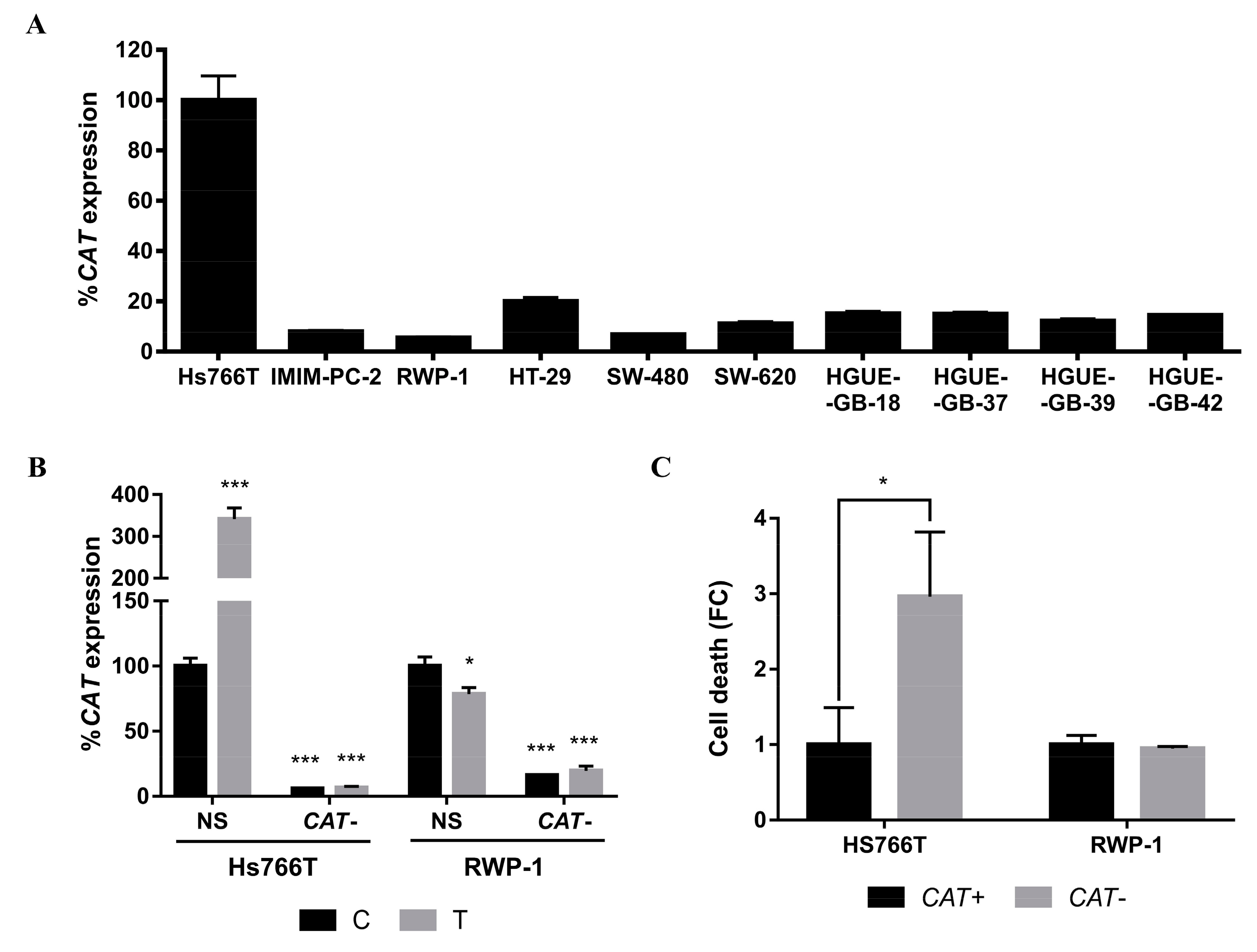
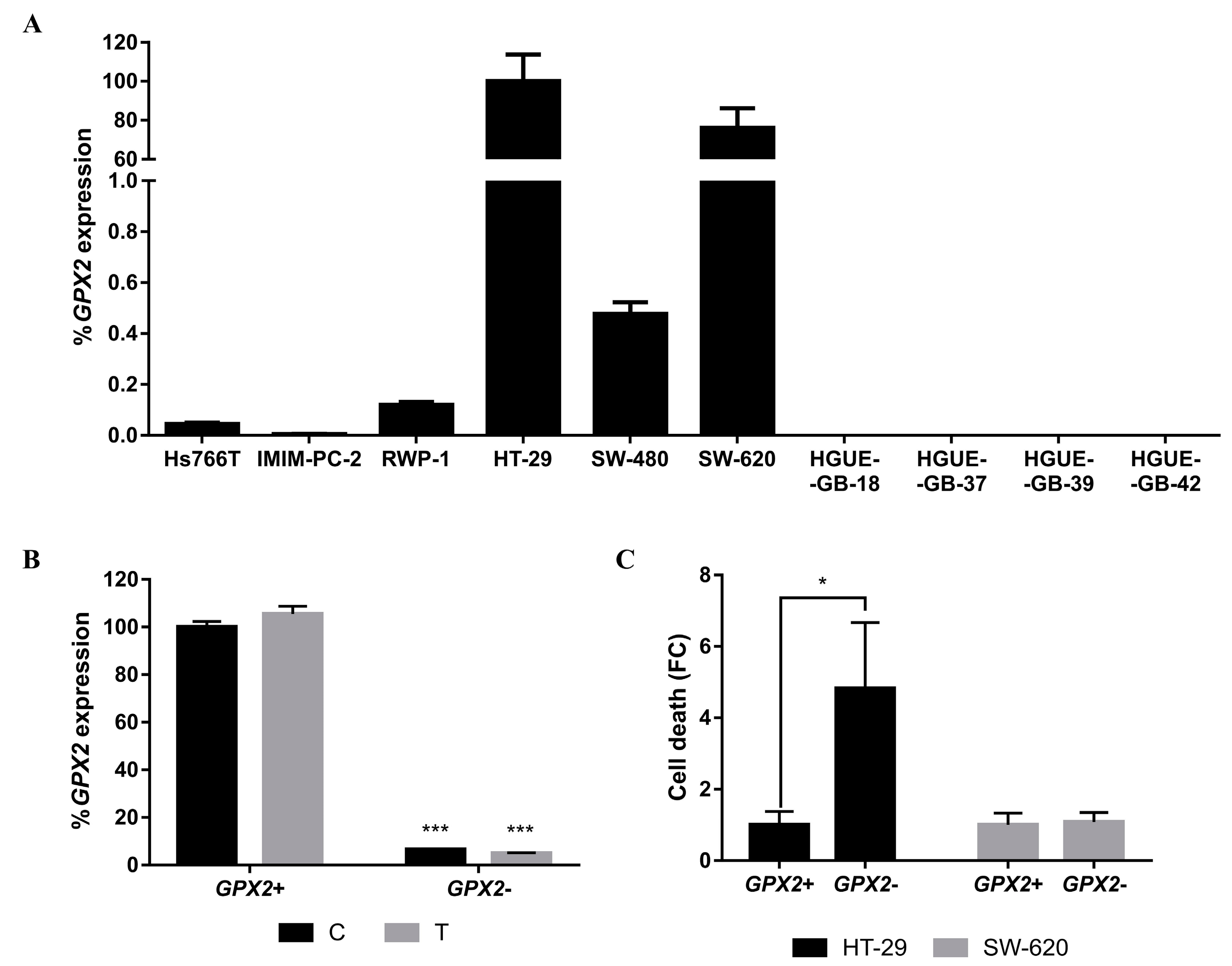
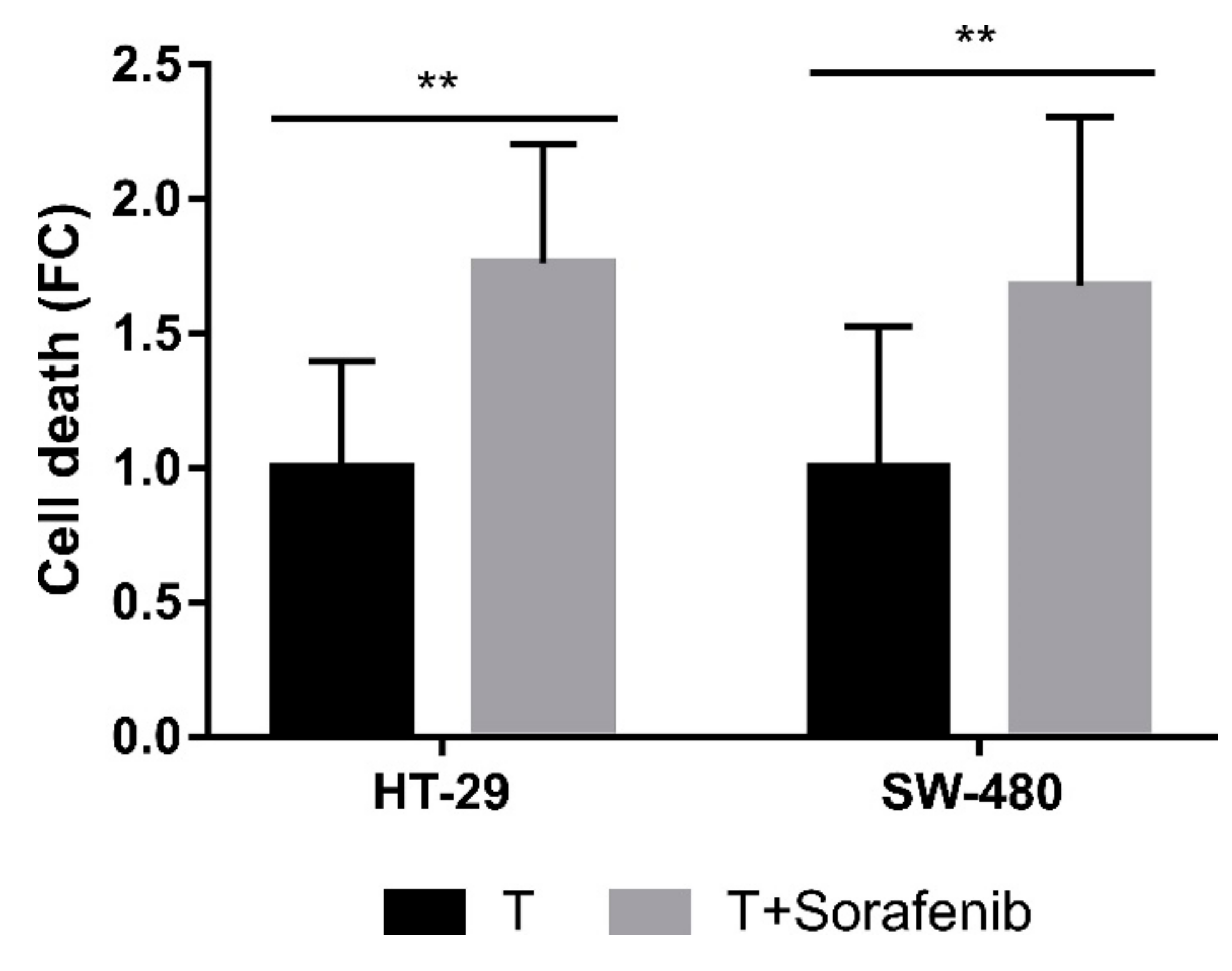
2.4. Molecular Mechanisms Involved in the Acquisition of Resistance to CLytA-DAAO-Induced Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemical Reagents
4.3. Cell Death
4.4. Mitochondrial Membrane Potential
4.5. Immunocytochemistry
4.6. ELISA Assay
4.7. Gene Expression Analysis
4.8. Small Interfering RNA (siRNA) Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DAAO | D-amino acid oxidase |
| CLytA | Choline-binding module of the amidase N-acetylmuramoyl-L-alanine |
| DEAE | Diethylaminoethanol |
| PARP-1 | Poly (ADP-ribose) polymerase 1 |
| AIF | Apoptosis-inducing factor |
| MMP | Mitochondrial membrane potential |
| MAPK | Mitogen-activated protein kinase |
| ERK | Extracellular signal-regulated kinase |
| JNK | c-Jun N-terminal kinase |
| P38 | P38 mitogen-activated protein kinase |
| ROS | Reactive oxygen species |
| NF-κB | Nuclear factor kappa B |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| CAT | Catalase |
| GPX2 | Glutathione peroxidase 2 |
| PTP | Permeability transition pore |
| ER | Endoplasmic reticulum |
| IP3R | Inositol-1,4,5-triphosphate receptor |
| DDR | DNA damage response |
| IRE1α | Inositol-requiring 1 alpha |
| PERK | Double-strand RNA-activated protein kinase-like ER kinase |
| ATF6 | Activating transcription factor 6 |
| XBP1 | X-box binding protein 1 |
| HGUE | Hospital General Universitario de Elche (General University Hospital of Elche) |
| BAPTA/AM | 1,2-Bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid tetrakis(acetoxymethyl ester) |
| EGTA | Ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid |
| DPQ | 3,4-Dihydro-5(4-(1-piperindinyl)butoxy)-1(2H)-isoquinoline |
| PD 169316 | 4-(4-Fluorophenyl)-2-(4-nitrophenyl)-5-(4-pyridyl)-1H-imidazole |
| BAY 11-7082 | (E)-3-(4-Methylphenylsulfonyl)-2-propenenitrile |
| Sorafenib | N-(4-Chloro-3-(trifluoromethyl)phenyl)-N’-(4-(2-[N-methylcarbamoyl]-4-pyridyloxy)phenyl)urea |
| DAPI | 4′,6-diamidino-2-phenylindole |
| qPCR | Quantitative polymerase chain reaction |
| SEM | Standard Error of the Mean |
References
- Degterev, A.; Yuan, J. Expansion and evolution of cell death programmes. Nat. Rev. Mol. Cell Biol. 2008, 9, 378–390. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Peng, Z.L. Programmed cell death and cancer. Postgrad. Med. J. 2009, 85, 134–140. [Google Scholar] [CrossRef]
- Panghal, A.; Sharma, H.; Flora, S.; Naqvi, S. Suicide gene therapy: A promising approach towards gene delivery. Front. Nanosci. Nanotechnol. 2019, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Niculescu-Duvaz, I.; Springer, C.J. Introduction to the Background, Principles, and State of the Art in Suicide Gene Therapy. Mol. Biotechnol. 2005, 30, 71–88. [Google Scholar] [CrossRef]
- Pollegioni, L.; Piubelli, L.; Sacchi, S.; Pilone, M.S.; Molla, G. Physiological functions of D-amino acid oxidases: From yeast to humans. Cell. Mol. Life Sci. 2007, 64, 1373–1394. [Google Scholar] [CrossRef]
- López, R.; García, E. Recent trends on the molecular biology of pneumococcal capsules, lytic enzymes, and bacteriophage. FEMS Microbiol. Rev. 2004, 28, 553–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Puelles, J.M.; Sanz, J.M.; Garcia, J.L.; Garcia, E. Immobilization and single-step purification of fusion proteins using DEAE-cellulose. Eur. J. Biochem. 1992, 203, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Baile, M.; Bello-Gil, D.; Pérez-Valenciano, E.; Sanz, J.M.; García-Morales, P.; Maestro, B.; Ventero, M.P.; Alenda, C.; Barberá, V.M.; Saceda, M. CLytA-DAAO, free and immobilized in magnetic nanoparticles, induces cell death in human cancer cells. Biomolecules 2020, 10, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Xie, R.; Munoz, F.M.; Lau, S.S.; Monks, T.J. PARP-1 hyperactivation and reciprocal elevations in intracellular Ca2+ during ROS-induced nonapoptotic cell death. Toxicol. Sci. 2014, 140, 118–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Song, R.; Pang, Q.; Liu, Y.; Zhuang, J.; Chen, Y.; Hu, J.; Hu, J.; Liu, Y.; Liu, Z.; et al. Propofol inhibits parthanatos via ROS–ER–calcium–mitochondria signal pathway in vivo and vitro. Cell Death Dis. 2018, 9, 932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (δψm) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.Y.; Ho, F.M.; Shiah, S.G.; Chang, Y.; Lin, W.W. Oxidative stress initiates DNA damager MNNG-induced poly(ADP-ribose) polymerase-1-dependent parthanatos cell death. Biochem. Pharmacol. 2011, 81, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Suto, M.J.; Turner, W.R.; Arundel-Suto, C.M.; Werbel, L.M.; Sebolt-Leopold, J.S. Dihydroisoquinolinones: The design and synthesis of a new series of potent inhibitors of poly(ADP-ribose) polymerase. Anticancer Drug Des. 1991, 6, 107–117. [Google Scholar] [PubMed]
- Eliasson, M.J.L.; Sampei, K.; Mandir, A.S.; Hurn, P.D.; Traystman, R.J.; Bao, J.; Pieper, A.; Wang, Z.-Q.; Dawson, T.M.; Snyder, S.H.; et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997, 3, 1089–1095. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochodrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann. N. Y. Acad. Sci. 2008, 1147, 233–241. [Google Scholar] [CrossRef]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, A messenger of death. Front. Biosci. 2009, 14, 1116–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugas, E.; Susin, S.A.; Zamzami, N.; Ferri, K.F.; Irinopoulou, T.; Larochette, N.; Prévost, M.; Leber, B.; Andrews, D.; Penninger, J.; et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000, 14, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akay, C.; Thomas, C.; Gazzit, Y. Arsenic Trioxide and Paclitaxel Induce Apoptosis by Different Mechanisms. Cell Cycle 2004, 3, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Daugas, E.; Ravagnan, L.; Samejima, K.; Zamzami, N.; Loeffler, M.; Costantini, P.; Ferri, K.F.; Irinopoulou, T.; Prévost, M.C.; et al. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 2000, 192, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Rezatabar, S.; Karimian, A.; Rameshknia, V.; Parsian, H.; Majidinia, M.; Kopi, T.A.; Bishayee, A.; Sadeghinia, A.; Yousefi, M.; Monirialamdari, M.; et al. RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. J. Cell. Physiol. 2019, 234, 14951–14965. [Google Scholar] [CrossRef]
- Ono, K.; Han, J. The p38 signal transduction pathway Activation and function. Cell. Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-B, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Emmink, B.L.; Laoukili, J.; Kipp, A.P.; Koster, J.; Govaert, K.M.; Fatrai, S.; Verheem, A.; Steller, E.J.A.; Brigelius-Flohé, R.; Jimenez, C.R.; et al. GPx2 suppression of H2O2 stress links the formation of differentiated tumor mass to metastatic capacity in colorectal cancer. Cancer Res. 2014, 74, 6717–6730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banning, A.; Kipp, A.; Schmitmeier, S.; Löwinger, M.; Florian, S.; Krehl, S.; Thalmann, S.; Thierbach, R.; Steinberg, P.; Brigelius-Flohé, R. Glutathione peroxidase 2 inhibits cyclooxygenase-2-mediated migration and invasion of HT-29 adenocarcinoma cells but supports their growth as tumors in nude mice. Cancer Res. 2008, 68, 9746–9753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Hattori, N.; Iida, N.; Yamashita, S.; Mori, A.; Kimura, K.; Yoshino, T.; Ushijima, T. Targeting of super-enhancers and mutant BRAF can suppress growth of BRAF-mutant colon cancer cells via repression of MAPK signaling pathway. Cancer Lett. 2017, 402, 100–109. [Google Scholar] [CrossRef]
- Roma, C.; Rachiglio, A.M.; Pasquale, R.; Fenizia, F.; Iannaccone, A.; Tatangelo, F.; Antinolfi, G.; Parrella, P.; Graziano, P.; Sabatino, L.; et al. BRAF V600E mutation in metastatic colorectal cancer: Methods of detection and correlation with clinical and pathologic features. Cancer Biol. Ther. 2016, 17, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Huang, M.; Gong, Y.; Lin, C.; Guo, W. BRAF and EGFR inhibitors synergize to increase cytotoxic effects and decrease stem cell capacities in BRAF(V600E)-mutant colorectal cancer cells. Acta Biochim. Biophys. Sin. 2018, 50, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Kroemer, G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 1997, 3, 614–620. [Google Scholar] [CrossRef]
- Kass, G.E.N.; Orrenius, S. Calcium Signaling and Cytotoxicity. Environ. Health Perspect. 1999, 107, 25–35. [Google Scholar]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Kerr, P.M.; Javadov, S.; Woodfield, K.Y. Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim. Biophys. Acta 1998, 1366, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Kruman, I.I.; Mattson, M.P. Pivotal role of mitochondrial calcium uptake in neural cell apoptosis and necrosis. J. Neurochem. 1999, 72, 529–540. [Google Scholar] [CrossRef]
- Koya, R.C.; Fujita, H.; Shimizu, S.; Ohtsu, M.; Takimoto, M.; Tsujimoto, Y.; Kuzumaki, N. Gelsolin inhibits apoptosis by blocking mitochondrial membrane potential loss and cytochrome c release. J. Biol. Chem. 2000, 275, 15343–15349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deniaud, A.; Sharaf El Dein, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef]
- Görlach, A.; Klappa, P.; Kietzmann, T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Santarpia, L.L.; Lippman, S.; El-Naggar, A. Targeting the Mitogen-Activated Protein Kinase RAS-RAF Signaling Pathway in Cancer Therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [Green Version]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—From inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [Green Version]
- Loukopoulos, P.; Kanetaka, K.; Takamura, M.; Shibata, T.; Sakamoto, M.; Hirohashi, S. Orthotopic transplantation models of pancreatic adenocarcinoma derived from cell lines and primary tumors and displaying varying metastatic activity. Pancreas 2004, 29, 193–203. [Google Scholar] [CrossRef]
- Hempen, P.M.; Kurpad, H.; Calhoun, E.S.; Abraham, S.; Kern, S.E. A double missense variation of the BUB1 gene and a defective mitotic spindle checkpoint in the pancreatic cancer cell line Hs766T. Hum. Mutat. 2003, 21, 445. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Lüschen, S.; Scherer, G.; Ussat, S.; Ungefroren, H.; Adam-Klages, S. Inhibition of p38 mitogen-activated protein kinase reduces TNF-induced activation of NF-κB, elicits caspase activity, and enhances cytotoxicity. Exp. Cell Res. 2004, 293, 196–206. [Google Scholar] [CrossRef]
- Cheng, Y.; Qiu, F.; Ye, Y.C.; Guo, Z.M.; Tashiro, S.I.; Onodera, S.; Ikejima, T. Autophagy inhibits reactive oxygen species-mediated apoptosis via activating p38-nuclear factor-kappa B survival pathways in oridonin-treated murine fibrosarcoma L929 cells. FEBS J. 2009, 276, 1291–1306. [Google Scholar] [CrossRef]
- Lee, J.S.; Surh, Y.J. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005, 224, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.M.; Lee, K.M.; Lee, H.J.; Yun, J.H.; Nho, C.W. Physalin A regulates the Nrf2 pathway through ERK and p38 for induction of detoxifying enzymes. BMC Complementary Altern. Med. 2019, 19, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Sisinni, L.; Pietrafesa, M.; Lepore, S.; Maddalena, F.; Condelli, V.; Esposito, F.; Landriscina, M. Endoplasmic reticulum stress and unfolded protein response in breast cancer: The balance between apoptosis and autophagy and its role in drug resistance. Int. J. Mol. Sci. 2019, 20, 857. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Wang, H.; Wei, S.; Wang, Z.; Ji, G. Inhibition of ER stress-related IRE1α/CREB/NLRP1 pathway promotes the apoptosis of human chronic myelogenous leukemia cell. Mol. Immunol. 2018, 101, 377–385. [Google Scholar] [CrossRef]
- Corazzari, M.; Rapino, F.; Ciccosanti, F.; Giglio, P.; Antonioli, M.; Conti, B.; Fimia, G.M.; Lovat, P.E.; Piacentini, M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015, 22, 946–958. [Google Scholar] [CrossRef]
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Caputo, F.; Santini, C.; Bardasi, C.; Cerma, K.; Casadei-Gardini, A.; Spallanzani, A.; Andrikou, K.; Cascinu, S.; Gelsomino, F. BRAF-mutated colorectal cancer: Clinical and molecular insights. Int. J. Mol. Sci. 2019, 20, 5369. [Google Scholar] [CrossRef] [Green Version]
- Ducreux, M.; Chamseddine, A.; Laurent-Puig, P.; Smolenschi, C.; Hollebecque, A.; Dartigues, P.; Samallin, E.; Boige, V.; Malka, D.; Gelli, M. Molecular Targeted therapy of BRAF-mutant colorectal cancer. Ther. Adv. Med. Oncol. 2019, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ventero, M.P.; Fuentes-Baile, M.; Quereda, C.; Perez-Valeciano, E.; Alenda, C.; Garcia-Morales, P.; Esposito, D.; Dorado, P.; Manuel Barbera, V.; Saceda, M. Radiotherapy resistance acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE 2019, 14, e0212581. [Google Scholar] [CrossRef] [PubMed] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuentes-Baile, M.; García-Morales, P.; Pérez-Valenciano, E.; Ventero, M.P.; Sanz, J.M.; Romero, C.d.J.; Barberá, V.M.; Alenda, C.; Saceda, M. Cell Death Mechanisms Induced by CLytA-DAAO Chimeric Enzyme in Human Tumor Cell Lines. Int. J. Mol. Sci. 2020, 21, 8522. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228522
Fuentes-Baile M, García-Morales P, Pérez-Valenciano E, Ventero MP, Sanz JM, Romero CdJ, Barberá VM, Alenda C, Saceda M. Cell Death Mechanisms Induced by CLytA-DAAO Chimeric Enzyme in Human Tumor Cell Lines. International Journal of Molecular Sciences. 2020; 21(22):8522. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228522
Chicago/Turabian StyleFuentes-Baile, María, Pilar García-Morales, Elizabeth Pérez-Valenciano, María P. Ventero, Jesús M. Sanz, Camino de Juan Romero, Víctor M. Barberá, Cristina Alenda, and Miguel Saceda. 2020. "Cell Death Mechanisms Induced by CLytA-DAAO Chimeric Enzyme in Human Tumor Cell Lines" International Journal of Molecular Sciences 21, no. 22: 8522. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228522