A New Twist in Protein Kinase B/Akt Signaling: Role of Altered Cancer Cell Metabolism in Akt-Mediated Therapy Resistance



Abstract
:1. Introduction
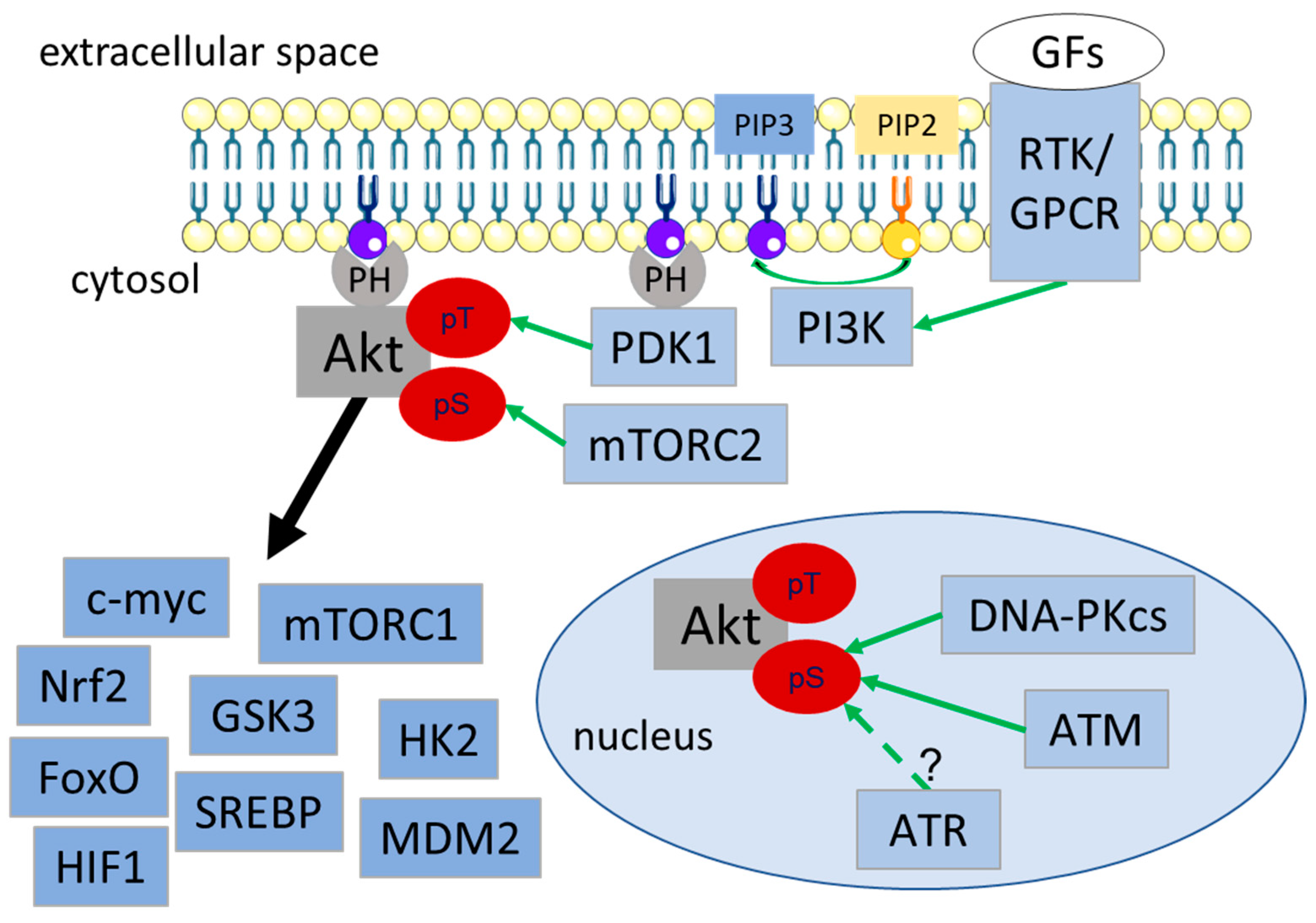
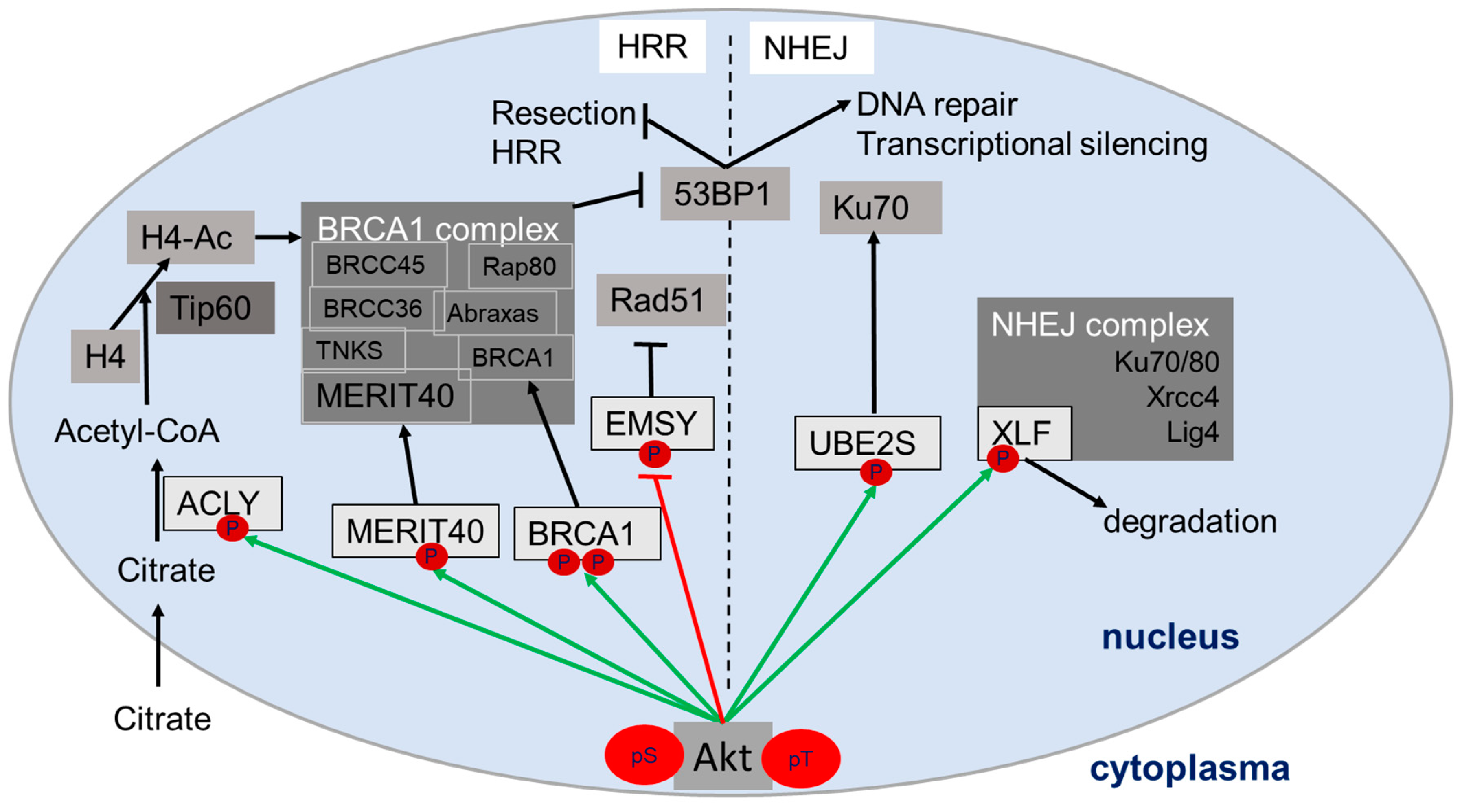
2. Role of Akt in DNA Damage Response and DSB Repair
3. Relevance of Metabolic Processes in the DDR
3.1. Reactive Oxygen Species and Antioxidant Defense
3.2. Nucleotide Synthesis
3.3. Basic Metabolites Required for the Modification of Proteins, DNA and Chromatin
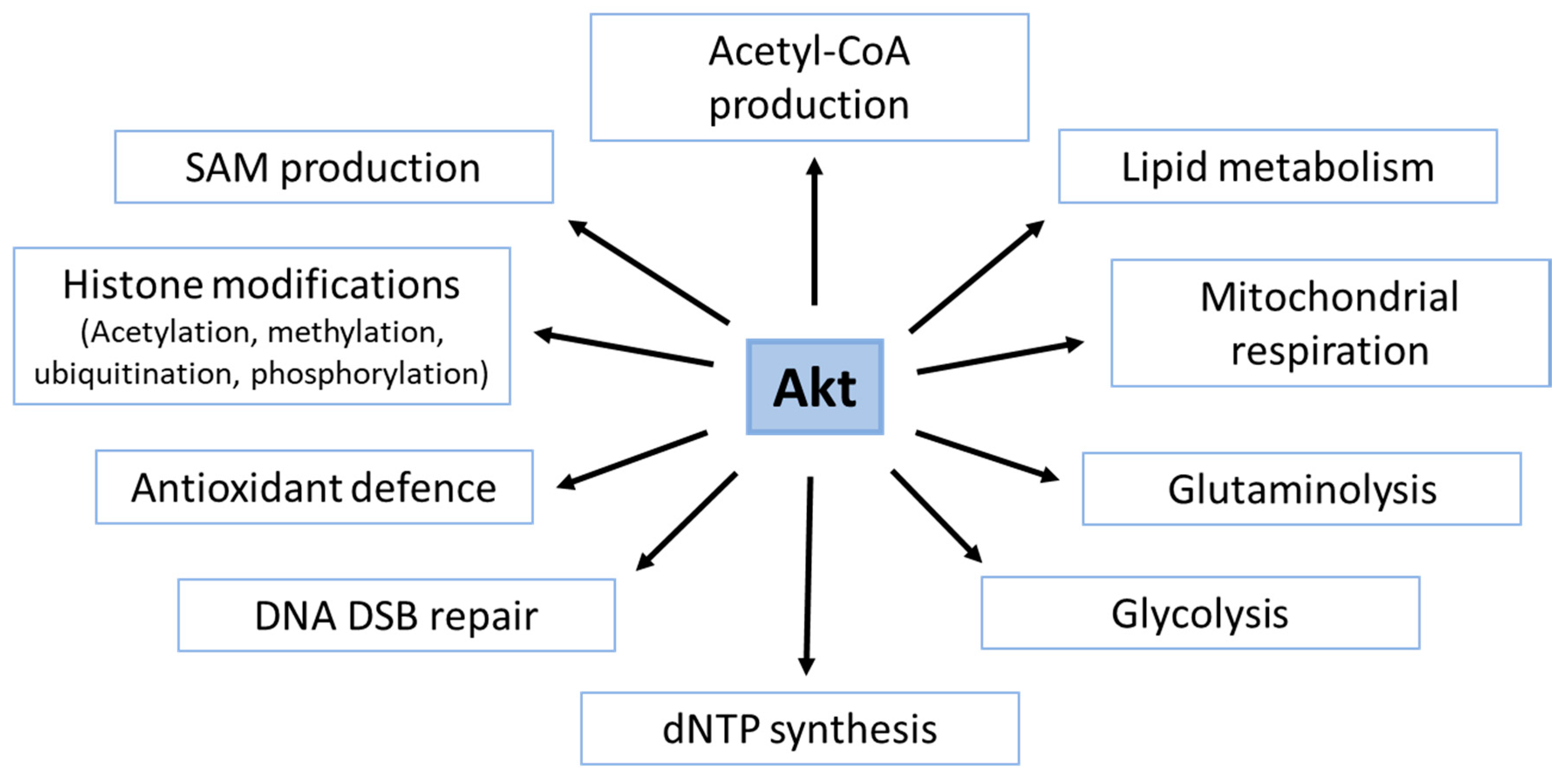
4. Role of Akt in the Generation of Basic Metabolites with Importance to DDR
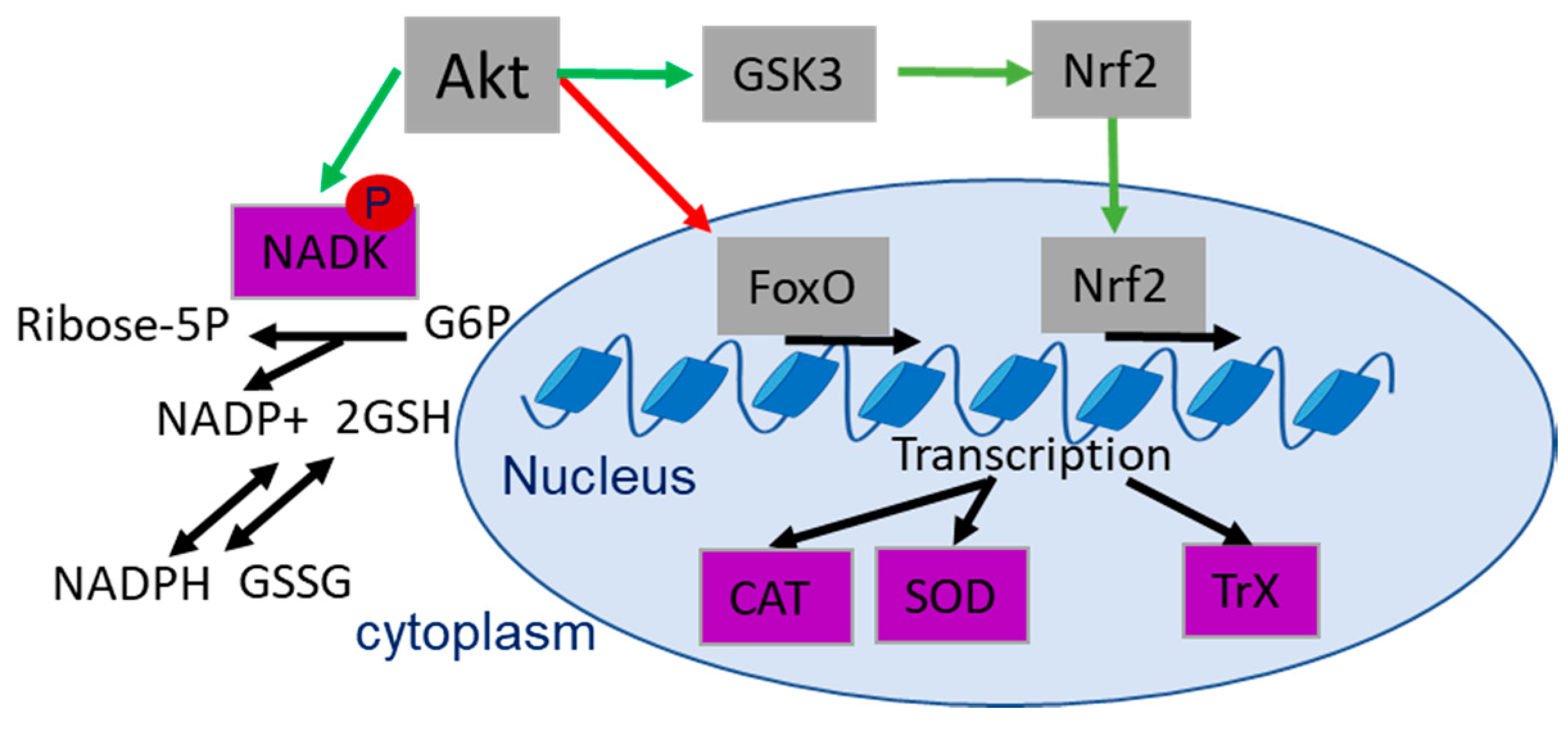
4.1. Role of Akt in Regulating the Antioxidant Defense
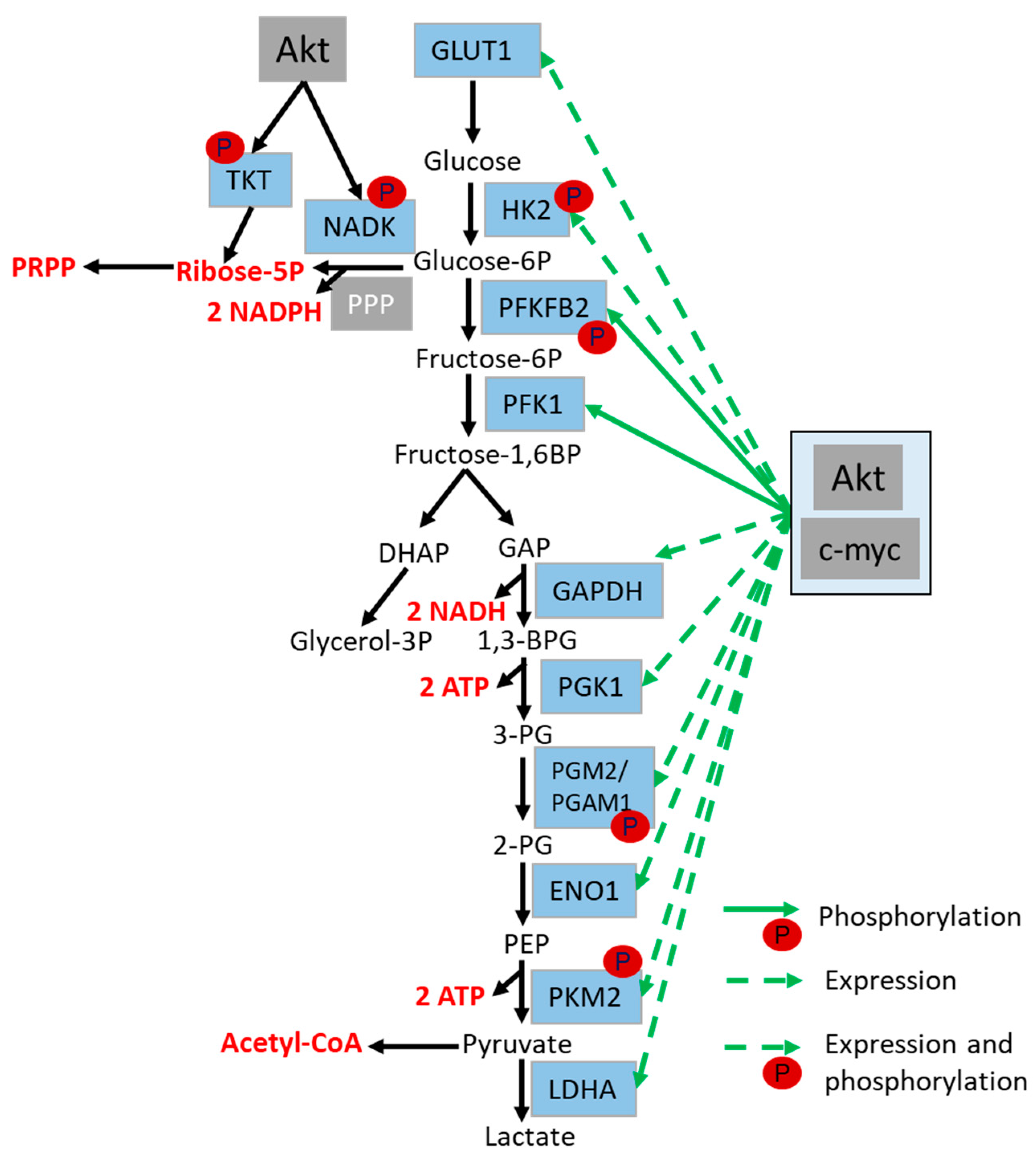
4.2. Role of Akt in the Regulation of Glycolysis and Glutaminolysis
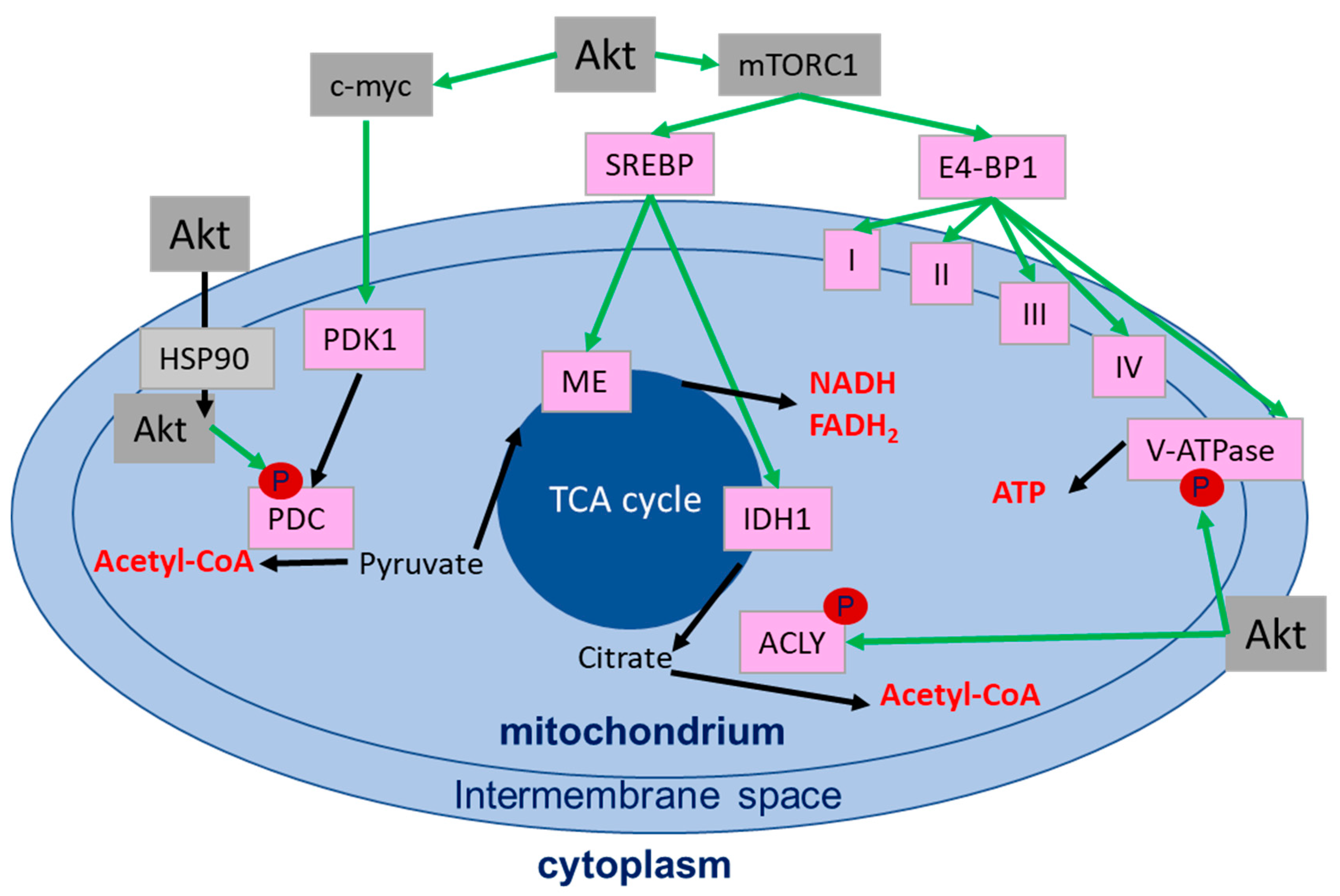
4.3. Role of Akt in the Regulation of the Mitochondrial Function
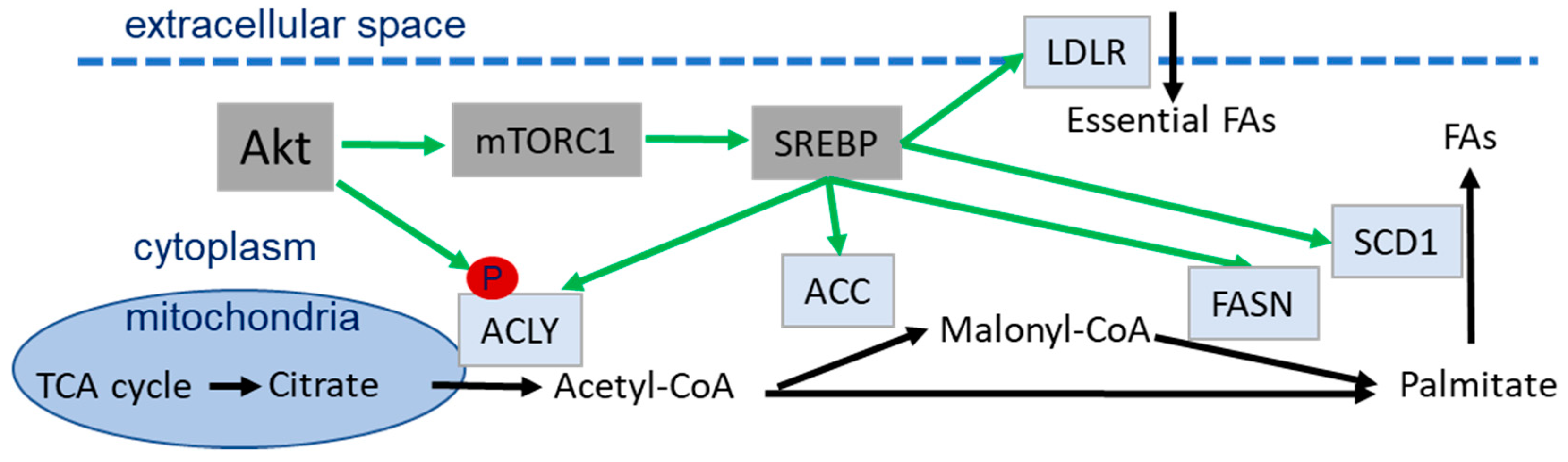
4.4. Role of Akt in Lipid Metabolism
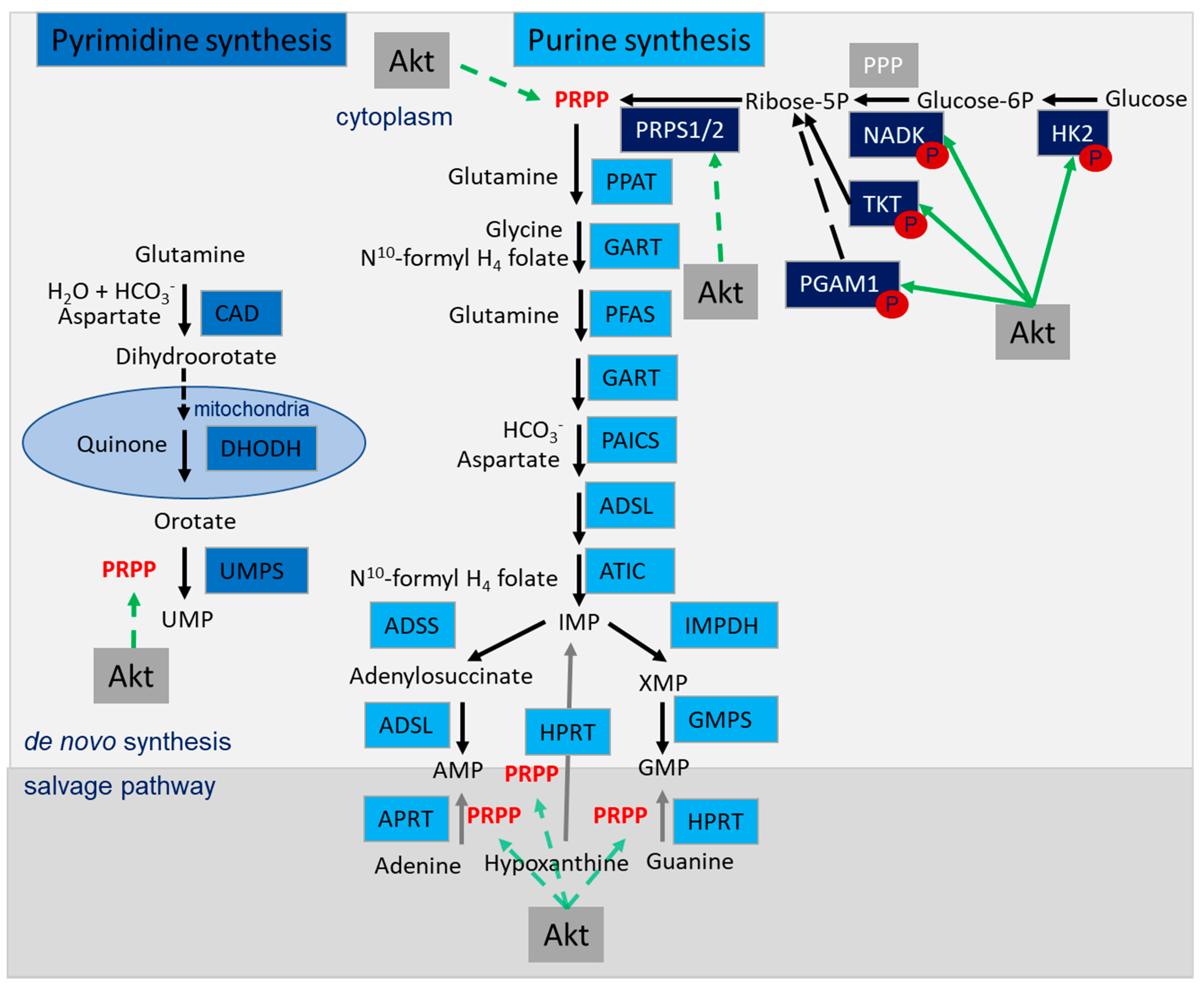
4.5. Role of Akt in the Regulation of Nucleotide Synthesis
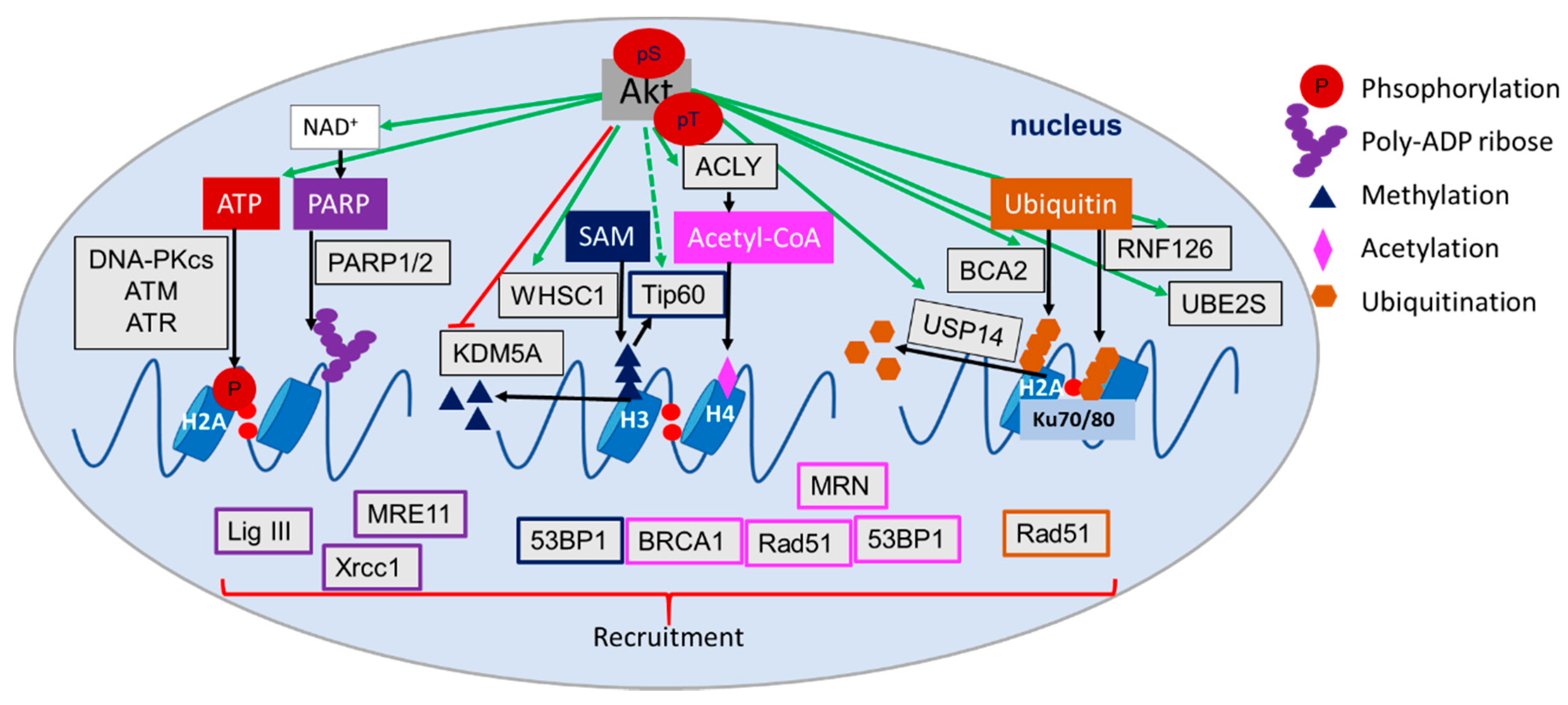
4.6. Role of Akt in the Regulation of (Histone) Acetylation, Methylation and Ubiquitination
5. Therapeutic Implications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Targets | References | |
|---|---|---|---|
| TCA & OxPhos | Respiratory chain | Complex I–V | [227,229] |
| Cytochrome bc1 complex | [228] | ||
| Cytochrome c oxidase | [228] | ||
| TCA cycle | IDH | [231,232] | |
| Citrate | [252] | ||
| Glycolysis | GLUTs | [222] | |
| HKs | [222] | ||
| PFK | [222] | ||
| PGAM1 | [222,224] | ||
| PKM2 | [222] | ||
| LDHA | [222] | ||
| MCTs | [222] | ||
| PDK | [222,225,226] | ||
| PDHA | [225,226] | ||
| Lipid metabolism | ACLY | [234,235] | |
| ACC | [191] | ||
| FASN | [191] | ||
| SCD1 | [191] | ||
| LDLR | [191] | ||
| Nucleotide pools | dNTP pool | [119,253] | |
| Antioxidant system | Thioredoxin system | TrX | [236,237] |
| TrxR | [236,237] | ||
| ASK1 | [237] | ||
| ROS | [237] | ||
| GSH | [237] | ||
| NADPH production | NADK | [239] | |
| NAMPT | [238] | ||
| Epigentic modifications | Acetylation | HATs | [246] |
| HDACs | [243,245,246] | ||
| Methylation | Histone (lys) methyltransferases | [246,247] | |
| protein arginine methyltransferases | [246,247] | ||
| histone lysine demethylases | [246,247] | ||
| DNA methyltransferases | [246] | ||
| Ubiquitination | E1 enzyme | [218,249,250] | |
| E2 enzyme | [218,249,250] | ||
| E3 ligases | [218,249,250] | ||
| Deubiquitinases (DUBs) | [218,220,248,249,250] | ||
| Parylation | PARP inhibitors | [240,241] | |
6. Conclusions and Outlook
7. Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1,3-BPG | 1,3-bisphosphogylcerate |
| 53BP1 | Tumor suppressor p53-binding protein 1 |
| ACC | acetyl-CoA carboxylase |
| ACLY | ATP citrate lyase |
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ASCT2 | amino acid transporter type 2 |
| ATM | ataxia telangiectasia mutated |
| ATP | Adenosine triphosphate |
| ATR | ataxia telangiectasia and Rad3 related |
| BCA2 | breast cancer associated gene 2 |
| BRCA1 | Breast cancer type 1 susceptibility protein |
| BRCC | BRCA1/BRCA2-containing complex |
| CAD | carbamoyl-phosphate synthetase |
| CAT | Catalase |
| CPT | carnitine palmitoyltransferase |
| CtIP | CTBP-interacting protein |
| DDR | DNA damage response |
| DNA-PKcs | DNA-dependent protein kinase, catalytic subunit |
| DNMT | DNA methyltransferase |
| DSB | Double strand break |
| DUB | deubiquitinase |
| EMSY | BRCA2-interacting transcriptional repressor |
| ENO1 | enolase |
| FADH2 | Flavin adenine dinucleotide |
| FAO | Fatty acid oxidation |
| FASN | fatty acid synthase |
| DHAP | dihydroxyacetone phosphate |
| FH | fumarate hydratase |
| FoxO | Forkhead-Box-Protein O3 |
| FTO | fat mass and obesity-associated protein |
| G6P | glucose-6-phosphate |
| GAP | Glycerine aldehyde phosphate |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GLS | Glutaminase |
| GLUT | Glucose transporter |
| GMP | guanine monophosphate |
| GPCR | G-protein coupled receptors |
| GrX | glutathione-glutaredoxin/glutathione peroxidase |
| GSH | reduced glutathione |
| GSK3 | glycogen synthase kinase 3 |
| GTP | Guanine triphosphate |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| HIF1 | Hypoxia inducible factor 1 |
| HK2 | Hexokinase 2 |
| HMT | Histone methyltransferase |
| HRR | homologous recombination repair |
| HSP90 | heat shock protein-90 |
| IDH | isocitrate dehydrogenase |
| INPP4B | Inositol polyphosphate 4-phosphatase type II |
| LDHA | lactate dehydrogenase |
| LDLR | low density lipoprotein receptor |
| MAT | methionine adenosyltransferase |
| MDM2 | Mouse double minute 2 homolog |
| MERIT40 | mediator of Rap80 interactions and targeting 40 kDa |
| MRN | Mre11, Rad50 and Nbs1 complex |
| MTHFD | formyltetrahydrofolate synthetase |
| MTHFD1/2 | Methylenetetrahydrofolate Dehydrogenase |
| mTOR | mechanistic target of rapamycin/mammalian target of rapamycin |
| NAAD | nicotinic acid adenine dinucleotide |
| NAD+/H | Nicotinamide adenine dinucleotide |
| NADK | NAD kinase |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NADS | NAD synthase |
| NAM | nicotinamide |
| NAMN | nicotinic acid mononucleotide |
| Nampt | nicotinamide phosphoribosyltransferase |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| NHEJ | non-homologous end joining |
| Nmnat | nicotinamide mononucleotide adenylyltransferase |
| Nqo1 | cytosolic flavoprotein, NAD(P)H Quinone Dehydrogenase 1 |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| NTP | Nucleotide triphosphate |
| OGDH | oxoglutarate dehydrogenase |
| OxPhos | oxidative phosphorylation |
| PARP | poly-(ADP-ribose) polymerase |
| PDC | pyruvate dehydrogenase complex |
| PDH | pyruvate dehydrogenase |
| PDK1 | Pyruvate dehydrogenase kinase 1 |
| PDK1 | phosphoinositide-dependent kinase 1 |
| PEP | Phosphoenolpyruvic acid |
| PFK | Phospho-fructo-kinase |
| PGAM1 | phosphoglycerate mutase 1 |
| PGK1 | phosphoglycerate kinase 1 |
| PH | pleckstrin homology |
| PHLPP1/2 | PH domain and leucine rich repeat protein phosphatase 1 |
| PI3K | Phosphatidyl-inositol-3-kinase |
| PKM2 | Pyruvate kinase M2 |
| PPP | pentose phosphate pathway |
| PRPP | phospho-ribose pyrophosphate |
| PRPS1/2 | ribose-phosphate pyrophosphokinase 1/2 |
| PTEN | phosphatase and tensin homologue |
| Rap80 | receptor-associated protein 80 |
| RNF126 | Ring finger protein 126 |
| ROS | reactive oxygen species |
| RTK | Receptor tyrosine kinase |
| S6K | S6 kinase |
| SAM | S-adenosylmethionine |
| SCD1 | stearoyl-coenzyme A desaturase 1 |
| SDH | succinate dehydrogenase |
| SIRT | Sirtuin |
| SOD | superoxide dismutase |
| SREBP | Sterol regulatory element binding proteins |
| TCA | tricarboxylic acid cycle |
| THF | tetrahydrofolate |
| TKT | transketolase |
| TNKS | tankerase |
| TrX | thioredoxin |
| TSC2 | tuberous sclerosis complex 2 |
| UBE2S | Ubiquitin conjugating enzyme E2 S |
| UMP | uridine monophosphate |
| XLF | XRCC4-like factor |
| XRCC1 | X-ray repair cross-complementing protein 1 |
References
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA Double-Strand Break Repair as Determinant of Cellular Radiosensitivity to Killing and Target in Radiation Therapy. Front. Oncol. 2013, 3, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing radiation and complex DNA damage: From prediction to detection challenges and biological significance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Gay, C.M.; Byers, L.A. Targeting DNA damage repair in small cell lung cancer and the biomarker landscape. Transl. Lung Cancer Res. 2018, 7, 50–68. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2011, 711, 61–72. [Google Scholar] [CrossRef]
- Jachimowicz, R.D.; Goergens, J.; Reinhardt, H.C. DNA double-strand break repair pathway choice—From basic biology to clinical exploitation. Cell Cycle 2019, 18, 1423–1434. [Google Scholar] [CrossRef]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef]
- Soria, G.; Polo, S.E.; Almouzni, G. Prime, Repair, Restore: The Active Role of Chromatin in the DNA Damage Response. Mol. Cell 2012, 46, 722–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ayrapetov, M.K.; Xu, C.; Gursoy-yuzugullu, O.; Hu, Y.; Price, B.D. H2AZ controls DSB repair. Mol. Cell 2012, 48, 723–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeenk, G.; van Attikum, H. The Chromatin Response to DNA Breaks: Leaving a Mark on Genome Integrity. Annu. Rev. Biochem. 2013, 82, 55–80. [Google Scholar] [CrossRef] [PubMed]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Shia, L.; Oberdoerffer, P. Chromatin dynamics in DNA double-strand break repair. Biochim. Biophys. Acta 2012, 1819, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Matschke, V.; Theiss, C.; Matschke, J. Oxidative stress: The lowest common denominator of multiple diseases. Neural Regen. Res. 2019, 14, 238–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Efimova, E.V.; Takahashi, S.; Shamsi, N.A.; Wu, D.; Labay, E.; Ulanovskaya, O.A.; Weichselbaum, R.R.; Kozmin, S.A.; Kron, S.J. Linking Cancer Metabolism to DNA Repair and Accelerated Senescence. Mol. Cancer Res. 2015, 14, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, M.O.; Perry, N.J.S.; Poulogiannis, G. DNA damage, repair, and cancer metabolism. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- van Vugt, M.A.T.M. Shutting down the power supply for DNA repair in cancer cells. J. Cell Biol. 2017, 216, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Xiang, K.; Jendrossek, V.; Matschke, J. Oncometabolites and the response to radiotherapy. Radiat. Oncol. 2020, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.B.; Lewis, A.L.; Baldwin, K.K.; Resnick, M.A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. USA 1993, 90, 5613–5617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matschke, J.; Riffkin, H.; Klein, D.; Handrick, R.; Lüdemann, L.; Metzen, E.; Shlomi, T.; Stuschke, M.; Jendrossek, V. Targeted Inhibition of Glutamine-Dependent Glutathione Metabolism Overcomes Death Resistance Induced by Chronic Cycling Hypoxia. Antioxid. Redox Signal. 2016, 25, 89–107. [Google Scholar] [CrossRef]
- Jutten, B.; Rouschop, K.M.A. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle 2014, 13, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wang, H.; De Ridder, M. Targeting antioxidant enzymes as a radiosensitizing strategy. Cancer Lett. 2018, 438, 154–164. [Google Scholar] [CrossRef]
- Toulany, M. Targeting DNA double-strand break repair pathways to improve radiotherapy response. Genes 2019, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Biau, J.; Chautard, E.; Verrelle, P.; Dutreix, M. Altering DNA repair to improve radiation therapy: Specific and multiple pathway targeting. Front. Oncol. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matschke, J.; Wiebeck, E.; Hurst, S.; Rudner, J.; Jendrossek, V. Role of SGK1 for fatty acid uptake, cell survival and radioresistance of NCI-H460 lung cancer cells exposed to acute or chronic cycling severe hypoxia. Radiat. Oncol. 2016, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hlouschek, J.; Hansel, C.; Jendrossek, V.; Matschke, J. The mitochondrial citrate carrier (SLC25A1) sustains redox homeostasis and mitochondrial metabolism supporting radioresistance of cancer cells with tolerance to cycling severe hypoxia. Front. Oncol. 2018, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kass, E.M.; Moynahan, M.E.; Jasin, M. When Genome Maintenance Goes Badly Awry. Mol. Cell 2016, 62, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, H.; McCabe, H.; Lord, C.J.; Tutt, A.H.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- McLornan, D.P.; List, A.; Mufti, G.J. Applying synthetic lethality for the selective targeting of cancer. N. Engl. J. Med. 2014, 371, 1725–1735. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: The First Synthetic Lethal Targeted Therapy. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Stadler, J.; Richly, H. Regulation of DNA repair mechanisms: How the chromatin environment regulates the DNA damage response. Int. J. Mol. Sci. 2017, 18, 1715. [Google Scholar] [CrossRef] [Green Version]
- Zernickel, E.; Sak, A.; Riaz, A.; Klein, D.; Groneberg, M.; Stuschke, M. Targeting of BRM sensitizes BRG1-mutant lung cancer cell lines to radiotherapy. Mol. Cancer Ther. 2019, 18, 656–666. [Google Scholar] [CrossRef] [Green Version]
- Riaz, M.A.; Sak, A.; Erol, Y.B.; Groneberg, M.; Thomale, J.; Stuschke, M. Metformin enhances the radiosensitizing effect of cisplatin in non-small cell lung cancer cell lines with different cisplatin sensitivities. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayo, J.; Tran, T.A.; Wang, L.; Peña-Llopis, S.; Das, A.K.; Martinez, E.D. Jumonji Inhibitors Overcome Radioresistance in Cancer through Changes in H3K4 Methylation at Double-Strand Breaks. Cell Rep. 2018, 25, 1040–1050.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scanlon, S.E.; Glazer, P.M. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair (Amst.) 2015, 32, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.R.; Glazer, P.M. Impact of hypoxia on DNA repair and genome integrity. Mutagenesis 2020, 35, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Koritzinsky, M.; Zhao, H.; Bindra, R.; Glazer, P.M.; Powell, S.; Belmaaza, A.; Wouters, B.; Bristow, R.G. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 2008, 68, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase-AKT pathway in humancancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2019, 20, 74–88. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulany, M.; Rodemann, H.P. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin. Cancer Biol. 2015, 35, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Narayan, R.; Giovannetti, E.; Peters, G.J. Role of Akt signaling in resistance to DNA-targeted therapy. World J. Clin. Oncol. 2016, 7, 352–369. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3421–3426. [Google Scholar] [CrossRef] [Green Version]
- Gol, T.M.; Rodemann, H.P.; Dittmann, K. Depletion of Akt1 and Akt2 impairs the repair of radiation-induced DNA double strand breaks via homologous recombination. Int. J. Mol. Sci. 2019, 20, 6316. [Google Scholar] [CrossRef] [Green Version]
- Mueck, K.; Rebholz, S.; Harati, M.D.; Rodemann, H.P.; Toulany, M. Akt1 stimulates homologous recombination repair of DNA double-strand breaks in a Rad51-dependent manner. Int. J. Mol. Sci. 2017, 18, 2473. [Google Scholar] [CrossRef] [Green Version]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKBα/Akt1 Acts Downstream of DNA-PK in the DNA Double-Strand Break Response and Promotes Survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Ther. 2008, 7, 1772–1781. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Huang, K.Y.; Yang, C.H.; Yang, Y.S.; Lee, W.Y.; Chiang, C.W. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55α regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J. Biol. Chem. 2008, 283, 1882–1892. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Bruhn, C.; Peretti, M.; Cassani, C.; Carotenuto, W.V.; Elgendy, M.; Shubassi, G.; Lucca, C.; Bermejo, R.; Varasi, M.; et al. PP2A Controls Genome Integrity by Integrating Nutrient-Sensing and Metabolic Pathways with the DNA Damage Response. Mol. Cell 2017, 67, 266–281.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor suppressor and metabolic regulator. Front. Endocrinol. (Lausanne) 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in Mice of the mTORC Components raptor, rictor, or mLST8 Reveals that mTORC2 Is Required for Signaling to Akt-FOXO and PKCα, but Not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persad, S.; Attwell, S.; Gray, V.; Mawji, N.; Deng, J.T.; Leung, D.; Yan, J.; Sanghera, J.; Walsh, M.P.; Dedhar, S. Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: Critical roles for kinase activity and amino acids arginine 211 and serine 343. J. Biol. Chem. 2001, 276, 27462–27469. [Google Scholar] [CrossRef] [Green Version]
- Oeck, S.; Al-Refae, K.; Riffkin, H.; Wiel, G.; Handrick, R.; Klein, D.; Iliakis, G.; Jendrossek, V. Activating Akt1 mutations alter DNA double strand break repair and radiosensitivity. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Guinea Viniegra, J.; Martínez, N.; Modirassari, P.; Hernández Losa, J.; Parada Cobo, C.; Sánchez-Arévalo Lobo, V.J.; Aceves Luquero, C.I.; Álvarez-Vallina, L.; Ramón, Y.; Cajal, S.; et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J. Biol. Chem. 2005, 280, 4029–4036. [Google Scholar] [CrossRef] [Green Version]
- Shiga, S.; Murata, Y.; Hashimoto, T.; Urushihara, Y.; Fujishima, Y.; Kudo, K.; Sonohara, Y.; Kurusu, M.; Takeda, K.; Jingu, K.; et al. DNA-PKcs is activated under nutrient starvation and activates Akt, MST1, FoxO3a, and NDR1. Biochem. Biophys. Res. Commun. 2020, 521, 668–673. [Google Scholar] [CrossRef]
- Khalil, A.; Morgan, R.N.; Adams, B.R.; Golding, S.E.; Dever, S.M.; Rosenberg, E.; Povirk, L.F.; Valerie, K. ATM-dependent ERK signaling via AKT in response to DNA double-strand breaks. Cell Cycle 2011, 10, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Maehama, T.; Dixon, J.E. The Tumor Suppressor, PTEN/MMAC1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agoulnik, I.U.; Hodgson, M.C.; Bowden, W.A.; Ittmann, M.M. INPP4B: The New Kid on the PI3K Block INPP4b structure ANd fuNctIoN. Oncotarget 2011, 2, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewinner, C.; Wang, Z.C.; Richardson, A.; Teruya-Feldstein, J.; Etemadmoghadam, D.; Bowtell, D.; Barretina, J.; Lin, W.M.; Rameh, L.; Salmena, L.; et al. Evidence that Inositol Polyphosphate 4-Phosphatase Type II Is a Tumor Suppressor that Inhibits PI3K Signaling. Cancer Cell 2009, 16, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, C.; Carlson, S.M.; White, F.M.; Davis, R.J. Mcl-1 Integrates the Opposing Actions of Signaling Pathways That Mediate Survival and Apoptosis. Mol. Cell. Biol. 2009, 29, 3845–3852. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Kops, G.J.P.L.; Burgering, B.M.T. Forkhead transcription factors: New insights into protein kinase B (c- akt) signaling. J. Mol. Med. 1999, 77, 656–665. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP Activity Is Regulated by mTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Guo, C.; Xie, A.; Gao, D.; Guo, J.; Zhang, J.; Willis, N.; Su, A.; Asara, J.M.; et al. Akt-mediated phosphorylation of XLF impairs non-homologous end-joining DNA repair. Mol. Cell 2015, 57, 648–661. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Li, X.; Liu, Q.; Xu, J.; Ge, H.; Wang, Z.; Wang, H.; Wang, Z.; Shi, C.; Xu, X.; et al. UBE2S, a novel substrate of Akt1, associates with Ku70 and regulates DNA repair and glioblastoma multiforme resistance to chemotherapy. Oncogene 2017, 36, 1145–1156. [Google Scholar] [CrossRef]
- Pham, H.T.; Nguyen, T.T.; Nguyen, L.P.; Han, S.S.; Lim, Y.S.; Hwang, S.B. Hepatitis C Virus Downregulates Ubiquitin-Conjugating Enzyme E2S Expression To Prevent Proteasomal Degradation. J. Virol. 2019, 93, 1–15. [Google Scholar]
- Paul, A.; Wang, B. RNF8- and Ube2S-Dependent Ubiquitin Lysine 11-Linkage Modification in Response to DNA Damage. Mol. Cell 2017, 66, 458–472.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altiok, S.; Batt, D.; Altiok, N.; Papautsky, A.; Downward, J.; Roberts, T.M.; Avrahamt, H. Heregulin induces phosphorylation of BRCA1 through phosphatidylinositol 3-kinase/AKT in breast cancer cells. J. Biol. Chem. 1999, 274, 32274–32278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, A.C.; Lyons, T.R.; Young, C.D.; Hansen, K.C.; Anderson, S.M.; Holt, J.T. AKT regulates BRCA1 stability in response to hormone signaling. Mol. Cell. Endocrinol. 2010, 319, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Montaser-Kouhsari, L.; Beck, A.H.; Toker, A. MERIT40 Is an Akt Substrate that Promotes Resolution of DNA Damage Induced by Chemotherapy. Cell Rep. 2015, 11, 1358–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Huang, J.; Chen, J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009, 23, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Shao, G.; Patterson-Fortin, J.; Messick, T.E.; Feng, D.; Shanbhag, N.; Wang, Y.; Greenberg, R.A. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009, 23, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Jelinic, P.; Eccles, L.A.; Tseng, J.; Cybulska, P.; Wielgos, M.; Powell, S.N.; Levine, D.A. The EMSY threonine 207 phospho-site is required for EMSYdriven suppression of DNA damage repair. Oncotarget 2017, 8, 13792–13804. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Wang, Z.; Yang, L.; Guo, X.; Yang, G. The function of EMSY in cancer development. Tumor Biol. 2014, 35, 5061–5066. [Google Scholar] [CrossRef]
- Holler, M.; Grottke, A.; Mueck, K.; Manes, J.; Jücker, M.; Rodemann, H.P.; Toulany, M. Dual targeting of Akt and mTORC1 impairs repair of DNA double-strand breaks and increases radiation sensitivity of human tumor cells. PLoS ONE 2016, 11, 1–19. [Google Scholar] [CrossRef]
- Iida, M.; Harari, P.M.; Wheeler, D.L.; Toulany, M. Targeting AKT/PKB to improve treatment outcomes for solid tumors. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2020, 819, 111690. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deberardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Ui, A.; Chiba, N.; Yasui, A. Relationship among DNA double-strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer Sci. 2020, 111, 1443–1451. [Google Scholar] [CrossRef]
- Dabin, J.; Fortuny, A.; Polo, S.E. Epigenome Maintenance in Response to DNA Damage. Mol. Cell 2016, 62, 712–727. [Google Scholar] [CrossRef] [Green Version]
- Lahtz, C.; Pfeifer, G.P. Epigenetic changes of DNA repair genes in cancer. J. Mol. Cell Biol. 2011, 3, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Mcdonald, J.T.; Kim, K.; Norris, A.; Vlashi, E.; Phillips, T.M.; Lagadec, C.; Donna, L.D.; Ratikan, J.; Szelag, H.; Hlatky, L.; et al. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010, 70, 8886–8895. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Ali, S.S.; Ahsan, H.; Zia, M.K.; Siddiqui, T.; Khan, F.H. Understanding oxidants and antioxidants: Classical team with new players. J. Food Biochem. 2020, 44, e13145. [Google Scholar] [CrossRef]
- Sies, H.; de Groot, H. Role of reactive oxygen species in cell toxicity. Toxicol. Lett. 1992, 64, 547–551. [Google Scholar] [CrossRef]
- Hlouschek, J.; Ritter, V.; Wirsdörfer, F.; Klein, D.; Jendrossek, V.; Matschke, J. Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018, 439, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P. ROS and radiotherapy: More we care. Oncotarget 2017, 8, 35482–35483. [Google Scholar] [CrossRef] [PubMed]
- Buj, R.; Aird, K.M. Deoxyribonucleotide triphosphate metabolism in cancer and metabolic disease. Front. Endocrinol. (Lausanne) 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Mitochondrial purine and pyrimidine metabolism and beyond. Nucleosides Nucleotides Nucleic Acids 2016, 35, 578–594. [Google Scholar] [CrossRef]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer cells tune the signaling pathways to empower de novo synthesis of nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef] [Green Version]
- Parker, W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef] [Green Version]
- Polo, S.E.; Jackson, S.P. POLO, JACKSON—2011—Prot reparo. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.C.; Burman, B.; Kruhlak, M.; Misteli, T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014, 9, 1703–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008, 4, e1000155. [Google Scholar] [CrossRef]
- Shanbhan, N.M.; Rafalska-Metcalf, I.U.; Balane- Bolivar, C.; Janicki, S.M.; Greenberg, R.A. An ATM-Dependent Transcriptional Silencing Program is Transmitted Through Chromatin in Cis to DNA Double Strand Breaks. Cell 2011, 141, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Kim, J.; Oberdoerffer, P. Metabolic modulation of chromatin: Implications for DNA repair and genomic integrity. Front. Genet. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuozzo, C.; Porcellini, A.; Angrisano, T.; Morano, A.; Lee, B.; Di Pardo, A.; Messina, S.; Iuliano, R.; Fusco, A.; Santillo, M.R.; et al. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007, 3, 1144–1162. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Sassone-Corsi, P. Metabolic signaling to chromatin. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–24. [Google Scholar] [CrossRef]
- Uckelmann, M.; Sixma, T.K. Histone ubiquitination in the DNA damage response. DNA Repair (Amst.) 2017, 56, 92–101. [Google Scholar] [CrossRef]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [Green Version]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Mailand, N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst.) 2010, 9, 1219–1228. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopfner, K.P. ATP puts the brake on DNA double-strand break repair: A new study shows that ATP switches the Mre11-Rad50-Nbs1 repair factor between signaling and processing of DNA ends Prospects & Overviews K.-P. Hopfner. BioEssays 2014, 36, 1170–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, M.M. Mismatch binding, ADP-ATP exchange and intramolecular signaling during mismatch repair. DNA Repair 2016, 38, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.E.; Hatzivassiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef] [Green Version]
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD metabolism: Implications in aging and longevity. Ageing Res. Rev. 2018, 47, 1–17. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Amé, J.C.; De Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Beneke, S. Regulation of chromatin structure by poly(ADP-ribosyl)ation. Front. Genet. 2012, 3, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Grillo, M.A.; Colombatto, S. S-adenosylmethionine and its products. Amino Acids 2008, 34, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Johnson, J.L.; Hanigan, C.L.; Locasale, J.W. Interactions between epigenetics and metabolism in cancers. Front. Oncol. 2012, 2, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.D.; Lum, M.A.; Xu, C.; Black, J.D.; Wang, X. Ubiquitin-dependent regulation of phospho-AKT Dynamics by the ubiquitin E3 LIGASE, NEDD4-1, in the insulin-like growth factor-1 response. J. Biol. Chem. 2013, 288, 1674–1684. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, L.Y.; Arsene, D.; Mato, J.M.; Lu, S.C. Methionine adenosyltransferases in cancers: Mechanisms of dysregulation and implications for therapy. Exp. Biol. Med. 2018, 243, 107–117. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D.; Cell, N.; Author, B. Histone H3 methylation links DNA damage detection to activation of the Tip60 tumor suppressor HHS Public Access Author manuscript. Nat. Cell Biol. 2009, 11, 1376–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penicud, K.; Behrens, A. DMAP1 is an essential regulator of ATM activity and function. Oncogene 2014, 33, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Lien, E.C.; Lyssiotis, C.A.; Juvekar, A.; Hu, H.; Asara, J.M.; Cantley, L.C.; Toker, A. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, V.; Patra, K.C.; Hay, N. Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. eLife 2018, 7, e32213. [Google Scholar] [CrossRef]
- Cerutti, P.; Ghosh, R.; Oya, Y.; Amstad, P. The Role of the Cellular Antioxidant Defense in Oxidant Carcinogenesis. Environ. Health Perspect. 1994, 102 (Suppl. 10), 123–129. [Google Scholar]
- Hoxhaj, G.; Ben-Sahra, I.; Lockwood, S.E.; Timson, R.C.; Byles, V.; Henning, G.T.; Gao, P.; Selfors, L.M.; Asara, J.M.; Manning, B.D. Direct stimulation of NADP + synthesis through Akt-mediated phosphorylation of NAD kinase. Science 2019, 363, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.; Niere, M.; Ziegler, M. NAD kinase levels control the NADPH concentration in human cells. J. Biol. Chem. 2007, 282, 33562–33571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Senapedis, W.T.; Kennedy, C.J.; Boyle, P.M.; Silver, P.A. Whole genome siRNA cell-based screen links mitochondria to Akt signaling network through uncoupling of electron transport chain. Mol. Biol. Cell 2011, 22, 1791–1805. [Google Scholar] [CrossRef]
- Weiss, C.N.; Ito, K. DNA damage response, redox status and hematopoiesis. Blood Cells, Mol. Dis. 2014, 52, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.K. Glucose transporters: Structure, function and consequences of deficiency. J. Inherit. Metab. Dis. 2000, 23, 237–246. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [Green Version]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Tozzi, M.G.; Camici, M.; Mascia, L.; Sgarrella, F.; Ipata, P.L. Pentose phosphates in nucleoside interconversion and catabolism. FEBS J. 2006, 273, 1089–1101. [Google Scholar] [CrossRef]
- Kim, J.W.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef]
- Fang, J.; Zhou, S.H.; Fan, J.; Yan, S.X. Roles of glucose transporter-1 and the phosphatidylinositol 3-kinase/protein kinase B pathway in cancer radioresistance (Review). Mol. Med. Rep. 2015, 11, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neary, C.L.; Pastorino, J.G. Nucleocytoplasmic shuttling of hexokinase II in a cancer cell. Biochem. Biophys. Res. Commun. 2010, 394, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef] [Green Version]
- Novellasdemunt, L.; Tato, I.; Navarro-Sabate, A.; Ruiz-Meana, M.; Méndez-Lucas, A.; Perales, J.C.; Garcia-Dorado, D.; Ventura, F.; Bartrons, R.; Luis Rosa, J. Akt-dependent activation of the heart 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase (PFKFB2) isoenzyme by amino acids. J. Biol. Chem. 2013, 288, 10640–10651. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Tan, X.; Liu, P.; Wu, Y.; Qian, S.; Zhang, X. Phosphoglycerate mutase 1 (PGAM1) promotes pancreatic ductal adenocarcinoma (PDAC) metastasis by acting as a novel downstream target of the PI3K/Akt/mTOR pathway. Oncol. Res. 2018, 26, 1123–1131. [Google Scholar] [CrossRef]
- Qu, J.; Sun, W.; Zhong, J.; Lv, H.; Zhu, M.; Xu, J.; Jin, N.; Xie, Z.; Tan, M.; Lin, S.H.; et al. Phosphoglycerate mutase 1 regulates dNTP pool and promotes homologous recombination repair in cancer cells. J. Cell Biol. 2017, 216, 409–424. [Google Scholar] [CrossRef]
- Ye, G.X.; Qin, Y.; Wang, S.; Pan, D.B.; Xu, S.Q.; Wu, C.J.; Wang, X.M.; Wang, J.; Ye, H.L.; Shen, H.J. Lamc1 promotes the Warburg effect in hepatocellular carcinoma cells by regulating PKM2 expression through AKT pathway. Cancer Biol. Ther. 2019, 20, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Liu, W.R.; Zhang, B.; Zhang, L.M.; Li, C.G.; Liu, C.; Zhang, H.; Huo, Y.S.; Ma, Y.C.; Tian, P.F.; et al. LncRNA H19 downregulation confers erlotinib resistance through upregulation of PKM2 and phosphorylation of AKT in EGFR-mutant lung cancers. Cancer Lett. 2020, 486, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Palmada, M.; Speil, A.; Jeyaraj, S.; Böhmer, C.; Lang, F. The serine/threonine kinases SGK1, 3 and PKB stimulate the amino acid transporter ASCT2. Biochem. Biophys. Res. Commun. 2005, 331, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ren, M.; Zeng, X.; He, P.; Ma, X.; Qiao, S. Leucine stimulates ASCT2 amino acid transporter expression in porcine jejunal epithelial cell line (IPEC-J2) through PI3K/Akt/mTOR and ERK signaling pathways. Amino Acids 2015, 46, 2633–2642. [Google Scholar] [CrossRef]
- Wise, D.R.; Deberardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.F.; Liu, Y.; Fan, Y.; Hua, S.N.; Qu, H.Y.; Dong, S.W.; Li, R.L.; Zhao, M.Y.; Zhen, Y.; Yu, X.L.; et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. J. Hematol. Oncol. 2015, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Lu, T.; Tian, K.; Zhou, D.; Yuan, J.; Wang, X.; Zhu, Z.; Wan, D.; Yao, Y.; Zhu, X.; et al. Alpha-enolase promotes gastric cancer cell proliferation and metastasis via regulating AKT signaling pathway. Eur. J. Pharmacol. 2019, 845, 8–15. [Google Scholar] [CrossRef]
- Dimri, M.; Humphries, A.; Laknaur, A.; Elattar, S.; Lee, T.J.; Sharma, A.; Kolhe, R.; Satyanarayana, A. NAD(P)H Quinone Dehydrogenase 1 Ablation Inhibits Activation of the Phosphoinositide 3-Kinase/Akt Serine/Threonine Kinase and Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Pathways and Blocks Metabolic Adaptation in Hepatocellular. Hepatology 2020, 71, 549–568. [Google Scholar] [CrossRef]
- Takeo, F.; Muhammad, R.K.; Courtney, D.D.; Johnique, T.A.F.J. The emerging role and targetability of the TCA cycle in cancer metabolism. Discov. Med. 2016, 21, 373–380. [Google Scholar] [CrossRef]
- Guo, R.; Gu, J.; Zong, S.; Wu, M.; Yang, M. Structure and mechanism of mitochondrial electron transport chain. Biomed. J. 2018, 41, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Barksdale, K.A.; Bijur, G.N. The basal flux of Akt in the mitochondria is mediated by heat shock protein-90. J. Neurochem. 2009, 108, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. MTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goo, C.K.; Lim, H.Y.; Ho, Q.S.; Too, H.P.; Clement, M.V.; Wong, K.P. PTEN/Akt Signaling Controls Mitochondrial Respiratory Capacity through 4E-BP1. PLoS ONE 2012, 7, 1–12. [Google Scholar] [CrossRef]
- McGuire, C.M.; Forgac, M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J. Biol. Chem. 2018, 293, 9113–9123. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.C.; Yang, J.C.; Chang, Y.C.; Chuang, J.G.; Lin, C.W.; Wu, M.S.; Chow, L.P. VCP Phosphorylation-Dependent Interaction Partners Prevent Apoptosis in Helicobacter pylori-Infected Gastric Epithelial Cells. PLoS ONE 2013, 8, e55724. [Google Scholar] [CrossRef]
- Cerniglia, G.J.; Dey, S.; Gallagher-Colombo, S.M.; Daurio, N.A.; Tuttle, S.; Busch, T.M.; Lin, A.; Sun, R.; Esipova, T.V.; Vinogradov, S.A.; et al. The PI3K/Akt pathway regulates oxygen metabolism via pyruvate dehydrogenase (PDH)-E1α phosphorylation. Mol. Cancer Ther. 2015, 14, 1928–1938. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Leu, H.B.; Chen, Y.; Chen, Y.H.; Epperson, C.M.; Juang, C.; Wang, P.H. Protein kinase b (pkb/akt1) formed signaling complexes with mitochondrial proteins and prevented glycolytic energy dysfunction in cultured cardiomyocytes during ischemia-reperfusion injury. Endocrinology 2014, 155, 1618–1628. [Google Scholar] [CrossRef]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of Acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [Green Version]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology-divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Rohlenova, K.; Neuzil, J.; Rohlena, J. The role of Her2 and other oncogenes of the PI3K/AKT pathway in mitochondria. Biol. Chem. 2016, 397, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.L. PI-3-K and AKT: Onto the mitochondria. Adv. Drug Deliv. Rev. 2009, 61, 1276–1282. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, L.; Xu, S.; Shen, A. zong ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: An updated review. Prog. Lipid Res. 2020, 77, 101006. [Google Scholar] [CrossRef]
- Saavedra-García, P.; Nichols, K.; Mahmud, Z.; Fan, L.Y.N.; Lam, E.W.F. Unravelling the role of fatty acid metabolism in cancer through the FOXO3-FOXM1 axis. Mol. Cell. Endocrinol. 2018, 462, 82–92. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, R.; Zhang, L.; Li, J.; Lou, K.; Shi, B. The lipid metabolism gene FTO influences breast cancer cell energy metabolism via the PI3K/AKT signaling pathway. Oncol. Lett. 2017, 13, 4685–4690. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; Brozzetti, S.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Ridgway, N.D. Substrate channeling in the glycerol-3-phosphate pathway regulates the synthesis, storage and secretion of glycerolipids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158438. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017, 67, 252–265.e6. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.X.; Peng, X.H.; Xiong, Y.; Liao, D.F.; Cao, D.; Li, L. Dual role of insulin-like growth factor-1 in acetyl-CoA carboxylase-alpha activity in human colon cancer cells HCT-8: Downregulating its expression and phosphorylation. Mol. Cell. Biochem. 2011, 357, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Hove-Jensen, B.; Andersen, K.R.; Kilstrup, M.; Martinussen, J.; Switzer, R.L.; Willemoes, M. Phosphoribosyl Diphosphate (PRPP): Biosynthesis, Enzymology, Utilization, and Metabolic Significanc. Microbiol. Mol. Biol. Rev. 2017, 81, 1–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, A.; Connelly, S.; Jiang, J.; Zhuang, S.; Amador, D.T.; Phan, T.; Pilz, R.B.; Boss, G.R. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol. Cell 2014, 55, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 2014, 157, 1088–1103. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Linder, S.J.; Mostoslavsky, R. Put Your Mark Where Your Damage Is: Acetyl-CoA Production by ACLY Promotes DNA Repair. Mol. Cell 2017, 67, 165–167. [Google Scholar] [CrossRef] [Green Version]
- Ghobashi, A.H.; Kamel, M.A. Tip60: Updates. J. Appl. Genet. 2018, 59, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, X.; Wang, C.; Zhang, S.; Wang, Z.; Li, M.; Wang, Y.; Wang, X.; Yang, X. HDAC6, modulated by miR-206, promotes endometrial cancer progression through the PTEN/AKT/mTOR pathway. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zheng, Y.; Zhang, W.; Hou, L.; Gao, Y. Scutellarin circumvents chemoresistance, promotes apoptosis, and represses tumor growth by HDAC/miR-34a-mediated down-modulation of Akt/mTOR and NF-κB-orchestrated signaling pathways in multiple myeloma. Int. J. Clin. Exp. Pathol. 2020, 13, 212–219. [Google Scholar] [PubMed]
- Chai, R.; Fu, H.; Zheng, Z.; Liu, T.; Ji, S.; Li, G. Resveratrol inhibits proliferation and migration through SIRT1 mediated post-translational modification of PI3K/AKT signaling in hepatocellular carcinoma cells. Mol. Med. Rep. 2017, 16, 8037–8044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Zhao, F.; Liu, J.; Liu, H. ASCT2 Is Involved in SARS-Mediated β-Casein Synthesis of Bovine Mammary Epithelial Cells with Methionine Supply. J. Agric. Food Chem. 2019. [Google Scholar] [CrossRef]
- Li, N.; Xue, W.; Yuan, H.; Dong, B.; Ding, Y.; Liu, Y.; Jiang, M.; Kan, S.; Sun, T.; Ren, J.; et al. AKT-mediated stabilization of histone methyltransferase WHSC1 promotes prostate cancer metastasis. J. Clin. Investig. 2017, 127, 1284–1302. [Google Scholar] [CrossRef] [Green Version]
- Spangle, J.M.; Dreijerink, K.M.; Groner, A.C.; Cheng, H.; Ohlson, C.E.; Reyes, J.; Lin, C.Y.; Bradner, J.; Zhao, J.J.; Roberts, T.M.; et al. PI3K/AKT Signaling Regulates H3K4 Methylation in Breast Cancer. Cell Rep. 2016, 15, 2692–2704. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Liu, W.; Sun, J.; Kong, Y.; Chen, C. Roles of RNF126 and BCA2 E3 ubiquitin ligases in DNA damage repair signaling and targeted cancer therapy. Pharmacol. Res. 2020, 155, 104748. [Google Scholar] [CrossRef]
- Liu, B.; Chen, J.; Zhang, S. Emerging role of ubiquitin-specific protease 14 in oncogenesis and development of tumor: Therapeutic implication. Life Sci. 2019, 239, 116875. [Google Scholar] [CrossRef]
- Sharma, A.; Alswillah, T.; Singh, K.; Chatterjee, P.; Willard, B.; Venere, M.; Summers, M.K.; Almasan, A. USP14 regulates DNA damage repair by targeting RNF168-dependent ubiquitination. Autophagy 2018, 14, 1976–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, H.; Tang, W. Glycolysis Inhibitors for Anticancer Therapy: A Review of Recent Patents. Recent Pat. Anticancer. Drug Discov. 2016, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sun, Q.; Li, H.; Li, K.; Ren, X. The role of phosphoglycerate mutase 1 in tumor aerobic glycolysis and its potential therapeutic implications. Int. J. Cancer 2014, 135, 1991–1996. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, X. Phosphoglycerate mutase 1: Its glycolytic and non-glycolytic roles in tumor malignant behaviors and potential therapeutic significance. Onco. Targets. Ther. 2020, 13, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A. Metabolic flexibility in cancer: Targeting the pyruvate dehydrogenase kinase:pyruvate dehydrogenase axis. Mol. Cancer Ther. 2019, 18, 1673–1681. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; Gillies McKenna, W.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [Green Version]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Emmings, E.; Mullany, S.; Chang, Z.; Landen, C.N.; Linder, S.; Bazzaro, M. Targeting mitochondria for treatment of chemoresistant ovarian cancer. Int. J. Mol. Sci. 2019, 20, 229. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Oeck, S.; West, A.P.; Mangalhara, K.C.; Sainz, A.G.; Newman, L.E.; Zhang, X.O.; Wu, L.; Yan, Q.; Bosenberg, M.; et al. Mitochondrial DNA stress signalling protects the nuclear genome. Nat. Metab. 2019, 1, 1209–1218. [Google Scholar] [CrossRef]
- Liu, X.; Gong, Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark. Res. 2019, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Khawaja, M.R.; DiNardo, C.D.; Atkins, J.T.; Janku, F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov. Med. 2016, 21, 373–380. [Google Scholar]
- Guo, D.; Bell, E.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven Lipid Metabolism to Treat Cancer. Curr. Pharm. Des. 2015, 20, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, X.-Y.; Zhang, Q.-H.; Liu, J.-H.; Cao, R.-X.; Zhong, J.; Yi, G.-H.; Quan, Z.-H.; Pizzorno, G. ATP Citrate Lyase Inhibitors as Novel Cancer Therapeutic Agents. Recent Pat. Anticancer. Drug Discov. 2012, 7, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 2018, 157, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, F.; Soltani, A.; Ghahremanloo, A.; Javid, H.; Hashemy, S.I. The thioredoxin system and cancer therapy: A review. Cancer Chemother. Pharmacol. 2019, 84, 925–935. [Google Scholar] [CrossRef]
- Jia, J.J.; Geng, W.S.; Wang, Z.Q.; Chen, L.; Zeng, X.S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Pramono, A.A.; Rather, G.M.; Herman, H.; Lestari, K.; Bertino, J.R. NAD- and NADPH-Contributing Enzymes as Therapeutic Targets in Cancer: An Overview. Biomolecules 2020, 10, 358. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, P.M.; Bansal, N.; Kerrigan, J.E.; Abali, E.E.; Scotto, K.W.; Bertino, J.R. NAD+ kinase as a therapeutic target in cancer. Clin. Cancer Res. 2016, 22, 5189–5195. [Google Scholar] [CrossRef] [Green Version]
- Tangutoori, S.; Baldwin, P.; Sridhar, S. PARP inhibitors: A new era of targeted therapy. Maturitas 2015, 81, 5–9. [Google Scholar] [CrossRef]
- Hou, W.H.; Chen, S.H.; Yu, X. Poly-ADP ribosylation in DNA damage response and cancer therapy. Mutat. Res. Rev. Mutat. Res. 2019, 780, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Gallyas, F.; Sumegi, B.; Szabo, C. Role of akt activation in PARP inhibitor resistance in cancer. Cancers 2020, 12, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; Volume 138. [Google Scholar]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Duan, Y.-T.; Lu, P.; Zhang, Z.-J.; Zheng, X.-K.; Wang, J.-L.; Feng, W.-S. Epigenetic Targets and their Inhibitors in Cancer Therapy. Curr. Top. Med. Chem. 2019, 18, 2395–2419. [Google Scholar] [CrossRef]
- McCabe, M.T.; Mohammad, H.P.; Barbash, O.; Kruger, R.G. Targeting Histone Methylation in Cancer. Cancer J. 2017, 23, 292–301. [Google Scholar] [CrossRef]
- Wertz, I.E.; Murray, J.M. Structurally-defined deubiquitinase inhibitors provide opportunities to investigate disease mechanisms. Drug Discov. Today Technol. 2019, 31, 109–123. [Google Scholar] [CrossRef]
- Liu, J.; Shaik, S.; Dai, X.; Wu, Q.; Zhou, X.; Wang, Z.; Wei, W. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2015, 1855, 50–60. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, P.; Wang, X.; Linder, S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015, 147, 32–54. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xu, L. UBE2S promotes the proliferation and survival of human lung adenocarcinoma cells. BMB Rep. 2018, 51, 642–647. [Google Scholar] [CrossRef]
- Huang, L.; Wang, C.; Xu, H.; Peng, G. Targeting citrate as a novel therapeutic strategy in cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188332. [Google Scholar] [CrossRef] [PubMed]
- Puigvert, J.C.; Sanjiv, K.; Helleday, T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 2016, 283, 232–245. [Google Scholar] [CrossRef] [PubMed]









| Antioxidant Defense | Nucleotide Synthesis | Glycolysis | Glutaminolysis | SAM Production | TCA & OxPhos | Lipid Metabolism |
|---|---|---|---|---|---|---|
| NADPH | PRPP | Pyruvate | αKG | Met | ATP | Acetyl-CoA |
| GSH | NADH | Lactate | ATP | SAM | NADH | Malonyl-CoA |
| NADPH | NADH | FADH2 | Palmitate | |||
| Pyrimidine | ATP | Acetyl-CoA | Fatty acids | |||
| Purine | ||||||
| dNTPs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Götting, I.; Jendrossek, V.; Matschke, J. A New Twist in Protein Kinase B/Akt Signaling: Role of Altered Cancer Cell Metabolism in Akt-Mediated Therapy Resistance. Int. J. Mol. Sci. 2020, 21, 8563. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228563
Götting I, Jendrossek V, Matschke J. A New Twist in Protein Kinase B/Akt Signaling: Role of Altered Cancer Cell Metabolism in Akt-Mediated Therapy Resistance. International Journal of Molecular Sciences. 2020; 21(22):8563. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228563
Chicago/Turabian StyleGötting, Isabell, Verena Jendrossek, and Johann Matschke. 2020. "A New Twist in Protein Kinase B/Akt Signaling: Role of Altered Cancer Cell Metabolism in Akt-Mediated Therapy Resistance" International Journal of Molecular Sciences 21, no. 22: 8563. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228563