Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease?



1
Department of Pathology, University of Pisa, 56100 Pisa, Italy
2
Department of New Biology, Daegu Gyeongbuk Institute of Science & Technology (DGIST), Daegu 42988, Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(22), 8672; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228672
Submission received: 3 November 2020
/
Revised: 14 November 2020
/
Accepted: 15 November 2020
/
Published: 17 November 2020
(This article belongs to the Special Issue Understanding Amyloid Structures and Disease: A Continuing Challenge in Health Research)
Abstract
:Transthyretin (TTR), previously named prealbumin is a plasma protein secreted mainly by the liver and choroid plexus (CP) that is a carrier for thyroid hormones (THs) and retinol (vitamin A). The structure of TTR, with four monomers rich in β-chains in a globular tetrameric protein, accounts for the predisposition of the protein to aggregate in fibrils, leading to a rare and severe disease, namely transthyretin amyloidosis (ATTR). Much effort has been made and still is required to find new therapeutic compounds that can stabilize TTR (“kinetic stabilization”) and prevent the amyloid genetic process. Moreover, TTR is an interesting therapeutic target for neurodegenerative diseases due to its recognized neuroprotective properties in the cognitive impairment context and interestingly in Alzheimer’s disease (AD). Much evidence has been collected regarding the neuroprotective effects in AD, including through in vitro and in vivo studies as well as a wide range of clinical series. Despite this supported hypothesis of neuroprotection for TTR, the mechanisms are still not completely clear. The aim of this review is to highlight the most relevant findings on the neuroprotective role of TTR, and to summarize the recent progress on the development of TTR tetramer stabilizers.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Transthyretin (TTR) is a plasma protein secreted mainly by the liver and choroid plexus (CP) [1]. It was identified in 1942 and initially denominated “prealbumin”, as it migrates just in front of the albumin band in electrophoresis gels [2]. Later it was found that prealbumin could bind thyroid hormones (THs) [3] and retinol-binding protein (RBP) [4], therefore biochemists adopted the name “transthyretin” (TTR) to indicate its role as a transporter for thyroid hormones and retinol (vitamin A). [5]
Although its function may vary, TTR is a highly-conserved 55-kDa homotetrameric protein present in several vertebrate species, including humans, and also observed in bacteria, nematodes, and plants [6,7,8]. The X-ray crystal structure of human TTR shows that the protein has a globular shape, comprising four identical 127-amino-acid β-sheet-rich subunits [9]. The four monomers in the tetramer interact with each other through non-covalent bonds. A strong interaction between two monomers generates dimers that assemble as a tetramer producing a central hydrophobic channel lined by amino acids from both dimers. This channel has two similar binding sites for THs [10] (Figure 1). These binding sites can also accommodate other small compounds present in plasma, such as metabolism derivatives, components of the diet, or even small molecules administered as drugs [11].
The predominance of the β-chain structure in the polypeptide chains of the TTR tetramer, and its organization as β-sheets contribute to the intrinsic propensity of the protein to aggregate, leading to the formation and deposition of fibrils under specific conditions (Figure 2), thus originating transthyretin amyloidosis (ATTR), a rare, yet underdiagnosed disease characterized by progressive impairment of neurologic and cardiac function [12,13]. Notably, ATTR includes two subtypes—wild-type (ATTRwt) and variant ATTR (ATTRv)—that differ regarding the pathogenesis of amyloidosis [14]. Variant TTR deposition causes autosomal-dominant hereditary ATTR amyloidosis. The three main phenotypes of hereditary ATTR amyloidosis are familial amyloid polyneuropathy (FAP), familial amyloid cardiomyopathy (FAC), and familial leptomeningeal amyloidosis. On the other hand, wild-type ATTR deposition leads to an acquired amyloid disease, senile systemic amyloidosis (SSA), which typically develops later than hereditary ATTR [15,16]. Despite great efforts, the poor prognosis for patients with both ATTRwt and ATTRv remains unchanged, thus the availability of effective and less invasive treatments compared to liver transplantation, the current first-line treatment for hereditary ATTR amyloidosis [17], is urgently required. Over the past two decades, much has been learned about the factors that influence the propensity of TTR to aggregate [18]. Advanced biophysical information led to the development of a therapeutic strategy, termed, ‘kinetic stabilization’ to prevent wt- and v-TTR amyloidogenesis. Several stabilizing compounds that predominantly bind to the most unoccupied TH binding sites were found, the most representative being diflunisal and tafamidis [19,20].
Besides its well-known role in the transport of THs and vitamin A, TTR is increasingly recognized as possessing neuroprotective properties in multiple contexts, including cerebral ischemia and Alzheimer’s disease (AD) [21,22].
The earliest description of a protective role for TTR in AD dates back to 1994 when Schwarzman and co-workers reported TTR to be the major Aβ-binding protein in cerebrospinal fluid (CSF) [23]. These authors described that TTR was able to inhibit Aβ aggregation and toxicity, suggesting that when TTR fails to sequester Aβ, amyloid formation occurs [23,24]. These results have been further confirmed in various subsequent studies. In particular, studies by Buxbaum and co-workers [25] on APP transgenic mice revealed that overexpression of human (h)TTR ameliorated AD features. Increased brain Aβ levels and deposition were observed by Oliveira et al. [26] in APP transgenic mice with genetic reduction of TTR. In a triple transgenic mouse model of AD (3xTg-AD), with CP dysfunction and defective CSF production, a diminished secretion of TTR was observed [27]. Notably, Alemi et al. [28] demonstrated that TTR can transport Aβ from, but not into, the brain. Moreover, the same authors also showed that TTR increased Aβ internalization by SAHep cells (human hepatoma cells) and by primary hepatocytes derived from TTR+/+ mice when compared to TTR−/− animals [28]. Since a lower low-density lipoprotein receptor-related protein 1 (LRP1) expression was found in brains and livers of TTR−/− mice and in cells incubated without TTR, the authors proposed that TTR acts as a carrier of Aβ at the blood–brain barrier and liver, using LRP1 [28].
Despite a large body of evidence supporting the idea of neuroprotection by TTR [29,30,31,32], substantially related to decreased levels of TTR in both CSF and plasma of AD patients, the cause of TTR reduction in AD is not known yet. The aim of this review is to highlight the most relevant findings on the neuroprotective role of TTR.
2. TTR Physiology and Metabolism
TTR is predominantly synthesized and secreted by the liver and CP to the plasma and cerebrospinal fluid (CSF), respectively [1]. TTR plasma concentration is age-dependent, and in healthy newborns, it is about half that in adults [33,34]. TTR values vary from 20 to 40 mg/dL [33]. In spite of the low TTR levels in CSF (~2 mg/dL), the CP is presented as the major site of TTR expression, and TTR represents approximately 20% of the total CSF protein content [35]. Besides the liver and the CP, TTR is produced in the retina, pancreas (α cells), and to a small extent in the heart, skeletal muscle, stomach, and spleen [1,36,37,38].
TTR binds and transports about 15% of serum thyroxine (T4) and up to 80% of T4 in the CNS [39,40]. The four monomers of the TTR tetramer, demarcate through the molecule an open channel that has two binding sites for THs [41]. These two binding sites present negative cooperativity [42], implying that once the first TH molecule occupies the first site, the binding affinity for the second molecule is profoundly reduced. Thus, just one molecule of T4 is transported by TTR.
In addition to transporting T4, TTR also transports vitamin A (retinol) from its main storage site in the liver to target tissues [4]. The transport of vitamin A in circulation occurs through retinol-binding protein (RBP) [43]. TTR associates to the RBP–retinol complex before secretion into the plasma, generating a very stable form of retinol transport which allows its delivery to cells while preventing renal filtration and subsequent degradation [44,45] Under physiological conditions, due to low RBP levels compared to those of TTR, just one molecule of RBP is transported by the TTR tetramer [46,47]. Notably, T4 binding to TTR is not affected by RBP binding [4].
Besides its role in the transport of T4 and vitamin A another important function of TTR is its proteolytic activity on several substrates [48,49], including the apoliprotein A-I (apoA-I) [50], neuropeptide Y (NPY) [51], and Aβ peptide [52], further corroborating the relevance of TTR under both physiological and pathological conditions.
3. The role of TTR in the Nervous System
Several reports have shown different roles for TTR in nervous system physiology [53]. Studies with TTR-null (TTR−/−) [31,54] mice revealed that these animals present reduced signs of depressive-like behavior and increased exploratory activity and anxiety, probably due to increased levels of noradrenaline in the limbic forebrain [55].
In addition, further evidence of the importance of TTR in the modulation of depressive behavior came after observing increased levels of NPY in the dorsal root ganglia (DRG), sciatic nerve, spinal cord, hippocampus, cortex, and CSF of TTR−/− mice [30,56]. Additionally, Sousa and coworkers also described that TTR−/− mice display memory impairment compared with wild-type (TTR+/+) animals, suggesting that the absence of TTR worsens cognitive deficits, a trait that is usually associated with aging [55].
In addition, studies by Fleming et al. demonstrated for the first time that TTR enhances nerve regeneration [30]. Indeed, under nerve crush conditions, the absence of TTR slowed nerve regeneration. Later, the same authors demonstrated that TTR delivery to crushed sciatic nerves rescued the regeneration phenotype in TTR-null animals, further confirming the role of TTR as a nerve regeneration enhancer [56] They also showed that the neurogenic activity of TTR is mediated by megalin-dependent internalization [56].
The neuroprotective role of TTR has been widely documented in animal models of cerebral ischemia [57,58,59]. Notably, in a mouse model of permanent middle cerebral artery occlusion (pMCAO), Santos and co-workers [29] showed that in conditions of a compromised heat-shock response, CSF TTR contributes to control neuronal cell death and cerebral edema and inflammation, thereby influencing brain protection and neuro-repair processes.
4. The Protective Role of TTR in Alzheimer’s Disease
Alzheimer’s disease is a neurodegenerative disorder that involves a progressive memory deficit, cognitive decline, and behavioral disturbances. The two main histopathological marks of AD are the intraneuronal presence of neurofibrillary tangles consisting of aggregates of hyper- phosphorylated tau protein, and the extracellular accumulation of senile plaques consisting of aggregates of amyloid-β peptide (Aβ) [60,61,62,63]. Lowering Aβ levels is a major therapeutic goal in AD, which might be achieved by interfering with the production, aggregation, or degradation of the peptide [64,65]
The knowledge of TTR involvement in the process of Aβ fibril formation dates back to the end of the last century [60] when it was found that TTR was able to bind Aβ40 and form stable complexes [23]. In 1986, Elovaara et al. [66] also reported the first description of decreased TTR levels in CSF of AD patients. At present it is commonly accepted by the scientific community that the neuroprotective effect of TTR in AD is linked to the following aspects: (1) TTR levels are decreased in the CSF of AD patients [67], (2) overexpressing human TTR wild type (hTTR-wt) in an AD mouse model normalizes cognition and memory [25], and (3) in vitro TTR reduces Aβ fibrillation [52,68,69].
TTR levels have been measured with different methods in plasma and in CSF of patients with AD and correlated with the development and prognosis of the disease. In 1997 Serot et al. obtained a total of 149 samples of CSF from control patients (n = 109 young, middle aged, and elderly controls) and patients with AD (n = 40). They measured TTR by the kinetic nephelemetric automated method and showed that TTR levels were correlated with age, but were significantly lower in patients with AD, compared with controls of the same age [67]. In 2008 Gloeckner et al. conducted a similar study on a total of 106 samples of CSF from patients with five types of dementia, that included AD and control healthy people. The levels of TTR, measured with the same nephelemetric method, were found to be significantly reduced in AD, with significantly-lower levels in patients with severe AD compared with mild forms; moreover a 15 ng/mL cut off value was established that was highly prognostic of the severity of the disease [70].
TTR levels were also measured in the plasma of patients with AD (n = 111) by ELISA (enzyme-linked immunosorbent assay) assay and they were confirmed to be significantly reduced, independently of age, when compared with controls (n = 90) [71]. Moreover, TTR levels have been found to correlate with AD stage and with female gender in a study by Ribeiro et al [72] and also to be predictive of cognitive decline in the ensuing months in a study by Velayudhan et al. [73] Taken together these data suggest a possible role for TTR as a peripheral biomarker for an early diagnosis of AD.
None of the previous studies prospectively evaluated the role of TTR as biomarker of initial dementia. Very recently Tien et al. reported the results of a 5-year longitudinal study that followed the progression of 184 patients with amnestic mild cognitive impairment (MCI) and 40 sex-matched controls. In this study, TTR was demonstrated to be an independent predictor for MCI conversion to AD (p = 0.023, 95% CI 1–1.007) [74].
In animal models Choi et al. crossed mice harbouring FAD-linked APPswe and PS1_E9 transgenes with TTR knockout mice and showed that in these animals the deposition of Aβ was higher and accelerated compared with the same model and normal TTR, suggesting a role for TTR in modulating the timing and the amount of Aβ deposition [75]. These data were confirmed in the same mouse model by Oliveira et al., who also found a possible role of TTR in the modulation of the major association between female gender and AD [26].
Finally, the group of Buxbaum et al. obtained a model of APP23 AD-like and increased expression of TTR, that displayed a significant improvement in the Barnes maze test for cognitive function and spatial learning, when compared with APP23 AD-like and normal TTR expression [76].
The reason for the hypothesized TTR role in AD neuroprotection has been partially elucidated by in vitro studies. Costa et al. performed competition binding assays using soluble Aβ peptide and recombinant 125I-TTR to test the interaction between TTR and Aβ; they showed that TTR could bind Aβ, with different mutated TTRs binding Aβ with different affinities. Moreover, they found that TTR was capable of interfering with Aβ fibrillization by both inhibiting and disrupting fibril formation, since co-incubation with the two molecules resulted in the abolishment of Aβ toxicity [52].
Despite extensive research that spanned about three decades, the exact mechanism by which TTR modulates the process of converting monomeric Aβ into amyloid fibrils, as well as the relevance of Aβ cleavage by TTR, still remains unknown.
Animal models of AD, such as Caenorhabditis elegans (CE) transgenic for human Aβ42 and TTR [77], and mice transgenic models for mutant forms of the amyloid β precursor protein (APP), expressing different levels of endogenous TTR or hTTR [78], have been widely used to dissect the mechanism behind the disease. Recently, using biophysical methods, Ghadami et al. [79] provided compelling evidence that the protective role exerted by TTR in AD, lies in the ability of TTR to inhibit the microscopic steps of both primary and secondary nucleation of Aβ aggregation, in turn limiting both the toxicity of Aβ oligomers and the ability of the fibrils to proliferate.
In addition to TTR's well-documented ability to bind Aβ peptides, TTR proteolytic activity has been also shown to impact on Aβ fibrillogenesis [21], neuronal-secreted Aβ degradation, and reduction of Aβ-induced toxicity in hippocampal neurons [52,68], thereby contributing to the neuroprotective effect of TTR in AD. Future in vivo studies will be required to address whether TTR proteolytic activity is therapeutically relevant in AD.
5. Transthyretin Stability in Alzheimer’s Disease
Alterations in TTR tetramer are observed in AD. In subjects with mild cognitive impairment (MCI) reduced levels of TTR are detected, with the decrease becoming more pronounced with disease progression [80]. Although the underlying cause is not fully established, it has been proposed that TTR tetramer instability might play a role [81], suggesting that for an optimal binding to Aβ the tetramer form of TTR is required [80,82].
Destabilization of TTR may results from gene mutations [82]. It is widely known that TTR protein destabilized by TTR gene mutation has a tendency to dissociate into monomers, which then misfold and aggregate into amyloid fibrils, leading to autosomal-dominant hereditary amyloidosis, including familial amyloid polyneuropathy (FAP), familial amyloid cardiomyopathy (FAC), and familial leptomeningeal amyloidosis. To date, more than 140 mutations in TTR with amyloidogenic potential have been reported. The V30M TTR variant is the most common amyloidogenic form in the pathology, which leads to FAP, but other clinically-aggressive mutants have been described, including V122I, which is the most-common mutation found in cardiac amyloidosis, and L55P [26,27]. Notably, it was shown that the TTR mutation Leu-55-Pro (L55P TTR) significantly altered tetramer stability and increased amyloidogenicity under physiological conditions [72,83]. The L55P TTR tetramer was revealed to also be very sensitive to acidic conditions, readily dissociating to form the monomeric amyloidogenic intermediate between pH 5.5–5.0, while wild-type TTR remains stable and nonamyloidogenic [83].
Furthermore, T119M TTR has been described as a non-amyloidogenic transthyretin variant. Longo Alves et al. compared the stability and clearance of V30M TTR and T119M TTR and described that the more stable properties of T119M variant could be involved in the protective clinical effect of the T119M mutation in FAP. Baures et al. and Oza et al., studied several small compounds that are structurally similar to the TTR natural ligand (T4) and share the same TTR binding sites as T4, proposing them as inhibitors of TTR fibril formation in vitro [13]. In addition, with regard to the stabilization of TTR, Costa et al. described that TTR mutations, such as T119M, Y78F, V30M, and L55P, bind differently to Aβ. The observation of an inverse relationship between the amyloidogenic potential of TTR and the affinity for Aβ peptide is suggestive of a direct correlation between TTR stability and its neuroprotective properties [6].
An association of TTR variants in AD patients has been observed in a limited number of studies [84,85], therefore further studies are required to confirm whether TTR gene variability does play a major role in AD. Other events, including acidification of the medium, failure of the folding system, or interaction with various components such as metal ions and carbohydrates, may affect TTR stability [86]. Consistently, plasma TTR from AD patients showed decreased ability to bind T4 and decreased folded/monomeric ratios [70,71,80]. Therefore, it has been proposed that the lower concentration levels of TTR observed in the plasma of AD patients may result from the fast clearance of altered TTR. The loss in TTR tetrameric structure decreases Aβ affinity and therefore does not allow TTR to exert its protective effect in AD. Taken together this evidence has led to the hypothesis that TTR stabilization would restore both its plasma levels and proper binding to Aβ, hence its neuroprotective role in AD.
Ultimately, it is important to mention that TTR expression, in the liver and CP, is regulated by 17β-estradiol [87]. Interestingly, it has been reported that in senescence, the AD incidence is higher in women compared to men, suggesting that estrogens may play a relevant role in this process [88]. Thus, decreased estrogens levels in AD women [80] may contribute to the decline in TTR concentration observed in AD.
In view of the fact that the amyloidogenic potential of TTR is inversely correlated with its stability, the use of drugs able to stabilize the TTR tetrameric fold could result in increased TTR/Aβ interaction.
6. Transthyretin Tetramer Stabilizers
The first evidence of TTR stabilization through binding of small molecules derived from the observation that when TTR is bound to T4 it is less prone to aggregation. In addition, since in both the CSF and plasma the two T4 binding sites within TTR are largely unoccupied (<1% T4 bound) [9], small compounds able to bind tightly to the TTR-T4 binding sites may confer TTR stabilization.
Several non-steroidal anti-inflammatory drugs (NSAIDs), such as salicylates, diclofenac, flufenamic acid, and diflunisal, have been known for a long time to compete with T4 for the binding to TTR, [89]. Among these, diflunisal was one of the most-promising compounds due to its affinity and specificity to bind TTR. In addition, several diflunisal derivatives have been synthetized to improve its affinity and selectivity to bind TTR in plasma [90,91]. Interestingly, in a revealing experiment using an AD transgenic mouse model with TTR genetic reduction (AD/TTR+/-), administration of the iodinated derivative of diflunisal, namely iododiflunisal (IDIF) resulted in decreased amyloid burden and total Aβ brain levels, along with improved cognitive function of the animals [92]. Notably, recent data showed that the TTR/IDIF complex exhibits improved Blood Brain Barrier (BBB) permeability compared to TTR and IDIF alone [93], providing higher Aβ sequestering capacity, and adding to the therapeutic potential of TTR in AD.
Tafamidis, a benzoxazole derivative that binds to T4-binding sites of TTR efficiently inhibiting the dissociation of tetramers, recently emerged as a very promising drug for the treatment of familial amyloid polyneuropathy [20] and TTR-mediated amyloid cardiomyopathy (ATTR-CM) [94].
A number of therapies to reduce the amount of transthyretin protein have been in the pipeline for years. Recent studies suggested the possibility of antisense oligonucleotides (ASO) and small interfering RNA (siRNA) as genetic therapeutic agents for blocking ATTR expression. Indeed, ASOs and siRNAs could cleave mRNA prior to protein synthesis and inhibit ATTR production.
Recently, positive Phase III data on inotersen, an antisense oligonucleotide against TTR developed by Ionis Pharmaceuticals and Akcea Therapeutics, have been published [95]. Meanwhile, Alnylam has developed two RNAi drugs against TTR, namely revusiran and patisiran. These two drugs differ in their delivery strategy [96,97] —revusiran is chemically conjugated to a sugar called N-acetylgalactosamine (GalNAc), whereas patisiran is encapsulated in a nanoparticle made of a synthetic lipid called DLin-MC3-DMA [98,99] Revusiran failed in a Phase III trial [100], whereas positive Phase III results were obtained for patisiran [101]. This resulted in patisiran being the first siRNA-based drug to be approved by the U.S. Food and Drug Administration for the treatment of hereditary transthyretin amyloidosis [102,103]. Nevertheless, additional investigations on novel treatment strategies are still needed to understand the pathological role of ATTR in amyloidoses.
In the recent years, nutraceutical strategies focused on investigating the ability of natural polyphenols, such as resveratrol, to inhibit amyloid fibril aggregation, revealing for many of them neuroprotective properties in a number of experimental settings [104]. Indeed, administration of resveratrol to AD mice with TTR genetic reduction produced a significant decrease of brain Aβ accumulation and a rise in plasma TTR concentration, confirming the stabilization hypothesis. Nevertheless, different mechanisms may be involved in resveratrol action on Aβ brain levels, as it is reported that this polyphenol promotes intracellular proteasomal degradation of Aβ, while not affecting the production of the peptide [105]. Cellular studies also indicated that both resveratrol and IDIF efficiently stabilized TTR, leading to an improved TTR-assisted Aβ transport at the BBB [22].
Other polyphenols, including nordihydroguaiaretic acid (NDGA), rosmarinic acid, caffeic acid, and epigallocatechin gallate (EGCG), have also been investigated in vitro for their interaction with TTR [106,107]. The results of a small pilot clinical study with EGCG administration revealed a reduction of myocardial mass in the case of cardiomyopathy, indicating an inhibitory effect of EGCG on TTR amyloid fibril formation [108,109].
In the last fifteen years, an increasing amount of evidence underlined that in mice transgenic for human TTR V30M, supplementation with curcumin, a natural phenol produced by turmeric plants, reduces TTR load and degrades amyloid deposits in tissues, therefore targeting multiple steps in the ATTR amyloidogenic cascade [110].
In AD mice with TTR genetic reduction, evidence of a thicker basement membrane, a hallmark of AD, has been shown [51], likely reflecting vascular alterations thought to occur early, and prior to Aβ deposition, during AD development. Therefore, taking into account that currently Aβ-based therapies do not meet much favor, the stabilization of TTR can offer a new therapeutic target in the early treatment of AD.
7. Conclusions
Several lines of evidence suggest that TTR has a neuroprotective role in AD, and the TTR/Aβ complex is emerging as a possible new target for AD. Taking into account that, to date, there are no effective disease-slowing or -modifying treatments for AD, the discovery that TTR stabilization, through the use of small-molecule compounds, enhances the TTR/Aβ interaction, opens a new avenue that relies on the recovery of TTR activity, without affecting gene expression. Additional research will be needed to validate TTR/Aβ as a target for AD, and to move active molecules addressing this target in preclinical studies towards clinical trials for AD patients.
Author Contributions
F.S., J.H.K. and G.C. wrote and reviewed the article. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Soprano, D.R.; Herbert, J.; Soprano, K.J.; Schon, E.A.; Goodman, D.S. Demonstration of transthyretin mRNA in the brain and other extrahepatic tissues in the rat. J. Biol. Chem. 1985, 260, 11793–11798. [Google Scholar] [PubMed]
- Vieira, M.; Saraiva, M.J. Transthyretin: A multifaceted protein. Biomol. Concepts 2014, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- INGBAR, S.H. Pre-albumin: A thyroxinebinding protein of human plasma. Endocrinology 1958. [Google Scholar] [CrossRef]
- Raz, A.; Goodman, D.S. The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J. Biol. Chem. 1969, 244, 3230–3237. [Google Scholar] [PubMed]
- Nomenclature Committee of the International Union of Biochemistry (NC-IUB). Enzyme Nomenclature. Recommendations 1978. Supplement 1: Corrections and additions. Eur. J. Biochem. 1980. [Google Scholar] [CrossRef]
- Schreiber, G.; Richardson, S.J.; Baldwin, J. The evolution of gene expression, structure and function of transthyretin. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997, 116, 137–160. [Google Scholar] [CrossRef]
- Power, D.M.; Elias, N.P.; Richardson, S.J.; Mendes, J.; Soares, C.M.; Santos, C.R.A. Evolution of the thyroid hormone-binding protein, transthyretin. Gen. Comp. Endocrinol. 2000, 119, 241–255. [Google Scholar] [CrossRef] [Green Version]
- Eneqvist, T.; Lundberg, E.; Nilsson, L.; Abagyan, R.; Sauer-Eriksson, A.E. The transthyretin-related protein family. Eur. J. Biochem. 2003. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A.; Benson, M.D. Transthyretin: A review from a structural perspective. Cell. Mol. Life Sci. 2001, 58, 1491–1521. [Google Scholar] [CrossRef]
- Wojtczak, A. Crystal structure of rat transthyretin at 2.5 Å resolution: First report on a unique tetrameric structure. Acta Biochim. Pol. 1997. [Google Scholar] [CrossRef]
- Palaninathan, S.K. Nearly 200 X-Ray Crystal Structures of Transthyretin: What Do They Tell Us About This Protein and the Design of Drugs for TTR Amyloidoses? Curr. Med. Chem. 2012, 19, 2324–2342. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Kincaid, J.C. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007, 36, 411–423. [Google Scholar] [CrossRef]
- Sekijima, Y. Transthyretin (ATTR) amyloidosis: Clinical spectrum, molecular pathogenesis and disease-modifying treatments. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y. Hereditary Transthyretin Amyloidosis Summary Genetic counseling GeneReview Scope Suggestive Findings. In GeneReviews NCBI Bookshelf; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2001; pp. 1–28. [Google Scholar]
- Cornwell, G.G.; Murdoch, W.L.; Kyle, R.A.; Westermark, P.; Pitkänen, P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 1983. [Google Scholar] [CrossRef]
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, G.; Steen, L.; Suhr, O.; Ericzon, B.G.; Groth, C.G.; Andersen, O.; Wallin, B.G.; Seymour, A.; Richardson, S.; Hawkins, P.N.; et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet 1993. [Google Scholar] [CrossRef]
- Sekijima, Y. Recent progress in the understanding and treatment of transthyretin amyloidosis. J. Clin. Pharm. Ther. 2014, 39, 225–233. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA-J. Am. Med. Assoc. 2013. [Google Scholar] [CrossRef] [Green Version]
- Coelho, T.; Maia, L.F.; Da Silva, A.M.; Cruz, M.W.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceição, I.M.; Schmidt, H.H.J.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012. [Google Scholar] [CrossRef]
- Silva, C.S.; Eira, J.; Ribeiro, C.A.; Oliveira, Â.; Sousa, M.M.; Cardoso, I.; Liz, M.A. Transthyretin neuroprotection in Alzheimer’s disease is dependent on proteolysis. Neurobiol. Aging 2017. [Google Scholar] [CrossRef]
- Alemi, M.; Silva, S.C.; Santana, I.; Cardoso, I. Transthyretin stability is critical in assisting beta amyloid clearance–Relevance of transthyretin stabilization in Alzheimer’s disease. CNS Neurosci. Ther. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K.; et al. Transthyretin sequesters amyloid β protein and prevents amyloid formation. Proc. Natl. Acad. Sci. USA 1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzman, A.L.; Goldgaber, D. Interaction of transthyretin with amyloid β-protein: Binding and inhibition of amyloid formation. CIBA Found. Symp. 1996. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Aβ toxicity. Proc. Natl. Acad. Sci. USA 2008. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.M.; Ribeiro, C.A.; Cardoso, I.; Saraiva, M.J. Gender-dependent transthyretin modulation of brain amyloid-β Levels: Evidence from a mouse model of alzheimer’s disease. J. Alzheimers Dis. 2011. [Google Scholar] [CrossRef] [Green Version]
- González-Marrero, I.; Giménez-Llort, L.; Johanson, C.E.; Carmona-Calero, E.M.; Castañeyra-Ruiz, L.; Brito-Armas, J.M.; Castañeyra-Perdomo, A.; Castro-Fuentes, R. Choroid plexus dysfunction impairs beta-amyloid clearance in a triple transgenic mouse model of alzheimer’s disease. Front. Cell. Neurosci. 2015. [Google Scholar] [CrossRef] [Green Version]
- Alemi, M.; Gaiteiro, C.; Ribeiro, C.A.; Santos, L.M.; Gomes, J.R.; Oliveira, S.M.; Couraud, P.O.; Weksler, B.; Romero, I.; Saraiva, M.J.; et al. Transthyretin participates in beta-amyloid transport from the brain to the liver-involvement of the low-density lipoprotein receptor-related protein 1? Sci. Rep. 2016. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.D.; Lambertsen, K.L.; Clausen, B.H.; Akinc, A.; Alvarez, R.; Finsen, B.; Saraiva, M.J. CSF transthyretin neuroprotection in a mouse model of brain ischemia. J. Neurochem. 2010. [Google Scholar] [CrossRef]
- Fleming, C.E.; Saraiva, M.J.; Sousa, M.M. Transthyretin enhances nerve regeneration. J. Neurochem. 2007. [Google Scholar] [CrossRef]
- Sousa, J.C.; Marques, F.; Dias-Ferreira, E.; Cerqueira, J.J.; Sousa, N.; Palha, J.A. Transthyretin influences spatial reference memory. Neurobiol. Learn. Mem. 2007. [Google Scholar] [CrossRef]
- Li, X.; Masliah, E.; Reixach, N.; Buxbaum, J.N. Neuronal production of transthyretin in human and murine alzheimer’s disease: Is it protective? J. Neurosci. 2011. [Google Scholar] [CrossRef] [Green Version]
- Stabilini, R.; Vergani, C.; Agostoni, A.; Agostoni, R.P.V. Influence of age and sex on prealbumin levels. Clin. Chim. Acta 1968. [Google Scholar] [CrossRef]
- Vahlquist, A.; Rask, L.; Peterson, P.A.; Berg, T. The concentrations of retinol-binding protein, prealbumin, and transferrin in the sera of newly delivered mothers and children of various ages. Scand. J. Clin. Lab. Invest. 1975. [Google Scholar] [CrossRef]
- Schreiber, G.; Aldred, A.R.; Jaworowski, A.; Nilsson, C.; Achen, M.G.; Segal, M.B. Thyroxine transport from blood to brain via transthyretin synthesis in choroid plexus. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1990. [Google Scholar] [CrossRef]
- Pfeffer, B.A.; Becerra, S.P.; Borst, D.E.; Won, P. Expression of transthyretin and retinol binding protein mRNAs and secretion of transthyretin by cultured monkey retinal pigment epithelium. Mol. Vis. 2004, 10, 23–30. [Google Scholar] [PubMed]
- Kato, M.; Kato, K.; Blaner, W.S.; Chertow, B.S.; Goodman, D.S. Plasma and cellular retinoid-binding proteins and transthyretin (prealbumin) are all localized in the islets of Langerhans in the rat. Proc. Natl. Acad. Sci. USA 1985. [Google Scholar] [CrossRef] [Green Version]
- Jacobsson, B.; Collins, V.P.; Grimelius, L.; Pettersson, T.; Sandstedt, B.; Carlstrom, A. Transthyretin immunoreactivity in human and porcine liver, choroid plexus, and pancreatic islets. J. Histochem. Cytochem. 1989. [Google Scholar] [CrossRef]
- Hagen, G.A.; Solberg, L.A. Brain and cerebrospinal fluid permeability to intravenous thyroid hormones. Endocrinology 1974. [Google Scholar] [CrossRef]
- Palha, J.A. Transthyretin as a thyroid hormone carrier: Function revisited. Clin. Chem. Lab. Med. 2002. [Google Scholar] [CrossRef]
- Blake, C.C.F.; Geisow, M.J.; Swan, I.D.A.; Rerat, C.; Rerat, B. Structure of human plasma prealbumin at 2.5 A resolution. A preliminary report on the polypeptide chain conformation, quaternary structure and thyroxine binding. J. Mol. Biol. 1974. [Google Scholar] [CrossRef]
- Tomar, D.; Khan, T.; Singh, R.R.; Mishra, S.; Gupta, S.; Surolia, A.; Salunke, D.M. Crystallographic Study of Novel Transthyretin Ligands Exhibiting Negative-Cooperativity between Two Thyroxine Binding Sites. PLoS ONE 2012. [Google Scholar] [CrossRef] [Green Version]
- Kanai, M.; Raz, A.; Goodman, D.S. Retinol-binding protein: The transport protein for vitamin A in human plasma. J. Clin. Invest. 1968. [Google Scholar] [CrossRef] [Green Version]
- Epstein, F.H.; Goodman, D.S. Vitamin A and Retinoids in Health and Disease. N. Engl. J. Med. 1984, 310, 1023–1031. [Google Scholar] [CrossRef]
- Noy, N.; Slosberg, E.; Scarlatal, S. Interactions of Retinol with Binding Proteins: Studies with Retinol-Binding Protein and with Transthyretin. Biochemistry 1992. [Google Scholar] [CrossRef]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 1995. [Google Scholar] [CrossRef]
- Van Bennekum, A.M.; Wei, S.; Gamble, M.V.; Vogel, S.; Piantedosi, R.; Gottesman, M.; Episkopoui, V.; Blaner, W.S. Biochemical basis for depressed serum retinol levels in transthyretin-deficient mice. J. Biol. Chem. 2001. [Google Scholar] [CrossRef] [Green Version]
- Liz, M.A.; Faro, C.J.; Saraiva, M.J.; Sousa, M.M. Transthyretin, a new cryptic protease. J. Biol. Chem. 2004. [Google Scholar] [CrossRef] [Green Version]
- Liz, M.A.; Gomes, C.M.; Saraiva, M.J.; Sousa, M.M. ApoA-I cleaved by transthyretin has reduced ability to promote cholesterol efflux and increased amyloidogenicity. J. Lipid Res. 2007. [Google Scholar] [CrossRef] [Green Version]
- Sousa, M.M.; Berglund, L.; Saraiva, M.J. Transthyretin in high density lipoproteins: Association with apolipoprotein A-I. J. Lipid Res. 2000, 41, 58–65. [Google Scholar]
- Liz, M.A.; Fleming, C.E.; Nunes, A.F.; Almeida, M.R.; Mar, F.M.; Choe, Y.; Craik, C.S.; Powers, J.C.; Bogyo, M.; Sousa, M.M. Substrate specificity of transthyretin: Identification of natural substrates in the nervous system. Biochem. J. 2009. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Ferreira-da-Silva, F.; Saraiva, M.J.; Cardoso, I. Transthyretin protects against A-beta peptide toxicity by proteolytic cleavage of the peptide: A mechanism sensitive to the kunitz protease inhibitor. PLoS ONE 2008. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.M.; Cardoso, I.; Saraiva, M.J. Transthyretin: Roles in the nervous system beyond thyroxine and retinol transport. Expert Rev. Endocrinol. Metab. 2012, 7, 181–189. [Google Scholar] [CrossRef]
- Brouillette, J.; Quirion, R. Transthyretin: A key gene involved in the maintenance of memory capacities during aging. Neurobiol. Aging 2008. [Google Scholar] [CrossRef]
- Carlos Sousa, J.; Grandela, C.; Fernández-Ruiz, J.; De Miguel, R.; De Sousa, L.; Magalhães, A.I.; João Saraiva, M.; Sousa, N.; Palha, J.A. Transthyretin is involved in depression-like behaviour and exploratory activity. J. Neurochem. 2004. [Google Scholar] [CrossRef] [Green Version]
- Fleming, C.E.; Mar, F.M.; Franquinho, F.; Saraiva, M.J.; Sousa, M.M. Transthyretin internalization by sensory neurons is megalin mediated and necessary for its neuritogenic activity. J. Neurosci. 2009. [Google Scholar] [CrossRef]
- Chen, R.; Vendrell, I.; Chen, C.P.; Cash, D.; O’Toole, K.G.; Williams, S.A.; Jones, C.; Preston, J.E.; Wheeler, J.X. Proteomic analysis of rat plasma following transient focal cerebral ischemia. Biomark. Med. 2011. [Google Scholar] [CrossRef]
- Suzuyama, K.; Shiraishi, T.; Oishi, T.; Ueda, S.; Okamoto, H.; Furuta, M.; Mineta, T.; Tabuchi, K. Combined proteomic approach with SELDI-TOF-MS and peptide mass fingerprinting identified the rapid increase of monomeric transthyretin in rat cerebrospinal fluid after transient focal cerebral ischemia. Mol. Brain Res. 2004. [Google Scholar] [CrossRef]
- Liverman, C.S.; Cui, L.; Yong, C.; Choudhuri, R.; Klein, R.M.; Welch, K.M.A.; Berman, N.E.J. Response of the brain to oligemia: Gene expression, c-Fos, and Nrf2 localization. Mol. Brain Res. 2004. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Funato, H.; Enya, M.; Yoshimura, M.; Morishima-Kawashima, M.; Ihara, Y. Presence of sodium dodecyl sulfate-stable amyloid β-protein dimers in the hippocampus CA1 not exhibiting neurofibrillary tangle formation. Am. J. Pathol. 1999. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Forman, M.S.; Trojanowski, J.Q.; Lee, V.M.Y. Neurodegenerative diseases: A decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004, 10, 1055–1063. [Google Scholar] [CrossRef]
- Elovaara, I.; Maury, C.P.J.; Palo, J. Serum amysoid A protein, albumin and prealbumin in Alzheimer’s disease and in demented patients with Down’s syndrome. Acta Neurol. Scand. 1986. [Google Scholar] [CrossRef]
- Serot, J.M.; Christmann, D.; Dubost, T.; Couturier, M. Cerebrospinal fluid transthyretin: Aging and late onset Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1997. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, X.; Ladiwala, A.R.A.; Du, D.; Yadav, J.K.; Tessier, P.M.; Wright, P.E.; Kelly, J.W.; Buxbaum, J.N. Mechanisms of transthyretin inhibition of β-amyloid aggregation in vitro. J. Neurosci. 2013. [Google Scholar] [CrossRef]
- Liu, L.; Murphy, R.M. Kinetics of inhibition of β-amyloid aggregation by transthyretin. Biochemistry 2006. [Google Scholar] [CrossRef]
- Gloeckner, S.F.; Meyne, F.; Wagner, F.; Heinemann, U.; Krasnianski, A.; Meissner, B.; Zerr, I. Quantitative analysis of transthyretin, tau and amyloid-β in patients with dementia. J. Alzheimers Dis. 2008. [Google Scholar] [CrossRef]
- Han, S.H.; Jung, E.S.; Sohn, J.H.; Hong, H.J.; Hong, H.S.; Kim, J.W.; Na, D.L.; Kim, M.; Kim, H.; Ha, H.J.; et al. Human serum transthyretin levels correlate inversely with Alzheimer’s disease. J. Alzheimers Dis. 2011. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, C.A.; Saraiva, M.J.; Cardoso, I. Stability of the Transthyretin Molecule as a Key Factor in the Interaction with A-Beta Peptide-Relevance in Alzheimer’s Disease. PLoS ONE 2012. [Google Scholar] [CrossRef] [Green Version]
- Velayudhan, L.; Killick, R.; Hye, A.; Kinsey, A.; Güntert, A.; Lynham, S.; Ward, M.; Leung, R.; Lourdusamy, A.; To, A.W.M.; et al. Plasma transthyretin as a candidate marker for Alzheimer’s disease. J. Alzheimers Dis. 2012. [Google Scholar] [CrossRef] [Green Version]
- Tien, Y.T.; Lee, W.J.; Liao, Y.C.; Wang, W.F.; Jhang, K.M.; Wang, S.J.; Fuh, J.L. Plasma Transthyretin as a Predictor of Amnestic Mild Cognitive Impairment Conversion to Dementia. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef]
- Kim, H.S.; Choi, Y.; Shin, K.Y.; Joo, Y.; Lee, Y.K.; Jung, S.Y.; Suh, Y.H.; Kim, J.H. Swedish amyloid precursor protein mutation increases phosphorylation of eIF2α in vitro and in vivo. J. Neurosci. Res. 2007. [Google Scholar] [CrossRef] [PubMed]
- Tagoe, C.E.; Reixach, N.; Friske, L.; Mustra, D.; French, D.; Gallo, G.; Buxbaum, J.N. In vivo stabilization of mutant human transthyretin in transgenic mice. Amyloid 2007. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D. Expression of human β-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995. [Google Scholar] [CrossRef] [Green Version]
- Se, H.C.; Leight, S.N.; Lee, V.M.Y.; Li, T.; Wong, P.C.; Johnson, J.A.; Saraiva, M.J.; Sisodia, S.S. Accelerated Aβ deposition in APPswe/PS1ΔE9 mice with hemizygous deletions of TTR (transthyretin). J. Neurosci. 2007. [Google Scholar] [CrossRef] [Green Version]
- Ghadami, S.A.; Chia, S.; Ruggeri, F.S.; Meisl, G.; Bemporad, F.; Habchi, J.; Cascella, R.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Transthyretin Inhibits Primary and Secondary Nucleations of Amyloid-β Peptide Aggregation and Reduces the Toxicity of Its Oligomers. Biomacromolecules 2020. [Google Scholar] [CrossRef]
- A. Ribeiro, C.; Santana, I.; Oliveira, C.; Baldeiras, I.; Moreira, J.; Joao Saraiva, M.; Cardoso, I. Transthyretin Decrease in Plasma of MCI and AD Patients: Investigation of Mechanisms for Disease Modulation. Curr. Alzheimer Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Murphy, R.M. Characterization of the interaction of β-Amyloid with Transthyretin monomers and tetramers. Biochemistry 2010. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Gonçalves, A.; Saraiva, M.J.; Cardoso, I. Transthyretin binding to A-Beta peptide-Impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008. [Google Scholar] [CrossRef] [Green Version]
- McCutchen, S.L.; Colon, W.; Kelly, J.W. Transthyretin Mutation Leu-55-Pro Significantly Alters Tetramer Stability and Increases Amyloidogenicity. Biochemistry 1993. [Google Scholar] [CrossRef]
- Xiang, Q.; Bi, R.; Xu, M.; Zhang, D.F.; Tan, L.; Zhang, C.; Fang, Y.; Yao, Y.G. Rare Genetic Variants of the Transthyretin Gene Are Associated with Alzheimer’s Disease in Han Chinese. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef]
- Sassi, C.; Ridge, P.G.; Nalls, M.A.; Gibbs, R.; Ding, J.; Lupton, M.K.; Troakes, C.; Lunnon, K.; Al-Sarraj, S.; Brown, K.S.; et al. Influence of coding variability in APP-Aβ metabolism genes in sporadic Alzheimer’s disease. PLoS ONE 2016. [Google Scholar] [CrossRef] [Green Version]
- Hurshman, A.R.; White, J.T.; Powers, E.T.; Kelly, J.W. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 2004. [Google Scholar] [CrossRef] [PubMed]
- Quintela, T.; Gonçalves, I.; Baltazar, G.; Alves, C.H.; Saraiva, M.J.; Santos, C.R.A. 17β-estradiol induces transthyretin expression in murine choroid plexus via an oestrogen receptor dependent pathway. Cell. Mol. Neurobiol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Candore, G.; Balistreri, C.R.; Grimaldi, M.P.; Vasto, S.; Listì, F.; Chiappelli, M.; Licastro, F.; Lio, D.; Caruso, C. Age-related inflammatory diseases: Role of genetics and gender in the pathophysiology of Alzheimer’s disease. Proc. Ann. N. Y. Acad. Sci. 2006, 10889, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.L.; Lim, C.F.; Hall, J.G.; Barlow, J.W.; Craik, D.J.; Topliss, D.J.; Stockigt, J.R. Drug competition for thyroxine binding to transthyretin (prealbumin): Comparison with effects on thyroxine-binding globulin. J. Clin. Endocrinol. Metab. 1989. [Google Scholar] [CrossRef]
- Adamski-Werner, S.L.; Palaninathan, S.K.; Sacchettini, J.C.; Kelly, J.W. Diflunisal Analogues Stabilize the Native State of Transthyretin. Potent Inhibition of Amyloidogenesis. J. Med. Chem. 2004. [Google Scholar] [CrossRef]
- Miller, S.R.; Sekijima, Y.; Kelly, J.W. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Investig. 2004. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, C.A.; Oliveira, S.M.; Guido, L.F.; Magalhães, A.; Valencia, G.; Arsequell, G.; Saraiva, M.J.; Cardoso, I. Transthyretin stabilization by iododiflunisal promotes amyloid-β peptide clearance, decreases its deposition, and ameliorates cognitive deficits in an Alzheimer’s disease mouse model. J. Alzheimers Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rios, X.; Gómez-Vallejo, V.; Martín, A.; Cossío, U.; Morcillo, M.Á.; Alemi, M.; Cardoso, I.; Quintana, J.; Jiménez-Barbero, J.; Cotrina, E.Y.; et al. Radiochemical examination of transthyretin (TTR) brain penetration assisted by iododiflunisal, a TTR tetramer stabilizer and a new candidate drug for AD. Sci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with Hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013. [Google Scholar] [CrossRef]
- Butler, J.S.; Chan, A.; Costelha, S.; Fishman, S.; Willoughby, J.L.S.; Borland, T.D.; Milstein, S.; Foster, D.J.; Gonçalves, P.; Chen, Q.; et al. Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid 2016. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin-Mediated Amyloidosis. J. Clin. Pharmacol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chemie-Int. Ed. 2012. [Google Scholar] [CrossRef]
- Garber, K. Alnylam terminates revusiran program, stock plunges. Nat. Biotechnol. 2016. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]
- Wood, H. FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat. Rev. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccone, L.; Tonali, N.; Nencetti, S.; Orlandini, E. Natural compounds as inhibitors of transthyretin amyloidosis and neuroprotective agents: Analysis of structural data for future drug design. J. Enzyme Inhib. Med. Chem. 2020, 335, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-β peptides. J. Biol. Chem. 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoungoure, V.L.N.; Schluesener, J.; Moundipa, P.F.; Schluesener, H. Natural polyphenols binding to amyloid: A broad class of compounds to treat different human amyloid diseases. Mol. Nutr. Food Res. 2015, 59, 8–20. [Google Scholar] [CrossRef]
- Ortore, G.; Orlandini, E.; Braca, A.; Ciccone, L.; Rossello, A.; Martinelli, A.; Nencetti, S. Targeting Different Transthyretin Binding Sites with Unusual Natural Compounds. ChemMedChem 2016. [Google Scholar] [CrossRef] [Green Version]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P.; Hardt, S.; Giannitsis, E.; Schreiner, R.; Haberkorn, U.; et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin. Res. Cardiol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Aus dem Siepen, F.; Bauer, R.; Aurich, M.; Buss, S.J.; Steen, H.; Altland, K.; Katus, H.A.; Kristen, A.V. Green tea extract as a treatment for patients with wild-type transthyretin amyloidosis: An observational study. Drug Des. Devel. Ther. 2015. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Gonçalves, N.P.; Saraiva, M.J.; Almeida, M.R. Curcumin: A multi-Target disease-modifying agent for late-stage transthyretin amyloidosis. Sci. Rep. 2016. [Google Scholar] [CrossRef]
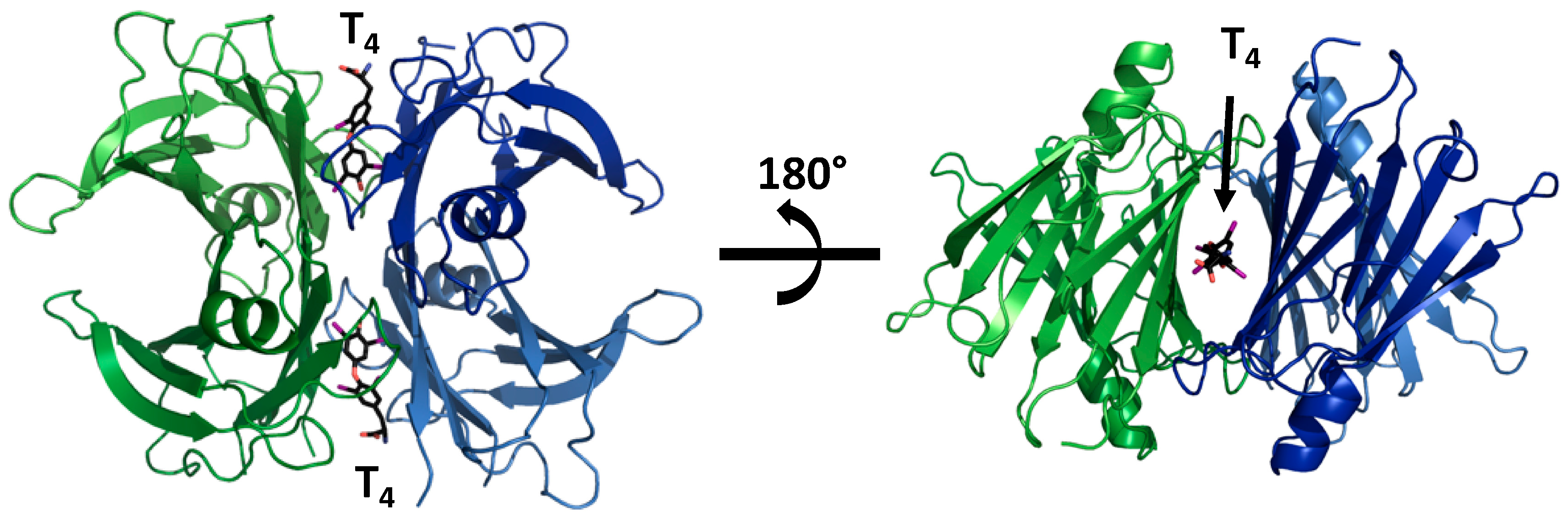
Figure 1.
Structural model of transthyretin (TTR) complexed with T4 (PDB 2rox). Upon forming a tetrameric complex, TTR constitutes two hydrophobic binding pockets, which are occupied by two T4 molecules in this model.
Figure 1.
Structural model of transthyretin (TTR) complexed with T4 (PDB 2rox). Upon forming a tetrameric complex, TTR constitutes two hydrophobic binding pockets, which are occupied by two T4 molecules in this model.

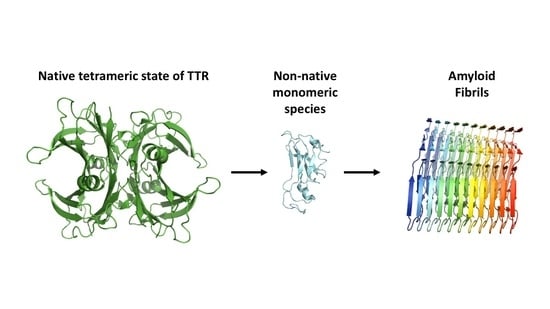
Figure 2.
The proposed aggregation mechanism of transthyretin (TTR). The native tetrameric state of TTR [PDB 4tlt] is stable and physiologically active, while dissociation of non-native monomeric species [PDB 2nbo] initiates the pathogenic aggregation pathway of TTR. This results in accumulation of amyloid fibrils [PDB 6sdz].
Figure 2.
The proposed aggregation mechanism of transthyretin (TTR). The native tetrameric state of TTR [PDB 4tlt] is stable and physiologically active, while dissociation of non-native monomeric species [PDB 2nbo] initiates the pathogenic aggregation pathway of TTR. This results in accumulation of amyloid fibrils [PDB 6sdz].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Saponaro, F.; Kim, J.H.; Chiellini, G. Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease? Int. J. Mol. Sci. 2020, 21, 8672. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228672
AMA Style
Saponaro F, Kim JH, Chiellini G. Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease? International Journal of Molecular Sciences. 2020; 21(22):8672. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228672
Chicago/Turabian StyleSaponaro, Federica, Jin Hae Kim, and Grazia Chiellini. 2020. "Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease?" International Journal of Molecular Sciences 21, no. 22: 8672. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228672
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.