The Role of Structure and Biophysical Properties in the Pleiotropic Effects of Statins


Abstract
:1. Introduction
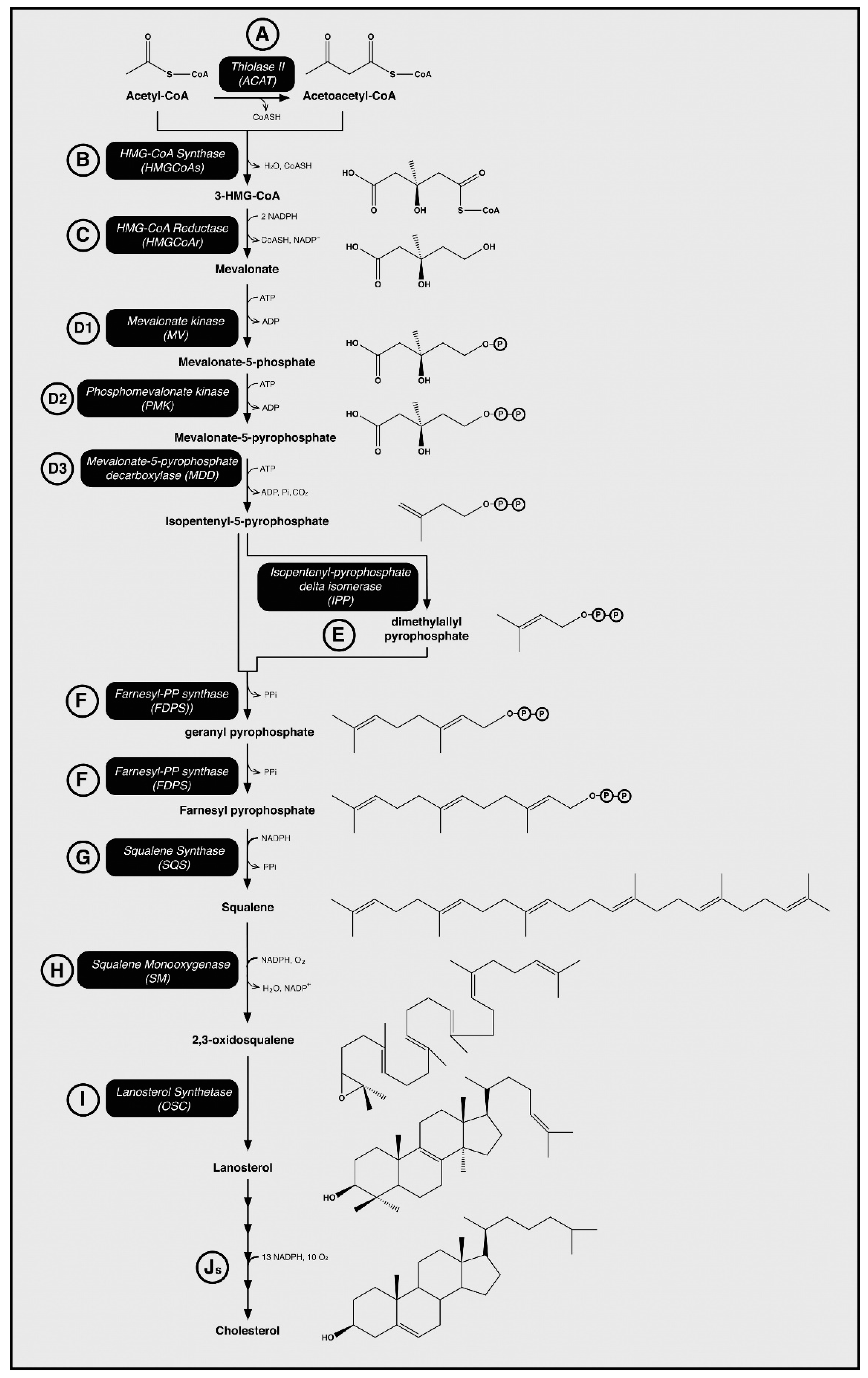
2. The Role of HMG-CoA Reductase in the Synthesis of Cholesterol
2.1. Cholesterol Biosynthesis
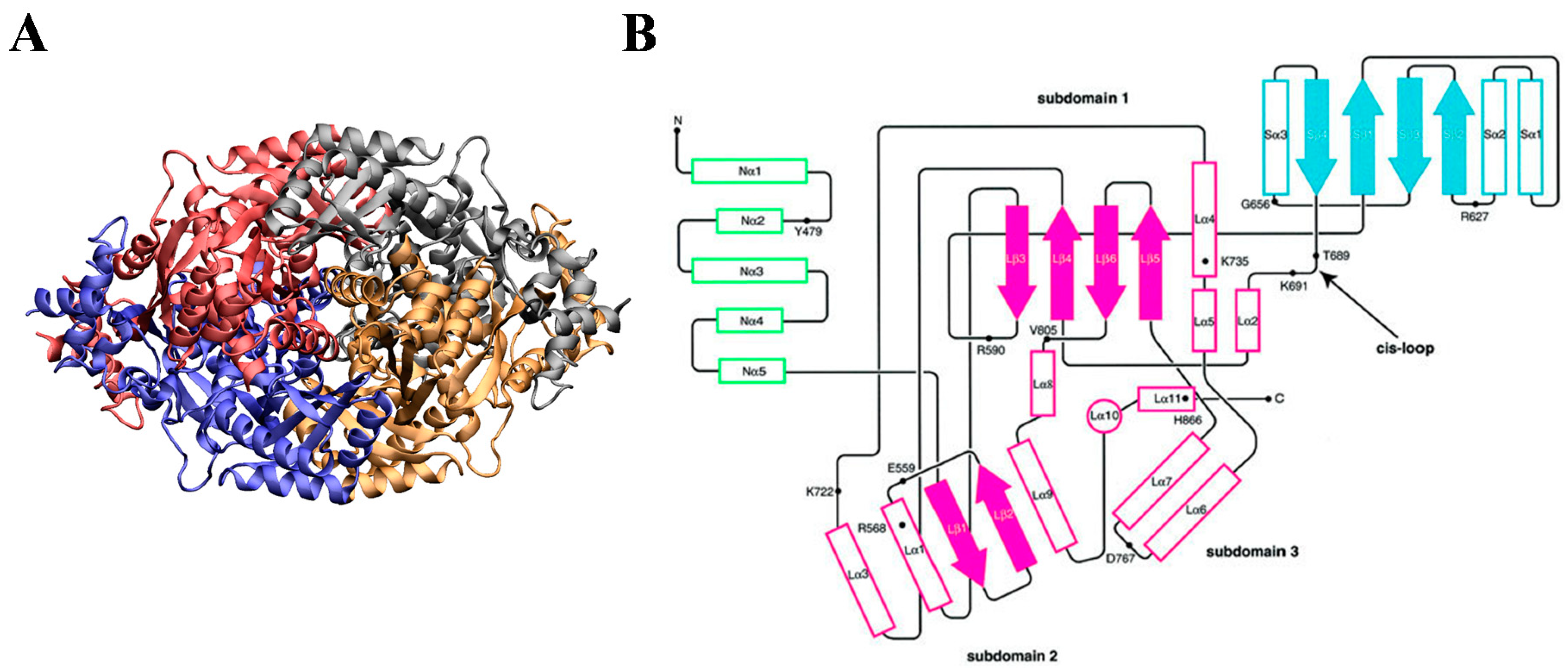
2.2. HMG-CoA Reductase

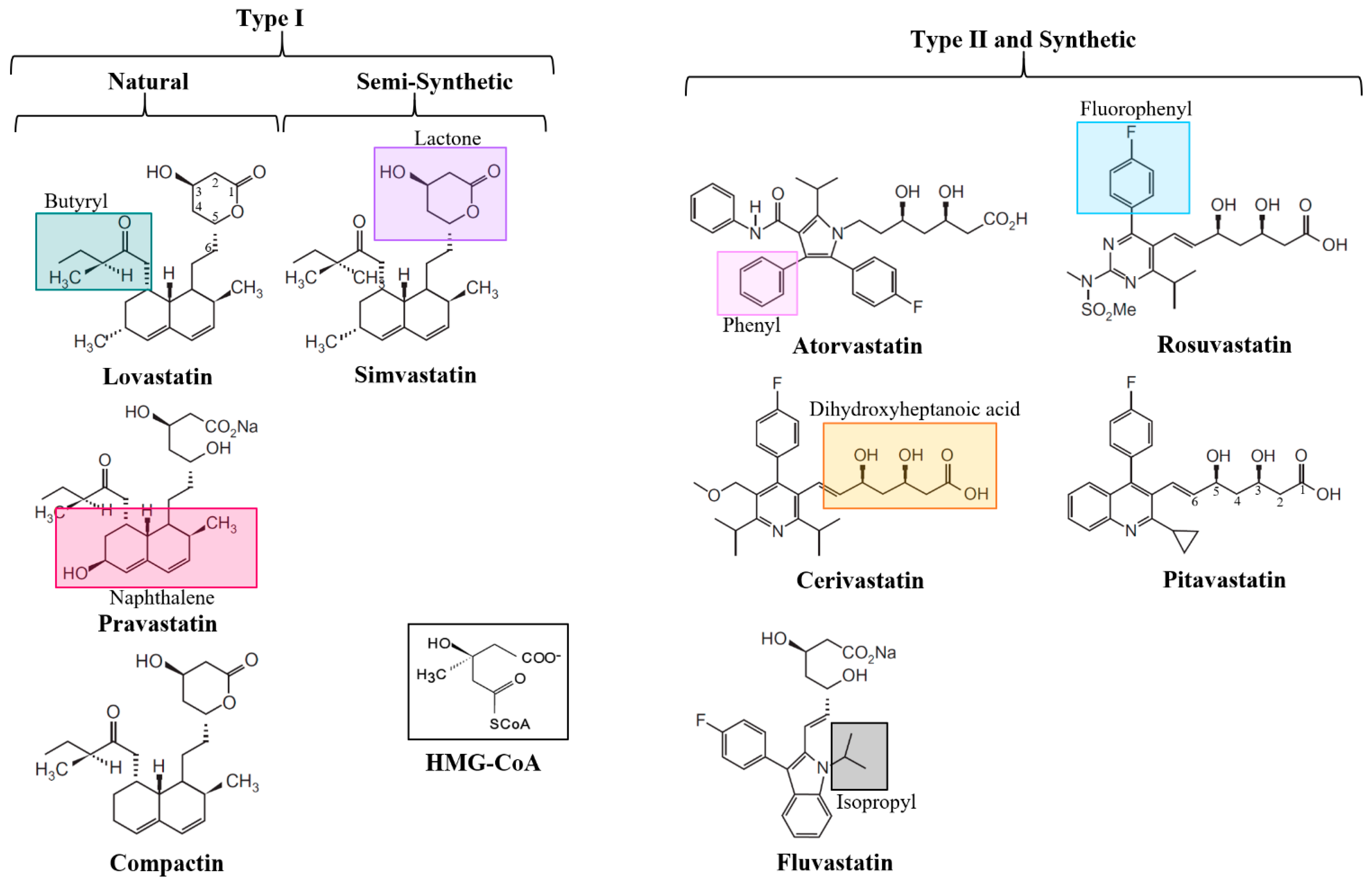
3. The Structure of Statins
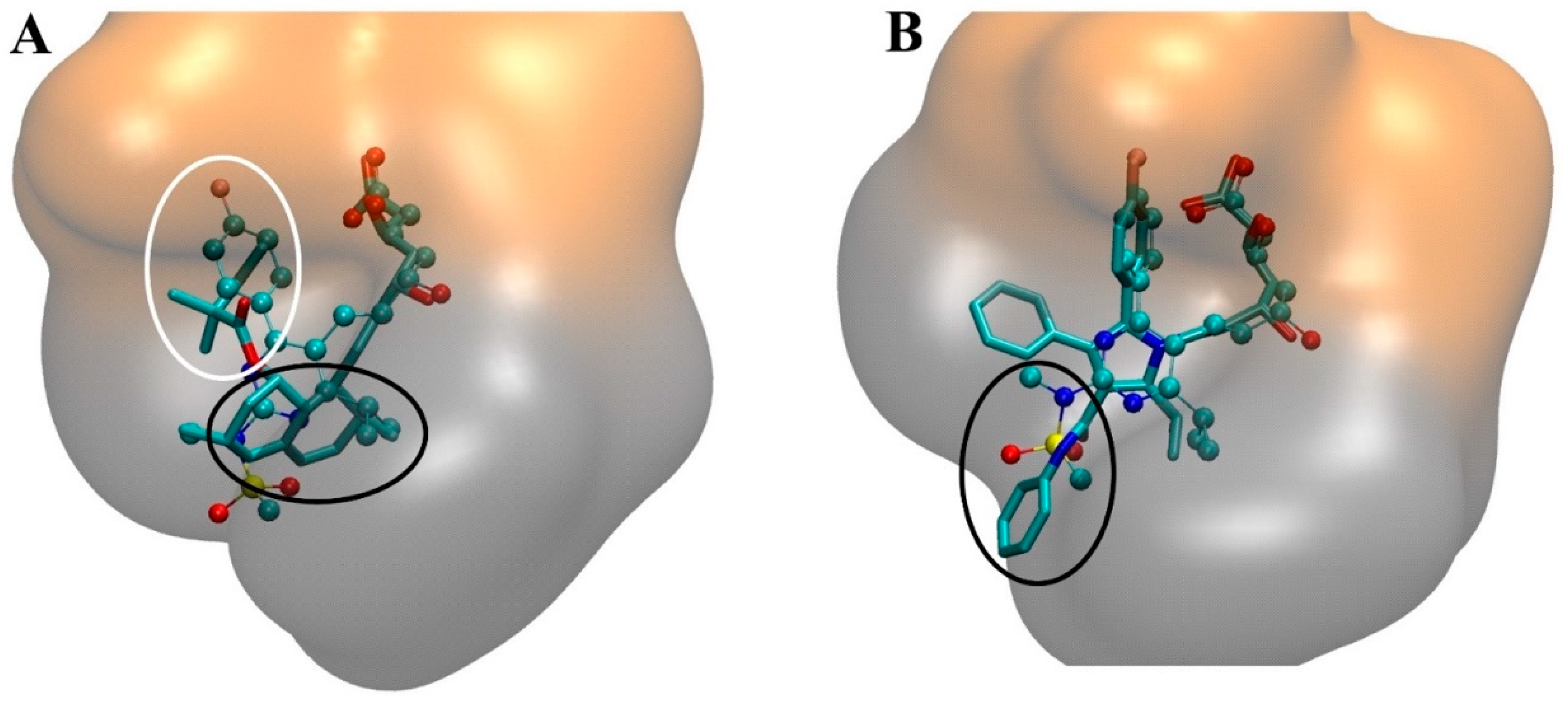
4. The Interaction of Statins with HMGR
4.1. Thermodynamics of Statin-HMGR Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type II | Type I | |||||||
|---|---|---|---|---|---|---|---|---|
| Atorvastatin | Cerivastatin | Rosuvastatin | Fluvastatin | Pitavastatin | Simvastatin | Pravastatin | Lovastatin | |
| Ki (nM) [76] | 8.0 (6.0–12.0) | 10.0 (7.0–15.0) | 3.5 (2.5–5.0) | 28.0 (18.0–42.0) | − | 11.0 (7.0–17.0) | 44.0 (29.0–66.0) | − |
| ΔG (kcal/mol) [77] | −10.9 ± 0.8 | −11.4 ± 0.4 | −12.3 ± 0.7 | −9.0 ± 0.4 | − | − | −9.7 ± 0.4 | − |
| ΔH (kcal/mol) [77] | −4.3 ± 0.1 | −3.3 ± 0.2 | −9.3 ± 0.1 | ~0 | − | − | −2.5 ± 0.1 | − |
| −TΔS (kcal/mol) [77] | −6.6 ± 0.6 | −8.1 ± 0.4 | −3.0 ± 0.7 | ~−9.0 | − | − | −7.2 ± 0.4 | |
| SASApolar (Å2) [77] | 323 | 304 | 341 | 291 | − | − | − | − |
| SASAnon-polar (Å2) [77] | 692 | 605 | 512 | 593 | − | − | − | − |
| log P (pH 7) [84,85] | 1.61 | 2.05 | 0.13 | 1.67 | − | 2.06 | −0.23 | 1.7 |
| log D (pH 7) [86] | 1.53 | 2.32 | − | 1.75 | 1.5 | 4.4 | −0.47 | 3.91 |
| LDL % Reduction at 40 mg dosage [87] | 48 | − | 60 | 27 | − | 41 | 34 | 34 |
4.2. Molecular Interactions in the Statin-HMGR Complex
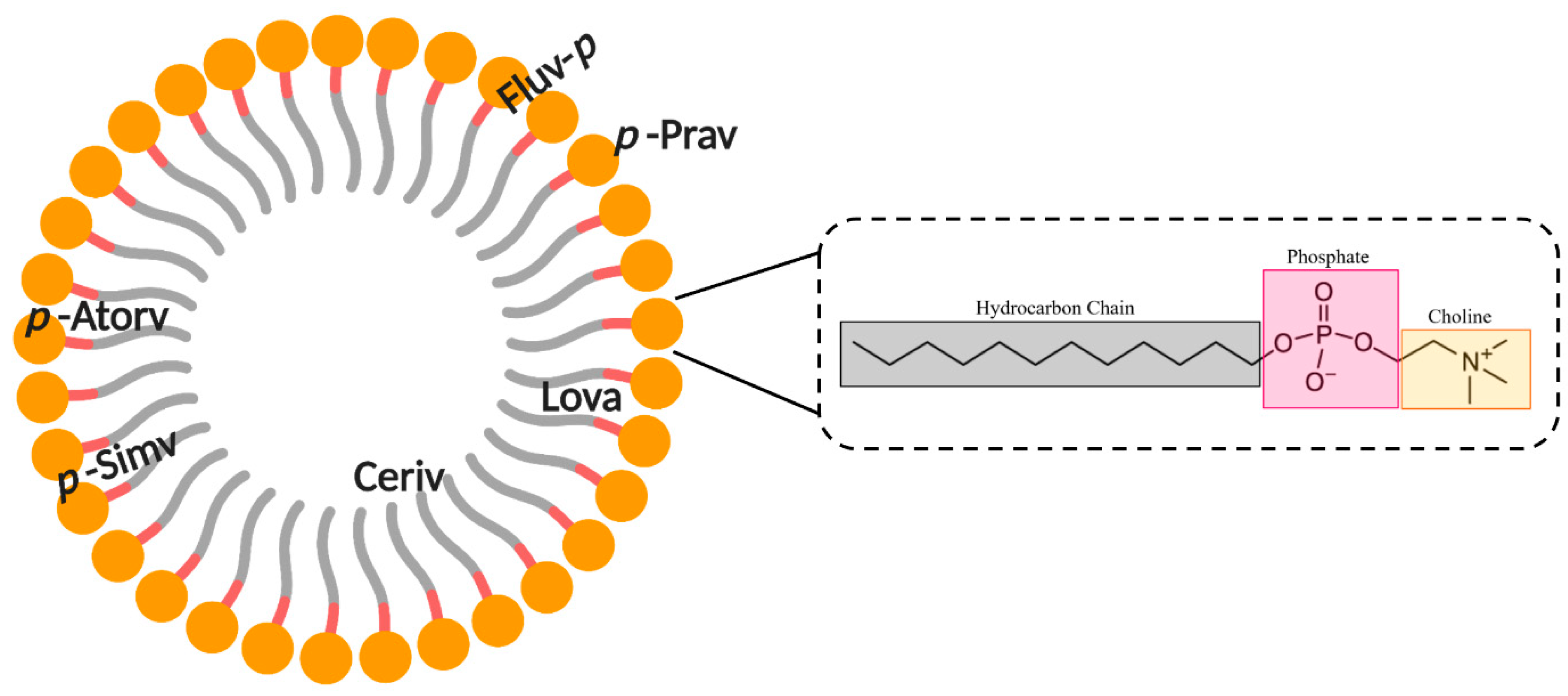
5. Interaction of Statins with Membranes
5.1. Lipophilicity of Statins
5.2. Statin–Membrane Interactions
6. Lipophilicity and Hepatoselectivity
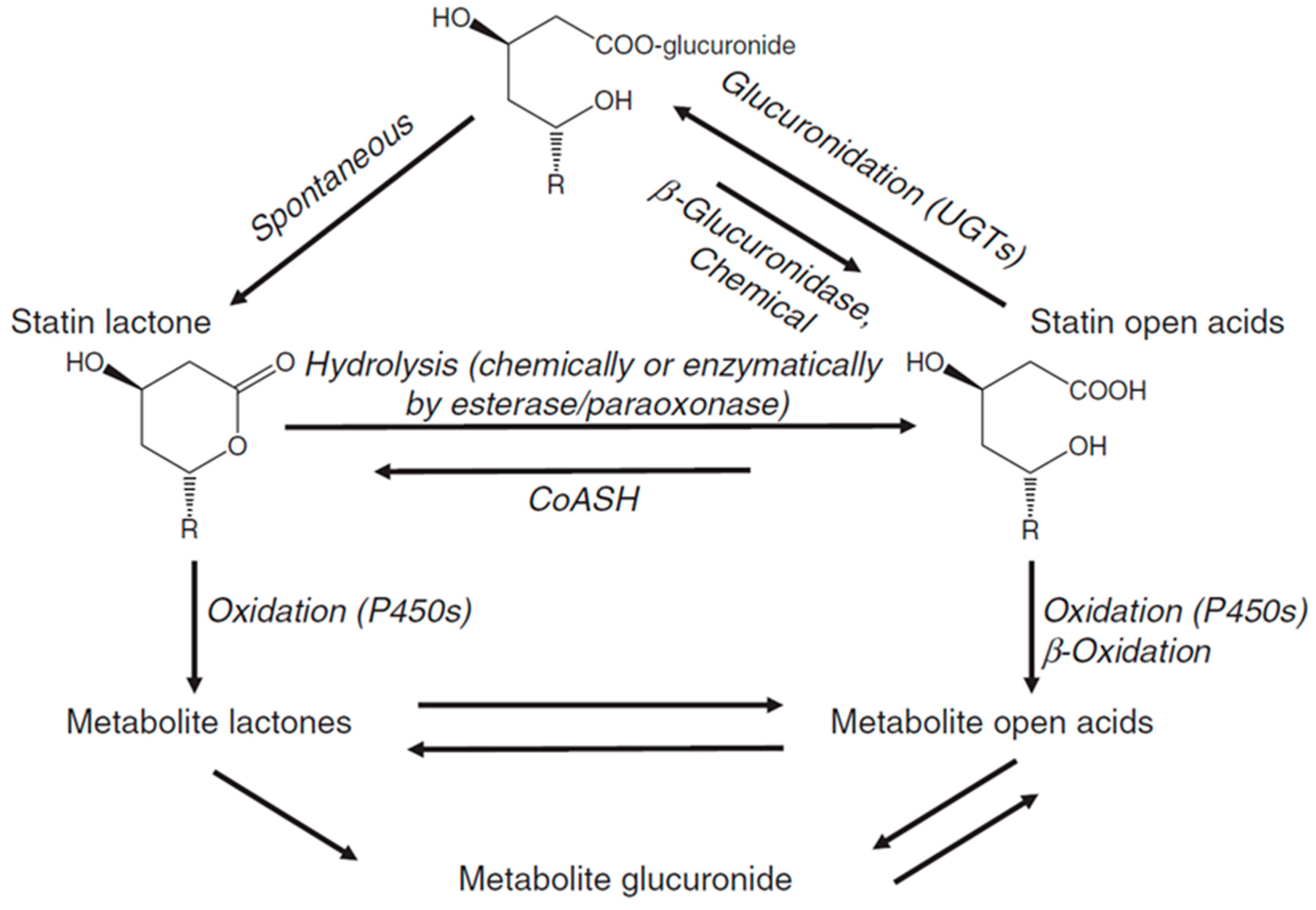
7. Statin Metabolism
7.1. Lactonization
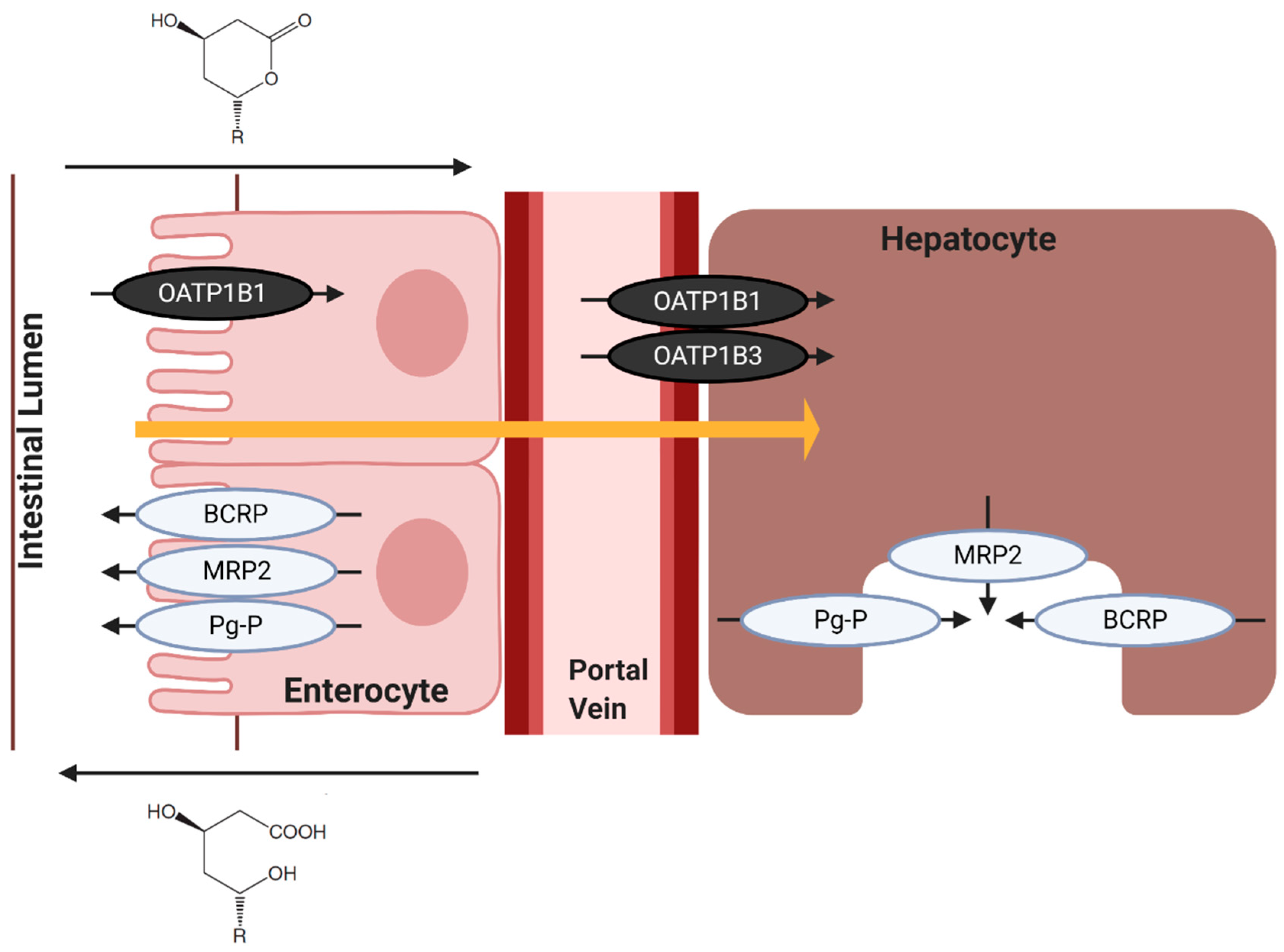
7.2. Hepatic Influx of Statins
7.3. Hepatic Efflux of Statins
8. Statin Transporters and Myopathy
9. Future Implications of Statins
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Endo, A. The origin of the statins. Atheroscler. Suppl. 2004, 5, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Monacolin, K. A new hypocholesterolemic agent produced by a Monascus species. J. Antibiot. Tokyo 1979, 32, 852–854. [Google Scholar] [CrossRef] [Green Version]
- Alberts, A.W.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E.; et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 1980, 77, 3957–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfarb, S.; Pitot, H.C. Improved assay of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Lipid. Res. 1971, 12, 512–515. [Google Scholar]
- Brown, A.G.; Smale, T.C.; King, T.J.; Hasenkamp, R.; Thompson, R.H. Crystal and molecular structure of compactin, a new antifungal metabolite from Penicillium brevicompactum. J. Chem. Soc. Perkin. 1976, 1, 1165–1170. [Google Scholar] [CrossRef]
- Endo, A.; Kuroda, M.; Tsujita, Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium. J. Antibiot. Tokyo 1976, 29, 1346–1348. [Google Scholar] [CrossRef] [Green Version]
- Endo, A. The discovery and development of HMG-CoA reductase inhibitors. J. Lipid. Res. 1992, 33, 1569–1582. [Google Scholar] [CrossRef]
- Bach, T.J.; Lichtenthaler, H.K. Mevinolin: A highly specific inhibitor of microsomal 3-hydroxy-3-methylglutaryl-coenzyme A reductase of radish plants. Z. Naturforsch. C Biosci. 1982, 37, 46–50. [Google Scholar] [CrossRef]
- Qiao, J.; Kontoyiannis, D.P.; Wan, Z.; Li, R.; Liu, W. Antifungal activity of statins against Aspergillus species. Med. Mycol. 2007, 45, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Cabral, M.E.; Delgado, O.D.; Sampietro, D.A.; Catalan, C.A.N.; Fariña, J.I. Antifungal activity and the potential correlation with Statin-Producing ability: An optimized screening applied to Filamentous fungi from Las yungas subtropical rainforest. Res. J. Microbiol. 2010, 5, 833–848. [Google Scholar] [CrossRef] [Green Version]
- Lima, W.G.; Alves-Nascimento, L.A.; Andrade, J.T.; Vieira, L.; de Azambuja Ribeiro, R.I.M.; Thome, R.G.; Dos Santos, H.B.; Ferreira, J.M.S.; Soares, A.C. Are the Statins promising antifungal agents against invasive candidiasis? Biomed. Pharmacother. 2019, 111, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Subhan, M.; Faryal, R.; Macreadie, I. Exploitation of Aspergillus terreus for the production of natural Statins. J. Fungi Basel 2016, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Demasi, M. Statin wars: Have we been misled about the evidence? A narrative review. Br. J. Sports Med. 2018, 52, 905–909. [Google Scholar] [CrossRef]
- Gazzerro, P.; Proto, M.C.; Gangemi, G.; Malfitano, A.M.; Ciaglia, E.; Pisanti, S.; Santoro, A.; Laezza, C.; Bifulco, M. Pharmacological actions of statins: A critical appraisal in the management of cancer. Pharmacol. Rev. 2012, 64, 102–146. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol biosynthesis: A mechanistic overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Andrews, T.C.; Hsia, J.A.; Kramer, J.H.; Shear, C.; Efficacy, A.S.G.A.C.C.; Safety, S. Correlation of non-high-density lipoprotein cholesterol with apolipoprotein B: Effect of 5 hydroxymethylglutaryl coenzyme A reductase inhibitors on non-high-density lipoprotein cholesterol levels. Am. J. Cardiol. 2001, 88, 265–269. [Google Scholar] [CrossRef]
- Jones, P.H.; Hunninghake, D.B.; Ferdinand, K.C.; Stein, E.A.; Gold, A.; Caplan, R.J.; Blasetto, J.W. Statin therapies for elevated lipid levels compared across doses to Rosuvastatin study, G. effects of rosuvastatin versus atorvastatin, simvastatin, and pravastatin on non-high-density lipoprotein cholesterol, apolipoproteins, and lipid ratios in patients with hypercholesterolemia: Additional results from the Stellar trial. Clin. Ther. 2004, 26, 1388–1399. [Google Scholar] [CrossRef]
- Jones, P.H.; Davidson, M.H.; Stein, E.A.; Bays, H.E.; McKenney, J.M.; Miller, E.; Cain, V.A.; Blasetto, J.W.; Group, S.S. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR*Trial). Am. J. Cardiol. 2003, 92, 152–160. [Google Scholar] [CrossRef]
- Ginsberg, H.N.; Le, N.A.; Short, M.P.; Ramakrishnan, R.; Desnick, R.J. Suppression of apolipoprotein B production during treatment of cholesteryl ester storage disease with lovastatin. Implications for regulation of apolipoprotein B synthesis. J. Clin. Investig. 1987, 80, 1692–1697. [Google Scholar] [CrossRef] [Green Version]
- Kostner, G.M.; Gavish, D.; Leopold, B.; Bolzano, K.; Weintraub, M.S.; Breslow, J.L. HMG COA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels. Circulation 1989, 80, 1313–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Sathasivam, S.; Lecky, B. Statin induced myopathy. BMJ 2008, 337, a2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, R.M.; Pirmohamed, M. Statin-Related Myotoxicity: A comprehensive review of pharmacokinetic, pharmacogenomic and muscle components. J. Clin. Med. 2019, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Bitzur, R.; Cohen, H.; Kamari, Y.; Harats, D. Intolerance to statins: Mechanisms and management. Diabetes Care 2013, 36 (Suppl. 2), S325–S330. [Google Scholar] [CrossRef] [Green Version]
- Selva-O’Callaghan, A.; Alvarado-Cardenas, M.; Pinal-Fernandez, I.; Trallero-Araguas, E.; Milisenda, J.C.; Martinez, M.A.; Marin, A.; Labrador-Horrillo, M.; Juarez, C.; Grau-Junyent, J.M. Statin-induced myalgia and myositis: An update on pathogenesis and clinical recommendations. Expert Rev. Clin. Immunol. 2018, 14, 215–224. [Google Scholar] [CrossRef]
- Mason, R.P.; Walter, M.F.; Day, C.A.; Jacob, R.F. Intermolecular differences of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors contribute to distinct pharmacologic and pleiotropic actions. Am. J. Cardiol. 2005, 96, 11F–23F. [Google Scholar] [CrossRef]
- Larocque, G.; Arnold, A.A.; Chartrand, E.; Mouget, Y.; Marcotte, I. Effect of sodium bicarbonate as a pharmaceutical formulation excipient on the interaction of fluvastatin with membrane phospholipids. Eur. Biophys. J. 2010, 39, 1637–1647. [Google Scholar] [CrossRef]
- Penkauskas, T.; Zentelyte, A.; Ganpule, S.; Valincius, G.; Pret, G. Pleiotropic effects of statins via interaction with the lipid bilayer: A combined approach. Biochim. Biophys. Acta Biomembr. 2020, 1862, 193306. [Google Scholar] [CrossRef]
- Knauer, M.J.; Urquhart, B.L.; Meyer zu Schwabedissen, H.E.; Schwarz, U.I.; Lemke, C.J.; Leake, B.F.; Kim, R.B.; Tirona, R.G. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ. Res. 2010, 106, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virani, S.S.; Smith, S.C., Jr.; Stone, N.J.; Grundy, S.M. Secondary prevention for atherosclerotic cardiovascular disease: Comparing recent US and European guidelines on Dyslipidemia. Circulation 2020, 141, 1121–1123. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S. Statins in the primary and secondary prevention of cardiovascular disease in women: Facts and myths. Tex. Heart Inst. J. 2013, 40, 288–289. [Google Scholar] [PubMed]
- Aday, A.W.; Everett, B.M. Statins in peripheral artery disease: What are we waiting for? Circulation 2018, 137, 1447–1449. [Google Scholar] [CrossRef] [PubMed]
- Jick, H.; Zornberg, G.L.; Jick, S.S.; Seshadri, S.; Drachman, D.A. Statins and the risk of dementia. Lancet 2000, 356, 1627–1631. [Google Scholar] [CrossRef]
- Wolozin, B.; Kellman, W.; Ruosseau, P.; Celesia, G.G.; Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 2000, 57, 1439–1443. [Google Scholar] [CrossRef] [Green Version]
- Scott, H.D.; Laake, K. Statins for the prevention of Alzheimer’s disease. Cochrane Database Syst. Rev. 2001, CD003160. [Google Scholar] [CrossRef]
- Fu, Y.C.; Wang, Y.H.; Chen, C.H.; Wang, C.K.; Wang, G.J.; Ho, M.L. Combination of calcium sulfate and simvastatin-controlled release microspheres enhances bone repair in critical-sized rat calvarial bone defects. Int. J. Nanomed. 2015, 10, 7231–7240. [Google Scholar] [CrossRef] [Green Version]
- Tai, I.C.; Wang, Y.H.; Chen, C.H.; Chuang, S.C.; Chang, J.K.; Ho, M.L. Simvastatin enhances Rho/actin/cell rigidity pathway contributing to mesenchymal stem cells’ osteogenic differentiation. Int. J. Nanomed. 2015, 10, 5881–5894. [Google Scholar] [CrossRef] [Green Version]
- Sigler, M.A.; Congdon, L.; Edwards, K.L. An evidence-based review of Statin use in patients with nonalcoholic fatty liver disease. Clin. Med. Insights Gastroenterol. 2018, 11, 1179552218787502. [Google Scholar] [CrossRef] [Green Version]
- Romana, B.; Batger, M.; Prestidge, C.A.; Colombo, G.; Sonvico, F. Expanding the therapeutic potential of statins by means of nanotechnology enabled drug delivery systems. Curr. Top. Med. Chem. 2014, 14, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Player, D.J.; Samanta, S.; Rangasami, V.K.; Hilborn, J.; Lewis, M.P.; Oommen, O.P.; Mudera, V. Hyaluronan derived nanoparticle for simvastatin delivery: Evaluation of simvastatin induced myotoxicity in tissue engineered skeletal muscle. Biomater. Sci. 2019, 8, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korani, S.; Bahrami, S.; Korani, M.; Banach, M.; Johnston, T.P.; Sahebkar, A. Parenteral systems for statin delivery: A review. Lipids Health Dis. 2019, 18, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, R.P. Cholesterol: Chemistry, Biochemistry, and Pathology; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Kumar, D.R.; Hanlin, E.; Glurich, I.; Mazza, J.J.; Yale, S.H. Virchow’s contribution to the understanding of thrombosis and cellular biology. Clin. Med. Res. 2010, 8, 168–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virchow, R. Cellular Pathology as Based upon Physiological and Pathological History, 2nd ed.; J. B. Lippincott & Co.: Philadelphia, PA, USA, 1863. [Google Scholar]
- Anitschkow, N. Uber die Veranderungen der Kannichenaorta bei experimentaeller Cholesterinsteatose. Beitrz. Path Anatalllg. Pathol. 1913, 56, 379–404. [Google Scholar]
- Steinberg, D.; Gotto, A.M., Jr. Preventing coronary artery disease by lowering cholesterol levels: Fifty years from bench to bedside. JAMA 1999, 282, 2043–2050. [Google Scholar] [CrossRef]
- Gofman, J.W.; Lindgren, F. The role of lipids and lipoproteins in atherosclerosis. Science 1950, 111, 166–171. [Google Scholar] [CrossRef] [Green Version]
- Gofman, J.W. Serum lipoproteins and the evaluation of atherosclerosis. Ann. N. Y. Acad. Sci. 1956, 64, 590–595. [Google Scholar] [CrossRef]
- Gofman, J.W.; Young, W.; Tandy, R. Ischemic heart disease, atherosclerosis, and longevity. Circulation 1966, 34, 679–697. [Google Scholar] [CrossRef] [Green Version]
- Keys, A.; Anderson, J.T.; Fidanza, F.; Keys, M.H.; Swahn, B. Effects of diet on blood lipids in man, particularly cholesterol and lipoproteins. Clin. Chem. 1955, 1, 34–52. [Google Scholar] [CrossRef]
- Keys, A.; Aravanis, C.; Blackburn, H.W.; Van Buchem, F.S.; Buzina, R.; Djordjevic, B.D.; Dontas, A.S.; Fidanza, F.; Karvonen, M.J.; Kimura, N.; et al. Epidemiological studies related to coronary heart disease: Characteristics of men aged 40–59 in seven countries. Acta Med. Scand. Suppl. 1966, 460, 1–392. [Google Scholar] [CrossRef] [PubMed]
- Keys, A.; Menotti, A.; Aravanis, C.; Blackburn, H.; Djordevic, B.S.; Buzina, R.; Dontas, A.S.; Fidanza, F.; Karvonen, M.J.; Kimura, N.; et al. The seven countries study: 2,289 deaths in 15 years. Prev. Med. 1984, 13, 141–154. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinitzer, F. Contributions to the knowledge of cholesterol. Liq. Cryst. 1989, 5, 7–18. [Google Scholar] [CrossRef]
- Goldstein, J.L. Cholesterol: A century of research. Howard Hughes Med. Inst. Bull. 2003, 16, 10–19. [Google Scholar]
- Bloch, K. The biological synthesis of cholesterol. Science 1965, 150, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Bucher, N.L.; Overath, P.; Lynen, F. beta-Hydroxy-beta-methyl-glutaryl coenzyme A reductase, cleavage and condensing enzymes in relation to cholesterol formation in rat liver. Biochim. Biophys. Acta 1960, 40, 491–501. [Google Scholar] [CrossRef]
- Tchen, T.T. Mevalonic kinase: Purification and properties. J. Biol. Chem. 1958, 233, 1100–1103. [Google Scholar] [PubMed]
- Agranoff, B.W.; Eggerer, H.; Henning, U.; Lynen, F. Biosynthesis of terpenes. VII. Isopentenyl pyrophosphate isomerase. J. Biol. Chem. 1960, 235, 326–332. [Google Scholar]
- Tada, M.; Lynen, F. On the biosynthesis of terpenes. XIV. On the determination of phosphomevalonic acid kinase and pyrophosphomevalonic acid decarboxylase in cell extracts. J. Biochem. 1961, 49, 758–764. [Google Scholar] [CrossRef]
- Summons, R.E.; Bradley, A.S.; Jahnke, L.L.; Waldbauer, J.R. Steroids, triterpenoids and molecular oxygen. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 951–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Osuga, J.; Tozawa, R.; Kitamine, T.; Yagyu, H.; Sekiya, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; Yahagi, N.; et al. Early embryonic lethality caused by targeted disruption of the 3-hydroxy-3-methylglutaryl-CoA reductase gene. J. Biol. Chem. 2003, 278, 42936–42941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochar, D.A.; Stauffacher, C.V.; Rodwell, V.W. Sequence comparisons reveal two classes of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mol. Genet. Metab. 1999, 66, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Friesen, J.A.; Rodwell, V.W. The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biol. 2004, 5, 248. [Google Scholar] [CrossRef] [Green Version]
- Istvan, E.S.; Palnitkar, M.; Buchanan, S.K.; Deisenhofer, J. Crystal structure of the catalytic portion of human HMG-CoA reductase: Insights into regulation of activity and catalysis. EMBO J. 2000, 19, 819–830. [Google Scholar] [CrossRef]
- Hampton, R.; Dimster-Denk, D.; Rine, J. The biology of HMG-CoA reductase: The pros of contra-regulation. Trends Biochem. Sci. 1996, 21, 140–145. [Google Scholar] [CrossRef]
- Burg, J.S.; Espenshade, P.J. Regulation of HMG-CoA reductase in mammals and yeast. Prog. Lipid. Res. 2011, 50, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Manzoni, M.; Rollini, M. Biosynthesis and biotechnological production of statins by filamentous fungi and application of these cholesterol-lowering drugs. Appl. Microbiol. Biotechnol. 2002, 58, 555–564. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Tang, Y. Efficient Synthesis of Simvastatin by Use of Whole-Cell Biocatalysis. Appl. Environ. Microb. 2007, 73, 2054–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrios-Gonzalez, J.; Miranda, R.U. Biotechnological production and applications of statins. Appl. Microbiol. Biotechnol. 2010, 85, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Holdgate, G.A.; Ward, W.H.; McTaggart, F. Molecular mechanism for inhibition of 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase by rosuvastatin. Biochem. Soc. Trans. 2003, 31, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonell, T.; Freire, E. Binding thermodynamics of statins to HMG-CoA reductase. Biochemistry 2005, 44, 11741–11748. [Google Scholar] [CrossRef] [PubMed]
- Corsini, A.; Bellosta, S.; Baetta, R.; Fumagalli, R.; Paoletti, R.; Bernini, F. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol. Ther. 1999, 84, 413–428. [Google Scholar] [CrossRef]
- Vega, S.; Kang, L.W.; Velazquez-Campoy, A.; Kiso, Y.; Amzel, L.M.; Freire, E. A structural and thermodynamic escape mechanism from a drug resistant mutation of the HIV-1 protease. Proteins 2004, 55, 594–602. [Google Scholar] [CrossRef]
- Ohtaka, H.; Muzammil, S.; Schon, A.; Velazquez-Campoy, A.; Vega, S.; Freire, E. Thermodynamic rules for the design of high affinity HIV-1 protease inhibitors with adaptability to mutations and high selectivity towards unwanted targets. Int. J. Biochem. Cell Biol. 2004, 36, 1787–1799. [Google Scholar] [CrossRef]
- Cozzini, P.; Fornabaio, M.; Marabotti, A.; Abraham, D.J.; Kellogg, G.E.; Mozzarelli, A. Free Energy of Ligand Binding to Protein: Evaluation of the Contribution of Water Molecules by Computational Methods. Curr. Med. Chem. 2004, 11, 3093–3118. [Google Scholar] [CrossRef] [Green Version]
- Kukic, P.; Nielsen, J.E. Electrostatics in proteins and protein-ligand complexes. Future Med. Chem. 2010, 2, 647–666. [Google Scholar] [CrossRef] [Green Version]
- Cabani, S.; Gianni, P.; Mollica, V.; Lepori, L. Group contributions to the thermodynamic properties of non-ionic organic solutes in dilute aqueous solution. J. Solut. Chem. 1981, 10, 563–595. [Google Scholar] [CrossRef]
- Joshi, H.N.; Fakes, M.G.; Serajuddin, A.T. Differentiation of 3-Hydroxy-3-methylglutaryl-coenzyme A Reductase Inhibitors by Their Relative Lipophilicity. Pharm. Pharmacol. Commun. 1999, 5, 269–271. [Google Scholar] [CrossRef]
- Rageh, A.H.; Atia, N.N.; Abdel-Rahman, H.M. Lipophilicity estimation of statins as a decisive physicochemical parameter for their hepato-selectivity using reversed-phase thin layer chromatography. J. Pharm. Biomed. Anal. 2017, 142, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Sugiyama, Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: Drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol. Ther. 2006, 112, 71–105. [Google Scholar] [CrossRef]
- Feingold, K.R. Cholesterol Lowering Drugs. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., et al., Eds.; MDText.com: South Dartmouth, MA, USA, 2020. [Google Scholar]
- Istvan, E. Statin inhibition of HMG-CoA reductase: A 3-dimensional view. Atheroscler. Suppl. 2003, 4, 3–8. [Google Scholar] [CrossRef]
- Lawrence, C.M.; Rodwell, V.W.; Stauffacher, C.V. Crystal structure of Pseudomonas mevalonii HMG-CoA reductase at 3.0 angstrom resolution. Science 1995, 268, 1758–1762. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, L.; Bochar, D.A.; Rodwell, V.W.; Stauffacher, C.V. Substrate-induced closure of the flap domain in the ternary complex structures provides insights into the mechanism of catalysis by 3-hydroxy-3-methylglutaryl-CoA reductase. Proc. Natl. Acad. Sci. USA 1999, 96, 7167–7171. [Google Scholar] [CrossRef] [Green Version]
- Endo, A.; Kuroda, M.; Tanzawa, K. Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase by ML-236A and ML-236B fungal metabolites, having hypocholesterolemic activity. FEBS Lett. 1976, 72, 323–326. [Google Scholar] [CrossRef] [Green Version]
- Fong, C.W. Statins in therapy: Understanding their hydrophilicity, lipophilicity, binding to 3-hydroxy-3-methylglutaryl-CoA reductase, ability to cross the blood brain barrier and metabolic stability based on electrostatic molecular orbital studies. Eur. J. Med. Chem. 2014, 85, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Sharp, K.A.; Nicholls, A.; Fine, R.F.; Honig, B. Reconciling the magnitude of the microscopic and macroscopic hydrophobic effects. Science 1991, 252, 106–109. [Google Scholar] [CrossRef]
- Zeichner, S.; Mihos, C.G.; Santana, O. The pleiotropic effects and therapeutic potential of the hydroxy-methyl-glutaryl-CoA reductase inhibitors in malignancies: A comprehensive review. J. Cancer Res. Ther. 2012, 8, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Bonsu, K.O.; Kadirvelu, A.; Reidpath, D.D. Lipophilic versus hydrophilic statin therapy for heart failure: A protocol for an adjusted indirect comparison meta-analysis. Syst. Rev. 2013, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.K. Isoprenoids as mediators of the biological effects of statins. J. Clin. Investig. 2002, 110, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T.; Ranadive, S.A.; Mahoney, E.M. Relative lipophilicities, solubilities, and structure-pharmacological considerations of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors pravastatin, lovastatin, mevastatin, and simvastatin. J. Pharm. Sci. 1991, 80, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Guillot, F.; Misslin, P.; Lemaire, M. Comparison of fluvastatin and lovastatin blood-brain barrier transfer using in vitro and in vivo methods. J. Cardiovasc. Pharmacol. 1993, 21, 339–346. [Google Scholar] [CrossRef]
- Bannan, C.C.; Calabro, G.; Kyu, D.Y.; Mobley, D.L. Calculating Partition Coefficients of Small Molecules in Octanol/Water and Cyclohexane/Water. J. Chem. Theory Comput. 2016, 12, 4015–4024. [Google Scholar] [CrossRef] [Green Version]
- Balaz, S. Modeling kinetics of subcellular disposition of chemicals. Chem. Rev. 2009, 109, 1793–1899. [Google Scholar] [CrossRef] [Green Version]
- Leo, A.; Hansch, C.; Elkins, D. Partition coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Klopman, G.; Zhu, H. Recent methodologies for the estimation of n-octanol/water partition coefficients and their use in the prediction of membrane transport properties of drugs. Mini Rev. Med. Chem. 2005, 5, 127–133. [Google Scholar] [CrossRef]
- Cumming, H.; Rucker, C. Octanol-Water Partition Coefficient Measurement by a Simple (1)H NMR Method. ACS Omega 2017, 2, 6244–6249. [Google Scholar] [CrossRef]
- Garcia, A.; Pochinda, S.; Elgaard-Jorgensen, P.N.; Khandelia, H.; Clarke, R.J. Evidence for ATP Interaction with Phosphatidylcholine Bilayers. Langmuir 2019, 35, 9944–9953. [Google Scholar] [CrossRef] [PubMed]
- Alghalayini, A.; Garcia, A.; Berry, T.; Cranfield, C.G. The Use of Tethered Bilayer Lipid Membranes to Identify the Mechanisms of Antimicrobial Peptide Interactions with Lipid Bilayers. Antibiotics (Basel) 2019, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, M.J.; Rusinova, R.; Gleason, N.J.; Koeppe, R.E., 2nd; Andersen, O.S. Interactions of drugs and amphiphiles with membranes: Modulation of lipid bilayer elastic properties by changes in acyl chain unsaturation and protonation. Faraday Discuss. 2013, 161, 461–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, D.; Charalambous, K.; Gee, A.; Law, R.V.; Ces, O. Amphiphilic drug interactions with model cellular membranes are influenced by lipid chain-melting temperature. J. R. Soc. Interf. 2014, 11, 20131062. [Google Scholar] [CrossRef]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Sanford, R.L.; Al’Aref, S.J.; Koeppe, R.E., 2nd; Andersen, O.S. Statins Modify Lipid Bilayer Properties. Biophys. J. 2013, 104, 86. [Google Scholar] [CrossRef] [Green Version]
- Redondo-Morata, L.; Lea Sanford, R.; Andersen, O.S.; Scheuring, S. Effect of Statins on the Nanomechanical Properties of Supported Lipid Bilayers. Biophys. J. 2016, 111, 363–372. [Google Scholar] [CrossRef] [Green Version]
- van Meer, G.; de Kroon, A.I. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Shurshalova, G.S.; Yulmetov, A.R.; Sharapova, D.A.; Aganov, A.V.; Klochkov, V.V. Interaction of Lovastatin with Model Membranes by NMR Data and from MD Simulations. Bionanoscience 2020, 10, 493–501. [Google Scholar] [CrossRef]
- Galiullina, L.F.; Scheidt, H.A.; Huster, D.; Aganov, A.; Klochkov, V. Interaction of statins with phospholipid bilayers studied by solid-state NMR spectroscopy. Biochim. Biophys. Acta Biomembr. 2019, 1861, 584–593. [Google Scholar] [CrossRef]
- Galiullina, L.F.; Aganova, O.V.; Latfullin, I.A.; Musabirova, G.S.; Aganov, A.V.; Klochkov, V.V. Interaction of different statins with model membranes by NMR data. Biochim. Biophys. Acta Biomembr. 2017, 1859, 295–300. [Google Scholar] [CrossRef]
- Galiullina, L.F.; Musabirova, G.S.; Latfullin, I.A.; Aganov, A.V.; Klochkov, V.V. Spatial structure of atorvastatin and its complex with model membrane in solution studied by NMR and theoretical calculations. J. Mol. Struct. 2018, 1167, 69–77. [Google Scholar] [CrossRef]
- Rakhmatullin, I.Z.; Galiullina, L.F.; Klochkova, E.A.; Latfullin, I.A.; Aganov, A.V.; Klochkov, V.V. Structural studies of pravastatin and simvastatin and their complexes with SDS micelles by NMR spectroscopy. J. Mol. Struct. 2016, 1105, 25–29. [Google Scholar] [CrossRef]
- Sariisik, E.; Kocak, M.; Kucuk Baloglu, F.; Severcan, F. Interaction of the cholesterol reducing agent simvastatin with zwitterionic DPPC and charged DPPG phospholipid membranes. Biochim. Biophys. Acta Biomembr. 2019, 1861, 810–818. [Google Scholar] [CrossRef]
- Custodio, J.B.; Almeida, L.M.; Madeira, V.M. The anticancer drug tamoxifen induces changes in the physical properties of model and native membranes. Biochim. Biophys. Acta 1993, 1150, 123–129. [Google Scholar] [CrossRef]
- Sahu, S.S.; Sarkar, P.; Shrivastava, S.; Chattopadhyay, A. Differential effects of simvastatin on membrane organization and dynamics in varying phases. Chem. Phys. Lipids 2019, 225, 104831. [Google Scholar] [CrossRef]
- Mouritsen, O.G. The liquid-ordered state comes of age. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1286–1288. [Google Scholar] [CrossRef] [Green Version]
- Thewalt, J.L.; Bloom, M. Phosphatidylcholine: Cholesterol phase diagrams. Biophys. J. 1992, 63, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; London, E. Structure and origin of ordered lipid domains in biological membranes. J. Membr. Biol. 1998, 164, 103–114. [Google Scholar] [CrossRef]
- Davidson, M.H.; Toth, P.P. Comparative effects of lipid-lowering therapies. Prog. Cardiovasc. Dis. 2004, 47, 73–104. [Google Scholar] [CrossRef]
- Lindahl, A.; Sandstrom, R.; Ungell, A.L.; Abrahamsson, B.; Knutson, T.W.; Knutson, L.; Lennernas, H. Jejunal permeability and hepatic extraction of fluvastatin in humans. Clin. Pharmacol. Ther. 1996, 60, 493–503. [Google Scholar] [CrossRef]
- Bellosta, S.; Paoletti, R.; Corsini, A. Safety of statins: Focus on clinical pharmacokinetics and drug interactions. Circulation 2004, 109, III-50–III-57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, K.C.E.; Pereira, B.M.V.; Rodrigues, A.C. An update on efflux and uptake transporters as determinants of statin response. Expert Opin. Drug Metab. Toxicol. 2018, 14, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Maeda, K.; Kondo, C.; Hirano, M.; Sasaki, M.; Suzuki, H.; Sugiyama, Y. Identification of the hepatic efflux transporters of organic anions using double-transfected Madin-Darby canine kidney II cells expressing human organic anion-transporting polypeptide 1B1 (OATP1B1)/multidrug resistance-associated protein 2, OATP1B1/multidrug resistance 1, and OATP1B1/breast cancer resistance protein. J. Pharmacol. Exp. Ther. 2005, 314, 1059–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsini, A. The safety of HMG-CoA reductase inhibitors in special populations at high cardiovascular risk. Cardiovasc. Drugs Ther. 2003, 17, 265–285. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Staffa, J.A.; Parks, M.; Green, L. Rhabdomyolysis with HMG-CoA reductase inhibitors and gemfibrozil combination therapy. Pharmacoepidemiol. Drug Saf. 2004, 13, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Goosen, T.C.; Bauman, J.N.; Davis, J.A.; Yu, C.; Hurst, S.I.; Williams, J.A.; Loi, C.M. Atorvastatin glucuronidation is minimally and nonselectively inhibited by the fibrates gemfibrozil, fenofibrate, and fenofibric acid. Drug Metab. Dispos. 2007, 35, 1315–1324. [Google Scholar] [CrossRef]
- Whitfield, L.R.; Porcari, A.R.; Alvey, C.; Abel, R.; Bullen, W.; Hartman, D. Effect of gemfibrozil and fenofibrate on the pharmacokinetics of atorvastatin. J. Clin. Pharmacol. 2011, 51, 378–388. [Google Scholar] [CrossRef]
- Koenen, A.; Kroemer, H.K.; Grube, M.; Meyer zu Schwabedissen, H.E. Current understanding of hepatic and intestinal OATP-mediated drug-drug interactions. Expert Rev. Clin. Pharmacol. 2011, 4, 729–742. [Google Scholar] [CrossRef]
- Furberg, C.D.; Pitt, B. Withdrawal of cerivastatin from the world market. Curr. Control. Trials Cardiovasc. Med. 2001, 2, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Muntean, D.M.; Thompson, P.D.; Catapano, A.L.; Stasiolek, M.; Fabis, J.; Muntner, P.; Serban, M.C.; Banach, M. Statin-associated myopathy and the quest for biomarkers: Can we effectively predict statin-associated muscle symptoms? Drug Discov. Today 2017, 22, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Park, W.K.; Kennedy, R.M.; Larsen, S.D.; Miller, S.; Roth, B.D.; Song, Y.; Steinbaugh, B.A.; Sun, K.; Tait, B.D.; Kowala, M.C.; et al. Hepatoselectivity of statins: Design and synthesis of 4-sulfamoyl pyrroles as HMG-CoA reductase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Subramanian, R.; Fang, X.; Ma, B.; Qiu, Y.; Lin, J.H.; Pearson, P.G.; Baillie, T.A. Glucuronidation of statins in animals and humans: A novel mechanism of statin lactonization. Drug Metab. Dispos. 2002, 30, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujino, H.; Yamada, I.; Shimada, S.; Yoneda, M.; Kojima, J. Metabolic fate of pitavastatin, a new inhibitor of HMG-CoA reductase: Human UDP-glucuronosyltransferase enzymes involved in lactonization. Xenobiotica 2003, 33, 27–41. [Google Scholar] [CrossRef]
- Hoffmann, C.; Nowosielski, M. DFT study on hydroxy acid–lactone interconversion of statins:the case of atorvastatin. Org. Biomol. Chem. 2008, 6, 3527–3531. [Google Scholar] [CrossRef]
- Kantola, T.; Kivisto, K.T.; Neuvonen, P.J. Effect of itraconazole on the pharmacokinetics of atorvastatin. Clin. Pharmacol. Ther. 1998, 64, 58–65. [Google Scholar] [CrossRef]
- Backman, J.T.; Kyrklund, C.; Neuvonen, M.; Neuvonen, P.J. Gemfibrozil greatly increases plasma concentrations of cerivastatin. Clin. Pharmacol. Ther. 2002, 72, 685–691. [Google Scholar] [CrossRef]
- Macwan, J.S.; Ionita, I.A.; Akhlaghi, F. A simple assay for the simultaneous determination of rosuvastatin acid, rosuvastatin-5S-lactone, and N-desmethyl rosuvastatin in human plasma using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Anal. Bioanal. Chem. 2012, 402, 1217–1227. [Google Scholar] [CrossRef]
- Kantola, T.; Kivisto, K.T.; Neuvonen, P.J. Erythromycin and verapamil considerably increase serum simvastatin and simvastatin acid concentrations. Clin. Pharmacol. Ther. 1998, 64, 177–182. [Google Scholar] [CrossRef]
- Neuvonen, P.J.; Jalava, K.M. Itraconazole drastically increases plasma concentrations of lovastatin and lovastatin acid. Clin. Pharmacol. Ther. 1996, 60, 54–61. [Google Scholar] [CrossRef]
- Wang, R.W.; Kari, P.H.; Lu, A.Y.; Thomas, P.E.; Guengerich, F.P.; Vyas, K.P. Biotransformation of lovastatin. IV. Identification of cytochrome P450 3A proteins as the major enzymes responsible for the oxidative metabolism of lovastatin in rat and human liver microsomes. Arch. Biochem. Biophys. 1991, 290, 355–361. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Gorham, L.M.; Ma, B.; Liu, L.; Yu, X.; Zhao, J.J.; Slaughter, D.E.; Arison, B.H.; Vyas, K.P. In vitro metabolism of simvastatin in humans [SBT]identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab. Dispos. 1997, 25, 1191–1199. [Google Scholar] [PubMed]
- Lennernas, H. Clinical pharmacokinetics of atorvastatin. Clin. Pharmacokinet. 2003, 42, 1141–1160. [Google Scholar] [CrossRef]
- Transon, C.; Leemann, T.; Vogt, N.; Dayer, P. In vivo inhibition profile of cytochrome P450TB (CYP2C9) by (±)-fluvastatin. Clin. Pharmacol. Ther. 1995, 58, 412–417. [Google Scholar] [CrossRef]
- Transon, C.; Leemann, T.; Dayer, P. In vitro comparative inhibition profiles of major human drug metabolising cytochrome P450 isozymes (CYP2C9, CYP2D6 and CYP3A4) by HMG-CoA reductase inhibitors. Eur. J. Clin. Pharmacol. 1996, 50, 209–215. [Google Scholar] [CrossRef]
- Fujino, H.; Saito, T.; Tsunenari, Y.; Kojima, J. Effect of gemfibrozil on the metabolism of pitavastatin—Determining the best animal model for human CYP and UGT activities. Drug Metab. Drug Interact. 2004, 20, 25–42. [Google Scholar] [CrossRef]
- Hatanaka, T. Clinical pharmacokinetics of pravastatin: Mechanisms of pharmacokinetic events. Clin. Pharmacokinet. 2000, 39, 397–412. [Google Scholar] [CrossRef]
- White, C.M. A review of the pharmacologic and pharmacokinetic aspects of rosuvastatin. J. Clin. Pharmacol. 2002, 42, 963–970. [Google Scholar] [CrossRef]
- Muck, W. Clinical pharmacokinetics of cerivastatin. Clin. Pharmacokinet. 2000, 39, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Mazzu, A.L.; Lasseter, K.C.; Shamblen, E.C.; Agarwal, V.; Lettieri, J.; Sundaresen, P. Itraconazole alters the pharmacokinetics of atorvastatin to a greater extent than either cerivastatin or pravastatin. Clin. Pharmacol. Ther. 2000, 68, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, K.; Nakajima, M.; Suzuki, M.; Shimada, N.; Yamazaki, H.; Yokoi, T. Inhibitory effects of amiodarone and its N-deethylated metabolite on human cytochrome P450 activities: Prediction of in vivo drug interactions. Br. J. Clin. Pharmacol. 2000, 49, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borders-Hemphill, V. Concurrent use of statins and amiodarone. Consult. Pharm. 2009, 24, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Fujita, Y.; Ieiri, I. An updated review of pharmacokinetic drug interactions and pharmacogenetics of statins. Expert Opin. Drug Metab. Toxicol. 2020, 16, 809–822. [Google Scholar] [CrossRef]
- Sissung, T.M.; Troutman, S.M.; Campbell, T.J.; Pressler, H.M.; Sung, H.; Bates, S.E.; Figg, W.D. Transporter Polymorphisms Affect Normal Physiology, Diseases, and Pharmacotherapy. Discov. Med. 2012, 13, 19–34. [Google Scholar]
- Rodrigues, A.C. Efflux and uptake transporters as determinants of statin response. Expert Opin. Drug Metab. Toxicol. 2010, 6, 621–632. [Google Scholar] [CrossRef]
- Fong, C.W. Permeability of the Blood-Brain Barrier: Molecular Mechanism of Transport of Drugs and Physiologically Important Compounds. J. Membr. Biol. 2015, 248, 651–669. [Google Scholar] [CrossRef]
- Sirtori, C.R. Tissue selectivity of hydroxymethylglutaryl coenzyme A (HMG CoA) reductase inhibitors. Pharmacol. Ther. 1993, 60, 431–459. [Google Scholar] [CrossRef]
- Sharma, P.; Butters, C.J.; Smith, V.; Elsby, R.; Surry, D. Prediction of the in vivo OATP1B1-mediated drug-drug interaction potential of an investigational drug against a range of statins. Eur. J. Pharm. Sci. 2012, 47, 244–255. [Google Scholar] [CrossRef]
- Chen, C.; Mireles, R.J.; Campbell, S.D.; Lin, J.; Mills, J.B.; Xu, J.J.; Smolarek, T.A. Differential interaction of 3-hydroxy-3-methylglutaryl-coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab. Dispos. 2005, 33, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Volpe, D.A.; Wang, Y.; Zhang, W.; Bode, C.; Owen, A.; Hidalgo, I.J. Use of transporter knockdown Caco-2 cells to investigate the in vitro efflux of statin drugs. Drug Metab. Dispos. 2011, 39, 1196–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 2013, 34, 45–78. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Akiyama, S.; Nishigaki, R.; Sugiyama, Y. Uptake is the rate-limiting step in the overall hepatic elimination of pravastatin at steady-state in rats. Pharm. Res. 1996, 13, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Kobayashi, K.; Sugiyama, Y. Primary active transport of pravastatin across the liver canalicular membrane in normal and mutant Eisai hyperbilirubinemic rats. Biopharm. Drug Dispos. 1996, 17, 607–621. [Google Scholar] [CrossRef]
- Watanabe, T.; Kusuhara, H.; Maeda, K.; Kanamaru, H.; Saito, Y.; Hu, Z.; Sugiyama, Y. Investigation of the rate-determining process in the hepatic elimination of HMG-CoA reductase inhibitors in rats and humans. Drug Metab. Dispos. 2010, 38, 215–222. [Google Scholar] [CrossRef]
- Sasaki, M.; Suzuki, H.; Ito, K.; Abe, T.; Sugiyama, Y. Transcellular transport of organic anions across a double-transfected Madin-Darby canine kidney II cell monolayer expressing both human organic anion-transporting polypeptide (OATP2/SLC21A6) and Multidrug resistance-associated protein 2 (MRP2/ABCC2). J. Biol. Chem. 2002, 277, 6497–6503. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, M.; Akiyama, S.; Ni’inuma, K.; Nishigaki, R.; Sugiyama, Y. Biliary excretion of pravastatin in rats: Contribution of the excretion pathway mediated by canalicular multispecific organic anion transporter. Drug Metab. Dispos. 1997, 25, 1123–1129. [Google Scholar]
- Niemi, M.; Arnold, K.A.; Backman, J.T.; Pasanen, M.K.; Godtel-Armbrust, U.; Wojnowski, L.; Zanger, U.M.; Neuvonen, P.J.; Eichelbaum, M.; Kivisto, K.T.; et al. Association of genetic polymorphism in ABCC2 with hepatic multidrug resistance-associated protein 2 expression and pravastatin pharmacokinetics. Pharmacogenet. Genom. 2006, 16, 801–808. [Google Scholar] [CrossRef]
- Bernsdorf, A.; Giessmann, T.; Modess, C.; Wegner, D.; Igelbrink, S.; Hecker, U.; Haenisch, S.; Cascorbi, I.; Terhaag, B.; Siegmund, W. Simvastatin does not influence the intestinal P-glycoprotein and MPR2, and the disposition of talinolol after chronic medication in healthy subjects genotyped for the ABCB1, ABCC2 and SLCO1B1 polymorphisms. Br. J. Clin. Pharmacol. 2006, 61, 440–450. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Maeda, K.; Matsushima, S.; Nozaki, Y.; Kusuhara, H.; Sugiyama, Y. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol. Pharmacol. 2005, 68, 800–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Wang, Y.; Grimm, S. ATP-dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab. Dispos. 2006, 34, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Keskitalo, J.E.; Zolk, O.; Fromm, M.F.; Kurkinen, K.J.; Neuvonen, P.J.; Niemi, M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2009, 86, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Staud, F.; Ceckova, M.; Micuda, S.; Pavek, P. Expression and function of p-glycoprotein in normal tissues: Effect on pharmacokinetics. Methods Mol. Biol. 2010, 596, 199–222. [Google Scholar] [CrossRef]
- Yamasaki, D.; Nakamura, T.; Okamura, N.; Kokudai, M.; Inui, N.; Takeuchi, K.; Watanabe, H.; Hirai, M.; Okumura, K.; Sakaeda, T. Effects of acid and lactone forms of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on the induction of MDR1 expression and function in LS180 cells. Eur. J. Pharm. Sci. 2009, 37, 126–132. [Google Scholar] [CrossRef]
- Rodrigues, A.C.; Curi, R.; Genvigir, F.D.; Hirata, M.H.; Hirata, R.D. The expression of efflux and uptake transporters are regulated by statins in Caco-2 and HepG2 cells. Acta Pharmacol. Sin. 2009, 30, 956–964. [Google Scholar] [CrossRef]
- Hirano, M.; Maeda, K.; Shitara, Y.; Sugiyama, Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J. Pharmacol. Exp. Ther. 2004, 311, 139–146. [Google Scholar] [CrossRef]
- Skottheim, I.B.; Gedde-Dahl, A.; Hejazifar, S.; Hoel, K.; Asberg, A. Statin induced myotoxicity: The lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur. J. Pharm. Sci. 2008, 33, 317–325. [Google Scholar] [CrossRef]
- Schirris, T.J.; Renkema, G.H.; Ritschel, T.; Voermans, N.C.; Bilos, A.; van Engelen, B.G.; Brandt, U.; Koopman, W.J.; Beyrath, J.D.; Rodenburg, R.J.; et al. Statin-Induced Myopathy is Associated with Mitochondrial Complex III Inhibition. Cell Metab. 2015, 22, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Westwood, F.R.; Bigley, A.; Randall, K.; Marsden, A.M.; Scott, R.C. Statin-induced muscle necrosis in the rat: Distribution, development, and fibre selectivity. Toxicol. Pathol. 2005, 33, 246–257. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.K. Molecular clues into the pathogenesis of statin-mediated muscle toxicity. Muscle Nerve 2005, 31, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.R., 3rd; Byington, R.P.; Furberg, C.D. HMG-CoA reductase inhibitor therapy and stroke risk reduction: An analysis of clinical trials data. Atherosclerosis 1998, 138, 11–24. [Google Scholar] [CrossRef]
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Presta, A.; Liu, J.; Sessa, W.C.; Stuehr, D.J. Substrate binding and calmodulin binding to endothelial nitric oxide synthase coregulate its enzymatic activity. Nitric. Oxide 1997, 1, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Liao, J.K. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J. Biol. Chem. 1998, 273, 24266–24271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, J.; Magenau, A.; Rodriguez, M.; Rentero, C.; Royo, T.; Enrich, C.; Thomas, S.R.; Grewal, T.; Gaus, K. Activation of Endothelial Nitric Oxide (eNOS) Occurs through Different Membrane Domains in Endothelial Cells. PLoS ONE 2016, 11, e0151556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kureishi, Y.; Luo, Z.; Shiojima, I.; Bialik, A.; Fulton, D.; Lefer, D.J.; Sessa, W.C.; Walsh, K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000, 6, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Laufs, U.; Huang, Z.; Nakamura, T.; Huang, P.; Moskowitz, M.A.; Liao, J.K. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1998, 95, 8880–8885. [Google Scholar] [CrossRef] [Green Version]
- Laufs, U.; Endres, M.; Stagliano, N.; Amin-Hanjani, S.; Chui, D.S.; Yang, S.X.; Simoncini, T.; Yamada, M.; Rabkin, E.; Allen, P.G.; et al. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J. Clin. Investig. 2000, 106, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Cardena, G.; Oh, P.; Liu, J.; Schnitzer, J.E.; Sessa, W.C. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: Implications for nitric oxide signaling. Proc. Natl. Acad. Sci. USA 1996, 93, 6448–6453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, P.; Xu, X.; Wang, Y.; Xia, Y.; Wang, D. Simvastatin increases the activity of endothelial nitric oxide synthase via enhancing phosphorylation. J. Huazhong Univ. Sci. Technol. Med. Sci. 2009, 29, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, C.R.; Gast, C.; Li, F.; Schafer, M.; Tillmanns, H.; Waldecker, B.; Wiecha, J. Cerivastatin activates endothelial calcium-activated potassium channels and thereby modulates endothelial nitric oxide production and cell proliferation. J. Am. Soc. Nephrol. 2004, 15, 868–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.Y.; DeVault, A.R.; Wang-Iverson, D.; Ivashkiv, E.; Swanson, B.N.; Sugerman, A.A. Comparative pharmacokinetics and pharmacodynamics of pravastatin and lovastatin. J. Clin. Pharmacol. 1990, 30, 1128–1135. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Li, H.; Tang, J.; Wang, J.; Luo, J.; Liu, B.; Wang, J.; Shi, X.; Cui, H.; Tang, J.; et al. Discovery of a potent HMG-CoA reductase degrader that eliminates statin-induced reductase accumulation and lowers cholesterol. Nat. Commun. 2018, 9, 5138. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, C.; Deplazes, E.; Cranfield, C.G.; Garcia, A. The Role of Structure and Biophysical Properties in the Pleiotropic Effects of Statins. Int. J. Mol. Sci. 2020, 21, 8745. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228745
Murphy C, Deplazes E, Cranfield CG, Garcia A. The Role of Structure and Biophysical Properties in the Pleiotropic Effects of Statins. International Journal of Molecular Sciences. 2020; 21(22):8745. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228745
Chicago/Turabian StyleMurphy, Christopher, Evelyne Deplazes, Charles G. Cranfield, and Alvaro Garcia. 2020. "The Role of Structure and Biophysical Properties in the Pleiotropic Effects of Statins" International Journal of Molecular Sciences 21, no. 22: 8745. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228745