The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
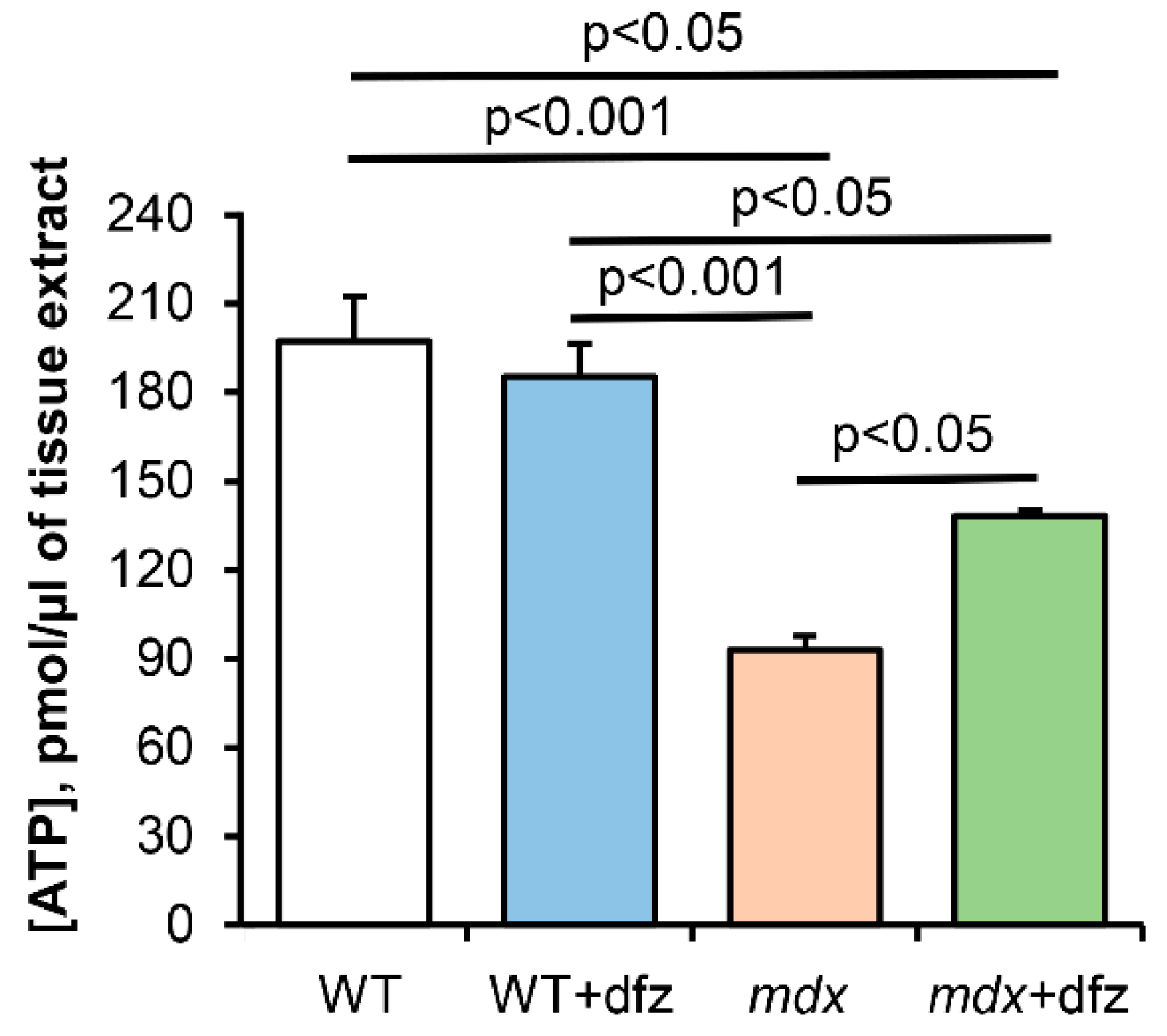
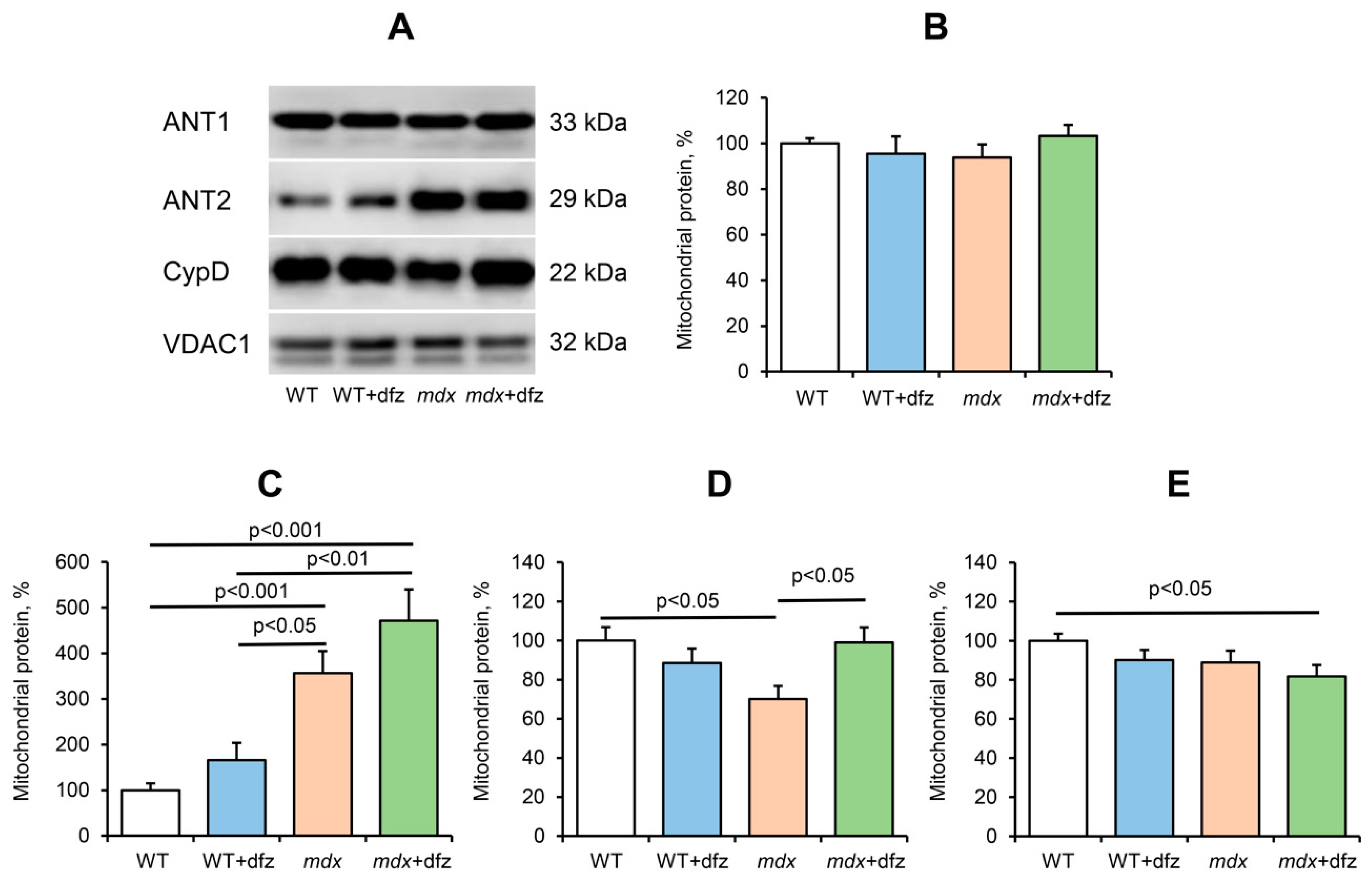
2.1. Effect of Deflazacort Administration on Respiration and Oxidative Phosphorylation of Skeletal Muscle Mitochondria of Dystrophin-Deficient and WT Mice
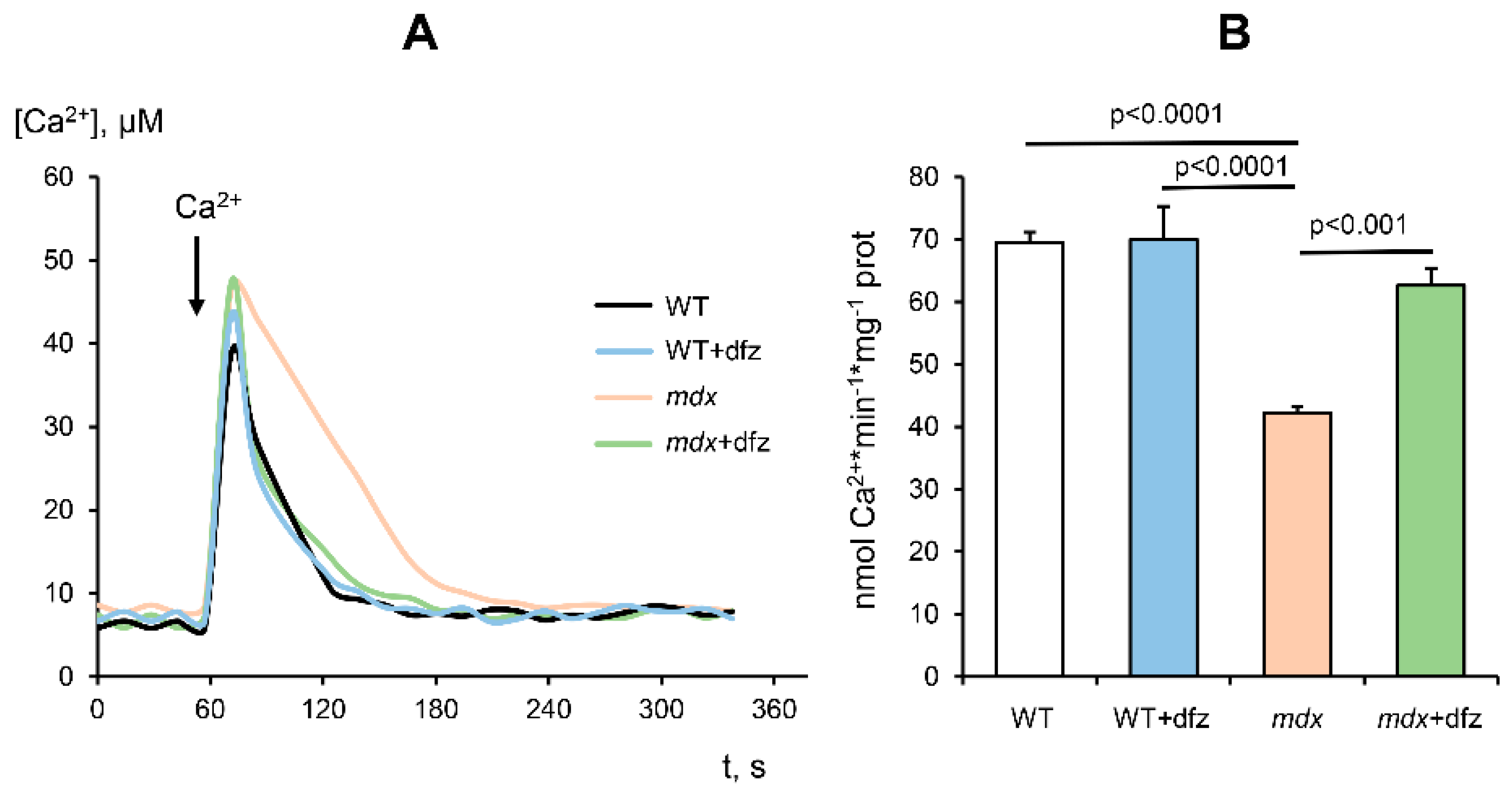
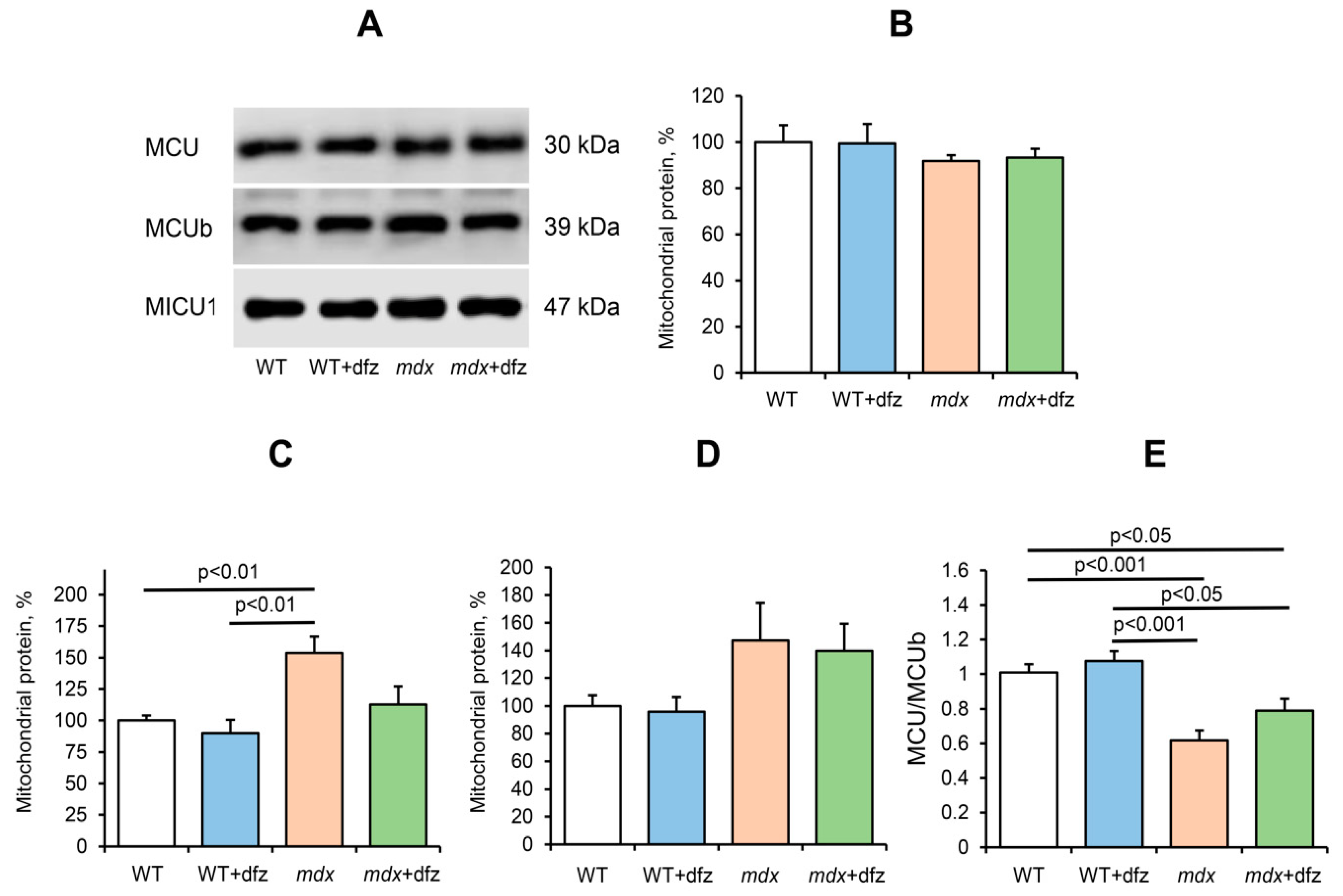
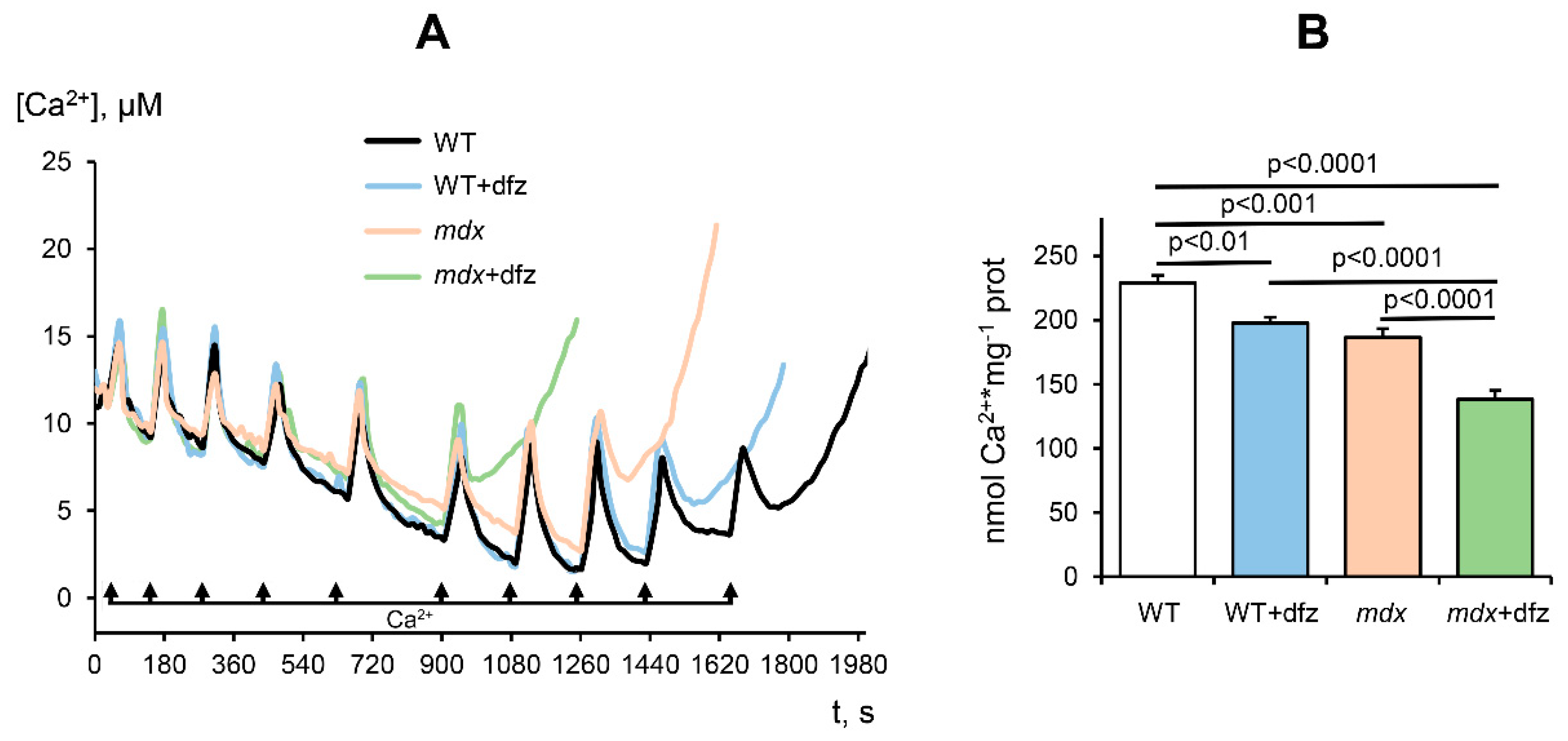
2.2. Effect of Deflazacort Administration on Ca2+ Uniport in the Skeletal-Muscle Mitochondria of Dystrophin-Deficient and WT Mice
2.3. Effect of Deflazacort Administration on MPT Pore Opening in the Skeletal-Muscle Mitochondria of Dystrophin-Deficient and WT Mice
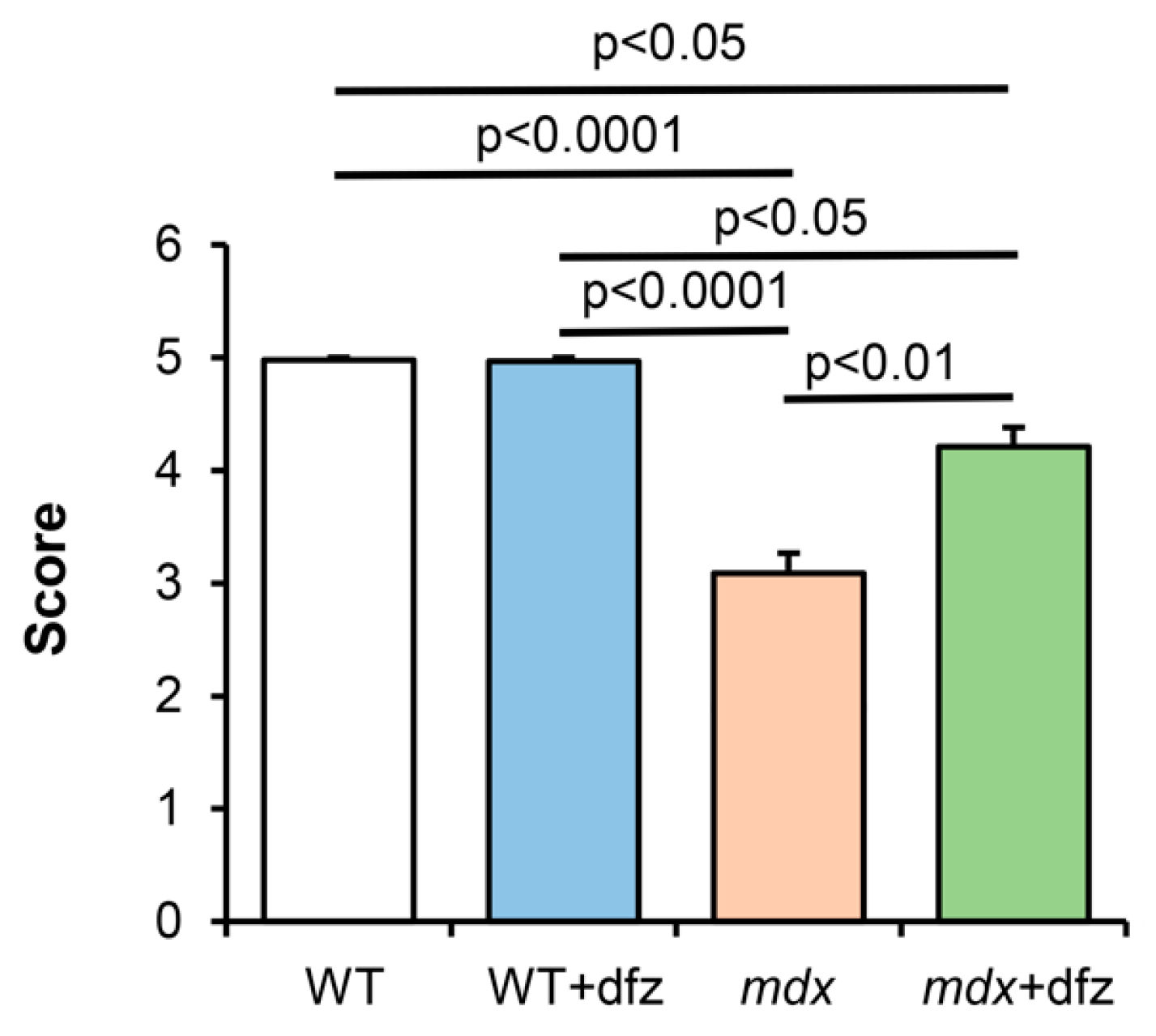
2.4. Deflazacort Improves the Muscle Function of Dystrophin-Deficient Mice
3. Discussion
4. Materials and Methods
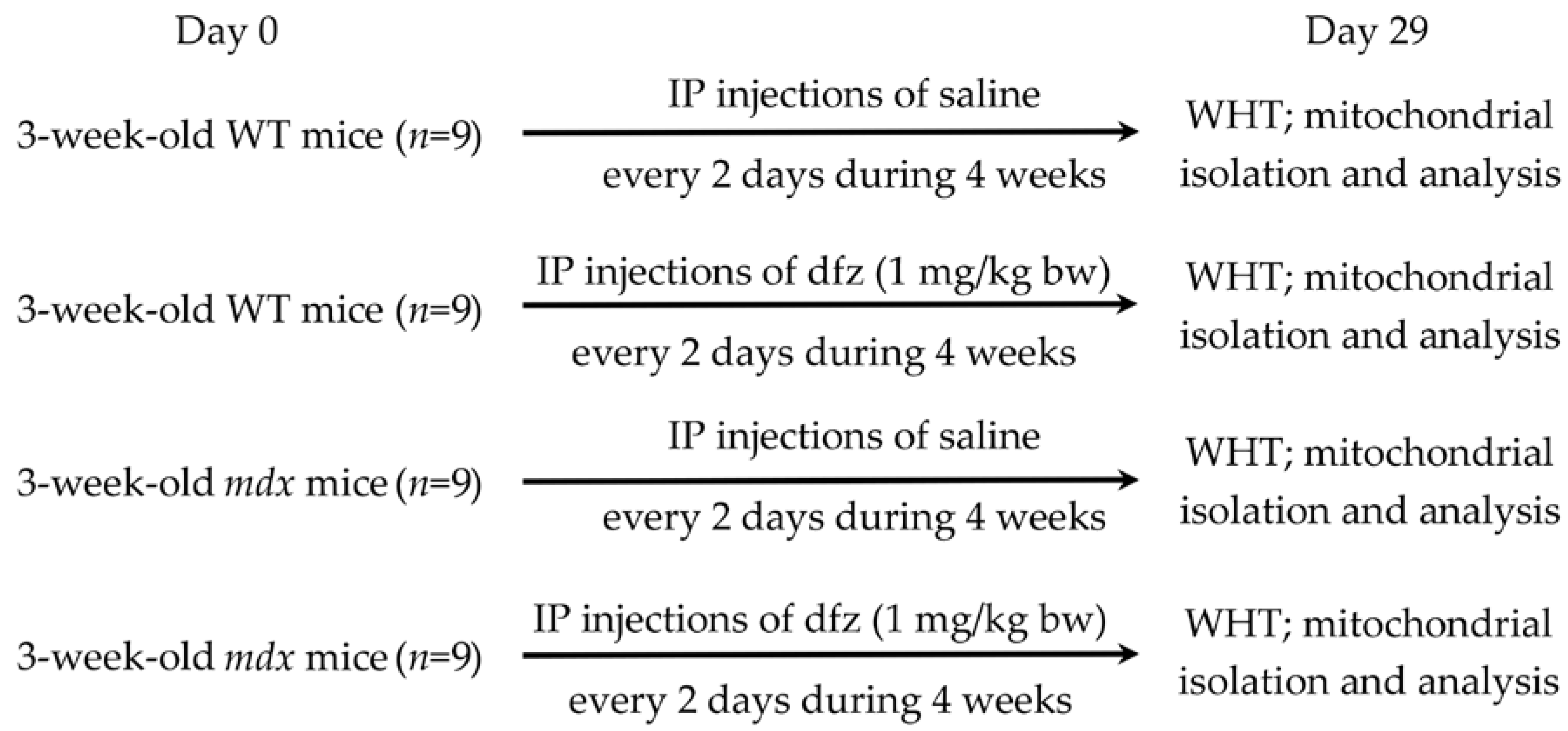
4.1. Animals
4.2. Deflazacort Administration
4.3. Physiological Test
4.4. Mitochondria Isolation
4.5. ATP Measurement in Skeletal Muscle
4.6. Determination of Mitochondrial Respiration and Oxidative Phosphorylation
4.7. Uptake and Release of Mitochondrial Calcium, Determination of Ca2+ Retention by Mitochondria
4.8. Electrophoresis and Immunoblotting of Mitochondrial Proteins
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DMD | Duchenne muscular dystrophy |
| MPT | Mitochondrial permeability transition |
| Dfz | Deflazacort |
References
- Emery, A.E. Population frequencies of inherited neuromuscular diseases-a world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Romitti, P.A.; Zhu, Y.; Puzhankara, S.; James, K.A.; Nabukera, S.K.; Zamba, G.K.; Ciafaloni, E.; Cunniff, C.; Druschel, C.M.; Mathews, K.D.; et al. Prevalence of Duchenne and Beckermuscular dystrophies in the United States. Pediatrics 2015, 135, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Ohlendieck, K.; Campbell, K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991, 115, 1685–1694. [Google Scholar] [CrossRef]
- Turner, P.R.; Westwood, T.; Regen, C.M.; Steinhardt, R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 1988, 335, 735–738. [Google Scholar] [CrossRef]
- Spencer, M.J.; Tidball, J.G. Calpain concentration is elevated although net calcium-dependent proteolysis is suppressed in dystrophin-deficient muscle. Exp. Cell Res. 1992, 203, 107–114. [Google Scholar] [CrossRef]
- Lindahl, M.; Backman, E.; Henriksson, K.G.; Gorospe, J.R.; Hoffman, E.P. Phospholipase A2 activity in dystrophinopathies. Neuromuscul. Disord. 1995, 5, 193–199. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of dystrophin disrupts skeletal muscle signaling: Rles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, J. Phosphorylase activity and glycogen, glucose-6-phosphate, and lactic acid content of human skeletal muscle in various myopathies. J. Lab. Clin. Med. 1965, 66, 452–463. [Google Scholar] [PubMed]
- Chi, M.M.Y.; Hintz, C.S.; McKee, D.; Felder, S.; Grant, N.; Kaiser, K.K.; Lowry, O.H. Effect of Duchenne muscular dystrophy on enzymes of energy metabolism in individual muscle fibers. Metabolism 1987, 36, 761–767. [Google Scholar] [CrossRef]
- Chinet, A.; Even, P.; Decrouy, A. Dystrophin-dependent efficiency of metabolic pathways in mouse skeletal muscles. Experientia 1994, 50, 602–605. [Google Scholar] [CrossRef]
- Van Bennekom, C.; Oerlemans, F.T.; Kulakowski, S.; De Bruyn, C.H. Enzymes of purine metabolism in muscle specimens from patients with Duchenne-type muscular dystrophy. Adv. Exp. Med. Biol. 1984, 165, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Molloy, M.P.; Chamberlain, J.S.; Andrews, P.C. Proteomic analysis of mdx skeletal muscle: Great reduction of adenylate kinase 1 expression and enzymatic activity. Proteomics 2003, 3, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Bonsett, C.; Rudman, A. Duchenne’s muscular dystrophy: A tissue culture perspective. Indiana Med. 1984, 77, 446–449. [Google Scholar] [PubMed]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.; Von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophindeficient Mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.F.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef] [Green Version]
- Ronzoni, E.; Wald, S.; Berg, L.; Ramsey, R. Distribution of high energy phosphate in normal and dystrophic muscle. Neurology 1958, 8, 359–368. [Google Scholar] [CrossRef]
- Vignos, P., Jr.; Warner, J. Glycogen, creatine and high energy phosphate in human muscle disease. J. Lab. Clin. Med. 1963, 62, 579–590. [Google Scholar]
- Cole, M.; Rafael, J.; Taylor, D.; Lodi, R.; Davies, K.; Styles, P. A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscul. Disord. 2002, 12, 247–257. [Google Scholar] [CrossRef]
- Shuttlewood, R.; Griffiths, J. The purine nucleotide profile in mouse, chicken and human dystrophic muscle: An abnormal ratio of inosine plus adenine nucleotides to guanine nucleotides. Clin. Sci. 1982, 62, 113–115. [Google Scholar] [CrossRef] [Green Version]
- Godin, R.; Daussin, F.; Matecki, S.; Li, T.; Petrof, B.J.; Burelle, Y. Peroxisome proliferator activated receptor γ coactivator 1-α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. J. Physiol. 2012, 590, 5487–5502. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.L.; Moir, L.; Hemming, S.; Edwards, B.; Squire, S.; Davies, K.; Guiraud, S. Utrophin influences mitochondrial pathology and oxidative stress in dystrophic muscle. Skelet. Muscle 2017, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; Van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.; Martins, A.S.; Niggli, E.; Shirokova, N. Dystrophic cardiomyopathy: Amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 2008, 77, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Rosiers, C.D.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H144–H153. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Morine, K.J.; Bish, L.T.; Pendrak, K.; Sleeper, M.M.; Barton, E.R.; Sweeney, H.L. Systemic myostatin inhibition via liver-targeted gene transfer in normal and dystrophic mice. PLoS ONE 2010, 5, e9176. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: A nonrandomized controlled trial. JAMA Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef]
- Markham, A.; Bryson, H.M. Deflazacort. A review of its pharmacological properties and therapeutic efficacy. Drugs 1995, 50, 317–333. [Google Scholar] [CrossRef]
- Bylo, M.; Farewell, R.; Coppenrath, V.A.; Yogaratnam, D. A review of deflazacort for patients with Duchenne muscular dystrophy. Ann. Pharmacother. 2020, 54, 788–794. [Google Scholar] [CrossRef]
- Bello, L.; Gordish-Dressman, H.; Morgenroth, L.P.; Henricson, E.K.; Duong, T.; Hoffman, E.P.; Cnaan, A.; McDonald, C.M. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne natural history study. Neurology 2015, 85, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griggs, R.C.; Miller, J.P.; Greenberg, C.R.; Fehlings, D.L.; Pestronk, A.; Mendell, J.R.; Moxley, R.T.; King, W.; Kissel, J.T.; Cwik, V.; et al. Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology 2016, 87, 2123–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Hanaoka, B.Y.; Peterson, C.A.; Horbinski, C.; Crofford, L.J. Implications of glucocorticoid therapy in idiopathic inflammatory myopathies. Nat. Rev. Rheumatol. 2012, 8, 448–457. [Google Scholar] [CrossRef]
- Schakman, O.; Gilson, H.; Kalista, S.; Thissen, J. Mechanisms of muscle atrophy induced by glucocorticoids. Hormone Res. 2009, 72 (Suppl. 1), 36–41. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Barefield, D.Y.; Warner, J.L.; Vo, A.H.; Hadhazy, M.; Earley, J.U.; Demonbreun, A.R.; McNally, E.M. Intermittent glucocorticoid steroid dosing enhances muscle repair without eliciting muscle atrophy. J. Clin. Investig. 2017, 127, 2418–2432. [Google Scholar] [CrossRef] [Green Version]
- Hammers, D.W.; Hart, C.C.; Patsalos, A.; Matheny, M.K.; Wright, L.A.; Nagy, L.; Sweeney, H.L. Glucocorticoids counteract hypertrophic effects of myostatin inhibition in dystrophic muscle. JCI Insight 2020, 5, e133276. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kame, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular etabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Paillard, M.; Csordás, G.; Szanda, G.; Golenár, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spät, A.; et al. Tissue-specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep. 2017, 18, 2291–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Talanov, E.Y.; Starinets, V.S.; Agafonov, A.V.; Dubinin, M.V.; Belosludtseva, N.V. Transport of Ca2+ and Ca2+-dependent permeability transition in rat liver mitochondria under the streptozotocin-induced type I diabetes. Cells 2019, 8, 1014. [Google Scholar] [CrossRef] [Green Version]
- Kaludercic, N.; Scorrano, L. MCUB hearts mitochondria in sickness, less in health. Circulation 2019, 140, 1734–1736. [Google Scholar] [CrossRef]
- Lambert, J.P.; Luongo, T.S.; Tomar, D.; Jadiya, P.; Gao, E.; Zhang, X.; Lucchese, A.M.; Kolmetzky, D.W.; Shah, N.S.; Elrod, J.W. MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation 2019, 140, 1720–1733. [Google Scholar] [CrossRef]
- Bhosale, G.; Sharpe, J.A.; Koh, A.; Kouli, A.; Szabadkai, G.; Duchen, M.R. Pathological consequences of MICU1 mutations on mitochondrial calcium signalling and bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1009–1017. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium. 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Jonas, E.A.; Porter, G.A., Jr.; Beutner, G.; Mnatsakanyan, N.; Alavian, K.N. Cell death disguised: The mitochondrial permeability transition pore as the c-subunit of the F(1)F(O) ATP synthase. Pharmacol. Res. 2015, 99, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial permeability transition: A molecular lesion with multiple drug targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef] [PubMed]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP synthase c-subunit-deficient mitochondria have a small cyclosporine A-sensitive channel, but lack the permeability transition pore. Cell Rep. 2019, 26, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Pinton, P. A New current for the mitochondrial permeability transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Archer, J.D.; Vargas, C.C.; Anderson, J.E. Persistent and improved functional gain in mdx dystrophic mice after treatment with L-arginine and deflazacort. FASEB J. 2006, 20, 738–740. [Google Scholar] [CrossRef]
- Symons, A.M.; Lewis, D.A.; Ancill, R.J. The actions of anti-inflammatory steroids on isolated rat liver mitochondrial function. J. Steroid Biochem. 1974, 5, 639–644. [Google Scholar] [CrossRef]
- Brouilly, N.; Lecroisey, C.; Martin, E.; Pierson, L.; Mariol, M.C.; Qadota, H.; Labouesse, M.; Streichenberger, N.; Mounier, N.; Gieseler, K. Ultra-structural time-course study in the C. elegans model for Duchenne muscular dystrophy highlights a crucial role for sarcomere-anchoring structures and sarcolemma integrity in the earliest steps of the muscle degeneration process. Hum. Mol. Genet. 2015, 24, 6428–6445. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, J.E.; Pollard, A.K.; Lesanpezeshki, L.; Deane, C.S.; Gaffney, C.J.; Etheridge, T.; Szewczyk, N.J.; Vanapalli, S.A. Muscle strength deficiency and mitochondrial dysfunction in a muscular dystrophy model of Caenorhabditis elegans and its functional response to drugs. Dis. Model Mech. 2018, 11, dmm036137. [Google Scholar] [CrossRef] [Green Version]
- Passaquin, A.C.; Lhote, P.; Rüegg, U.T. Calcium influx inhibition by steroids and analogs in C2C12 skeletal muscle cells. Br. J. Pharmacol. 1998, 124, 1751–1759. [Google Scholar] [CrossRef]
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597. [Google Scholar] [CrossRef] [Green Version]
- Koshkin, V.; Bikopoulos, G.; Chan, C.B.; Wheeler, M.B. The characterization of mitochondrial permeability transition in clonal pancreatic beta-cells. Multiple modes and regulation. J. Biol. Chem. 2004, 279, 41368–41376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Hemendinger, R.; Greenamyre, J.T.; Rosenfeld, J. Species- and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am. J. Phys. Cell Physiol. 2007, 292, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endlicher, R.; Kriváková, P.; Lotkova, H.; Milerová, M.; Drahota, Z.; Cervinková, Z. Tissue specific sensitivity of mitochondrial permeability transition pore to Ca2+ ions. Acta Med. (Hradec Kralove) 2009, 52, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Laker, R.C.; Taddeo, E.P.; Akhtar, Y.N.; Zhang, M.; Hoehn, K.L.; Yan, Z. The mitochondrial permeability transition pore regulator cyclophilin D exhibits tissuespecific control of metabolic homeostasis. PLoS ONE 2016, 11, e0167910. [Google Scholar] [CrossRef]
- Graham, B.H.; Waymire, K.G.; Cottrell, B.; Trounce, I.A.; MacGregor, G.R.; Wallace, D.C. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat. Genet. 1997, 16, 226–234. [Google Scholar] [CrossRef]
- Wu, S.; Sampson, M.J.; Decker, W.K.; Craigen, W.J. Each mammalian mitochondrial outer membrane porin protein is dispensable: Effects on cellular respiration. Biochim. Biophys. Acta 1999, 1452, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Hyzewicz, J.; Tanihata, J.; Kuraoka, M.; Ito, N.; Miyagoe-Suzuki, Y.; Takeda, S. Low intensity training of mdx mice reduces carbonylation and increases expression levels of proteins involved in energy metabolism and muscle contraction. Free Radic. Biol. Med. 2015, 82, 122–136. [Google Scholar] [CrossRef] [Green Version]
- Neginskaya, M.A.; Strubbe, J.O.; Amodeo, G.F.; West, B.A.; Yakar, S.; Bazil, J.N.; Pavlov, E.V. The very low number of calcium-induced permeability transition pores in the single mitochondrion. J. Gen. Physiol. 2020, 152, e202012631. [Google Scholar] [CrossRef]
- Colell, A.; Garcia-Ruiz, C.; Lluis, M.; Coll, O.; Mari, M.; Fernandez-Chaca, J.C. Cholesterol impairs the adenine nucleotide translocator-mediated mitochondrial permeability transition through altered membrane fluidity. J. Biol. Chem. 2003, 278, 33928–33935. [Google Scholar] [CrossRef] [Green Version]
- Guiraud, S.; Davies, K.E. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef]
- Peterson, J.M.; Bakkar, N.; Guttridge, D.C. NF-kappaB signaling in skeletal muscle health and disease. Curr. Top. Dev. Biol. 2011, 96, 85–119. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Vil-lalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.M.; Wang, D.J.; Shettigar, V.; Roof, S.R.; Canan, B.D.; Bakkar, N.; Shintaku, J.; Gu, J.M.; Little, S.C.; Ratnam, N.M.; et al. NF-κB inhibition rescues cardiac function by remodeling calcium genes in a Duchenne muscular dystrophy model. Nat. Commun. 2018, 9, 3431. [Google Scholar] [CrossRef] [PubMed]
- Guseva, N.V.; Taghiyev, A.F.; Sturm, M.T.; Rokhlin, O.W.; Cohen, M.B. Tumor necrosis factor-related apoptosis-inducing ligand-mediated activation of mitochondria-associated nuclear factor-kappaB in prostatic carcinoma cell lines. Mol. Cancer Res. 2004, 2, 574–584. [Google Scholar] [PubMed]
- Cogswell, P.C.; Kashatus, D.F.; Keifer, J.A.; Guttridge, D.C.; Reuther, J.Y.; Bristow, C.; Roy, S.; Nicholson, D.W.; Baldwin, A.S., Jr. NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J. Biol. Chem. 2003, 278, 2963–2968. [Google Scholar] [CrossRef] [Green Version]
- Nan, J.; Hu, H.; Sun, Y.; Zhu, L.; Wang, Y.; Zhong, Z.; Zhao, J.; Zhang, N.; Wang, Y.; Wang, Y.; et al. TNFR2 stimulation promotes mitochondrial fusion via Stat3- and NF-kB dependent activation of OPA1 expression. Circ. Res. 2017, 121, 392–410. [Google Scholar] [CrossRef]
- Liu, H.; Ma, Y.; Pagliari, L.J.; Perlman, H.; Yu, C.; Lin, A.; Pope, R.M. TNF-alpha-induced apoptosis of macrophages following inhibition of NF-kappa B: A central role for disruption of mitochondria. J. Immunol. 2004, 172, 1907–1915. [Google Scholar] [CrossRef] [Green Version]
- Psarra, A.M.; Sekeris, C.E. Nuclear receptors and other nuclear transcription factors in mitochondria: Regulatory molecules in a new environment. Biochim. Biophys. Acta 2008, 1783, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef]
- Deacon, R.M. Measuring motor coordination in mice. J. Vis. Exp. 2013, 75, e2609. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem. 1955, 217, 383–393. [Google Scholar] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228763
Dubinin MV, Talanov EY, Tenkov KS, Starinets VS, Belosludtseva NV, Belosludtsev KN. The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2020; 21(22):8763. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228763
Chicago/Turabian StyleDubinin, Mikhail V., Eugeny Yu. Talanov, Kirill S. Tenkov, Vlada S. Starinets, Natalia V. Belosludtseva, and Konstantin N. Belosludtsev. 2020. "The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 21, no. 22: 8763. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228763