Plasmin-Induced Activation of Human Platelets Is Modulated by Thrombospondin-1, Bona Fide Misfolded Proteins and Thiol Isomerases

 and
and 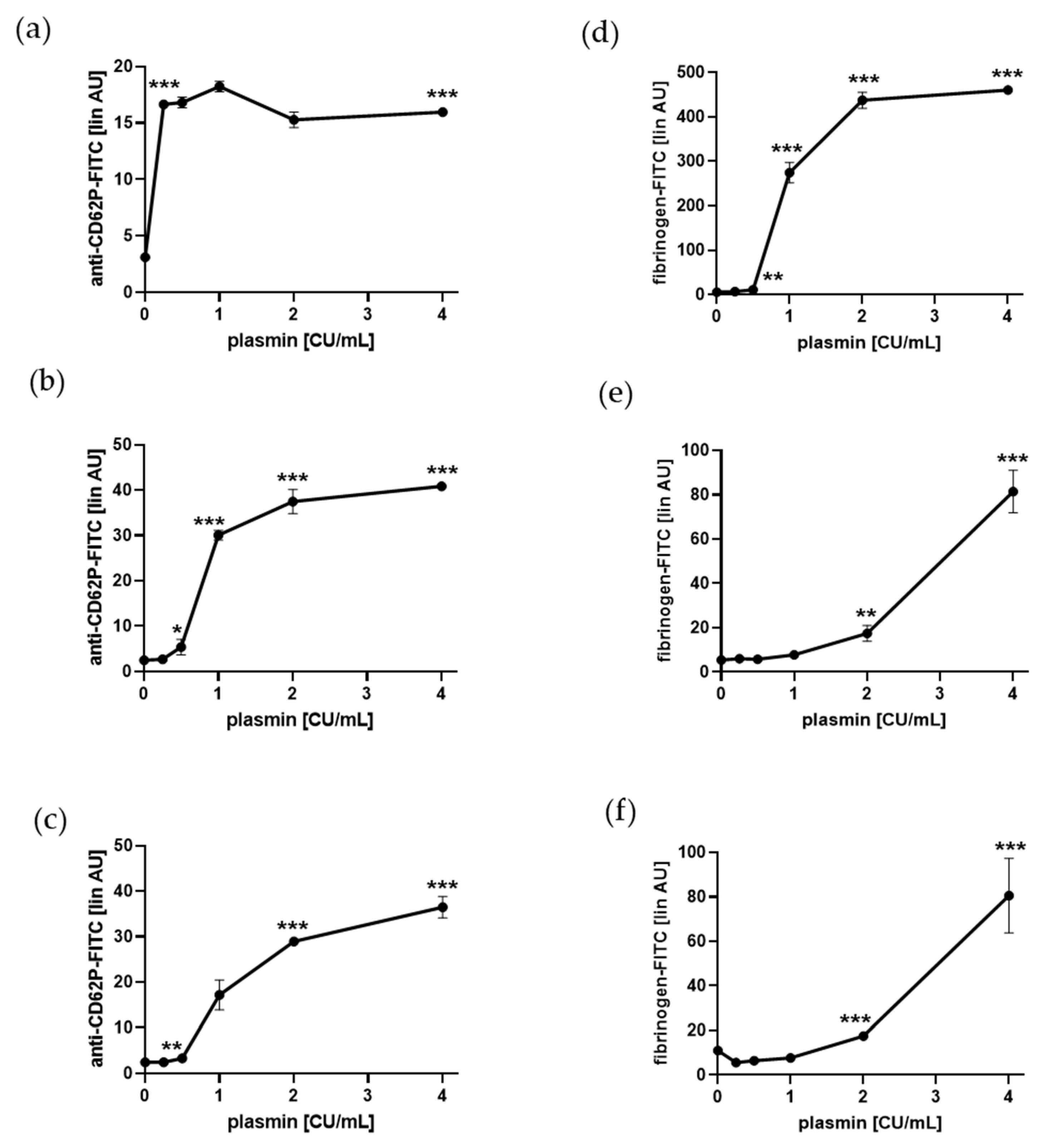{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Plasmin Induces a-Granule Exocytosis and Adhesion Protein Binding of Human Platelets
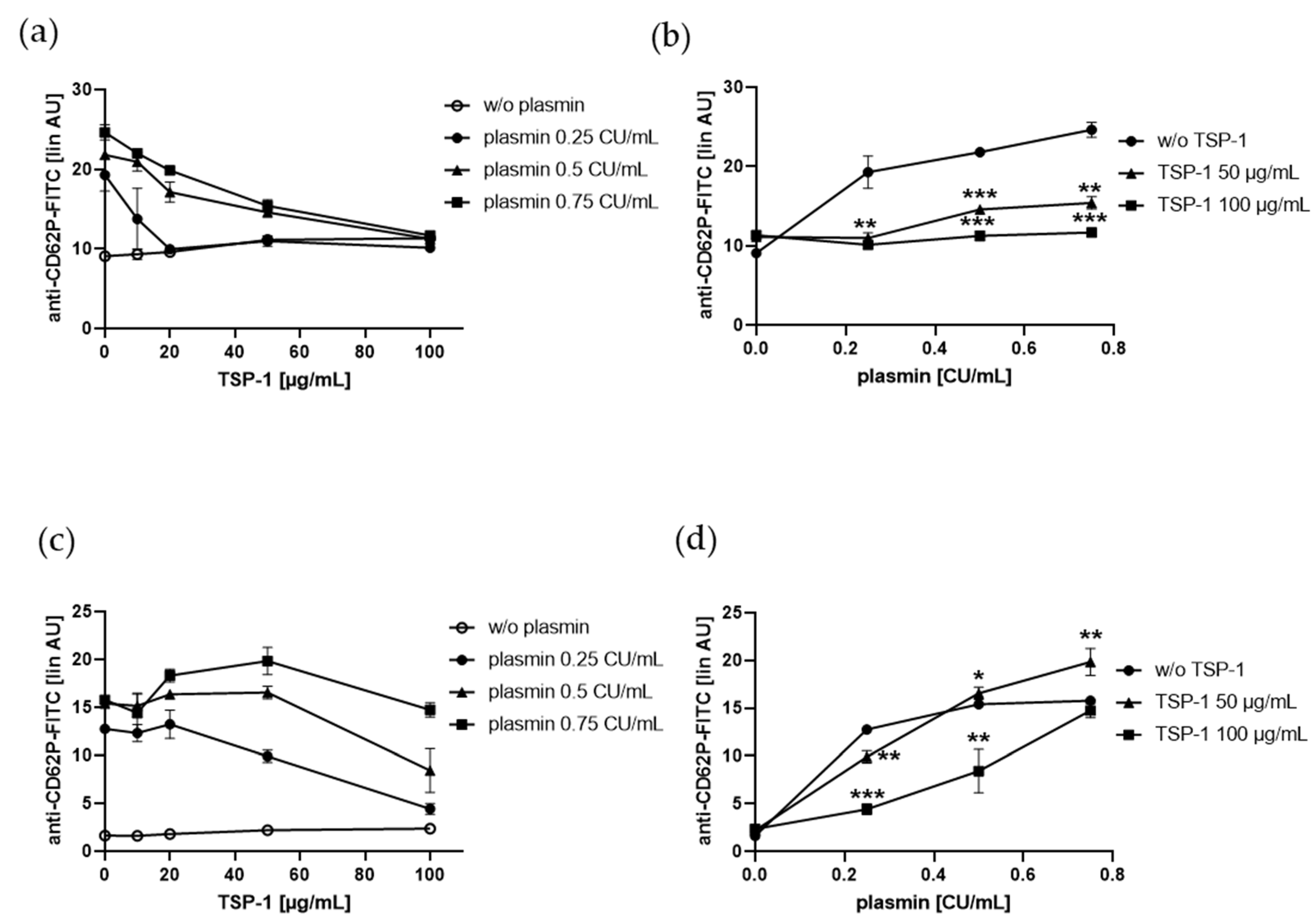
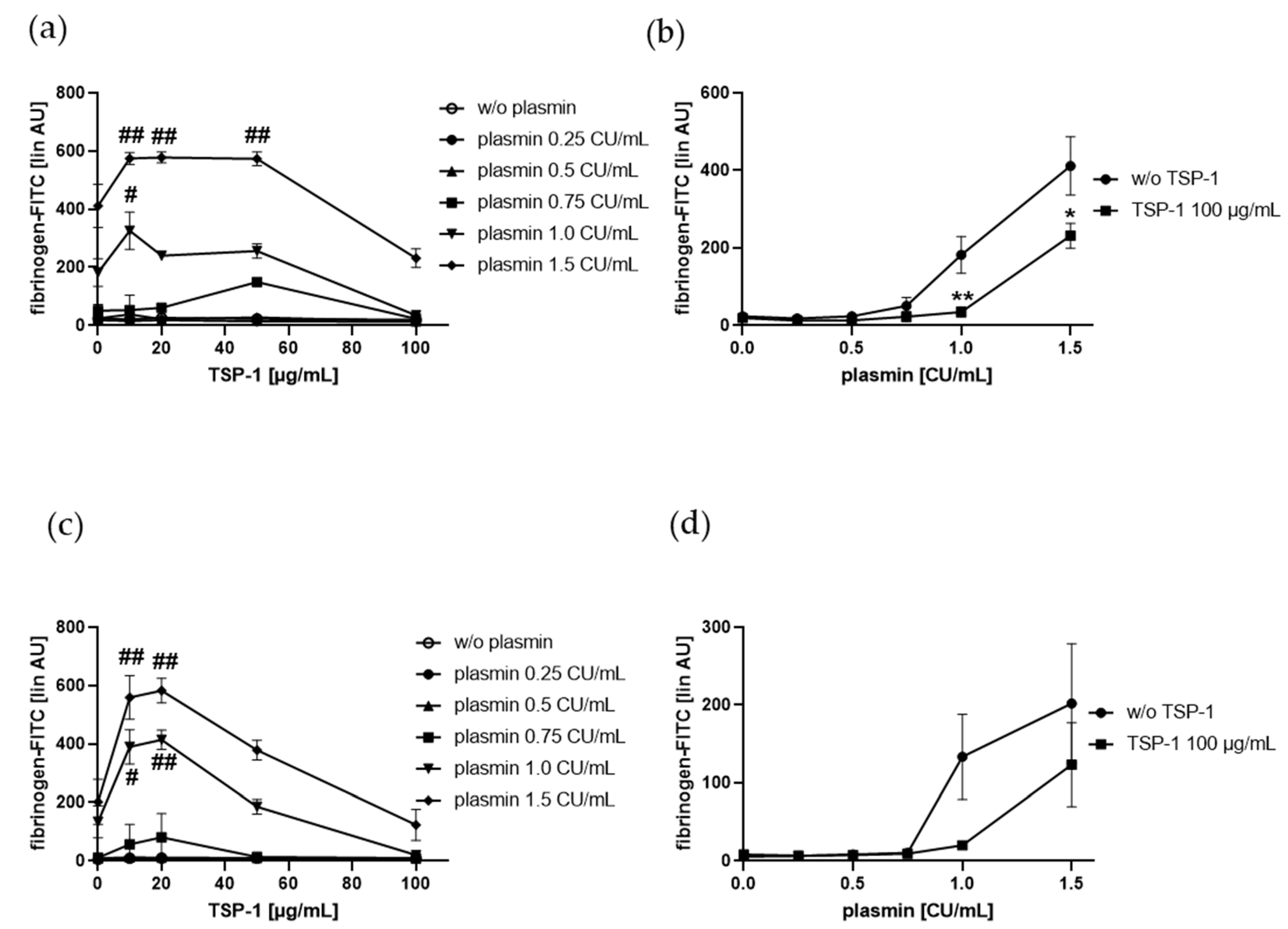
2.2. TSP-1 Acts As a Promoter and Inhibitor of Plasmin-Induced Platelet α-Granule Exocytosis
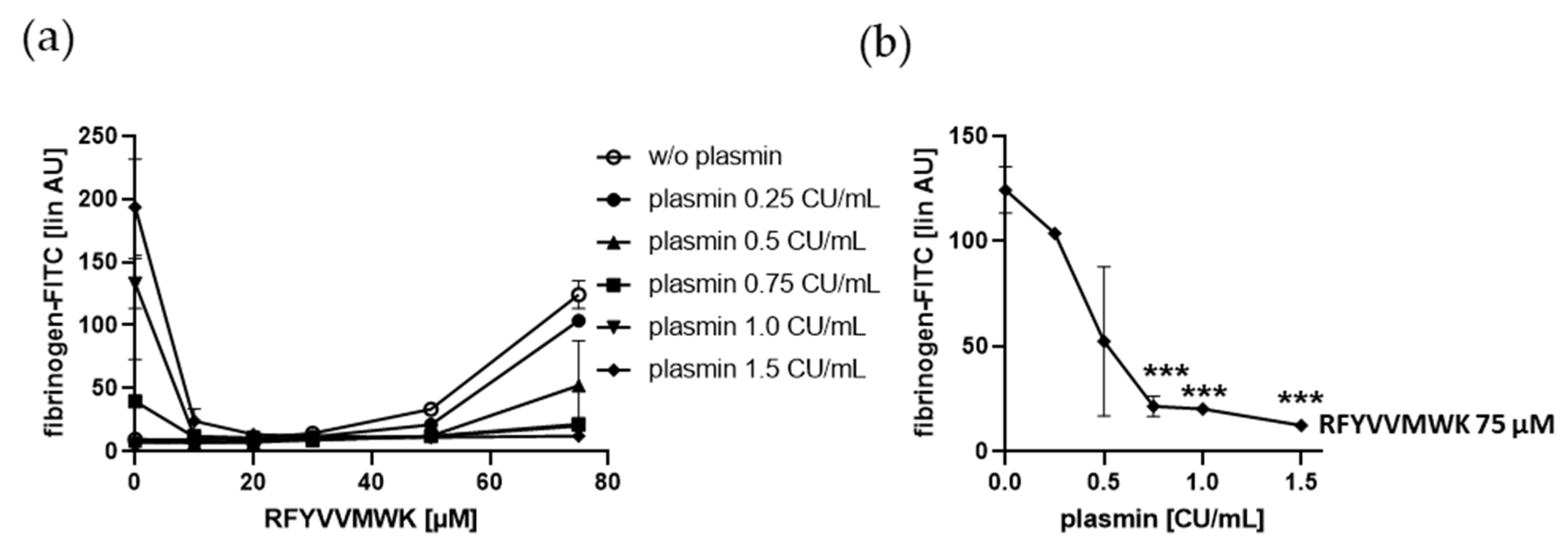
2.3. Plasmin-Induced Fibrinogen Binding to Human Platelets Is Diminished by the C-Terminal TSP-1 Peptide RFYVVMWK, and Plasmin Reduced RFYVVMWK-Induced Fibrinogen Binding Vice Versa
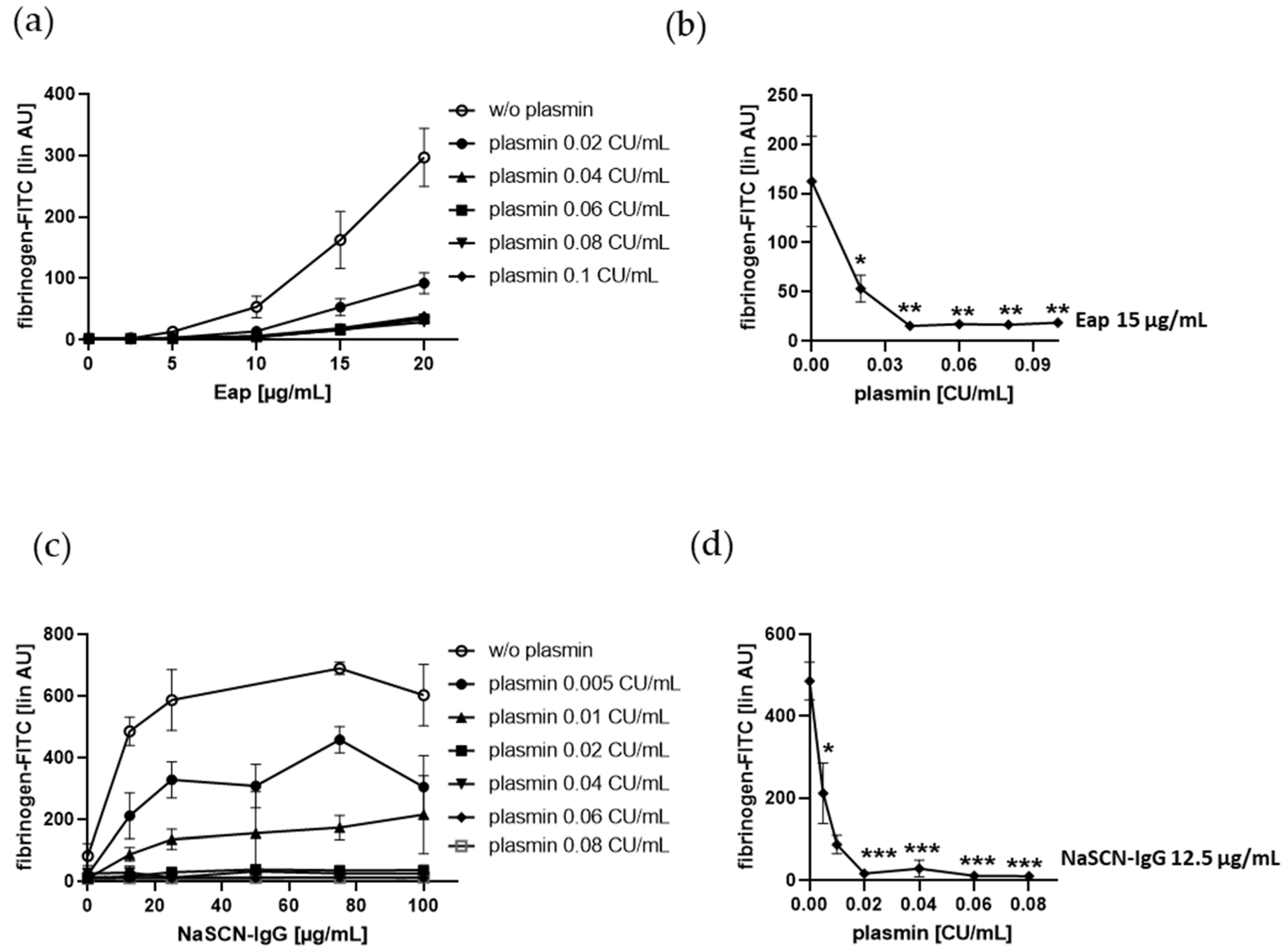
2.4. Fibrinogen Binding to Human Platelets Induced by the Bona Fide Amyloid-Like Eap or NaSCN-Modified IgG is Diminshed by Plasmin at Low Doses
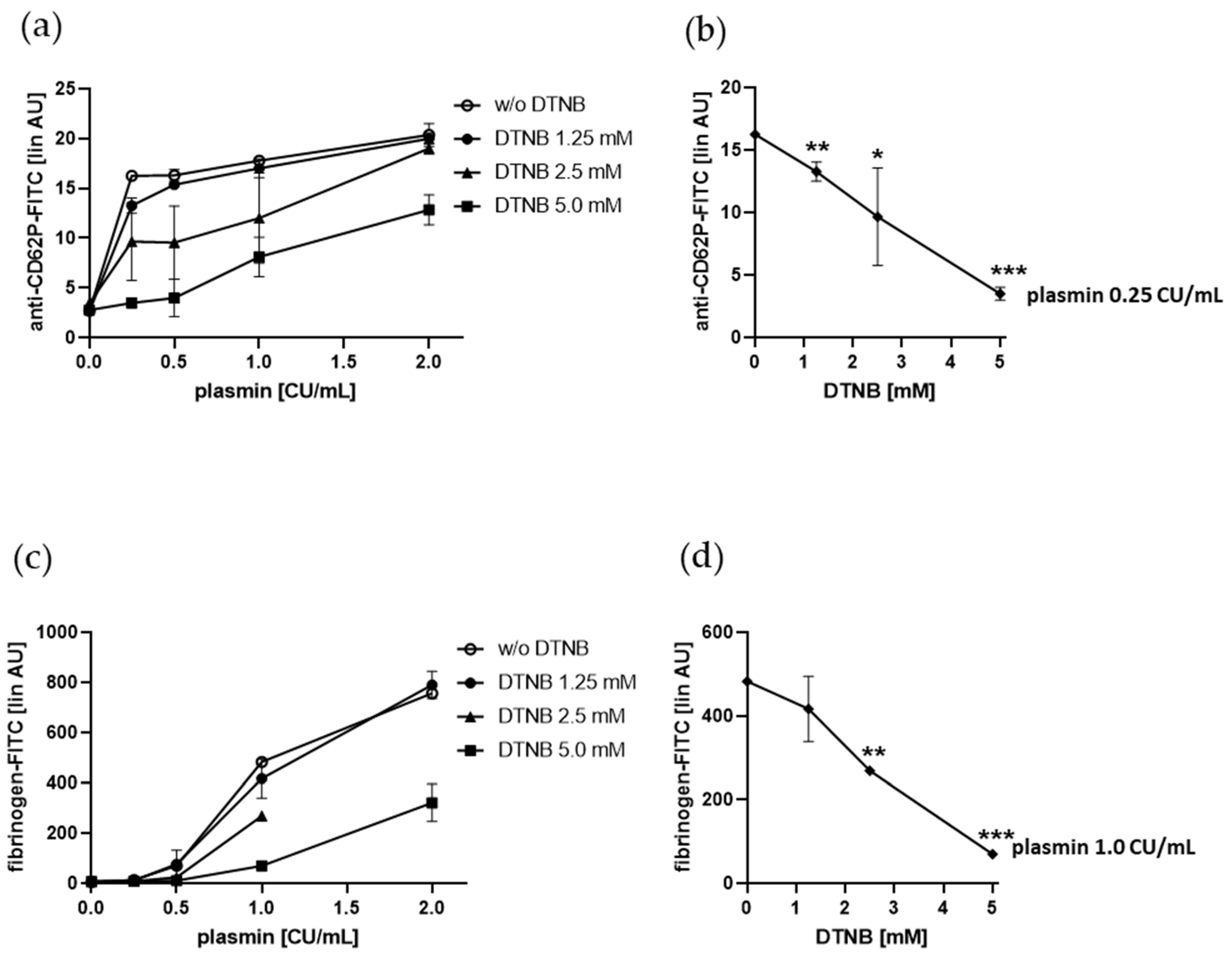
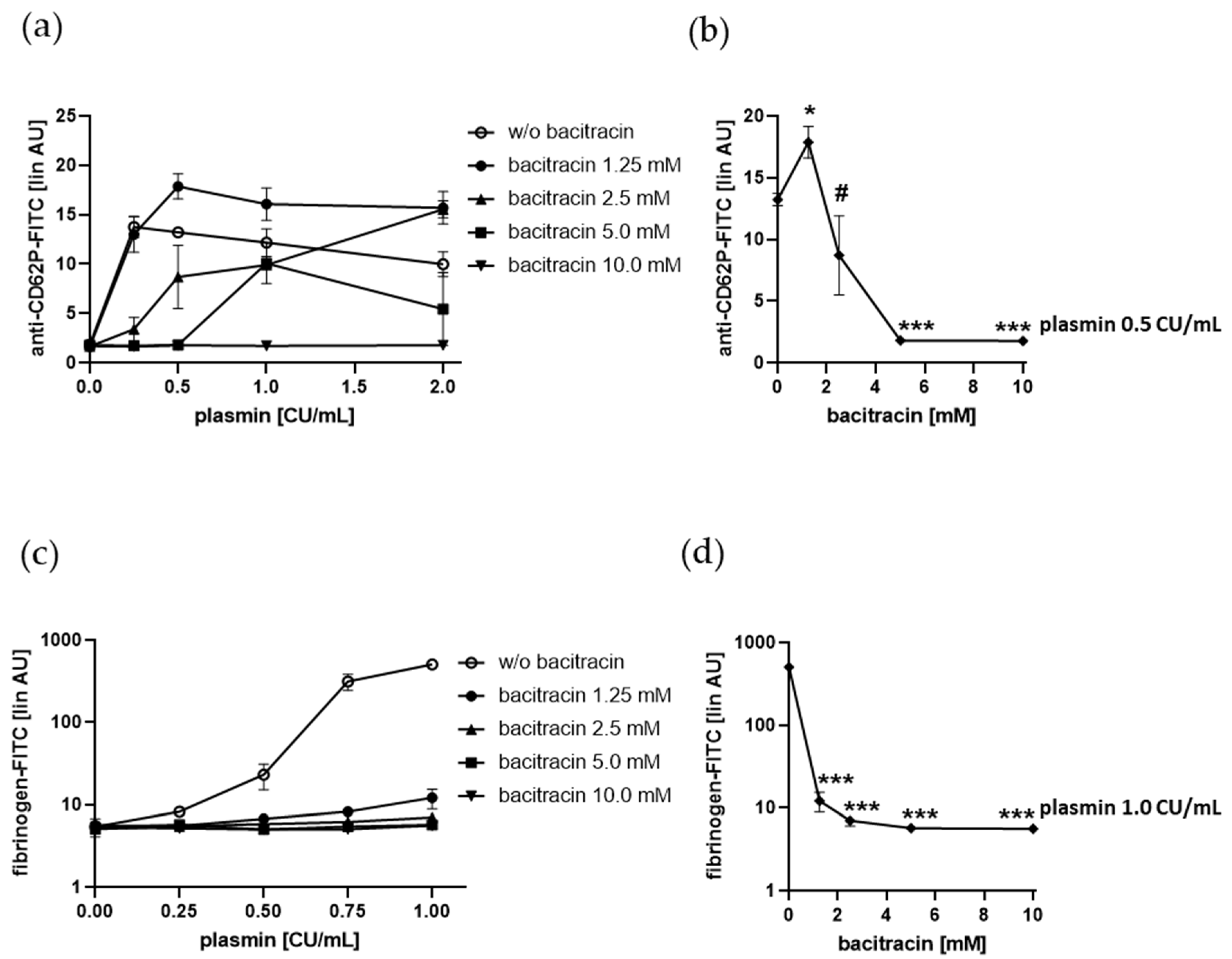
2.5. Plasmin-Induced Platelet P-selectin Expression and Fibrinogen Binding Depend on Enzymatically Catalyzed Disulfide Exchange on the Platelet Surface
3. Discussion
4. Materials and Methods
4.1. Blood Collection
4.2. Preparation of Human Platelets
4.3. Preparation of Denatured Human IgG by NaSCN Treatment
4.4. Analysis of P-selectin (CD62P) Expression on Isolated Platelets by Flow Cytometry
4.5. Analysis of P-selectin Expression on Platelets in Whole Blood by Flow Cytometry
4.6. Flow Cytometric Analysis of Exogenous Fibrinogen and Thrombospondin-1 Binding to Platelets
4.7. t-PA-dependent Plasminogen Conversion to Plasmin
4.8. Data Analysis and Statistics
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AU | arbitrary units |
| DTNB | 5,5’-dithiobis-(2-nitrobenzoic acid) |
| Eap | extracellular adherence protein |
| FDP | fibrin degradation products |
| PDI | protein disulfide isomerase |
| PRP | platelet-rich plasma |
| RT | room temperature |
| tPA | tissue-type plasminogen activator |
| TSP-1 | thrombospondin-1 |
| uPA | urokinase-type plasminogen activator |
References
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Colucci, M.S.N.; Semeraro, F. Platelets and Fibrinolysis. In Platelets in Thrombotic and Non-Thrombotic Disorders; Gresele, P., Kleiman, N.S., Lopez, J.A., Page, C.P., Eds.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 463–487. [Google Scholar]
- Miles, L.A.; Ginsberg, M.H.; White, J.G.; Plow, E.F. Plasminogen interacts with human platelets through two distinct mechanisms. J. Clin. Investig. 1986, 77, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- Whyte, C.S.; Morrow, G.B.; Baik, N.; Booth, N.A.; Jalal, M.M.; Parmer, R.J.; Miles, L.A.; Mutch, N.J. Exposure of plasminogen and the novel plasminogen receptor, Plg-RKT, on activated human and murine platelets. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Leung, L.L.; Harpel, P.C.; Nachman, R.L. Complex formation of platelet thrombospondin with plasminogen. Modulation of activation by tissue activator. J. Clin. Investig. 1984, 74, 1625–1633. [Google Scholar] [CrossRef]
- Mosher, D.F.; Misenheimer, T.M.; Stenflo, J.; Hogg, P.J. Modulation of fibrinolysis by thrombospondin. Ann. N. Y. Acad. Sci. 1992, 667, 64–69. [Google Scholar] [CrossRef]
- Maynard, D.M.; Heijnen, H.F.; Gahl, W.A.; Gunay-Aygun, M. The alpha-granule proteome: Novel proteins in normal and ghost granules in gray platelet syndrome. J. Thromb. Haemost. 2010, 8, 1786–1796. [Google Scholar] [CrossRef] [Green Version]
- Kehrel, B.; Kronenberg, A.; Schwippert, B.; Niesing-Bresch, D.; Niehues, U.; Tschope, D.; van de Loo, J.; Clemetson, K.J. Thrombospondin binds normally to glycoprotein IIIb deficient platelets. Biochem. Biophys. Res. Commun. 1991, 179, 985–991. [Google Scholar] [CrossRef]
- Bonnefoy, A.; Moura, R.; Hoylaerts, M.F. The evolving role of thrombospondin-1 in hemostasis and vascular biology. Cell Mol. Life Sci. 2008, 65, 713–727. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Leung, L.L.; Harpel, P.C.; Nachman, R.L. Platelet thrombospondin forms a trimolecular complex with plasminogen and histidine-rich glycoprotein. J. Clin. Investig. 1985, 75, 2065–2073. [Google Scholar] [CrossRef]
- Gebbink, M.F.; Bouma, B.; Maas, C.; Bouma, B.N. Physiological responses to protein aggregates: Fibrinolysis, coagulation and inflammation (new roles for old factors). FEBS Lett. 2009, 583, 2691–2699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinescu, P.; Brown, R.A.; Wyatt, A.R.; Ranson, M.; Wilson, M.R. Amorphous protein aggregates stimulate plasminogen activation, leading to release of cytotoxic fragments that are clients for extracellular chaperones. J. Biol. Chem. 2017, 292, 14425–14437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, H.M.; Kihiko, M.; Caldwell, J.N.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.P.; Rydel, R.E.; et al. The plasmin system is induced by and degrades amyloid-beta aggregates. J. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [CrossRef] [Green Version]
- Van Nostrand, W.E.; Porter, M. Plasmin cleavage of the amyloid beta-protein: Alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry 1999, 38, 11570–11576. [Google Scholar] [CrossRef]
- Samson, A.L.; Knaupp, A.S.; Sashindranath, M.; Borg, R.J.; Au, A.E.; Cops, E.J.; Saunders, H.M.; Cody, S.H.; McLean, C.A.; Nowell, C.J. Nucleocytoplasmic coagulation: An injury-induced aggregation event that disulfide crosslinks proteins and facilitates their removal by plasmin. Cell Rep. 2012, 2, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Korporaal, S.J.; Gorter, G.; van Rijn, H.J.; Akkerman, J.W. Effect of oxidation on the platelet-activating properties of low-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 867–872. [Google Scholar] [CrossRef]
- Herczenik, E.; Bouma, B.; Korporaal, S.J.; Strangi, R.; Zeng, Q.; Gros, P.; Van Eck, M.; Van Berkel, T.J.; Gebbink, M.F.; Akkerman, J.W. Activation of human platelets by misfolded proteins. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1657–1665. [Google Scholar] [CrossRef]
- McDonald, J.F.; Dimitry, J.M.; Frazier, W.A. An amyloid-like C-terminal domain of thrombospondin-1 displays CD47 agonist activity requiring both VVM motifs. Biochemistry 2003, 42, 10001–10011. [Google Scholar] [CrossRef]
- Dorahy, D.J.; Thorne, R.F.; Fecondo, J.V.; Burns, G.F. Stimulation of platelet activation and aggregation by a carboxyl-terminal peptide from thrombospondin binding to the integrin-associated protein receptor. J. Biol. Chem. 1997, 272, 1323–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulasne, D.; Judd, B.A.; Johansen, M.; Asazuma, N.; Best, D.; Brown, E.J.; Kahn, M.; Koretzky, G.A.; Watson, S.P. C-terminal peptide of thrombospondin-1 induces platelet aggregation through the Fc receptor gamma-chain-associated signaling pathway and by agglutination. Blood 2001, 98, 3346–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voit, S.; Udelhoven, M.; Lill, G.; Aktas, B.; Nieswandt, B.; Schror, K.; Weber, A.A. The C-terminal peptide of thrombospondin-1 stimulates distinct signaling pathways but induces an activation-independent agglutination of platelets and other cells. FEBS Lett. 2003, 544, 240–245. [Google Scholar] [CrossRef] [Green Version]
- Bertling, A.; Niemann, S.; Hussain, M.; Holbrook, L.; Stanley, R.G.; Brodde, M.F.; Pohl, S.; Schifferdecker, T.; Roth, J.; Jurk, K.; et al. Staphylococcal extracellular adherence protein induces platelet activation by stimulation of thiol isomerases. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1979–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Vorm, L.N.; Remijn, J.A.; de Laat, B.; Huskens, D. Effects of Plasmin on von Willebrand Factor and Platelets: A Narrative Review. TH Open 2018, 2, e218–e228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, W.; Magwenzi, S.; Aburima, A.; Naseem, K.M. Thrombospondin-1 induces platelet activation through CD36-dependent inhibition of the cAMP/protein kinase A signaling cascade. Blood 2010, 116, 4297–4306. [Google Scholar] [CrossRef]
- Kuijpers, M.J.; de Witt, S.; Nergiz-Unal, R.; van Kruchten, R.; Korporaal, S.J.; Verhamme, P.; Febbraio, M.; Tjwa, M.; Voshol, P.J.; Hoylaerts, M.F. Supporting roles of platelet thrombospondin-1 and CD36 in thrombus formation on collagen. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Jurk, K.; Clemetson, K.J.; de Groot, P.G.; Brodde, M.F.; Steiner, M.; Savion, N.; Varon, D.; Sixma, J.J.; Van Aken, H.; Kehrel, B.E. Thrombospondin-1 mediates platelet adhesion at high shear via glycoprotein Ib (GPIb): An alternative/backup mechanism to von Willebrand factor. FASEB J. 2003, 17, 1490–1492. [Google Scholar] [CrossRef]
- Hogg, P.J.; Owensby, D.A.; Mosher, D.F.; Misenheimer, T.M.; Chesterman, C.N. Thrombospondin is a tight-binding competitive inhibitor of neutrophil elastase. J. Biol. Chem. 1993, 268, 7139–7146. [Google Scholar]
- Lahav, J.; Jurk, K.; Hess, O.; Barnes, M.J.; Farndale, R.W.; Luboshitz, J.; Kehrel, B.E. Sustained integrin ligation involves extracellular free sulfhydryls and enzymatically catalyzed disulfide exchange. Blood 2002, 100, 2472–2478. [Google Scholar] [CrossRef] [Green Version]
- Jurk, K.; Lahav, J.; Van Aken, H.; Brodde, M.F.; Nofer, J.R.; Kehrel, B.E. Extracellular protein disulfide isomerase regulates feedback activation of platelet thrombin generation via modulation of coagulation factor binding. J. Thromb. Haemost. 2011, 9, 2278–2290. [Google Scholar] [CrossRef] [PubMed]
- Niewiarowski, S.; Senyi, A.F.; Gillies, P. Plasmin-induced platelet aggregation and platelet release reaction. Effects on hemostasis. J. Clin. Investig. 1973, 52, 1647–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, A.I.; Maas, A.K.; Ware, J.A.; Johnson, P.C.; Rittenhouse, S.E.; Salzman, E.W. Platelet protein phosphorylation, elevation of cytosolic calcium, and inositol phospholipid breakdown in platelet activation induced by plasmin. J. Clin. Investig. 1986, 78, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Blockmans, D.; Deckmyn, H.; Hove, L.V.; Vermylen, J. The effect of plasmin on platelet function. Platelets 1996, 7, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Soria, C.; Cramer, E.M.; Soria, J.; Maclouf, J.; Perrot, J.Y.; Li, H.; Commin, P.L.; Schumann, F.; Regnier, O.; et al. Temperature dependence of plasmin-induced activation or inhibition of human platelets. Blood 1991, 77, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Shigeta, O.; Kojima, H.; Jikuya, T.; Terada, Y.; Atsumi, N.; Sakakibara, Y.; Nagasawa, T.; Mitsui, T. Aprotinin inhibits plasmin-induced platelet activation during cardiopulmonary bypass. Circulation 1997, 96, 569–574. [Google Scholar] [CrossRef]
- Quinton, T.M.; Kim, S.; Derian, C.K.; Jin, J.; Kunapuli, S.P. Plasmin-mediated activation of platelets occurs by cleavage of protease-activated receptor 4. J. Biol. Chem. 2004, 279, 18434–18439. [Google Scholar] [CrossRef] [Green Version]
- Cramer, E.M.; Lu, H.; Caen, J.P.; Soria, C.; Berndt, M.C.; Tenza, D. Differential redistribution of platelet glycoproteins Ib and IIb-IIIa after plasmin stimulation. Blood 1991, 77, 694–699. [Google Scholar] [CrossRef] [Green Version]
- Schafer, A.I.; Adelman, B. Plasmin inhibition of platelet function and of arachidonic acid metabolism. J. Clin. Investig. 1985, 75, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selim, T.E.; Ghoneim, H.R.; Uknis, A.B.; Colman, R.W.; DeLa Cadena, R.A. High-molecular-mass and low-molecular-mass kininogens block plasmin-induced platelet aggregation by forming a complex with kringle 5 of plasminogen/plasmin. Eur. J. Biochem. 1997, 250, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Paparella, D.; Brister, S.J.; Buchanan, M.R. Coagulation disorders of cardiopulmonary bypass: A review. Intensive Care Med. 2004, 30, 1873–1881. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Nachman, R.L.; Leung, L.L.; Harpel, P.C. Activation of immobilized plasminogen by tissue activator. Multimolecular complex formation. J. Biol. Chem. 1985, 260, 10346–10352. [Google Scholar]
- Anonick, P.K.; Yoo, J.K.; Webb, D.J.; Gonias, S.L. Characterization of the antiplasmin activity of human thrombospondin-1 in solution. Biochem. J. 1993, 289, 903–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, P.J.; Stenflo, J.; Mosher, D.F. Thrombospondin is a slow tight-binding inhibitor of plasmin. Biochemistry 1992, 31, 265–269. [Google Scholar] [CrossRef]
- Kranenburg, O.; Bouma, B.; Kroon-Batenburg, L.M.; Reijerkerk, A.; Wu, Y.P.; Voest, E.E.; Gebbink, M.F. Tissue-type plasminogen activator is a multiligand cross-beta structure receptor. Curr. Biol. 2002, 12, 1833–1839. [Google Scholar] [CrossRef] [Green Version]
- Michaeli, A.; Finci-Yeheskel, Z.; Dishon, S.; Linke, R.P.; Levin, M.; Urieli-Shoval, S. Serum amyloid A enhances plasminogen activation: Implication for a role in colon cancer. Biochem. Biophys. Res. Commun. 2008, 368, 368–373. [Google Scholar] [CrossRef]
- Galantai, R.; Modos, K.; Fidy, J.; Kolev, K.; Machovich, R. Structural basis of the cofactor function of denatured albumin in plasminogen activation by tissue-type plasminogen activator. Biochem. Biophys. Res. Commun. 2006, 341, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Maas, C.; Schiks, B.; Strangi, R.D.; Hackeng, T.M.; Bouma, B.N.; Gebbink, M.F.; Bouma, B. Identification of fibronectin type I domains as amyloid-binding modules on tissue-type plasminogen activator and three homologs. Amyloid 2008, 15, 166–180. [Google Scholar] [CrossRef]
- Gebbink, M.F. Tissue-type plasminogen activator-mediated plasminogen activation and contact activation, implications in and beyond haemostasis. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 174–181. [Google Scholar] [CrossRef] [PubMed]
- Döhrmann, M.; Makhoul, S.; Gross, K.; Krause, M.; Pillitteri, D.; von Auer, C.; Walter, U.; Lutz, J.; Volf, I.; Kehrel, B.E.; et al. CD36-fibrin interaction propagates FXI-dependent thrombin generation of human platelets. FASEB J. 2020, 34, 9337–9357. [Google Scholar] [CrossRef]
- Horn, M.; Bertling, A.; Brodde, M.F.; Muller, A.; Roth, J.; Van Aken, H.; Jurk, K.; Heilmann, C.; Peters, G.; Kehrel, B.E. Human neutrophil alpha-defensins induce formation of fibrinogen and thrombospondin-1 amyloid-like structures and activate platelets via glycoprotein IIb/IIIa. J. Thromb. Haemost. 2012, 10, 647–661. [Google Scholar] [CrossRef]
- Dörmann, D.; Clemetson, K.J.; Kehrel, B.E. The GPIb thrombin-binding site is essential for thrombin-induced platelet procoagulant activity. Blood 2000, 96, 2469–2478. [Google Scholar] [CrossRef]
- Hussain, M.; Becker, K.; von Eiff, C.; Peters, G.; Herrmann, M. Analogs of Eap protein are conserved and prevalent in clinical Staphylococcus aureus isolates. Clin. Diagn Lab. Immunol. 2001, 8, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Jurk, K.; Schulz, A.S.; Kehrel, B.E.; Rapple, D.; Schulze, H.; Mobest, D.; Friedrich, W.W.; Omran, H.; Deak, E.; Henschler, R.; et al. Novel integrin-dependent platelet malfunction in siblings with leukocyte adhesion deficiency-III (LAD-III) caused by a point mutation in FERMT3. Thromb. Haemost. 2010, 103, 1053–1064. [Google Scholar] [CrossRef]
- Jurk, K.; Ritter, M.A.; Schriek, C.; Van Aken, H.; Droste, D.W.; Ringelstein, E.B.; Kehrel, B.E. Activated monocytes capture platelets for heterotypic association in patients with severe carotid artery stenosis. Thromb. Haemost. 2010, 103, 1193–1202. [Google Scholar]
- Xia, Z.; Wong, T.; Liu, Q.; Kasirer-Friede, A.; Brown, E.; Frojmovic, M.M. Optimally functional fluorescein isothiocyanate-labelled fibrinogen for quantitative studies of binding to activated platelets and platelet aggregation. Br. J. Haematol. 1996, 93, 204–214. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pielsticker, C.; Brodde, M.F.; Raum, L.; Jurk, K.; Kehrel, B.E. Plasmin-Induced Activation of Human Platelets Is Modulated by Thrombospondin-1, Bona Fide Misfolded Proteins and Thiol Isomerases. Int. J. Mol. Sci. 2020, 21, 8851. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228851
Pielsticker C, Brodde MF, Raum L, Jurk K, Kehrel BE. Plasmin-Induced Activation of Human Platelets Is Modulated by Thrombospondin-1, Bona Fide Misfolded Proteins and Thiol Isomerases. International Journal of Molecular Sciences. 2020; 21(22):8851. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228851
Chicago/Turabian StylePielsticker, Claudia, Martin F. Brodde, Lisa Raum, Kerstin Jurk, and Beate E. Kehrel. 2020. "Plasmin-Induced Activation of Human Platelets Is Modulated by Thrombospondin-1, Bona Fide Misfolded Proteins and Thiol Isomerases" International Journal of Molecular Sciences 21, no. 22: 8851. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228851