Gap Junctions between Endothelial Cells Are Disrupted by Circulating Extracellular Vesicles from Sickle Cell Patients with Acute Chest Syndrome



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Properties of Circulating EVs from Subjects with SCD
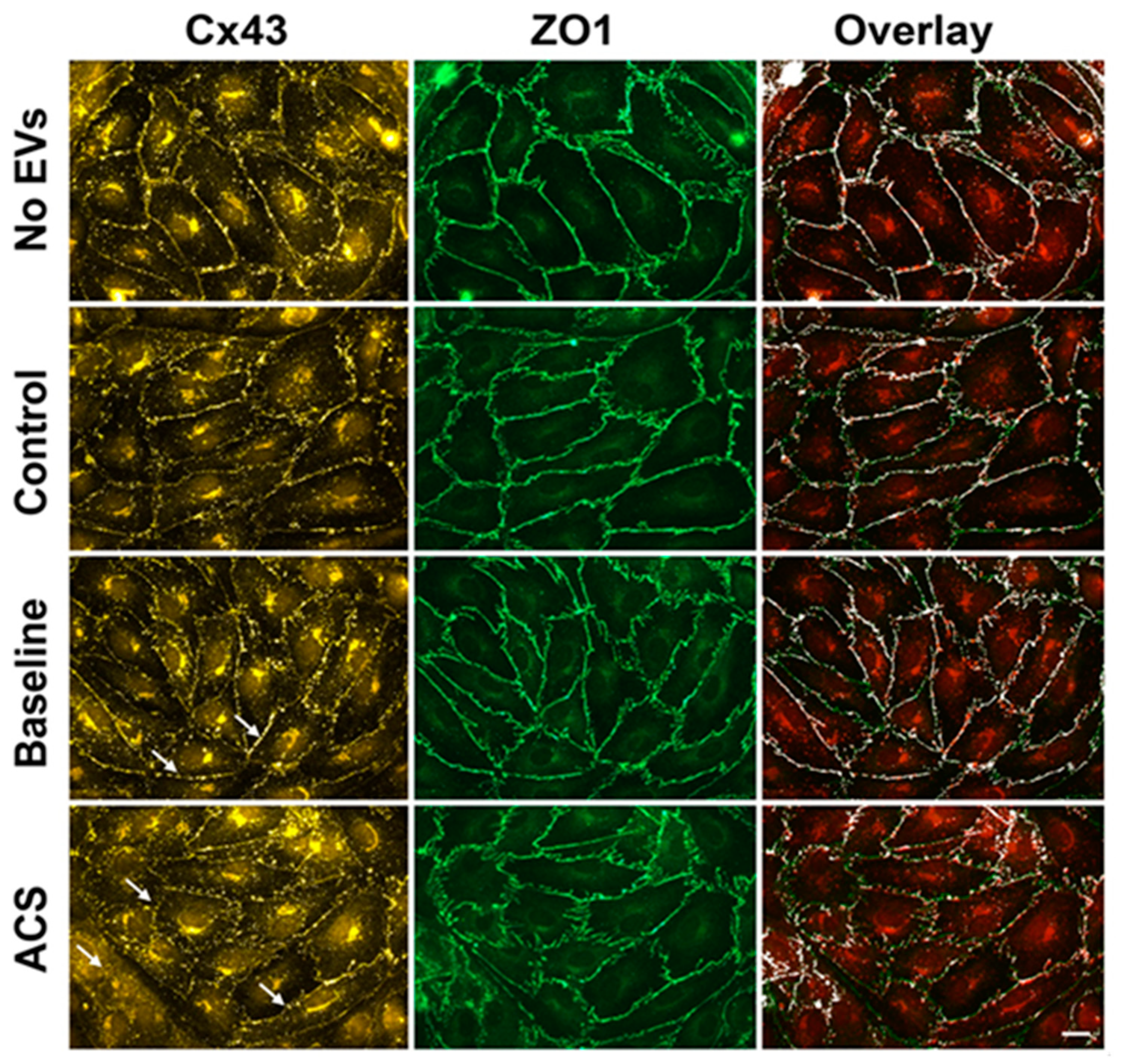
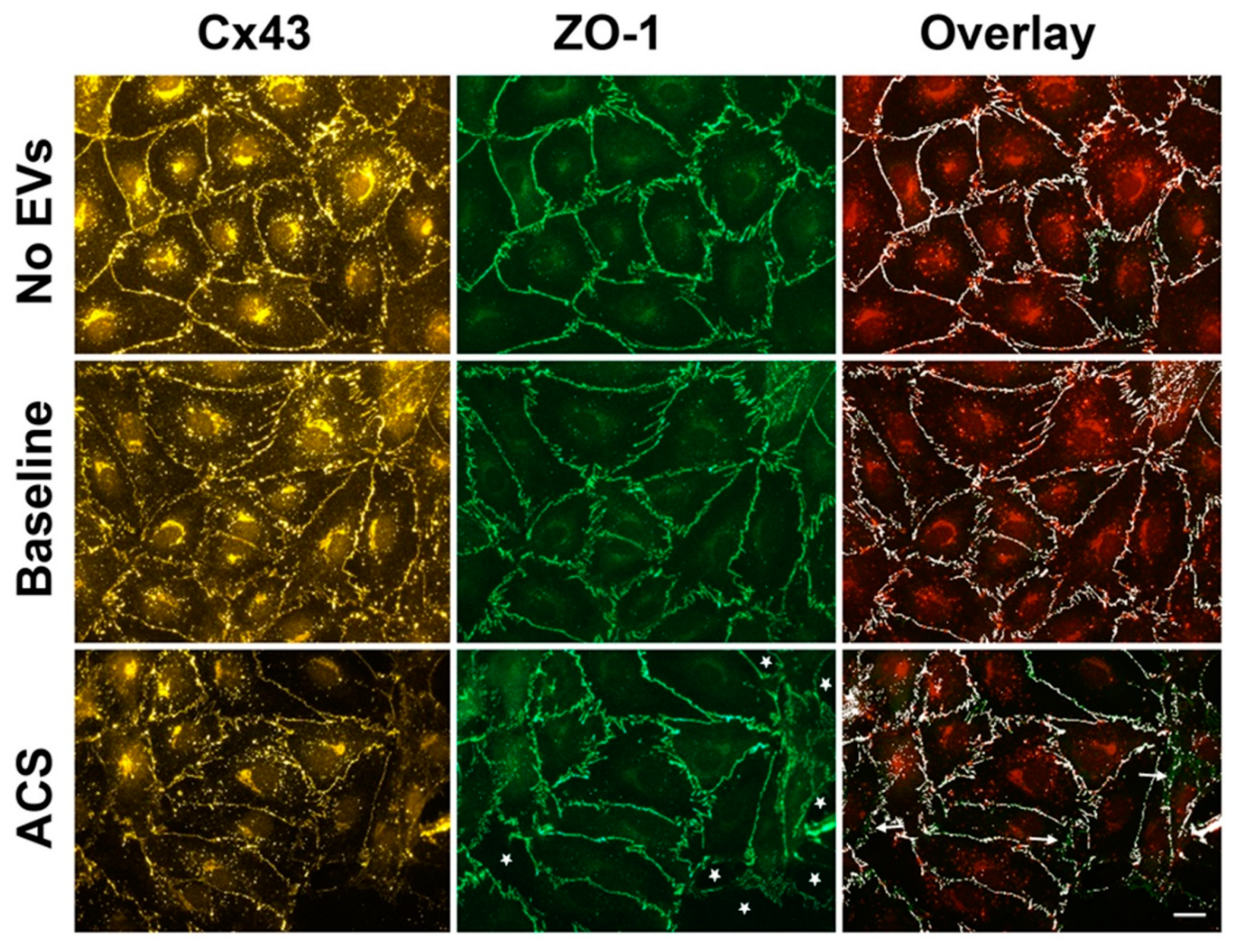
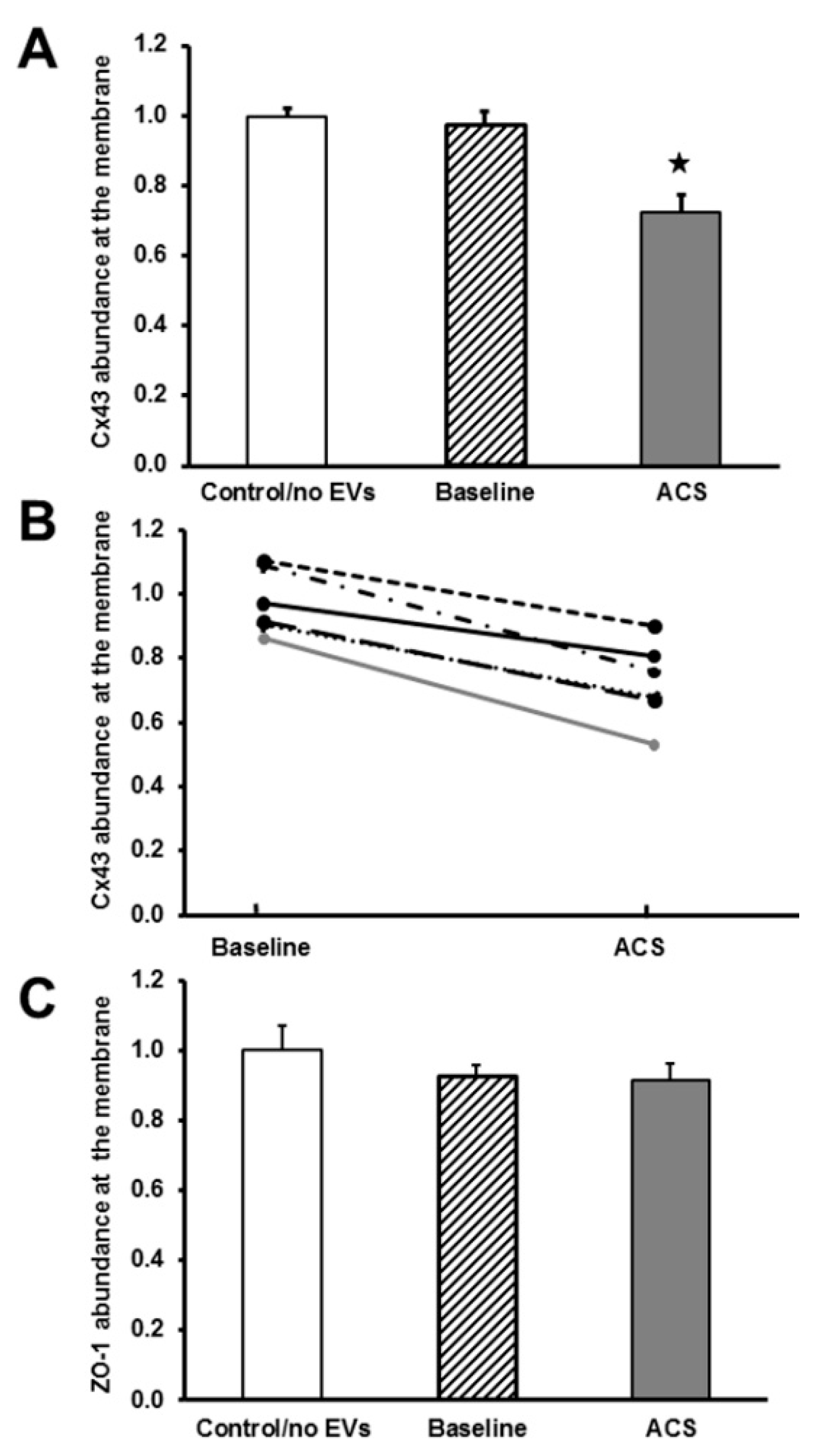
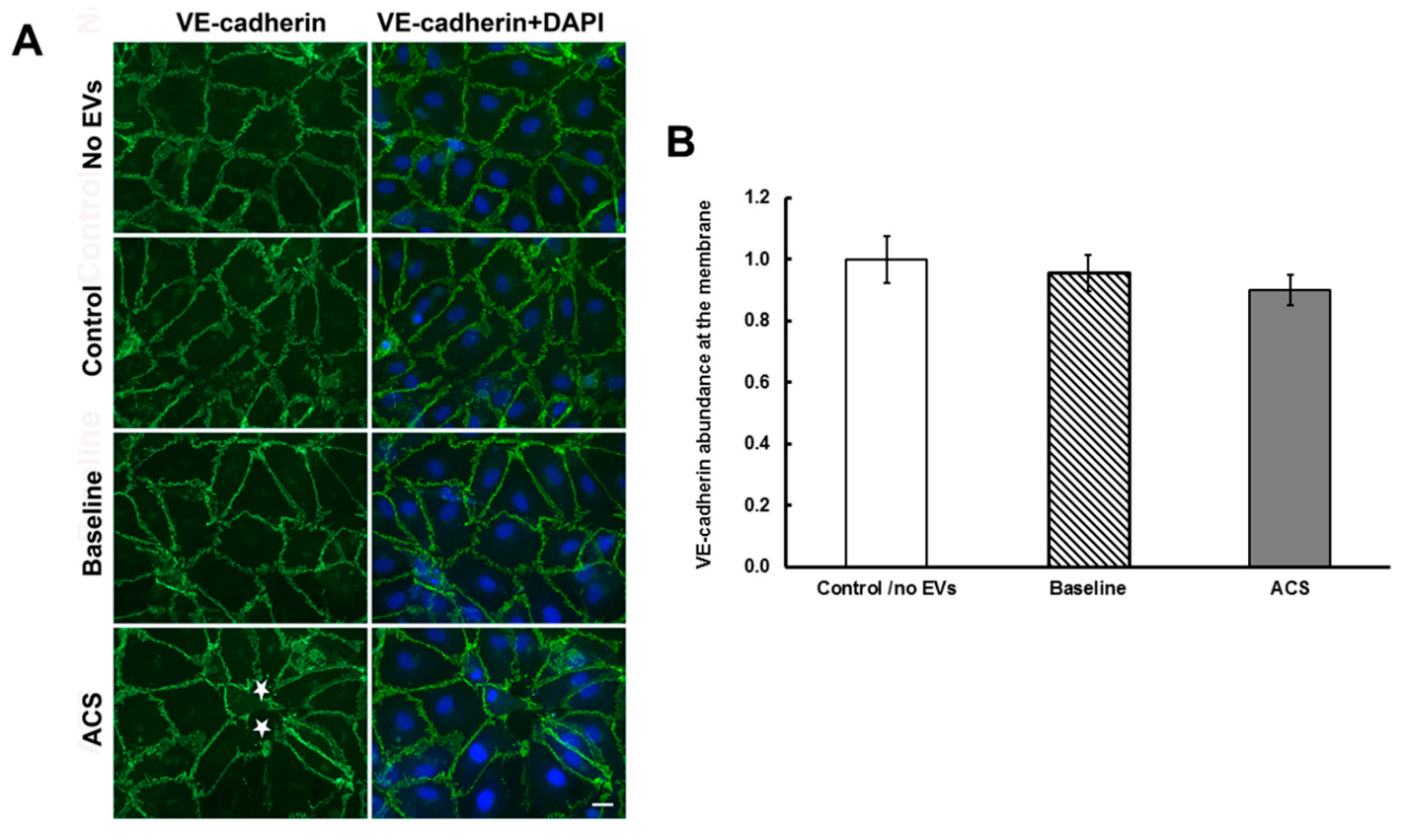
2.2. EVs Isolated during an Episode of ACS Reduce the Abundance of Cx43 (But Not Other Junctional Proteins) at Appositional Membranes
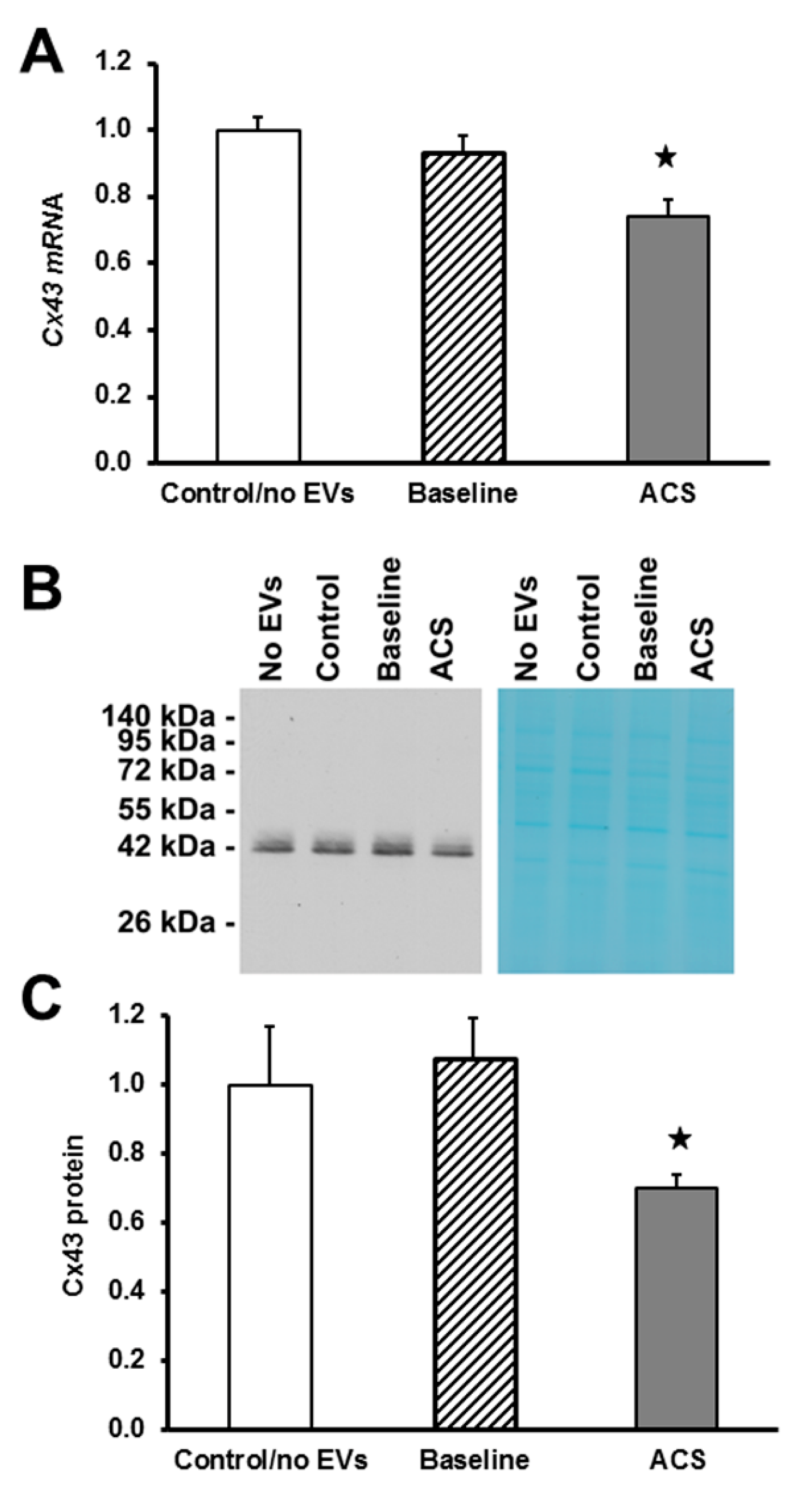
2.3. EVs Isolated during an Episode of ACS Cause Decreases in Cx43 mRNA and Protein Levels
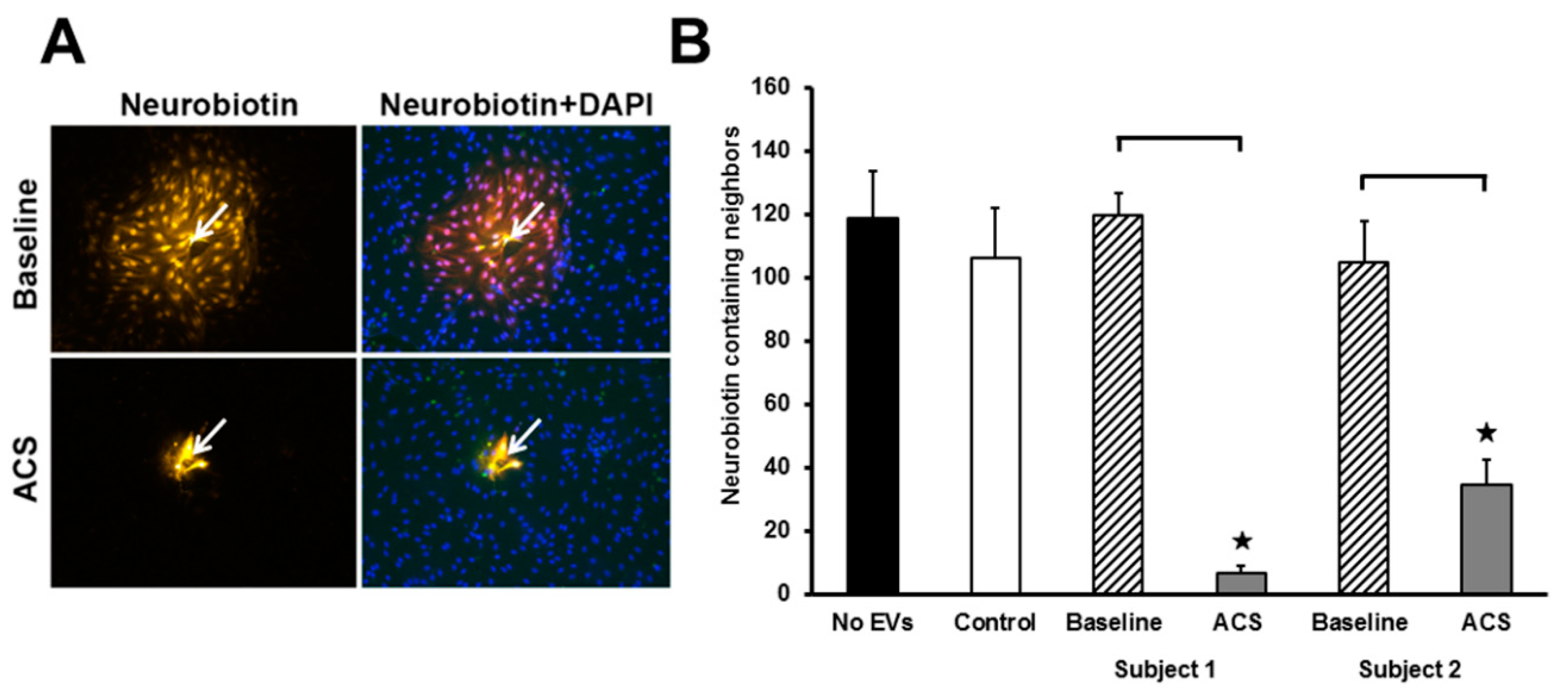
2.4. ACS EVs Reduce the Extent of Gap Junction-Mediated Intercellular Communication
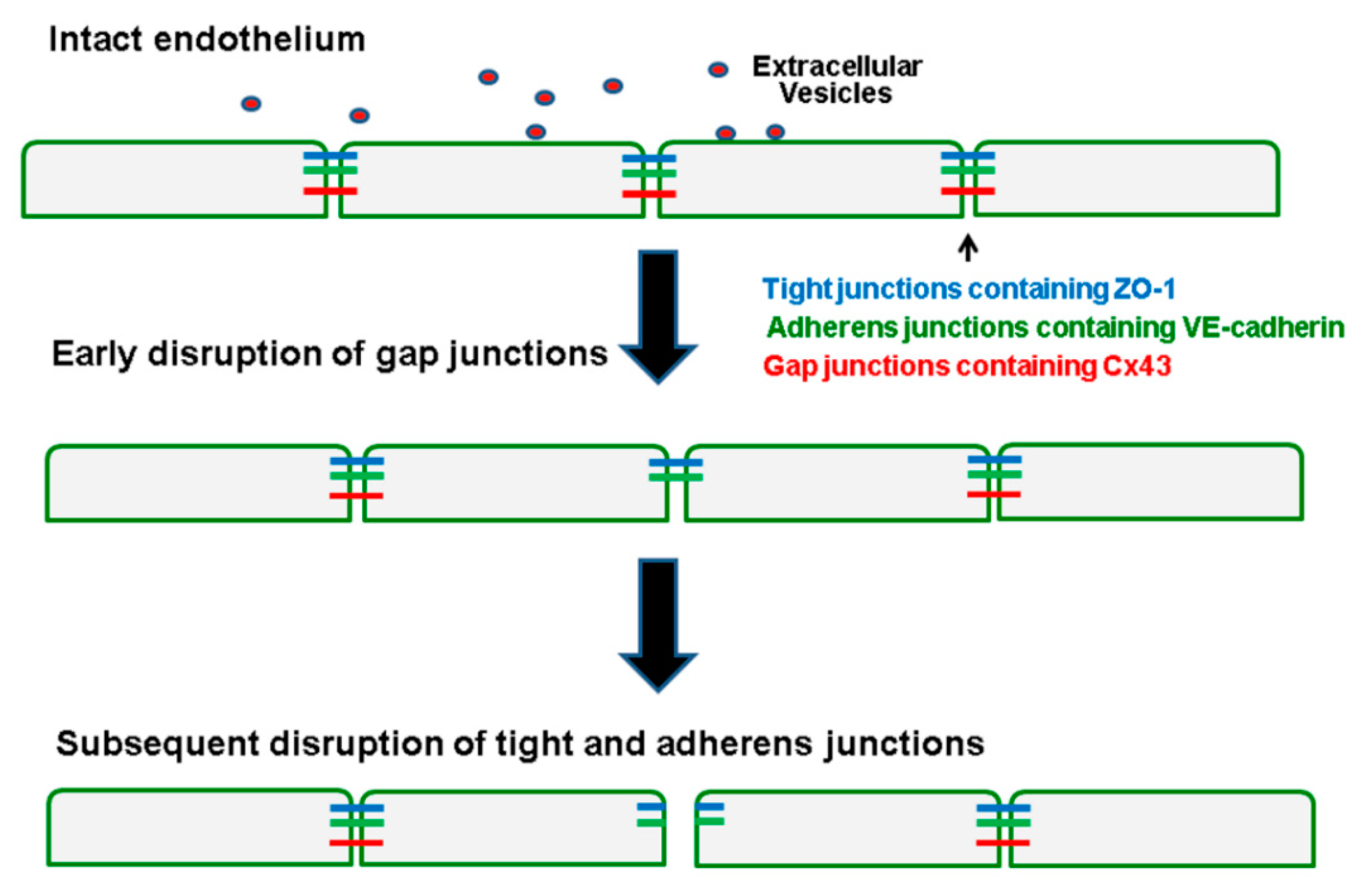
3. Discussion
4. Materials and Methods
4.1. Subjects with Sickle Cell Disease and Controls
4.2. Isolation of Small Extracellular Vesicles
4.3. Primary Endothelial Cell Culture
4.4. Antibodies
4.5. Immunohistochemistry of Endothelial Cells
4.6. Assay of Intercellular Communication Using Micro-Injected Neurobiotin
4.7. Immunoblotting of EVs and Endothelial Cell Lysates
4.8. Isolation of RNA and Quantification of mRNA Levels
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACS | Acute Chest Syndrome |
| Cx37 | Connexin37 |
| Cx40 | Connexin40 |
| Cx43 | Connexin43 |
| EVs | Extracellular vesicles |
| HMVEC-D | Human dermal microvascular endothelial cells |
| RPL13A J | Human ribosomal protein L13a |
| RT | Room Temperature |
| SCD | Sickle Cell Disease |
| ZO-1 | Zonula occludens-1 |
References
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [Green Version]
- Harris, E.S.; Nelson, W.J. VE-cadherin: At the front, center, and sides of endothelial cell organization and function. Curr. Opin. Cell Biol. 2010, 22, 651–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta Biomembr. 2008, 1778, 794–809. [Google Scholar] [CrossRef] [Green Version]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. 2018, 222, e12860. [Google Scholar] [CrossRef]
- Schmidt, K.; Windler, R.; De Wit, C. Communication through gap junctions in the endothelium. Adv. Pharmacol. 2016, 77, 209–240. [Google Scholar] [CrossRef]
- Larson, D.M.; Sheridan, J.D. Intercellular junctions and transfer of small molecules in primary vascular endothelial cultures. J. Cell Biol. 1982, 92, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, X.F.; Duling, B.R. Gap Junctions in the control of vascular function. Antioxid. Redox Signal. 2009, 11, 251–266. [Google Scholar] [CrossRef] [Green Version]
- Soon, A.S.C.; Chua, J.W.; Becker, D.L. Connexins in endothelial barrier function—Novel therapeutic targets countering vascular hyperpermeability. Thromb. Haemost. 2016, 116, 852–867. [Google Scholar] [CrossRef] [Green Version]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of sickle cell disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 263–292. [Google Scholar] [CrossRef]
- Ridger, V.C.; Boulanger, C.M.; Angelillo-Scherrer, A.; Badimon, L.; Blanc-Brude, O.; Bochaton-Piallat, M.-L.; Boilard, E.; Buzás, E.; Caporali, A.; Dignat-George, F.; et al. Microvesicles in vascular homeostasis and diseases. Position paper of the European Society of Cardiology (ESC) Working Group on Atherosclerosis and Vascular Biology. Thromb. Haemost. 2017, 117, 1296–1316. [Google Scholar] [CrossRef]
- Oggero, S.; Austin-Williams, S.; Norling, L.V. The contrasting role of extracellular vesicles in vascular inflammation and tissue repair. Front. Pharmacol. 2019, 10, 1479. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P.; Key, N.S. Microparticles in sickle cell anaemia: Promise and pitfalls. Br. J. Haematol. 2016, 174, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Lapping-Carr, G.; Gemel, J.; Mao, Y.; Beyer, E.C. Circulating Extracellular Vesicles and Endothelial Damage in Sickle Cell Disease. Front. Physiol. 2020, 11, 1063. [Google Scholar] [CrossRef] [PubMed]
- Khalyfa, A.; Khalyfa, A.A.; Akbarpour, M.; Connes, P.; Romana, M.; Lapping-Carr, G.; Zhang, C.; Andrade, J.; Gozal, D. Extracellular microvesicle microRNAs in children with sickle cell anaemia with divergent clinical phenotypes. Br. J. Haematol. 2016, 174, 786–798. [Google Scholar] [CrossRef]
- Lapping-Carr, G.; Khalyfa, A.; Rangel, S.; Darlington, W.; Beyer, E.C.; Peddinti, R.; Cunningham, J.M.; Gozal, D. Exosomes contribute to endothelial integrity and acute chest syndrome risk: Preliminary findings. Pediatr. Pulmonol. 2017, 52, 1478–1485. [Google Scholar] [CrossRef]
- Lapping-Carr, G.; Gemel, J.; Mao, Y.; Sparks, G.; Harrington, M.; Peddinti, R.; Beyer, E.C. Circulating extracellular vesicles from patients with acute chest syndrome disrupt adherens junctions between endothelial cells. Pediatr. Res. 2020. [Google Scholar] [CrossRef]
- Ludwig, A.-K.; Giebel, B. Exosomes: Small vesicles participating in intercellular communication. Int. J. Biochem. Cell Biol. 2012, 44, 11–15. [Google Scholar] [CrossRef]
- Momen-Heravi, F.; Balaj, L.; Alian, S.; Etigges, J.; Etoxavidis, V.; Ericsson, M.; Distel, R.J.; Ivanov, A.R.; Skog, J.; Kuo, W.P. Alternative methods for characterization of extracellular vesicles. Front. Physiol. 2012, 3, 354. [Google Scholar] [CrossRef] [Green Version]
- Musil, L.S.; Goodenough, D.A. Gap junctional intercellular communication and the regulation of connexin expression and function. Curr. Opin. Cell Biol. 1990, 2, 875–880. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of Connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Mitra, S.; Falk, M.M.; Caplan, S.H.; Wheelock, M.J.; Johnson, K.R.; Mehta, P.P. E-cadherin differentially regulates the assembly of Connexin43 and Connexin32 into gap junctions in human squamous carcinoma cells. J. Biol. Chem. 2010, 285, 10761–10776. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, R.; Chakraborty, S.; Johnson, K.E.; Falk, M.M.; Wheelock, M.J.; Johnson, K.R.; Mehta, P.P. Assembly of Connexin43 into gap junctions is regulated differentially by E-Cadherin and N-Cadherin in rat liver epithelial cells. Mol. Biol. Cell 2010, 21, 4089–4107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Li, W.E.; Huang, G.Y.; Meyer, R.; Chen, T.; Luo, Y.; Thomas, M.P.; Radice, G.L.; Lo, C.W. Modulation of mouse neural crest cell motility by N-cadherin and connexin 43 gap junctions. J. Cell Biol. 2001, 154, 217–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Patel, V.V.; Kostetskii, I.; Xiong, Y.; Chu, A.F.; Jacobson, J.T.; Yu, C.; Morley, G.E.; Molkentin, J.D.; Radice, G.L. Cardiac-specific loss of N-Cadherin leads to alteration in connexins with conduction sSlowing and arrhythmogenesis. Circ. Res. 2005, 97, 474–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, H.S.; Delmar, M.; Spray, D.C. Formation of the gap junction nexus: Binding partners for connexins. J. Physiol. 2002, 96, 243–249. [Google Scholar] [CrossRef]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef]
- Johnson, A.M.; Roach, J.P.; Hu, A.; Stamatovic, S.M.; Zochowski, M.R.; Keep, R.F.; Andjelkovic, A.V. Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. 2018, 32, 2615–2629. [Google Scholar] [CrossRef] [Green Version]
- Young, J.A.; Ting, K.K.; Li, J.; Moller, T.; Dunn, L.; Lu, Y.; Lay, A.J.; Moses, J.; Prado-Lourenço, L.; Khachigian, L.M.; et al. Regulation of vascular leak and recovery from ischemic injury by general and VE-cadherin-restricted miRNA antagonists of miR-27. Blood 2013, 122, 2911–2919. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Zhan, H.; Zhou, X.-Y.; Yao, L.; Yan, M.; Chen, A.; Liu, J.; Ren, X.; Zhang, X.; Liu, J.-X.; et al. MicroRNA-22 regulates inflammation and angiogenesis via targeting VE-cadherin. FEBS Lett. 2017, 591, 513–526. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, F.; Kidoya, H.; Naito, H.; Sakimoto, S.; Takakura, N. microRNA-125b inhibits tube formation of blood vessels through translational suppression of VE-cadherin. Oncogene 2013, 32, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena-Philippides, J.C.; Gardiner, A.S.; Caballero-Garrido, E.; Pan, R.; Zhu, Y.; Roitbak, T. Inhibition of MicroRNA-155 supports endothelial tight junction integrity following oxygen-glucose deprivation. J. Am. Heart Assoc. 2018, 7, e009244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichon, C.; Sabharwal, H.; Rüter, C.; Schmidt, M.A. MicroRNAs regulate tight junction proteins and modulate epithelial/endothelial barrier functions. Tissue Barriers 2014, 2, e944446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Y.; Peng, H.; Mastej, V.; Chen, W. MicroRNA Regulation of endothelial junction proteins and clinical consequence. Mediat. Inflamm. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Osbourne, A.; Calway, T.; Broman, M.; McSharry, S.; Earley, J.; Kim, G.H. Downregulation of connexin43 by microRNA-130a in cardiomyocytes results in cardiac arrhythmias. J. Mol. Cell. Cardiol. 2014, 74, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Zhou, T.-Y.; Cao, J.-N.; Feng, Q.-T.; Fu, Y.-J.; Xu, X.; Yang, C.-J. MicroRNA-206 downregulates Connexin43 in cardiomyocytes to induce cardiac arrhythmias in a transgenic mouse model. Hear. Lung Circ. 2019, 28, 1755–1761. [Google Scholar] [CrossRef]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef]
- Segal, S.S.; Duling, B.R. Conduction of vasomotor responses in arterioles: A role for cell-to-cell coupling? Am. J. Physiol. Heart Circ. Physiol. 1989, 256, H838–H845. [Google Scholar] [CrossRef]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in cardiovascular and neurovascular health and disease: Pharmacological Implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef]
- De Bock, M.; Culot, M.; Wang, N.; Bol, M.; Decrock, E.; De Vuyst, E.; Da Costa, A.; Dauwe, I.; Vinken, M.; Simon, A.M.; et al. Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood—brain barrier permeability. J. Cereb. Blood Flow Metab. 2011, 31, 1942–1957. [Google Scholar] [CrossRef] [Green Version]
- Gaete, P.S.; Lillo, M.A.; Figueroa, X.F. Functional role of connexins and pannexins in the interaction between vascular and nervous system. J. Cell. Physiol. 2014, 229, 1336–1345. [Google Scholar] [CrossRef]
- Okamoto, T.; Suzuki, K. The role of gap junction-mediated endothelial cell-cell interaction in the crosstalk between inflammation and blood coagulation. Int. J. Mol. Sci. 2017, 18, 2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoorelbeke, D.; Decrock, E.; De Smet, M.; De Bock, M.; Descamps, B.; Van Haver, V.; Delvaeye, T.; Krysko, D.V.; Vanhove, C.; Bultynck, G.; et al. Cx43 channels and signaling via IP3/Ca2+, ATP, and ROS/NO propagate radiation-induced DNA damage to non-irradiated brain microvascular endothelial cells. Cell Death Dis. 2020, 11, 194. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Murakami, K.; Akaogi, R.; Akanuma, S.-I.; Terasaki, T.; Hosoya, K.-I. Polarized hemichannel opening of pannexin 1/connexin 43 contributes to dysregulation of transport function in blood-brain barrier endothelial cells. Neurochem. Int. 2020, 132, 104600. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.M.; Haudenschild, C.C.; Beyer, E.C. Gap junction messenger RNA expression by vascular wall cells. Circ. Res. 1990, 66, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Little, T.L.; Beyer, E.C.; Duling, B.R. Connexin 43 and connexin 40 gap junctional proteins are present in arteriolar smooth muscle and endothelium in vivo. Am. J. Physiol. Heart Circ. Physiol. 1995, 268, H729–H739. [Google Scholar] [CrossRef] [PubMed]
- Gabriels, J.E.; Paul, D.L. Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed. Circ. Res. 1998, 83, 636–643. [Google Scholar] [CrossRef]
- Gemel, J.; Levy, A.E.; Simon, A.R.; Bennett, K.B.; Ai, X.; Akhter, S.; Beyer, E.C. Connexin40 abnormalities and atrial fibrillation in the human heart. J. Mol. Cell. Cardiol. 2014, 76, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Lin, X.; Gemel, J.; Glass, A.; Zemlin, C.W.; Beyer, E.C.; Veenstra, R.D. Connexin40 and connexin43 determine gating properties of atrial gap junction channels. J. Mol. Cell. Cardiol. 2010, 48, 238–245. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gemel, J.; Mao, Y.; Lapping-Carr, G.; Beyer, E.C. Gap Junctions between Endothelial Cells Are Disrupted by Circulating Extracellular Vesicles from Sickle Cell Patients with Acute Chest Syndrome. Int. J. Mol. Sci. 2020, 21, 8884. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238884
Gemel J, Mao Y, Lapping-Carr G, Beyer EC. Gap Junctions between Endothelial Cells Are Disrupted by Circulating Extracellular Vesicles from Sickle Cell Patients with Acute Chest Syndrome. International Journal of Molecular Sciences. 2020; 21(23):8884. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238884
Chicago/Turabian StyleGemel, Joanna, Yifan Mao, Gabrielle Lapping-Carr, and Eric C. Beyer. 2020. "Gap Junctions between Endothelial Cells Are Disrupted by Circulating Extracellular Vesicles from Sickle Cell Patients with Acute Chest Syndrome" International Journal of Molecular Sciences 21, no. 23: 8884. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238884