Morquio B Disease. Disease Characteristics and Treatment Options of a Distinct GLB1-Related Dysostosis Multiplex


Abstract
:1. Introduction
2. Dysostosis Multiplex in MBD
3. Other Organ and System Involvements
4. Overlaps of MBD with Morquio A and GM1 Gangliosidosis
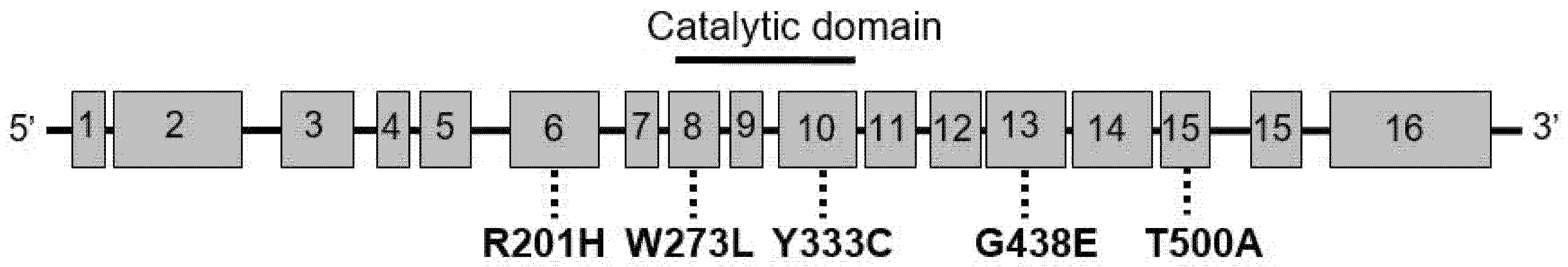
5. GLB1 Variants Associated with MBD
6. Biomarkers
7. Cytokines
8. Treatment
9. Research
Author Contributions
Funding
Conflicts of Interest
References
- Baehner, F.; Schmiedeskamp, C.; Krummenauer, F.; Miebach, E.; Bajbouj, M.; Whybra, C.; Kohlschütter, A.; Kampmann, C.; Beck, M. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J. Inherit. Metab. Dis. 2005, 28, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Brailsford, J.F. The classics: Chondro-osteo-dystrophy. Roentgenographic and clinical features of a child with dislocation of vertebrae. Clin. Orthop. Relat. Res. 1929, 7, 4–9. [Google Scholar]
- AbuMansour, I.S.; Yuskiv, N.; Paschke, E.; Stockler, S. Morquio-B disease: Clinical and genetic characteristics of a distinct GLB1 -related dysostosis multiplex. JIMD Rep. 2019, 51, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleier, M.; Yuskiv, N.; Priest, T.; Popurs, M.A.M.; Stockler-Ipsiroglu, S. Morquio B patient/caregiver survey: First insight into the natural course of a rare GLB1 related condition. Mol. Genet. Metab. Rep. 2018, 16, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; Tomatsu, S.; Patel, P.; Shimada, T.; Xie, L.; Yasuda, E.; Mason, R.; MacKenzie, W.G.; Theroux, M.; Bober, M.B.; et al. Non-invasive pulmonary function test on Morquio patients. Mol. Genet. Metab. 2015, 115, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Doherty, C.; Kubaski, F.; Tomatsu, S.; Shaffer, T.H. Non-invasive pulmonary function test on Morquio patients. Rare Dis. Res. Treat. 2018, 2, 55–62. [Google Scholar]
- Da Silveira, M.R.M.; Buriti, A.K.L.; Martins, A.M.; Gil, D.; De Azevedo, M.F. Audiometric evaluation in individuals with mucopolysaccharidosis. Clinics 2018, 73, 523. [Google Scholar] [CrossRef]
- Stockler-Ipsiroglu, S.; Yazdanpanah, N.; Yazdanpanah, M.; Popurs, M.M.; Yuskiv, N.; Schmitz Ferreira Santos, M.L.; Kim, C.A.; Fischinger Moura de Souza, C.; Lourenco, C.M.; Steiner, C.E.; et al. Morquio-like dysostosis multiplex presenting with neuronopathic features is a distinct GLB1-related phenotype. JIMD Res. Rep. 2020. submitted. [Google Scholar]
- Beck, M.; Petersen, E.M.; Spranger, J.; Beighton, P. Morquio’s disease type B (beta-galactosidase deficiency) in three siblings. S. Afr. Med. J. 1987, 72, 704–707. [Google Scholar]
- Holzgreve, W.; Gröbe, H.; Von Figura, K.; Kresse, H.; Beck, H.; Mattei, J.F. Morquio syndrome: Clinical findings in 11 patients with MPS IVA and 2 patients with MPS IVB. Qual. Life Res. 1981, 57, 360–365. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nanba, E.; Matsuda, J.; Higaki, K.; Oshima, A. β-Galactosidase Deficiency (β-Galactosidosis): GM1 Gangliosidosis and Morquio B Disease. In The Online Metabolic and Molecular Bases of Inherited Disease; Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Gibson, K.M., Mitchell, G., Eds.; The McGraw-Hill Companies, Inc.: New York, NY, USA, 2014. [Google Scholar]
- Ohto, U.; Usui, K.; Ochi, T.; Satow, Y.; Shimizu, T. Crystal Structure of Human beta -Galactosidase. J. Biol. Chem. 2012, 287, 1801–1812. [Google Scholar] [CrossRef] [Green Version]
- Privitera, S.; Prody, C.A.; Callahan, J.W.; Hinek, A. The 67-kDa Enzymatically Inactive Alternatively Spliced Variant of Beta-Galactosidase Is Identical to the Elastin/Laminin-binding Protein. J. Biol. Chem. 1998, 273, 6319–6326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschke, E.; Milos, I.; Kreimer-Erlacher, H.; Hoefler, G.; Beck, M.; Hoeltzenbein, M.; Kleijer, W.; Levade, T.; Michelakakis, H.; Radeva, B. Mutation analyses in 17 patients with deficiency in acid β-galactosidase: Three novel point mutations and high correlation of mutation W273L with Morquio disease type B. Hum. Genet. 2001, 109, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.W. Molecular basis of GM1 gangliosidosis and Morquio disease, type B. Structure–function studies of lysosomal β-galactosidase and the non-lysosomal β-galactosidase-like protein. Biochim. Biophys. Acta Mol. Basis Dis. 1999, 1455, 85–103. [Google Scholar] [CrossRef] [Green Version]
- Hofer, D.; Paul, K.; Fantur, K.; Beck, M.; Roubergue, A.; Vellodi, A.; Poorthuis, B.; Michelakakis, H.; Plecko, B.; Paschke, E. Phenotype determining alleles in GM1 gangliosidosis patients bearing novel GLB1 mutations. Clin. Genet. 2010, 78, 236–246. [Google Scholar] [CrossRef]
- Bagshaw, R.D.; Zhang, S.; Hinek, A.; Skomorowski, M.A.; Whelan, D.; Clarke, J.T.R.; Callahan, J.W. Novel mutations (Asn 484 Lys, Thr 500 Ala, Gly 438 Glu) in Morquio B disease. Biochim. Biophys. Acta Bioenerg. 2002, 1588, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Hofer, D.; Paul, K.; Fantur, K.; Beck, M.; Bürger, F.; Caillaud, C.; Fumić, K.; Ledvinová, J.; Ługowska, A.; Michelakakis, H.; et al. GM1 gangliosidosis and Morquio B disease: Expression analysis of missense mutations affecting the catalytic site of acid β-galactosidase. Hum. Mutat. 2009, 30, 1214–1221. [Google Scholar] [CrossRef]
- Mayer, F.Q.; Pereira, F.D.S.; Fensom, A.H.; Slade, C.; Matte, U.D.S.; Giugliani, R. New GLB1 mutation in siblings with Morquio type B disease presenting with mental regression. Mol. Genet. Metab. 2009, 96, 148. [Google Scholar] [CrossRef]
- Przybilla, M.J.; Ou, L.; Tăbăran, A.-F.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; O’Sullivan, M.G.; Whitley, C.B. Comprehensive behavioral and biochemical outcomes of novel murine models of GM1-gangliosidosis and Morquio syndrome type B. Mol. Genet. Metab. 2019, 126, 139–150. [Google Scholar] [CrossRef]
- Paschke, E.; Kresse, H. Morquio disease, type B: Activation of GM1-β-galactosidase by GM1-activator protein. Biochem. Biophys. Res. Commun. 1982, 109, 568–575. [Google Scholar] [CrossRef]
- Okumiya, T.; Sakuraba, H.; Kase, R.; Sugiura, T. Imbalanced substrate specificity of mutant β-galactosidase in patients with Morquio B disease. Mol. Genet. Metab. 2003, 78, 51–58. [Google Scholar] [CrossRef]
- Khan, S.A.; Mason, R.W.; Giugliani, R.; Orii, K.; Fukao, T.; Suzuki, Y.; Yamaguchi, S.; Kobayashi, H.; Orii, T.; Tomatsu, S. Glycosaminoglycans analysis in blood and urine of patients with mucopolysaccharidosis. Mol. Genet. Metab. 2018, 125, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Simonaro, C.M.; D’Angelo, M.; Haskins, M.E.; Schuchman, E.H. Joint and Bone Disease in Mucopolysaccharidoses VI and VII: Identification of New Therapeutic Targets and BioMarkers Using Animal Models. Pediatr. Res. 2005, 57, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonaro, C.M. Lysosomes, Lysosomal Storage Diseases, and Inflammation. J. Inborn Errors Metab. Screen. 2016, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Fujitsuka, H.; Sawamoto, K.; Peracha, H.; Mason, R.W.; MacKenzie, W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol. Genet. Metab. Rep. 2019, 19, 100455. [Google Scholar] [CrossRef]
- Latour, Y.L.; Yoon, R.; Thomas, S.E.; Grant, C.; Li, C.; Sena-Esteves, M.; Allende, M.L.; Proia, R.L.; Tifft, C.J. Human GLB1 knockout cerebral organoids: A model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol. Genet. Metab. Rep. 2019, 21, 100513. [Google Scholar] [CrossRef]
- Chen, J.C.; Luu, A.R.; Wise, N.; De Angelis, R.; Agrawal, V.; Mangini, L.; Vincelette, J.; Handyside, B.; Sterling, H.J.; Lo, M.J.; et al. Intracerebroventricular enzyme replacement therapy with β-galactosidase reverses brain pathologies due to GM1 gangliosidosis in mice. J. Biol. Chem. 2020, 295, 13532–13555. [Google Scholar] [CrossRef] [Green Version]
- Fischetto, R.; Palladino, V.; Mancardi, M.M.; Giacomini, T.; Palladino, S.; Gaeta, A.; Di Rocco, M.; Zampini, L.; Lassandro, G.; Favia, V.; et al. Substrate reduction therapy with Miglustat in pediatric patients with GM1 type 2 gangliosidosis delays neurological involvement: A multicenter experience. Mol. Genet. Genom. Med. 2020, 8, 1–10. [Google Scholar] [CrossRef]
- Front, S.; Biela-Banaś, A.; Burda, P.; Ballhausen, D.; Higaki, K.; Caciotti, A.; Morrone, A.; Charollais-Thoenig, J.; Gallienne, E.; Demotz, S.; et al. (5aR)-5a-C-Pentyl-4-epi-isofagomine: A powerful inhibitor of lysosomal β-galactosidase and a remarkable chaperone for mutations associated with GM1-gangliosidosis and Morquio disease type B. Eur. J. Med. Chem. 2017, 126, 160–170. [Google Scholar] [CrossRef]
- Kishnani, P.; Tarnopolsky, M.; Roberts, M.; Sivakumar, K.; Dasouki, M.; Dimachkie, M.M.; Finanger, E.; Goker-Alpan, O.; Guter, K.A.; Mozaffar, T.; et al. Duvoglustat HCl Increases Systemic and Tissue Exposure of Active Acid α-Glucosidase in Pompe Patients Co-administered with Alglucosidase α. Mol. Ther. 2017, 25, 1199–1208. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| System/Feature | Symptoms/Signs |
|---|---|
| Skeletal | Kyphoscoliosis, short trunk, pectus carinatum, short neck, coxa and genua valga, flat feet, joint laxity, and progressive growth impairment. |
| Radiological | Platyspondyly, odontoid hypoplasia, spinal canal narrowing, hip dysplasia, dysplasia of the carpal and tarsal bones, shortening and epi- and metaphyseal dysplasia of long bones. |
| Neurological (MBD plus) | Dystonia, dysarthria, dysphagia, ataxia, cognitive delay and epilepsy, spinal cord compression due to spinal canal narrowing. |
| Other organs | Corneal clouding, cardiac valve pathology, tracheal stenosis, adeno-tonsillar hypertrophy, dental problems. Hepatosplenomegaly is rare. |
| Functional outcomes | Pain in joints and limbs, limited mobility, limitation in self-care activities, abnormal pulmonary function (restrictive/obstructive lung disease, mainly due to chest structural abnormalities), sleep disturbances. |
| Reported surgeries | On average 3 orthopedic surgeries reported by the second decade of life, most frequently hip and knee replacement. |
| Biochemical | Accumulation of keratan sulfate in bone and cartilage, elevation of TNF-alpha, IL-1β, IL-6 and inflammatory proteases. |
| β-galactosidase activity | From 2 to 11.5% (pure skeletal presentation), and from 4.6 to 14.1% (MBD with neuronopathic presentation) |
| Reported GLB1 mutations | W273L, T500A, R210H, G438E, Y333C, G438E, T384S, Y333H (not invariably associated with MBD). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuskiv, N.; Higaki, K.; Stockler-Ipsiroglu, S. Morquio B Disease. Disease Characteristics and Treatment Options of a Distinct GLB1-Related Dysostosis Multiplex. Int. J. Mol. Sci. 2020, 21, 9121. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239121
Yuskiv N, Higaki K, Stockler-Ipsiroglu S. Morquio B Disease. Disease Characteristics and Treatment Options of a Distinct GLB1-Related Dysostosis Multiplex. International Journal of Molecular Sciences. 2020; 21(23):9121. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239121
Chicago/Turabian StyleYuskiv, Nataliya, Katsumi Higaki, and Sylvia Stockler-Ipsiroglu. 2020. "Morquio B Disease. Disease Characteristics and Treatment Options of a Distinct GLB1-Related Dysostosis Multiplex" International Journal of Molecular Sciences 21, no. 23: 9121. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239121