1. Introduction
Sjögren syndrome (SS) is a chronic autoimmune disease that primarily targets lacrimal and salivary glands leading to progressive exocrine gland dysfunction and debilitating ocular and oral dryness [
1]. The ensuing poor ocular and oral health may lead to defects in vision and difficulty with normal oral function such as talking and swallowing. Beyond these glandular manifestations, many individuals with SS develop autoimmunity affecting other organs, chronic musculoskeletal pain, and profound fatigue. The chronic immune stimulation is believed to contribute to the markedly increased risk of developing lymphoma. Current treatments largely aim to replace or augment tears and saliva but fail to adequately halt or reverse the chronic inflammation to prevent damage or progressive exocrine gland dysfunction. The lack of well-established therapeutics to modulate the immune response may relate to the gap in understanding of the early immunopathogenic mechanisms driving the exocrine gland inflammation.
Toll-like receptors (TLRs) are pattern recognition receptors that recognize a wide range of pathogen and damage associated molecules [
2]. TLR signals in innate and adaptive immune cells play early roles in immune responses and have been linked to infections, cancer, and autoimmunity [
3,
4]. Studies of human salivary gland tissue, peripheral blood mononuclear cells (PBMCs), and mouse models of SS have identified pathogenic roles for several cell surface and intracellular TLRs in salivary gland autoimmunity [
5]. TLRs are expressed at the ocular surface [
6], but evidence for the role of TLRs in lacrimal gland autoimmunity is limited. Administration of a TLR3 agonist resulted in lacrimal gland inflammation in a non-autoimmune-prone mouse strain (C57BL/6) [
7]. In an autoimmune-prone strain of mouse that spontaneously develops SS-like manifestations (NOD. B10Sn-
H2b/J) lacrimal gland inflammation decreased when
Myd88, which encodes a key adaptor in the signaling pathways of multiple TLRs, was disrupted [
8] suggesting a pathogenic role for MyD88-dependent TLRs. TLR7 is MyD88-dependent, and a role for TLR7 was suggested by development of spontaneous lacrimal gland inflammation in autoimmune-prone mice (BXSB/MpJ-
Yaa) in which males have increased expression of TLR7 [
9].
TLR7, an endosomal TLR, has been implicated in multiple autoimmune diseases including SS and type 1 diabetes (T1D) [
10,
11,
12,
13]. TLR7 recognizes uridine-rich ssRNA and guanosine [
14] and is expressed in monocytes, macrophages, dendritic cells, and B cells. Signaling through TLR7 is MyD88-dependent and uses NF-κB and interferon (IFN) regulatory factor pathways to drive expression of type I IFN and other inflammatory cytokines. In SS,
TLR7 expression was increased in PBMCs, and this increased expression positively correlated with IFN signatures in SS patients [
15,
16]. TLR7 has been detected in labial minor salivary glands of SS patients with expression noted in ductal cells and infiltrating immune cells [
16,
17]. A role for TLR7 has not been evaluated in nonobese diabetic (NOD) mice, which spontaneously develop SS-like manifestations and are a well-established model for the study of SS-like lacrimal and salivary gland autoimmunity [
18].
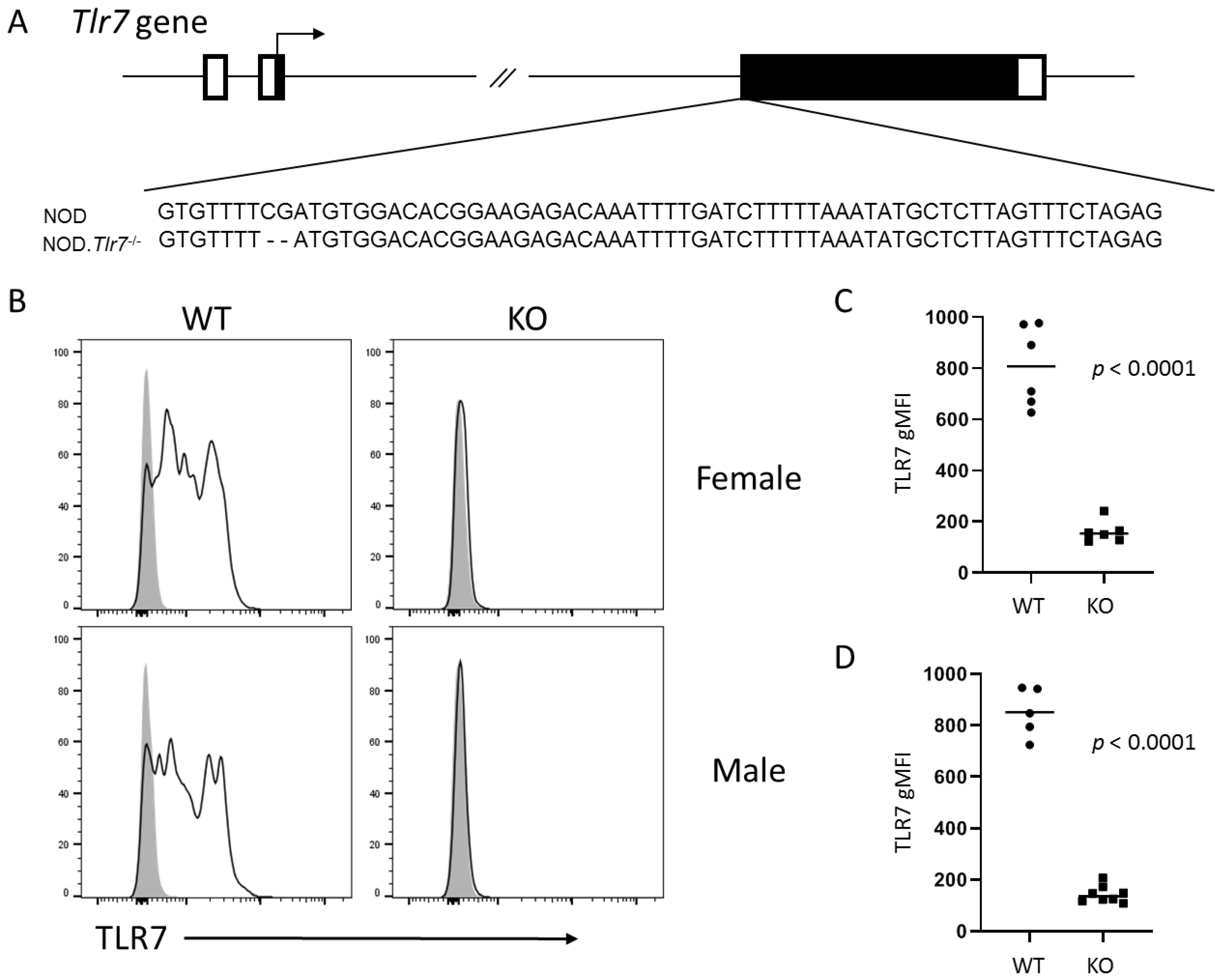
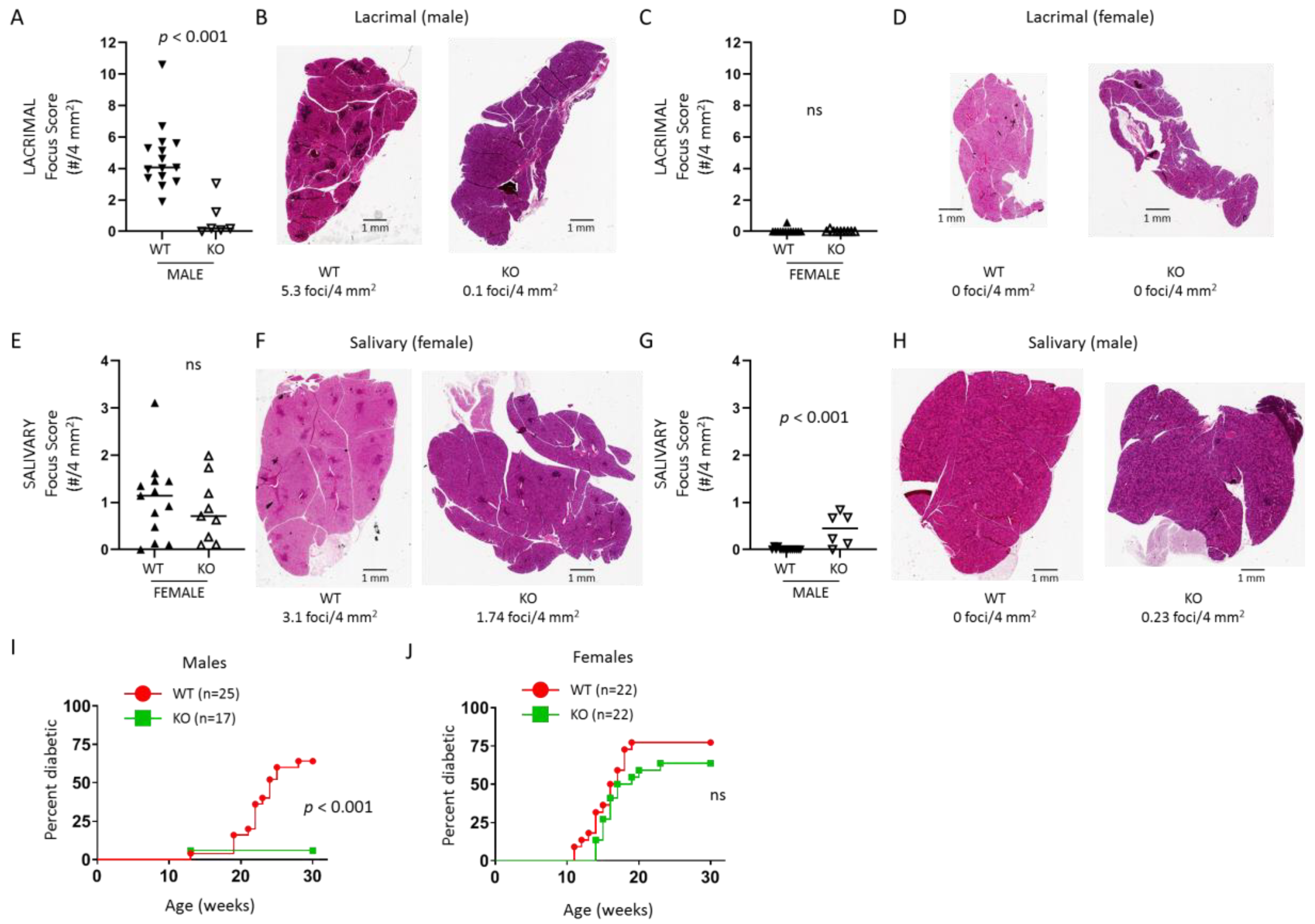
The purpose of this study was to define the role of TLR7 in the spontaneous development of SS-like autoimmunity in NOD mice. We developed gene-edited NOD mice lacking Tlr7 expression and found that male mice lacking TLR7 were protected from the spontaneous development of lacrimal gland inflammation, whereas females were not protected from spontaneous salivary gland inflammation. NOD mice are also a model of T1D given the spontaneous development of T1D in both sexes. In the absence of TLR7, male NOD mice were protected from T1D development, but females were not protected. Through RNA sequencing studies, we identified genes and pathways up-regulated in lacrimal glands of wild-type NOD mice compared to TLR7-deficient NOD mice and compared this gene set to a set of genes up-regulated in lacrimal gland disease in a type I IFN-dependent manner. Together, our findings suggest TLR7 plays a key pathogenic role in the development of lacrimal gland autoimmunity associated with a type I IFN response and with B cell responses in situ in a type I IFN-independent manner.
3. Discussion
TLR7 has been implicated in multiple autoimmune diseases including SS and T1D [
10,
11,
12,
13]. As an X-linked gene,
TLR7 may escape X-linked inactivation leading to increased TLR7 levels that have been associated with increased systemic autoimmunity (such as SS) in individuals with more than one X chromosome [
24,
27,
28]. Here, we evaluated the role of TLR7 in the spontaneous autoimmune manifestations in NOD mice by creating
Tlr7 KO NOD mice. We found that male NOD mice were protected from development of lacrimal gland inflammation and T1D development, while female NOD mice were not protected from developing salivary gland inflammation or T1D. This was not due to an X-linked phenomenon as TLR7 protein was not detected in either male or female
Tlr7 KO mice. Surprisingly, TLR7-deficiency resulted in development of salivary gland inflammation in some males, but no females (WT or
Tlr7 KO) developed lacrimal gland inflammation. These data suggest different disease mechanisms in the development of SS and T1D manifestations in male and female NOD mice with a role for TLR7 in promoting or protecting from disease manifestations in males but not females.
For the SS manifestations, we have previously demonstrated that male-specific lacrimal gland inflammation required intact type I IFN signaling [
23]. In contrast, female salivary gland inflammation required intact type II IFN signaling as
Ifng or
Ifngr KO NOD mice failed to develop salivary gland inflammation [
29]. Interestingly, male type II IFN signaling-deficient NOD mice were not protected from development of lacrimal gland inflammation [
29], and female type I IFN signaling-deficient NOD mice were not protected from developing salivary gland inflammation (our unpublished observation). Thus, the two sex-dependent SS manifestations in NOD mice are dependent on different IFN signaling pathways. Studies in humans have demonstrated that a positive IFN-signature in individuals with SS may be dominated by type I IFN, type II IFN, or a combination of type I and type II IFN [
30]. Together, these findings suggest that spontaneous SS-like disease in NOD mice may represent two different mechanisms of disease found in humans, with lacrimal gland disease in male NOD mice representing the type I IFN-dependent disease in humans and salivary gland disease in female NOD mice representing the type II IFN-dependent disease in humans. This is supported in part by recent demonstration of similarities in gene expression profiles in lacrimal glands of male NOD mice and salivary glands of humans, though that study did not specifically limit the human data to type I IFN-dominant disease [
31].
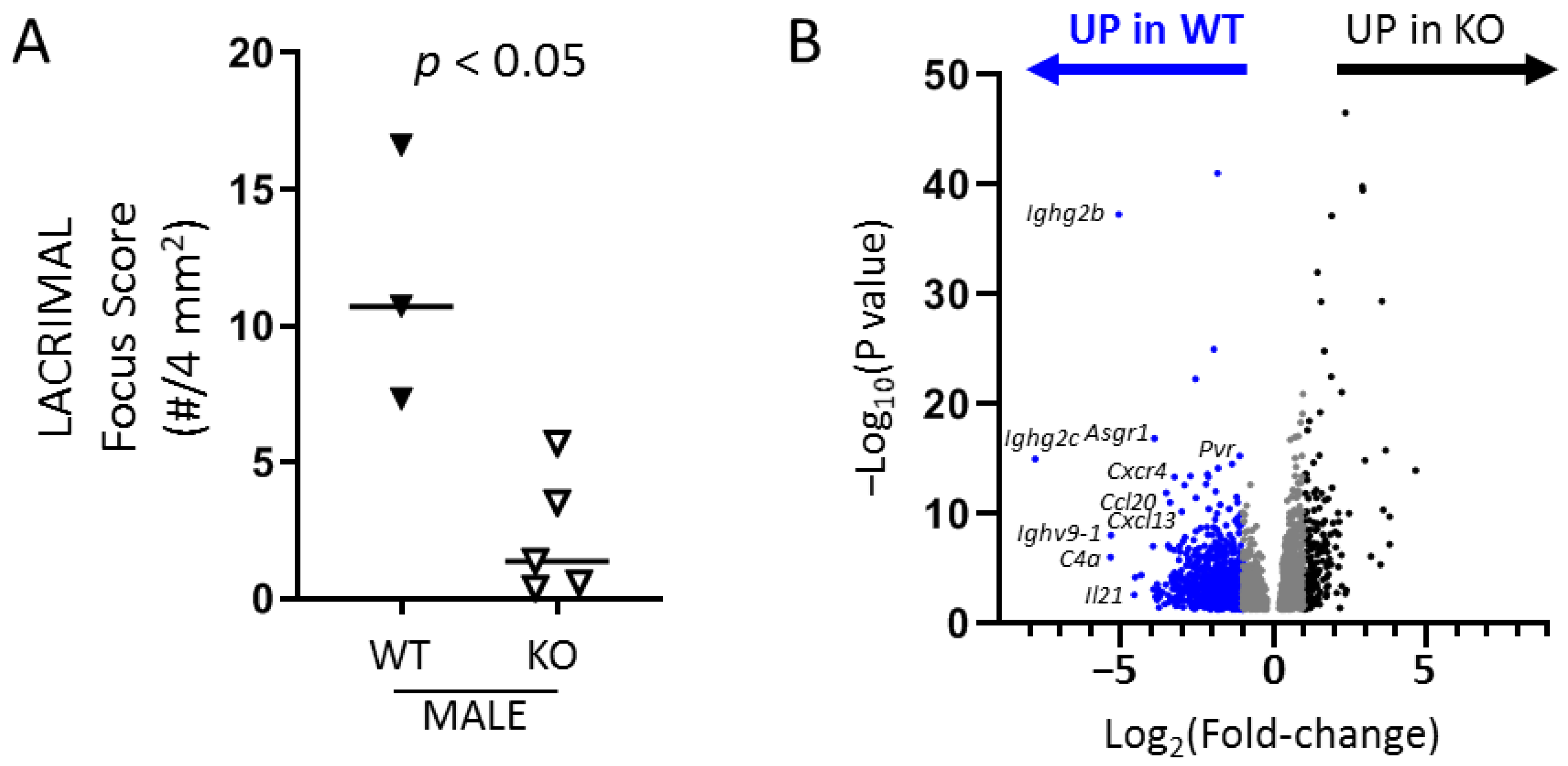
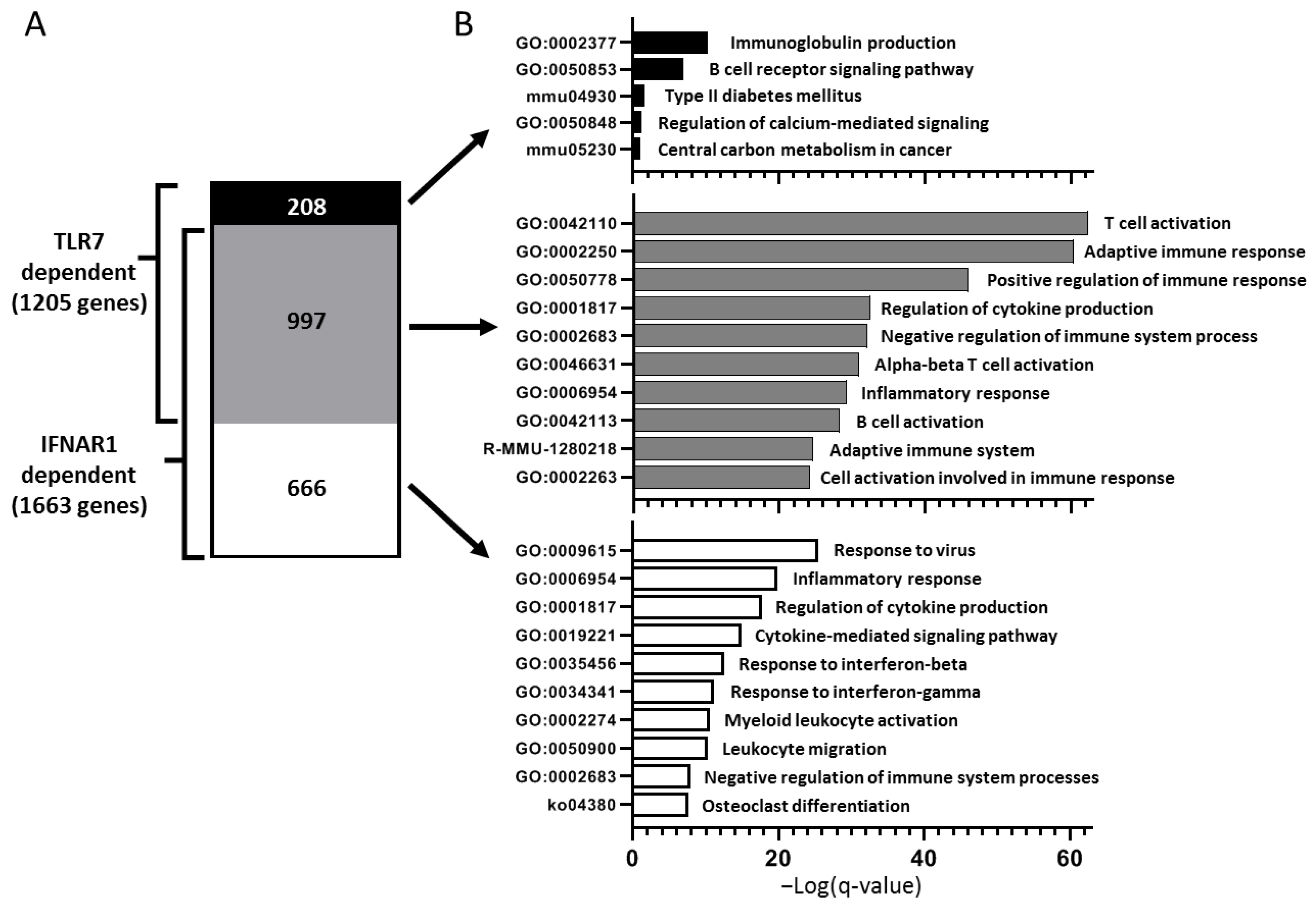
TLR7 signaling is a well-established type I IFN-driving stimulus, which is in accordance with our findings of the requirement of TLR7 for male-specific lacrimal gland inflammation in NOD mice but not for female-specific salivary gland disease. In support of this, a large majority (>80%) of genes up-regulated in lacrimal glands of WT NOD mice in a TLR7-dependent manner overlap with those genes up-regulated in a type I IFN-signaling-dependent manner. These genes were enriched for pathways involved in the adaptive immune response. Interestingly, though, the smaller portion of genes up-regulated in a TLR7- but not IFNAR1-dependent manner enriched for pathways related to B cell responses suggesting a unique role for TLR7 signaling in B cell responses within lacrimal glands. In SS, B cells may play pathogenic roles through several mechanisms including production of autoantibodies, presentation of autoantigens to T cells, and production of inflammatory cytokines [
32]. In B cell-deficient NOD mice, the initiation of lacrimal and salivary gland inflammation was not prevented [
33]. However, in WT NOD mice, B cells are present within gland infiltrates and accumulate in the lacrimal glands over time [
31,
34]. Together with our data here, these studies suggest that B cells are not required for initiation of exocrine gland inflammation but may contribute to the continued inflammation within the glands in a TLR7-dependent manner. In support of a pathogenic role for TLR7 in lacrimal gland inflammation, male BXSB/MpJ-
Yaa mice develop spontaneous lacrimal gland inflammation [
9]. These mice express higher levels of TLR7 due to translocation of X chromosome genes including
Tlr7 to the Y chromosome resulting in increased TLR7 protein expression [
35]. Notably, the lacrimal gland infiltrates in these mice were dominated by B cells [
9]. In B cells from humans with SS,
TLR7 up-regulation has also been reported [
16,
36]. TLR7 is expressed by many cells beyond B cells, and additional studies are necessary to determine if the pathogenic role of TLR7 in driving lacrimal gland inflammation in NOD mice is B cell-intrinsic and/or dependent on other TLR7-expressing cells.
While the different IFN-dependencies in the sex-specific SS manifestations in NOD mice may explain the male-specific protection from lacrimal gland disease in
Tlr7 KO mice, this does not explain the sex differences in T1D protection. Studies of IFN signaling-deficient NOD mice suggested a more complex role for type I and II IFN signaling in T1D development. Female NOD mice lacking either
Ifnar1 or
Ifngr1 showed some (but incomplete) protection, but males were protected only when both IFN receptor genes were disrupted [
22]. Notably, other studies reported no change in T1D development in female NOD mice deficient in type I IFN signaling [
37] or type II IFN signaling [
38], but these studies did not assess disease in males. In contrast to SS manifestations in NOD mice that occur only in one sex (lacrimal disease in males, salivary gland disease in females), T1D may develop in both males and females, though with different incidence generally favoring increased disease in females. The female T1D bias related to differences in microbiota and was lost when NOD mice were kept in germ free conditions [
39,
40]. MyD88-dependent signaling contributed to the role of microbiota on T1D development in NOD mice. Both male and female MyD88-deficient NOD mice were protected from diabetes but only when gut microbiota were present [
41]. TLR7 signaling is MyD88-dependent, but our findings here demonstrate a role for TLR7 signaling in T1D development only in males suggesting other MyD88-dependent TLRs may compensate for the loss of TLR7 in females in our studies. TLR2-deficient female NOD mice were partially protected from T1D development, and this also required the presence of microbiota [
42]. Both male and female TLR9-deficient NOD mice were protected from developing T1D [
43]. Together these data suggest that multiple TLRs play pathogenic roles in T1D development in NOD mice, but these roles differ between sexes with TLR7 providing a non-redundant role in T1D development specifically in male NOD mice. Whether these sex differences in the role of TLR7 in T1D development also depend on microbiota remains to be determined.
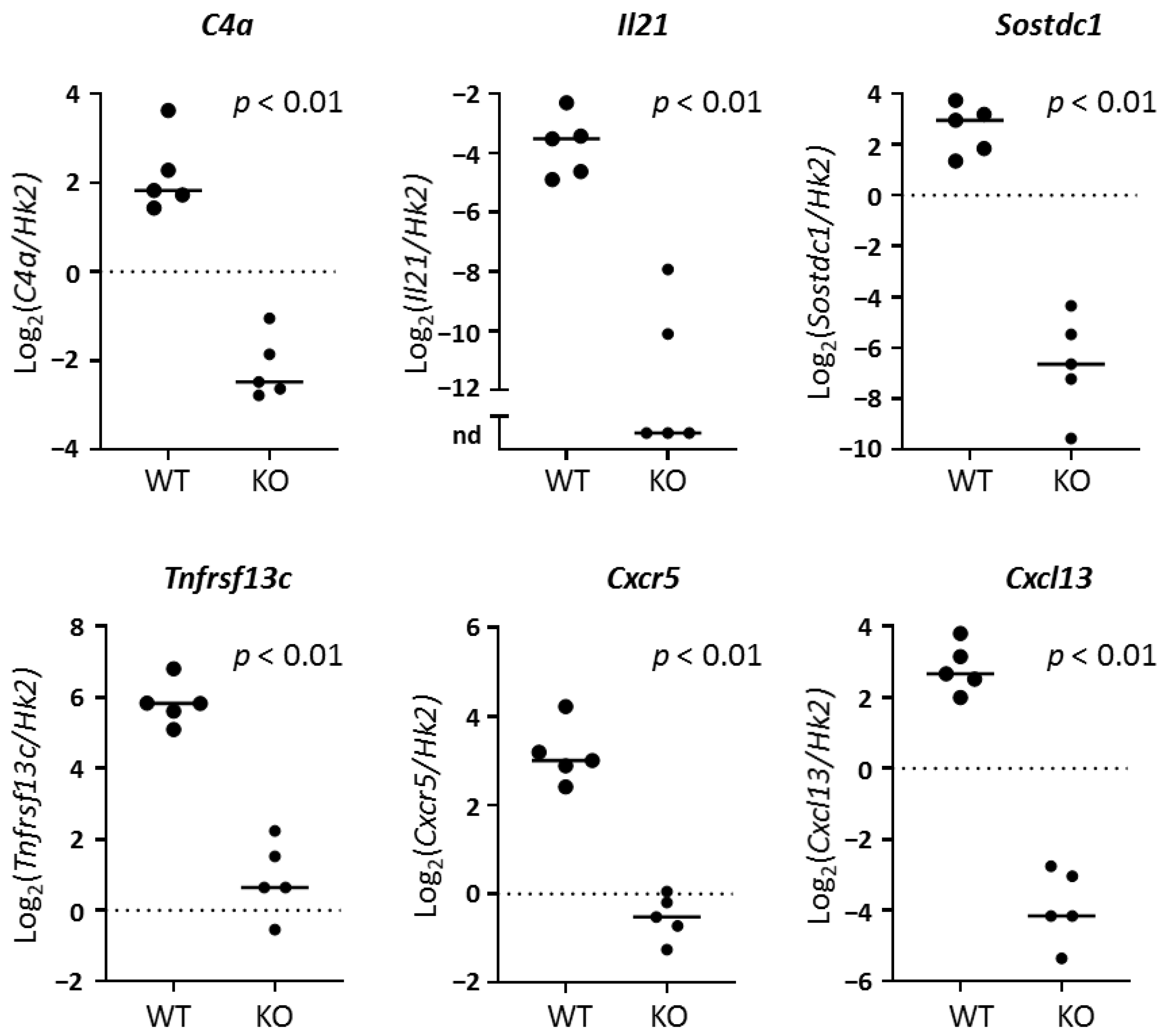
To better understand the role of TLR7 in lacrimal gland autoimmunity, we performed RNA sequencing studies on lacrimal glands and identified both common and unique immune pathways up-regulated in TLR7 or type I IFN-signaling-dependent manners. TLR7-dependent (but type I IFN-independent) pathways suggested a role for TLR7 in driving B cell responses in the lacrimal glands. Given the role of TLR7 in B cell biology, it is not surprising that three of the top five DE genes in WT lacrimal glands were immunoglobulin-related genes. The remaining genes in the top 5 DE in WT lacrimal glands in a TLR7-dependent manner (
C4a and
Il21) also play roles in B cell responses.
C4a encodes complement protein 4a, which plays a key role in both the classical and lectin pathways of the complement cascade [
44]. Deficiency in complement C4 protein has been associated with systemic autoimmunity in humans including the recent finding that copy number variations in the
C4A and
C4B genes were associated with increased risk of SS [
45]. In humans, though, a decrease in copy number resulting in lower protein levels was associated with higher risk of autoimmunity, which is contrary to our findings of an increase in
C4a expression associated with lacrimal gland inflammation in WT mice. Since complement proteins are involved in the clearance of cell debris, the up-regulation of
C4a may be a consequence of the epithelial cell damage in the context of inflammation rather than a cause in NOD mice, and the lack of C4 proteins due to decreased copy number variations in the associated genes in humans may contribute to ongoing inflammation by failing to adequately clear the debris. Given that NOD mice are deficient in C5, which is downstream of C4, they may have a similar deficiency in clearing cell debris through complement-mediated mechanisms [
46]. Despite the association of deficiencies in C4 with autoimmunity, laser capture microdissection coupled with gene expression studies demonstrated an increase in
C4A expression in ductal cells isolated from minor salivary glands of individuals with SS compared to healthy controls [
47]. Together, these results suggest that C4 plays a role in the immune regulation and/or dysregulation in the development of SS autoimmunity, but additional studies are needed to further define these mechanisms.
Interleukin-21 (IL-21) is an inflammatory cytokine produced by innate and adaptive immune cells that promotes B and T cell responses and may affect innate immune and non-immune cell functions. The role of IL-21 in SS has been reviewed [
48] and is further discussed in our recent report demonstrating decreased lacrimal gland inflammation in
Il21 KO NOD mice [
26]. IL-21 may be produced by follicular helper CD4 T cells (Tfh), which are characterized by expression of chemokine receptor CXCR5 and transcription factor BCL6. In NOD lacrimal glands, another IL-21-producing CD4 population that lacks these classic Tfh markers has been identified [
31]. This population of CD4 T cells expressed high levels of PD1, ICOS, CD73, and CD200 by flow cytometry, and transcriptional analyses demonstrated increased expression of
Tbx1,
Tnf, and
Ifng suggesting a Th1-type profile. Among the genes up-regulated in these IL-21-producing CD4 T cells was
Sostdc1, which we have also identified here as being highly DE in lacrimal glands of WT NOD mice in a TLR7-dependent manner.
Sostdc1 encodes Sclerostin domain-containing protein 1, a secreted protein that antagonizes both Wnt/β-catenin and bone morphogenetic protein signaling.
Sostdc1 plays many roles in development of tissues (including development of the eye [
49]) and in promoting cancer. Recent study using SOSTDC1-reporter mice identified a potential regulatory role for Tfh-secreted SOSTDC1 in promoting the development of regulatory follicular T cells in germinal centers following viral infection or immunization [
50]. Notably, these SOSTDC1-secreting Tfh cells expressed lower levels of
Il21 and lost the ability to help B cells. Whether the up-regulated
Sostdc1 in WT NOD lacrimal glands plays a pathogenic or regulatory role remains to be determined.
In summary, TLR7 is required for the spontaneous autoimmune manifestations that develop in WT male NOD mice including lacrimal gland inflammation and T1D development but is dispensable for the salivary gland inflammation and T1D that spontaneously develops in females. Moreover, TLR7 may provide a protective role in salivary gland disease in male NOD mice. In lacrimal glands, TLR7 is associated with many adaptive immune response genes common to a type I IFN response but also promotes B cell activity within the lacrimal glands in a type I IFN-independent manner. Previous microarray studies have identified increased
Tlr7 expression (~3-fold) in lacrimal glands from WT male compared to female NOD mice [
51], which may be dependent on type I IFN signaling as
Tlr7 was expressed ~4-fold greater in WT male NOD lacrimal glands compared to
Ifnar1-deficient male NOD lacrimal glands [
26]. Thus, the pathogenic roles of TLR7 and type I IFN signaling in lacrimal gland autoimmunity in NOD mice are complex (
Figure 6). Additional functional studies are required to determine the direct roles of TLR7 signaling in driving the gene expression changes reported here or, instead, to determine if additional complex interactions downstream of TLR7 are required. One key unanswered question that remains is what TLR7 ligands trigger the TLR7 response in lacrimal glands in the context of SS-like autoimmunity. While the possibility of an exogenous viral trigger for TLR7 signaling was not formally assessed, we favor the possibilities of either an endogenous retrovirus or an endogenous nonviral ligand released from dying epithelial or immune cells during lacrimal gland inflammation. In these regards, the recent identification of a new endogenous TLR7 ligand, U11snRNA [
52], may warrant further study in NOD mice. Ultimately, identifying the innate signals that drive lacrimal gland inflammation will provide targets for novel diagnostic tests and therapeutic modalities.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}