Adipokine-Modulated Immunological Homeostasis Shapes the Pathophysiology of Inflammatory Bowel Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Pathophysiology of Inflammatory Colon Disease
1.2. The Effect of Obesity on the Development of IBD
1.3. The Interaction of Adipose Tissue and the Intestinal Immune System
2. Adipokine-Modulated Immunological Regulation in the Pathophysiology of Inflammatory Colon Disorders
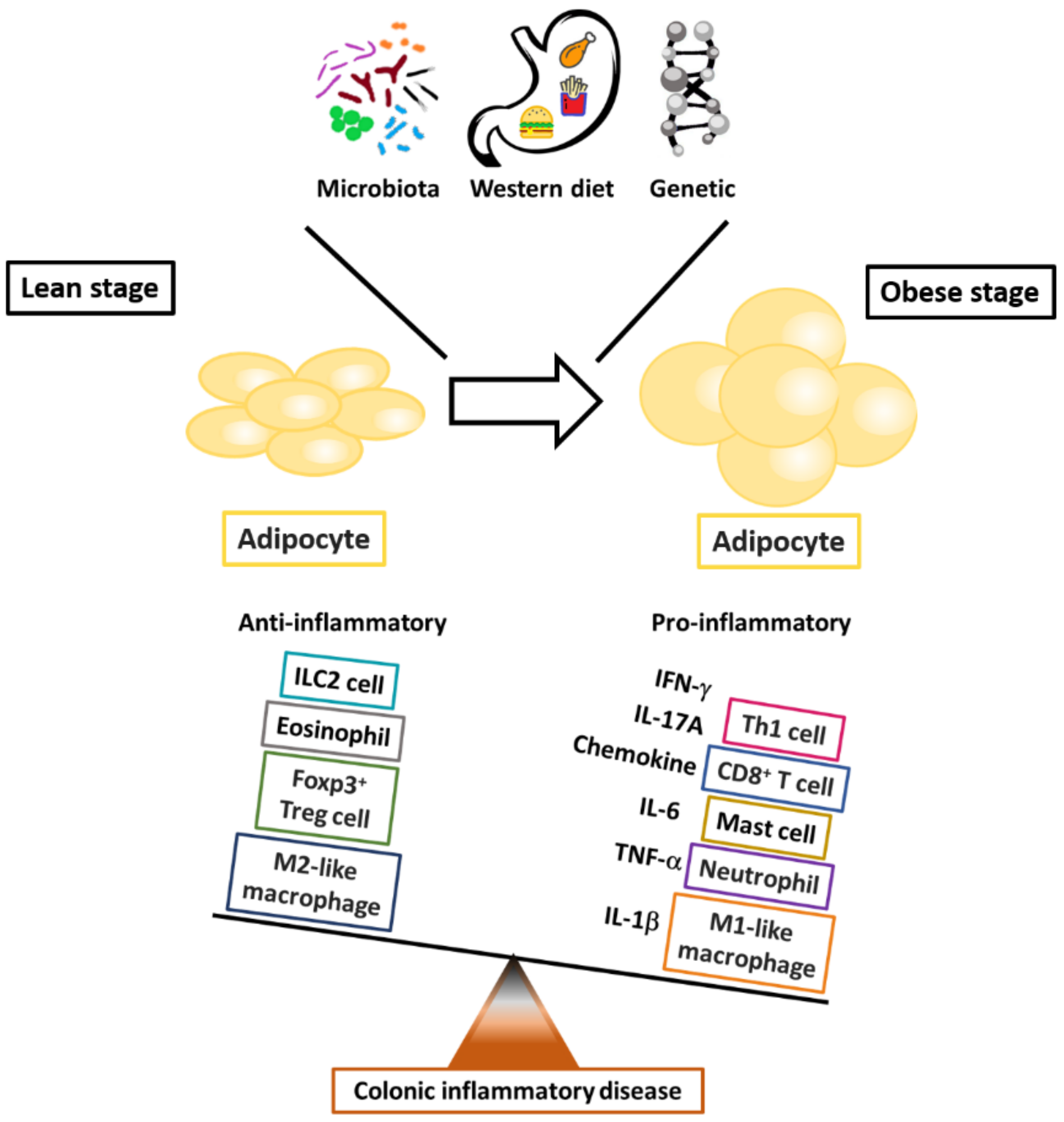
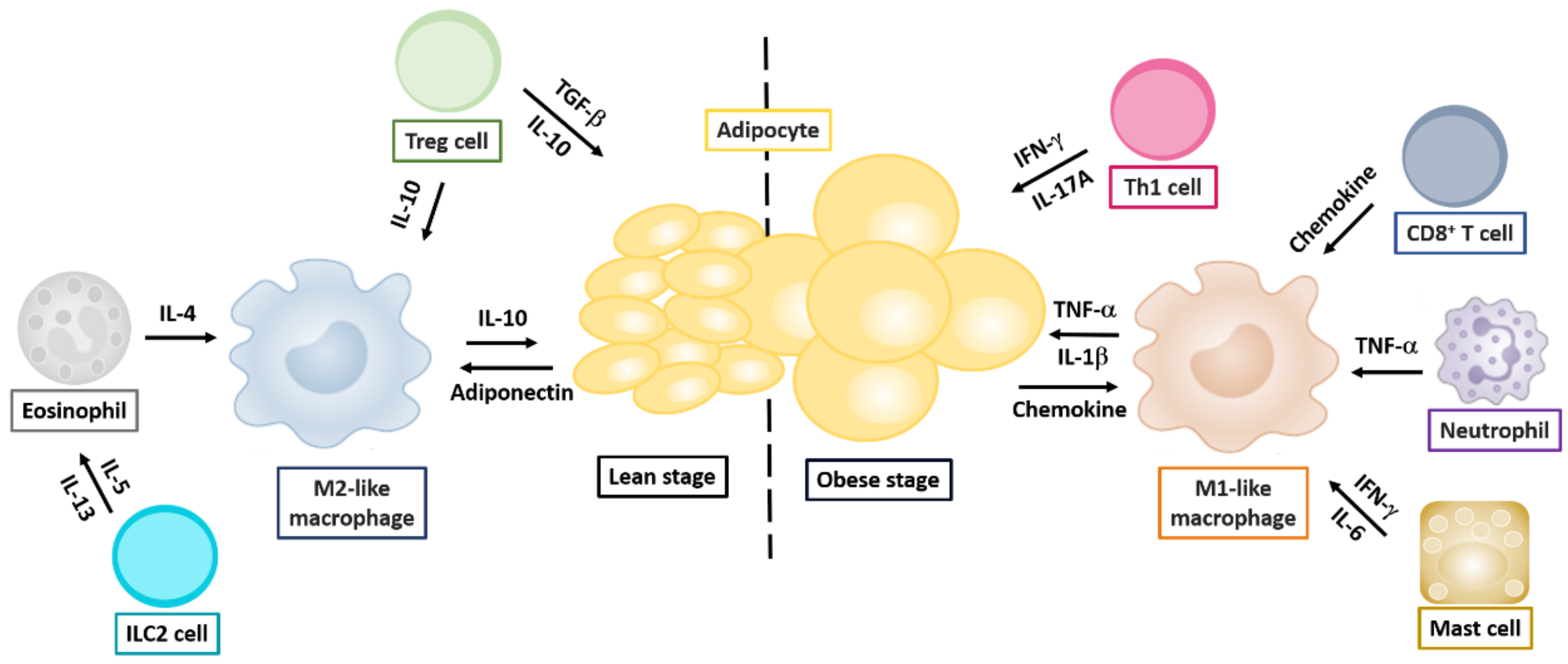
2.1. Obesity and Alteration of Immune Responses in Adipose Tissue
2.1.1. Macrophages
2.1.2. Effector T Helper Cells
2.1.3. Regulatory T Helper Cells
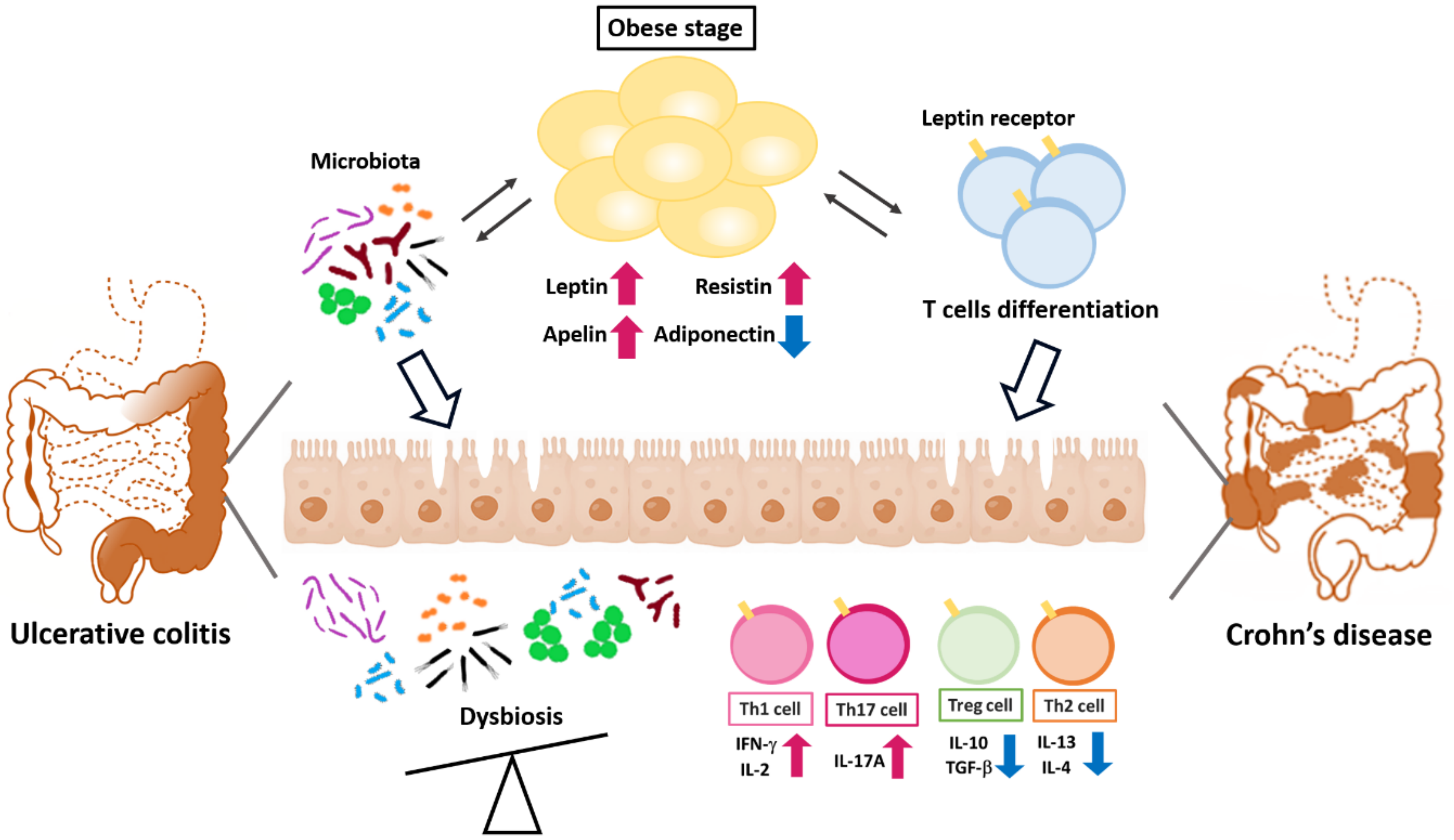
2.2. Adipokines and the Pathogenesis of Inflammatory Colon Diseases
2.2.1. Adiponectin
2.2.2. Leptin
2.2.3. Apelin
2.2.4. Resistin
2.2.5. Discussion
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Windsor, J.W.; Kaplan, G.G. Evolving Epidemiology of IBD. Curr. Gastroenterol. Rep. 2019, 21, 40. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Lees, C.W.; Barrett, J.C.; Parkes, M.; Satsangi, J. New IBD genetics: Common pathways with other diseases. Gut 2011, 60, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Becker, C.; Ivanov, I.I.; Hirth, S.; Wirtz, S.; Neufert, C.; Pouly, S.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 2009, 136, 257–267. [Google Scholar] [CrossRef] [PubMed]
- McAlindon, M.E.; Hawkey, C.J.; Mahida, Y.R. Expression of interleukin 1 beta and interleukin 1 beta converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 1998, 42, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.; Fu, S.H.; Chien, M.W.; Liu, Y.W.; Chen, S.J.; Sytwu, H.K. Post-Translational Modifications of Transcription Factors Harnessing the Etiology and Pathophysiology in Colonic Diseases. Int. J. Mol. Sci. 2020, 21, 207. [Google Scholar] [CrossRef]
- Fu, S.H.; Chien, M.W.; Hsu, C.Y.; Liu, Y.W.; Sytwu, H.K. Interplay between Cytokine Circuitry and Transcriptional Regulation Shaping Helper T Cell Pathogenicity and Plasticity in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 379. [Google Scholar] [CrossRef]
- Imam, T.; Park, S.; Kaplan, M.H.; Olson, M.R. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front. Immunol. 2018, 9, 1212. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.H.; Lin, M.H.; Yeh, L.T.; Wang, Y.L.; Chien, M.W.; Lin, S.H.; Chang, D.M.; Sytwu, H.K. Targeting tumour necrosis factor receptor 1 assembly reverses Th17-mediated colitis through boosting a Th2 response. Gut 2015, 64, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.H.; Braegger, C.P.; Walker-Smith, J.A.; MacDonald, T.T. Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease. Gut 1993, 34, 1705–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol. Rev. 2006, 212, 256–271. [Google Scholar] [CrossRef]
- Bouma, G.; Strober, W. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 2003, 3, 521–533. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42. [Google Scholar] [CrossRef] [Green Version]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Zhai, H.; Liu, A.; Huang, W.; Liu, X.; Feng, S.; Wu, J.; Yao, Y.; Wang, C.; Li, Q.; Hao, Q.; et al. Increasing rate of inflammatory bowel disease: A 12-year retrospective study in NingXia, China. BMC Gastroenterol. 2016, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Ananthakrishnan, A.N. Environmental risk factors for inflammatory bowel diseases: A review. Dig. Dis. Sci. 2015, 60, 290–298. [Google Scholar] [CrossRef]
- Zubrzycki, A.; Cierpka-Kmiec, K.; Kmiec, Z.; Wronska, A. The role of low-calorie diets and intermittent fasting in the treatment of obesity and type-2 diabetes. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Kujawska-Luczak, M.; Szulinska, M.; Skrypnik, D.; Musialik, K.; Swora-Cwynar, E.; Kregielska-Narozna, M.; Markuszewski, L.; Grzymislawska, M.; Bogdanski, P. The influence of orlistat, metformin and diet on serum levels of insulin-like growth factor-1 in obeses women with and without insulin resistance. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Deiuliis, J.; Shah, Z.; Shah, N.; Needleman, B.; Mikami, D.; Narula, V.; Perry, K.; Hazey, J.; Kampfrath, T.; Kollengode, M.; et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS ONE 2011, 6, e16376. [Google Scholar] [CrossRef] [Green Version]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Dandona, P.; Aljada, A.; Chaudhuri, A.; Mohanty, P.; Garg, R. Metabolic syndrome: A comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 2005, 111, 1448–1454. [Google Scholar] [CrossRef] [Green Version]
- Rocha, V.Z.; Folco, E.J. Inflammatory concepts of obesity. Int. J. Inflam. 2011, 2011, 529061. [Google Scholar] [CrossRef] [Green Version]
- Barbarroja, N.; Lopez-Pedrera, R.; Mayas, M.D.; Garcia-Fuentes, E.; Garrido-Sanchez, L.; Macias-Gonzalez, M.; El Bekay, R.; Vidal-Puig, A.; Tinahones, F.J. The obese healthy paradox: Is inflammation the answer? Biochem. J. 2010, 430, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Long, M.D.; Crandall, W.V.; Leibowitz, I.H.; Duffy, L.; del Rosario, F.; Kim, S.C.; Integlia, M.J.; Berman, J.; Grunow, J.; Colletti, R.B.; et al. Prevalence and epidemiology of overweight and obesity in children with inflammatory bowel disease. Inflamm. Bowel Dis. 2011, 17, 2162–2168. [Google Scholar] [CrossRef] [Green Version]
- Steed, H.; Walsh, S.; Reynolds, N. A brief report of the epidemiology of obesity in the inflammatory bowel disease population of Tayside, Scotland. Obes. Facts 2009, 2, 370–372. [Google Scholar] [CrossRef]
- Petersen, C.; Bell, R.; Klag, K.A.; Lee, S.H.; Soto, R.; Ghazaryan, A.; Buhrke, K.; Ekiz, H.A.; Ost, K.S.; Boudina, S.; et al. T cell-mediated regulation of the microbiota protects against obesity. Science 2019, 365. [Google Scholar] [CrossRef]
- Wang, Y.; Hooper, L.V. Immune control of the microbiota prevents obesity. Science 2019, 365, 316–317. [Google Scholar] [CrossRef]
- Nam, S.Y. Obesity-Related Digestive Diseases and Their Pathophysiology. Gut Liver 2017, 11, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Principi, M.; Iannone, A.; Losurdo, G.; Mangia, M.; Shahini, E.; Albano, F.; Rizzi, S.F.; La Fortezza, R.F.; Lovero, R.; Contaldo, A.; et al. Nonalcoholic Fatty Liver Disease in Inflammatory Bowel Disease: Prevalence and Risk Factors. Inflamm. Bowel Dis. 2018, 24, 1589–1596. [Google Scholar] [CrossRef]
- Saroli Palumbo, C.; Restellini, S.; Chao, C.Y.; Aruljothy, A.; Lemieux, C.; Wild, G.; Afif, W.; Lakatos, P.L.; Bitton, A.; Cocciolillo, S.; et al. Screening for Nonalcoholic Fatty Liver Disease in Inflammatory Bowel Diseases: A Cohort Study Using Transient Elastography. Inflamm. Bowel Dis. 2019, 25, 124–133. [Google Scholar] [CrossRef]
- Bartelt, A.; Heeren, J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 2014, 10, 24–36. [Google Scholar] [CrossRef]
- Yokota, T.; Oritani, K.; Takahashi, I.; Ishikawa, J.; Matsuyama, A.; Ouchi, N.; Kihara, S.; Funahashi, T.; Tenner, A.J.; Tomiyama, Y.; et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood 2000, 96, 1723–1732. [Google Scholar] [CrossRef]
- Lord, G.M.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998, 394, 897–901. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Mol. Asp. Med. 2013, 34, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.T.; Favelyukis, S.; Nguyen, A.K.; Reichart, D.; Scott, P.A.; Jenn, A.; Liu-Bryan, R.; Glass, C.K.; Neels, J.G.; Olefsky, J.M. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007, 282, 35279–35292. [Google Scholar] [CrossRef] [Green Version]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Caspar-Bauguil, S.; Cousin, B.; Galinier, A.; Segafredo, C.; Nibbelink, M.; Andre, M.; Casteilla, L.; Penicaud, L. Adipose tissues as an ancestral immune organ: Site-specific change in obesity. FEBS Lett. 2005, 579, 3487–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, T.J.; Ni Choileain, N.; Zang, Y.; Mannick, J.A.; Lederer, J.A. CD4+CD25+ regulatory T cells control innate immune reactivity after injury. J. Immunol. 2005, 174, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uko, V.; Vortia, E.; Achkar, J.P.; Karakas, P.; Fiocchi, C.; Worley, S.; Kay, M.H. Impact of abdominal visceral adipose tissue on disease outcome in pediatric Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 2286–2291. [Google Scholar] [CrossRef]
- Kredel, L.I.; Siegmund, B. Adipose-tissue and intestinal inflammation—Visceral obesity and creeping fat. Front. Immunol. 2014, 5, 462. [Google Scholar] [CrossRef] [Green Version]
- Kredel, L.; Batra, A.; Siegmund, B. Role of fat and adipokines in intestinal inflammation. Curr. Opin. Gastroenterol. 2014, 30, 559–565. [Google Scholar] [CrossRef]
- Connelly, T.M.; Juza, R.M.; Sangster, W.; Sehgal, R.; Tappouni, R.F.; Messaris, E. Volumetric fat ratio and not body mass index is predictive of ileocolectomy outcomes in Crohn’s disease patients. Dig. Surg. 2014, 31, 219–224. [Google Scholar] [CrossRef]
- Holt, D.Q.; Moore, G.T.; Strauss, B.J.; Hamilton, A.L.; De Cruz, P.; Kamm, M.A. Visceral adiposity predicts post-operative Crohn’s disease recurrence. Aliment. Pharmacol. Ther. 2017, 45, 1255–1264. [Google Scholar] [CrossRef]
- Van Der Sloot, K.W.; Joshi, A.D.; Bellavance, D.R.; Gilpin, K.K.; Stewart, K.O.; Lochhead, P.; Garber, J.J.; Giallourakis, C.; Yajnik, V.; Ananthakrishnan, A.N.; et al. Visceral Adiposity, Genetic Susceptibility, and Risk of Complications Among Individuals with Crohn’s Disease. Inflamm. Bowel Dis. 2017, 23, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Bryant, R.V.; Schultz, C.G.; Ooi, S.; Goess, C.; Costello, S.P.; Vincent, A.D.; Schoeman, S.; Lim, A.; Bartholomeusz, F.D.; Travis, S.P.L.; et al. Visceral Adipose Tissue Is Associated With Stricturing Crohn’s Disease Behavior, Fecal Calprotectin, and Quality of Life. Inflamm. Bowel Dis. 2019, 25, 592–600. [Google Scholar] [CrossRef]
- Rosenwald, M.; Wolfrum, C. The origin and definition of brite versus white and classical brown adipocytes. Adipocyte 2014, 3, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Kim, W.K.; Bae, K.H. Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J. Stem Cells 2014, 6, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Smorlesi, A.; Frontini, A.; Barbatelli, G.; Cinti, S. White, brown and pink adipocytes: The extraordinary plasticity of the adipose organ. Eur. J. Endocrinol. 2014, 170, R159–R171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.H. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, C.; Ziegler, J.F.; Letizia, M.; Schmidt, F.; Siegmund, B. Adipokines and Their Role in Intestinal Inflammation. Front. Immunol. 2018, 9, 1974. [Google Scholar] [CrossRef] [Green Version]
- Abella, V.; Scotece, M.; Conde, J.; Pino, J.; Gonzalez-Gay, M.A.; Gomez-Reino, J.J.; Mera, A.; Lago, F.; Gomez, R.; Gualillo, O. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017, 13, 100–109. [Google Scholar] [CrossRef]
- Frezza, E.E.; Wachtel, M.S.; Chiriva-Internati, M. Influence of obesity on the risk of developing colon cancer. Gut 2006, 55, 285–291. [Google Scholar] [CrossRef]
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 2009, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilski, J.; Mazur-Bialy, A.; Wojcik, D.; Surmiak, M.; Magierowski, M.; Sliwowski, Z.; Pajdo, R.; Kwiecien, S.; Danielak, A.; Ptak-Belowska, A.; et al. Role of Obesity, Mesenteric Adipose Tissue, and Adipokines in Inflammatory Bowel Diseases. Biomolecules 2019, 9, 780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desreumaux, P.; Ernst, O.; Geboes, K.; Gambiez, L.; Berrebi, D.; Muller-Alouf, H.; Hafraoui, S.; Emilie, D.; Ectors, N.; Peuchmaur, M.; et al. Inflammatory alterations in mesenteric adipose tissue in Crohn’s disease. Gastroenterology 1999, 117, 73–81. [Google Scholar] [CrossRef]
- Sheehan, A.L.; Warren, B.F.; Gear, M.W.; Shepherd, N.A. Fat-wrapping in Crohn’s disease: Pathological basis and relevance to surgical practice. Br. J. Surg. 1992, 79, 955–958. [Google Scholar] [CrossRef]
- Guedj, K.; Abitbol, Y.; Cazals-Hatem, D.; Morvan, M.; Maggiori, L.; Panis, Y.; Bouhnik, Y.; Caligiuri, G.; Corcos, O.; Nicoletti, A. Adipocytes orchestrate the formation of tertiary lymphoid organs in the creeping fat of Crohn’s disease affected mesentery. J. Autoimmun. 2019, 103, 102281. [Google Scholar] [CrossRef]
- Li, P.; Lu, M.; Nguyen, M.T.; Bae, E.J.; Chapman, J.; Feng, D.; Hawkins, M.; Pessin, J.E.; Sears, D.D.; Nguyen, A.K.; et al. Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. J. Biol Chem. 2010, 285, 15333–15345. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Deyoung, S.M.; Bodzin, J.L.; Saltiel, A.R. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 2007, 56, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Molofsky, A.B.; Liang, H.E.; Ricardo-Gonzalez, R.R.; Jouihan, H.A.; Bando, J.K.; Chawla, A.; Locksley, R.M. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011, 332, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisaka, S.; Usui, I.; Bukhari, A.; Ikutani, M.; Oya, T.; Kanatani, Y.; Tsuneyama, K.; Nagai, Y.; Takatsu, K.; Urakaze, M.; et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes 2009, 58, 2574–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisaka, S.; Usui, I.; Kanatani, Y.; Ikutani, M.; Takasaki, I.; Tsuneyama, K.; Tabuchi, Y.; Bukhari, A.; Yamazaki, Y.; Suzuki, H.; et al. Telmisartan improves insulin resistance and modulates adipose tissue macrophage polarization in high-fat-fed mice. Endocrinology 2011, 152, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Lackey, D.E.; Olefsky, J.M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 2016, 12, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.T.; Welch, J.S.; Ricote, M.; Binder, C.J.; Willson, T.M.; Kelly, C.; Witztum, J.L.; Funk, C.D.; Conrad, D.; Glass, C.K. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 1999, 400, 378–382. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Karrasch, T.; Schaeffler, A. Adipokines and the role of visceral adipose tissue in inflammatory bowel disease. Ann. Gastroenterol. 2016, 29, 424–438. [Google Scholar] [CrossRef]
- Batra, A.; Okur, B.; Glauben, R.; Erben, U.; Ihbe, J.; Stroh, T.; Fedke, I.; Chang, H.D.; Zeitz, M.; Siegmund, B. Leptin: A critical regulator of CD4+ T-cell polarization in vitro and in vivo. Endocrinology 2010, 151, 56–62. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Bottcher, C.; Letizia, M.; Yerinde, C.; Wu, H.; Freise, I.; Rodriguez-Sillke, Y.; Stoyanova, A.K.; Kreis, M.E.; Asbach, P.; et al. Leptin induces TNFalpha-dependent inflammation in acquired generalized lipodystrophy and combined Crohn’s disease. Nat. Commun. 2019, 10, 5629. [Google Scholar] [CrossRef] [Green Version]
- Reis, B.S.; Lee, K.; Fanok, M.H.; Mascaraque, C.; Amoury, M.; Cohn, L.B.; Rogoz, A.; Dallner, O.S.; Moraes-Vieira, P.M.; Domingos, A.I.; et al. Leptin receptor signaling in T cells is required for Th17 differentiation. J. Immunol. 2015, 194, 5253–5260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, D.; Zhu, J. Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J. Exp. Med. 2017, 214, 1861–1876. [Google Scholar] [CrossRef]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, A.; Chisanga, D.; Blume, J.; Gloury, R.; Britt, K.; Henstridge, D.C.; Zhan, Y.; Torres, S.V.; Liene, S.; Collins, N.; et al. Sex-specific adipose tissue imprinting of regulatory T cells. Nature 2020, 579, 581–585. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Procaccini, C.; Cali, G.; Pirozzi, G.; Fontana, S.; Zappacosta, S.; La Cava, A.; Matarese, G. A key role of leptin in the control of regulatory T cell proliferation. Immunity 2007, 26, 241–255. [Google Scholar] [CrossRef] [Green Version]
- Singh, U.P.; Singh, N.P.; Guan, H.; Busbee, B.; Price, R.L.; Taub, D.D.; Mishra, M.K.; Fayad, R.; Nagarkatti, M.; Nagarkatti, P.S. Leptin antagonist ameliorates chronic colitis in IL-10(-)/(-) mice. Immunobiology 2013, 218, 1439–1451. [Google Scholar] [CrossRef] [Green Version]
- Grassmann, S.; Wirsching, J.; Eichelmann, F.; Aleksandrova, K. Association Between Peripheral Adipokines and Inflammation Markers: A Systematic Review and Meta-Analysis. Obesity 2017, 25, 1776–1785. [Google Scholar] [CrossRef]
- Lopez-Jaramillo, P.; Gomez-Arbelaez, D.; Lopez-Lopez, J.; Lopez-Lopez, C.; Martinez-Ortega, J.; Gomez-Rodriguez, A.; Triana-Cubillos, S. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Horm. Mol. Biol. Clin. Investig. 2014, 18, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Wulster-Radcliffe, M.C.; Ajuwon, K.M.; Wang, J.; Christian, J.A.; Spurlock, M.E. Adiponectin differentially regulates cytokines in porcine macrophages. Biochem. Biophys. Res. Commun. 2004, 316, 924–929. [Google Scholar] [CrossRef]
- Ouchi, N.; Kihara, S.; Arita, Y.; Okamoto, Y.; Maeda, K.; Kuriyama, H.; Hotta, K.; Nishida, M.; Takahashi, M.; Muraguchi, M.; et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation 2000, 102, 1296–1301. [Google Scholar] [CrossRef]
- Kumada, M.; Kihara, S.; Ouchi, N.; Kobayashi, H.; Okamoto, Y.; Ohashi, K.; Maeda, K.; Nagaretani, H.; Kishida, K.; Maeda, N.; et al. Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation 2004, 109, 2046–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, A.; Chumanevich, A.; Fletcher, E.; Larsen, B.; Lattwein, K.; Kaur, K.; Fayad, R. Adiponectin deficiency: Role in chronic inflammation induced colon cancer. Biochim. Biophys. Acta 2012, 1822, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fayad, R.; Pini, M.; Sennello, J.A.; Cabay, R.J.; Chan, L.; Xu, A.; Fantuzzi, G. Adiponectin deficiency protects mice from chemically induced colonic inflammation. Gastroenterology 2007, 132, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kiyohara, T.; Murayama, Y.; Kihara, S.; Okamoto, Y.; Funahashi, T.; Ito, T.; Nezu, R.; Tsutsui, S.; Miyagawa, J.I.; et al. Production of adiponectin, an anti-inflammatory protein, in mesenteric adipose tissue in Crohn’s disease. Gut 2005, 54, 789–796. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Beales, I.L. Adiponectin stimulates proliferation and cytokine secretion in colonic epithelial cells. Regul. Pept. 2006, 134, 105–113. [Google Scholar] [CrossRef]
- Cioffi, J.A.; Shafer, A.W.; Zupancic, T.J.; Smith-Gbur, J.; Mikhail, A.; Platika, D.; Snodgrass, H.R. Novel B219/OB receptor isoforms: Possible role of leptin in hematopoiesis and reproduction. Nat. Med. 1996, 2, 585–589. [Google Scholar] [CrossRef]
- Scotece, M.; Conde, J.; Lopez, V.; Lago, F.; Pino, J.; Gomez-Reino, J.J.; Gualillo, O. Adiponectin and leptin: New targets in inflammation. Basic Clin. Pharmacol. Toxicol. 2014, 114, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Mackey-Lawrence, N.M.; Petri, W.A., Jr. Leptin and mucosal immunity. Mucosal Immunol. 2012, 5, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Santos-Alvarez, J.; Goberna, R.; Sanchez-Margalet, V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell Immunol. 1999, 194, 6–11. [Google Scholar] [CrossRef]
- Sanchez-Margalet, V.; Martin-Romero, C.; Gonzalez-Yanes, C.; Goberna, R.; Rodriguez-Bano, J.; Muniain, M.A. Leptin receptor (Ob-R) expression is induced in peripheral blood mononuclear cells by in vitro activation and in vivo in HIV-infected patients. Clin. Exp. Immunol. 2002, 129, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.K.; Lord, G.M.; Matarese, G.; Vendetti, S.; Ghatei, M.A.; Ritter, M.A.; Lechler, R.I.; Bloom, S.R. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J. Clin. Investig. 1999, 104, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Liu, Y.; Shi, F.D.; Zou, H.; Matarese, G.; La Cava, A. Cutting edge: Leptin-induced RORgammat expression in CD4+ T cells promotes Th17 responses in systemic lupus erythematosus. J. Immunol. 2013, 190, 3054–3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genser, L.; Aguanno, D.; Soula, H.A.; Dong, L.; Trystram, L.; Assmann, K.; Salem, J.E.; Vaillant, J.C.; Oppert, J.M.; Laugerette, F.; et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J. Pathol. 2018, 246, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, B.; Lehr, H.A.; Fantuzzi, G. Leptin: A pivotal mediator of intestinal inflammation in mice. Gastroenterology 2002, 122, 2011–2025. [Google Scholar] [CrossRef]
- Paul, G.; Schaffler, A.; Neumeier, M.; Furst, A.; Bataillle, F.; Buechler, C.; Muller-Ladner, U.; Scholmerich, J.; Rogler, G.; Herfarth, H. Profiling adipocytokine secretion from creeping fat in Crohn’s disease. Inflamm. Bowel Dis. 2006, 12, 471–477. [Google Scholar] [CrossRef]
- Barbier, M.; Vidal, H.; Desreumaux, P.; Dubuquoy, L.; Bourreille, A.; Colombel, J.F.; Cherbut, C.; Galmiche, J.P. Overexpression of leptin mRNA in mesenteric adipose tissue in inflammatory bowel diseases. Gastroenterol. Clin. Biol. 2003, 27, 987–991. [Google Scholar] [CrossRef]
- Barbier, M.; Cherbut, C.; Aube, A.C.; Blottiere, H.M.; Galmiche, J.P. Elevated plasma leptin concentrations in early stages of experimental intestinal inflammation in rats. Gut 1998, 43, 783–790. [Google Scholar] [CrossRef]
- Tuzun, A.; Uygun, A.; Yesilova, Z.; Ozel, A.M.; Erdil, A.; Yaman, H.; Bagci, S.; Gulsen, M.; Karaeren, N.; Dagalp, K. Leptin levels in the acute stage of ulcerative colitis. J. Gastroenterol. Hepatol. 2004, 19, 429–432. [Google Scholar] [CrossRef]
- Karmiris, K.; Koutroubakis, I.E.; Xidakis, C.; Polychronaki, M.; Voudouri, T.; Kouroumalis, E.A. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 100–105. [Google Scholar] [CrossRef]
- Sitaraman, S.; Liu, X.; Charrier, L.; Gu, L.H.; Ziegler, T.R.; Gewirtz, A.; Merlin, D. Colonic leptin: Source of a novel proinflammatory cytokine involved in IBD. FASEB J. 2004, 18, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Masri, B.; Daviaud, D.; Gesta, S.; Guigne, C.; Mazzucotelli, A.; Castan-Laurell, I.; Tack, I.; Knibiehler, B.; Carpene, C.; et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology 2005, 146, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Bing, C.; Wood, I.S. Adipose tissue and adipokines—Energy regulation from the human perspective. J. Nutr. 2006, 136, 1935S–1939S. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Wang, G.; Qiu, S.; de la Motte, C.; Wang, H.Q.; Gomez, G.; Englander, E.W.; Greeley, G.H., Jr. Increased colonic apelin production in rodents with experimental colitis and in humans with IBD. Regul. Pept. 2007, 142, 131–137. [Google Scholar] [CrossRef]
- O’Carroll, A.M.; Lolait, S.J.; Harris, L.E.; Pope, G.R. The apelin receptor APJ: Journey from an orphan to a multifaceted regulator of homeostasis. J. Endocrinol. 2013, 219, R13–R35. [Google Scholar] [CrossRef]
- Kwon, H.B.; Wang, S.; Helker, C.S.; Rasouli, S.J.; Maischein, H.M.; Offermanns, S.; Herzog, W.; Stainier, D.Y. In vivo modulation of endothelial polarization by Apelin receptor signalling. Nat. Commun. 2016, 7, 11805. [Google Scholar] [CrossRef] [Green Version]
- Berta, J.; Hoda, M.A.; Laszlo, V.; Rozsas, A.; Garay, T.; Torok, S.; Grusch, M.; Berger, W.; Paku, S.; Renyi-Vamos, F.; et al. Apelin promotes lymphangiogenesis and lymph node metastasis. Oncotarget 2014, 5, 4426–4437. [Google Scholar] [CrossRef] [Green Version]
- Sawane, M.; Kidoya, H.; Muramatsu, F.; Takakura, N.; Kajiya, K. Apelin attenuates UVB-induced edema and inflammation by promoting vessel function. Am. J. Pathol. 2011, 179, 2691–2697. [Google Scholar] [CrossRef]
- Masoud, A.G.; Lin, J.; Azad, A.K.; Farhan, M.A.; Fischer, C.; Zhu, L.F.; Zhang, H.; Sis, B.; Kassiri, Z.; Moore, R.B.; et al. Apelin directs endothelial cell differentiation and vascular repair following immune-mediated injury. J. Clin. Investig. 2020, 130, 94–107. [Google Scholar] [CrossRef]
- Jiang, S.; Park, D.W.; Tadie, J.M.; Gregoire, M.; Deshane, J.; Pittet, J.F.; Abraham, E.; Zmijewski, J.W. Human resistin promotes neutrophil proinflammatory activation and neutrophil extracellular trap formation and increases severity of acute lung injury. J. Immunol. 2014, 192, 4795–4803. [Google Scholar] [CrossRef] [Green Version]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Kaser, S.; Kaser, A.; Sandhofer, A.; Ebenbichler, C.F.; Tilg, H.; Patsch, J.R. Resistin messenger-RNA expression is increased by proinflammatory cytokines in vitro. Biochem. Biophys. Res. Commun. 2003, 309, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Al-Suhaimi, E.A.; Shehzad, A. Leptin, resistin and visfatin: The missing link between endocrine metabolic disorders and immunity. Eur. J. Med. Res. 2013, 18, 12. [Google Scholar] [CrossRef] [Green Version]
- Konrad, A.; Lehrke, M.; Schachinger, V.; Seibold, F.; Stark, R.; Ochsenkuhn, T.; Parhofer, K.G.; Goke, B.; Broedl, U.C. Resistin is an inflammatory marker of inflammatory bowel disease in humans. Eur. J. Gastroenterol. Hepatol. 2007, 19, 1070–1074. [Google Scholar] [CrossRef]
- Karmiris, K.; Koutroubakis, I.E.; Xidakis, C.; Polychronaki, M.; Kouroumalis, E.A. The effect of infliximab on circulating levels of leptin, adiponectin and resistin in patients with inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2007, 19, 789–794. [Google Scholar] [CrossRef]
- Bokarewa, M.; Nagaev, I.; Dahlberg, L.; Smith, U.; Tarkowski, A. Resistin, an adipokine with potent proinflammatory properties. J. Immunol. 2005, 174, 5789–5795. [Google Scholar] [CrossRef]
- Nishihara, T.; Matsuda, M.; Araki, H.; Oshima, K.; Kihara, S.; Funahashi, T.; Shimomura, I. Effect of adiponectin on murine colitis induced by dextran sulfate sodium. Gastroenterology 2006, 131, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Arsenescu, V.; Narasimhan, M.L.; Halide, T.; Bressan, R.A.; Barisione, C.; Cohen, D.A.; de Villiers, W.J.; Arsenescu, R. Adiponectin and plant-derived mammalian adiponectin homolog exert a protective effect in murine colitis. Dig. Dis. Sci. 2011, 56, 2818–2832. [Google Scholar] [CrossRef]
- Ge, Y.; Li, Y.; Chen, Q.; Zhu, W.; Zuo, L.; Guo, Z.; Gong, J.; Cao, L.; Gu, L.; Li, J. Adipokine apelin ameliorates chronic colitis in Il-10(-/-) mice by promoting intestinal lymphatic functions. Biochem. Pharmacol. 2018, 148, 202–212. [Google Scholar] [CrossRef]
- Singh, S.; Heien, H.C.; Sangaralingham, L.R.; Schilz, S.R.; Kappelman, M.D.; Shah, N.D.; Loftus, E.V., Jr. Comparative Effectiveness and Safety of Anti-Tumor Necrosis Factor Agents in Biologic-Naive Patients With Crohn’s Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 1120–1129.e6. [Google Scholar] [CrossRef] [Green Version]
- Dotan, I.; Ron, Y.; Yanai, H.; Becker, S.; Fishman, S.; Yahav, L.; Ben Yehoyada, M.; Mould, D.R. Patient factors that increase infliximab clearance and shorten half-life in inflammatory bowel disease: A population pharmacokinetic study. Inflamm. Bowel Dis. 2014, 20, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Rosario, M.; Dirks, N.L.; Gastonguay, M.R.; Fasanmade, A.A.; Wyant, T.; Parikh, A.; Sandborn, W.J.; Feagan, B.G.; Reinisch, W.; Fox, I. Population pharmacokinetics-pharmacodynamics of vedolizumab in patients with ulcerative colitis and Crohn’s disease. Aliment. Pharmacol. Ther. 2015, 42, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Klaasen, R.; Wijbrandts, C.A.; Gerlag, D.M.; Tak, P.P. Body mass index and clinical response to infliximab in rheumatoid arthritis. Arthritis Rheum. 2011, 63, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Eder, L.; Thavaneswaran, A.; Chandran, V.; Cook, R.J.; Gladman, D.D. Obesity is associated with a lower probability of achieving sustained minimal disease activity state among patients with psoriatic arthritis. Ann. Rheum. Dis. 2015, 74, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Luck, H.; Tsai, S.; Winer, S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab. 2016, 23, 413–426. [Google Scholar] [CrossRef] [Green Version]
- Mudter, J.; Neurath, M.F. Il-6 signaling in inflammatory bowel disease: Pathophysiological role and clinical relevance. Inflamm. Bowel Dis. 2007, 13, 1016–1023. [Google Scholar] [CrossRef]
- Johnson, A.M.; Costanzo, A.; Gareau, M.G.; Armando, A.M.; Quehenberger, O.; Jameson, J.M.; Olefsky, J.M. High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS ONE 2015, 10, e0122195. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, Y.-W.; Fu, S.-H.; Dong, J.-L.; Chien, M.-W.; Liu, Y.-W.; Hsu, C.-Y.; Sytwu, H.-K. Adipokine-Modulated Immunological Homeostasis Shapes the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 9564. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249564
Tsai Y-W, Fu S-H, Dong J-L, Chien M-W, Liu Y-W, Hsu C-Y, Sytwu H-K. Adipokine-Modulated Immunological Homeostasis Shapes the Pathophysiology of Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2020; 21(24):9564. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249564
Chicago/Turabian StyleTsai, Yi-Wen, Shin-Huei Fu, Jia-Ling Dong, Ming-Wei Chien, Yu-Wen Liu, Chao-Yuan Hsu, and Huey-Kang Sytwu. 2020. "Adipokine-Modulated Immunological Homeostasis Shapes the Pathophysiology of Inflammatory Bowel Disease" International Journal of Molecular Sciences 21, no. 24: 9564. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249564