Urea-Peptide Hybrids as VEGF-A165/NRP-1 Complex Inhibitors with Improved Receptor Affinity and Biological Properties

 , , and
, , and 
Abstract
:1. Introduction
2. Results and Discussion
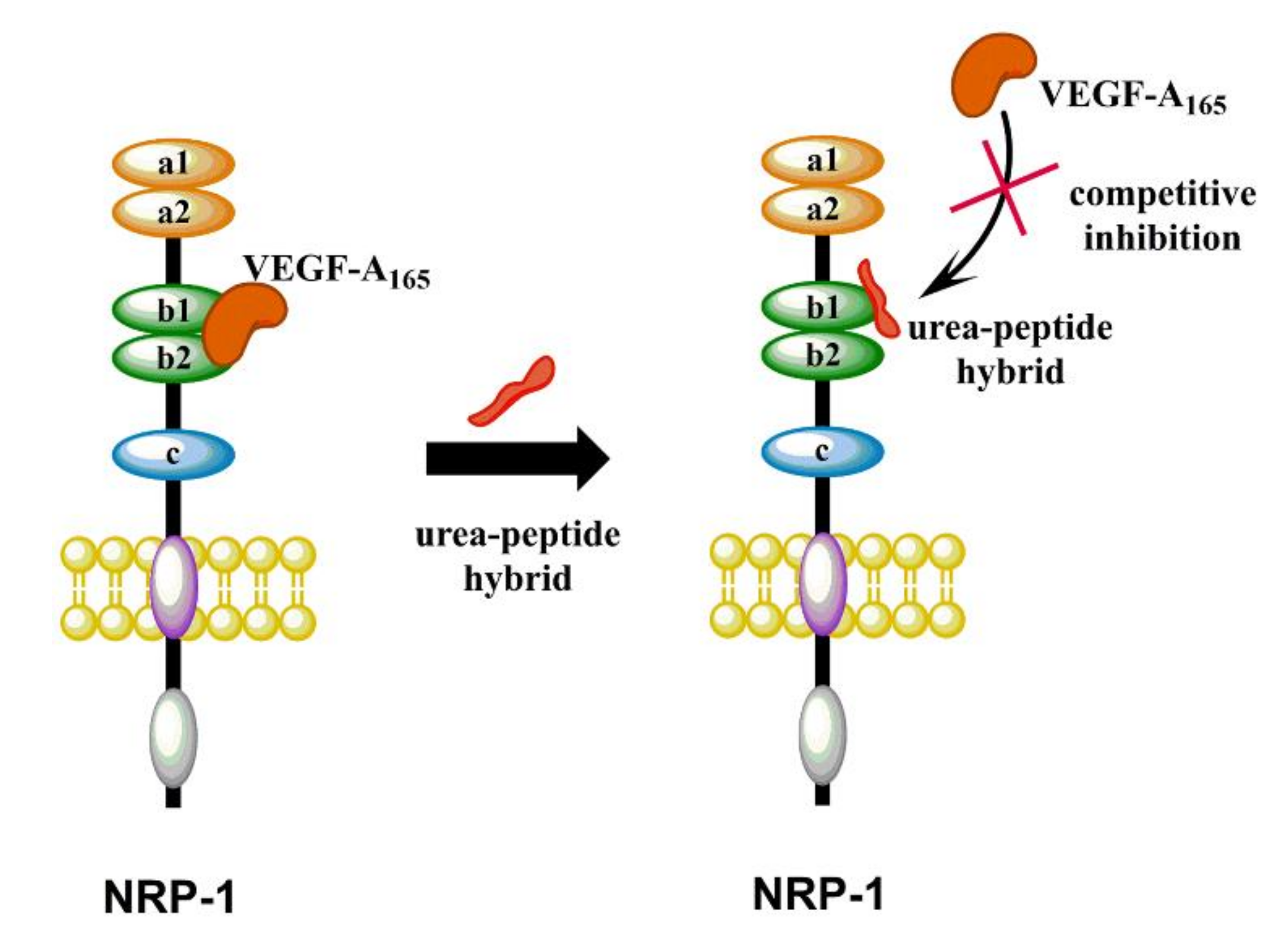
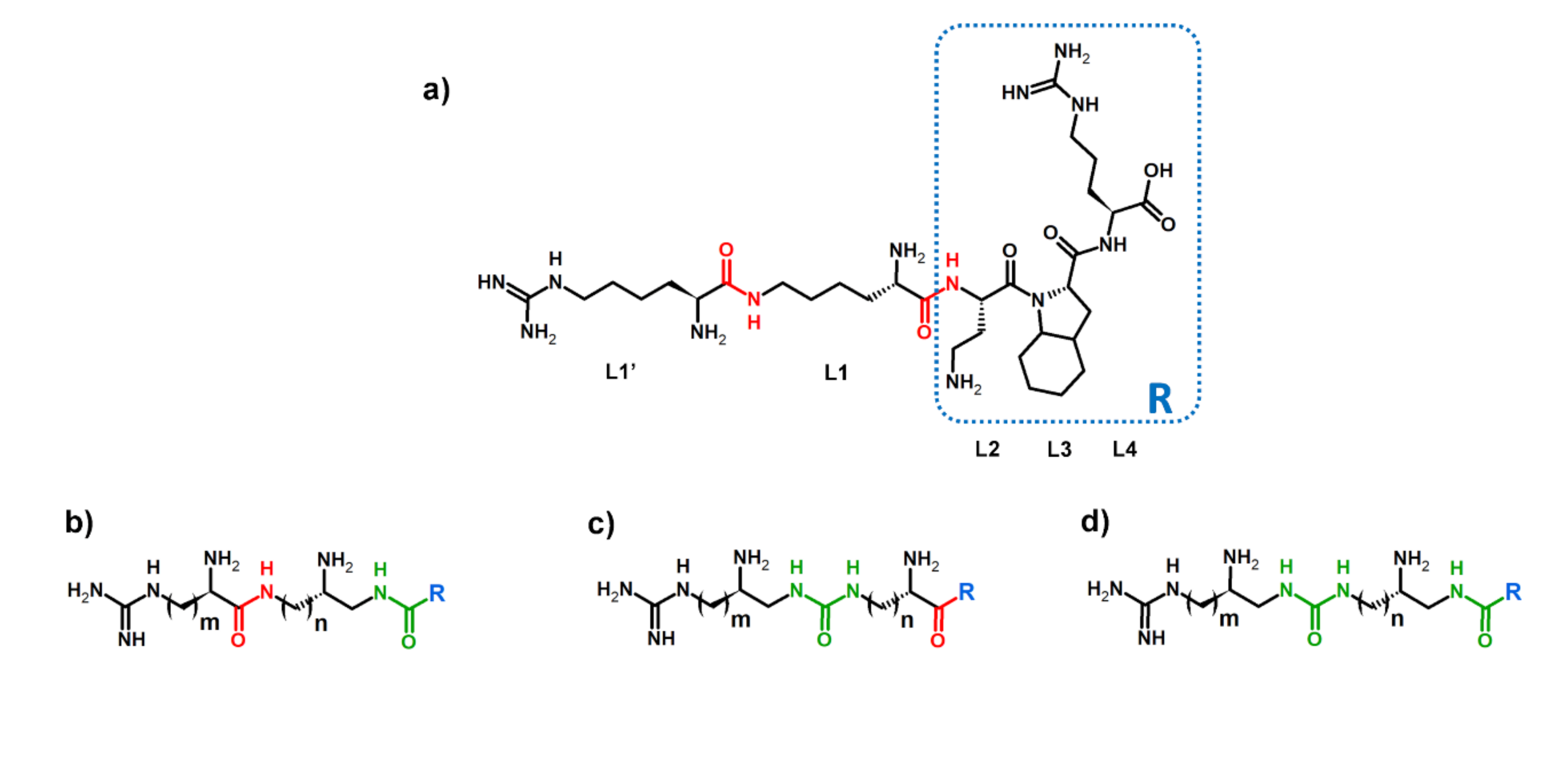
2.1. Design Strategy
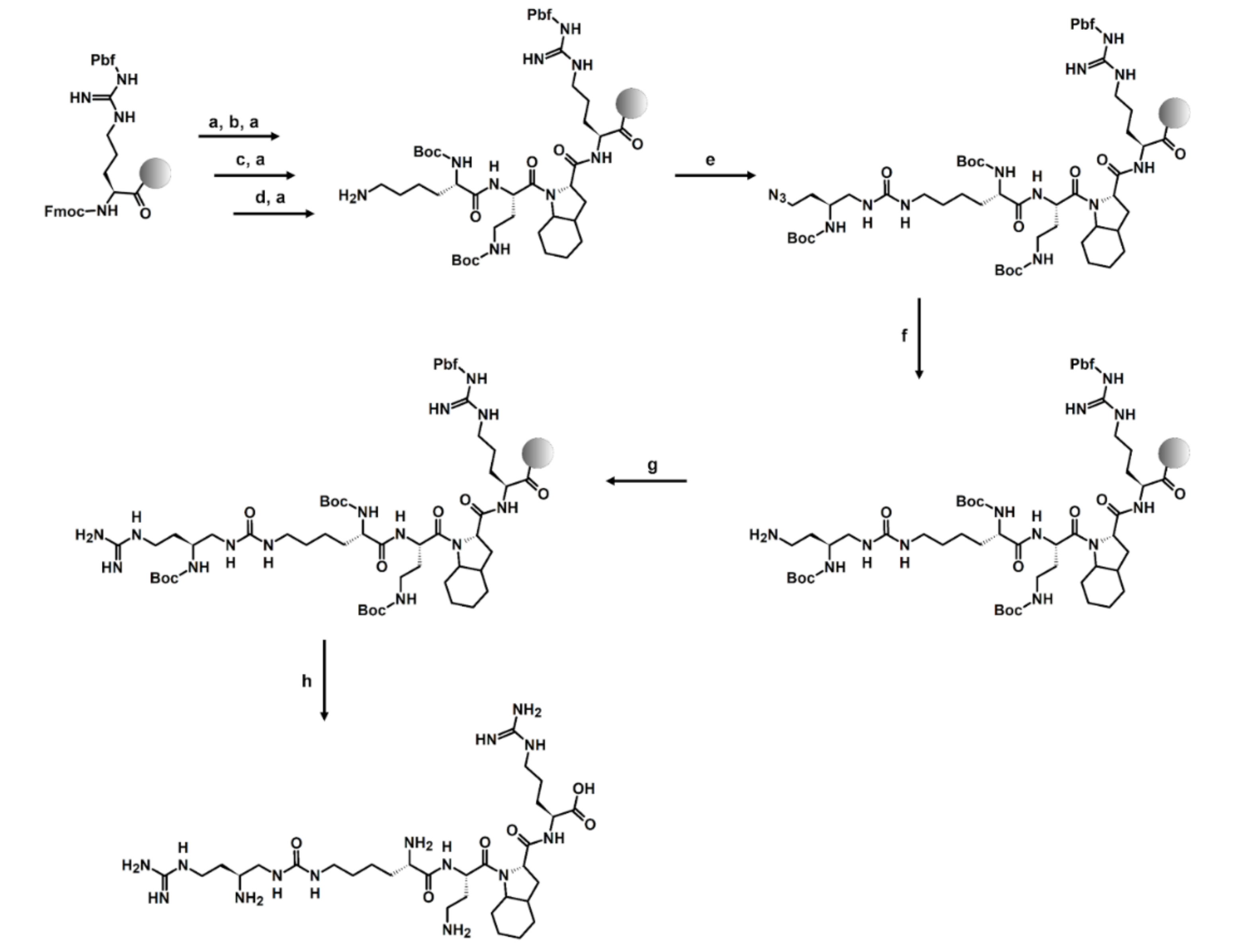
2.2. Synthesis
2.3. Receptor Binding Studies
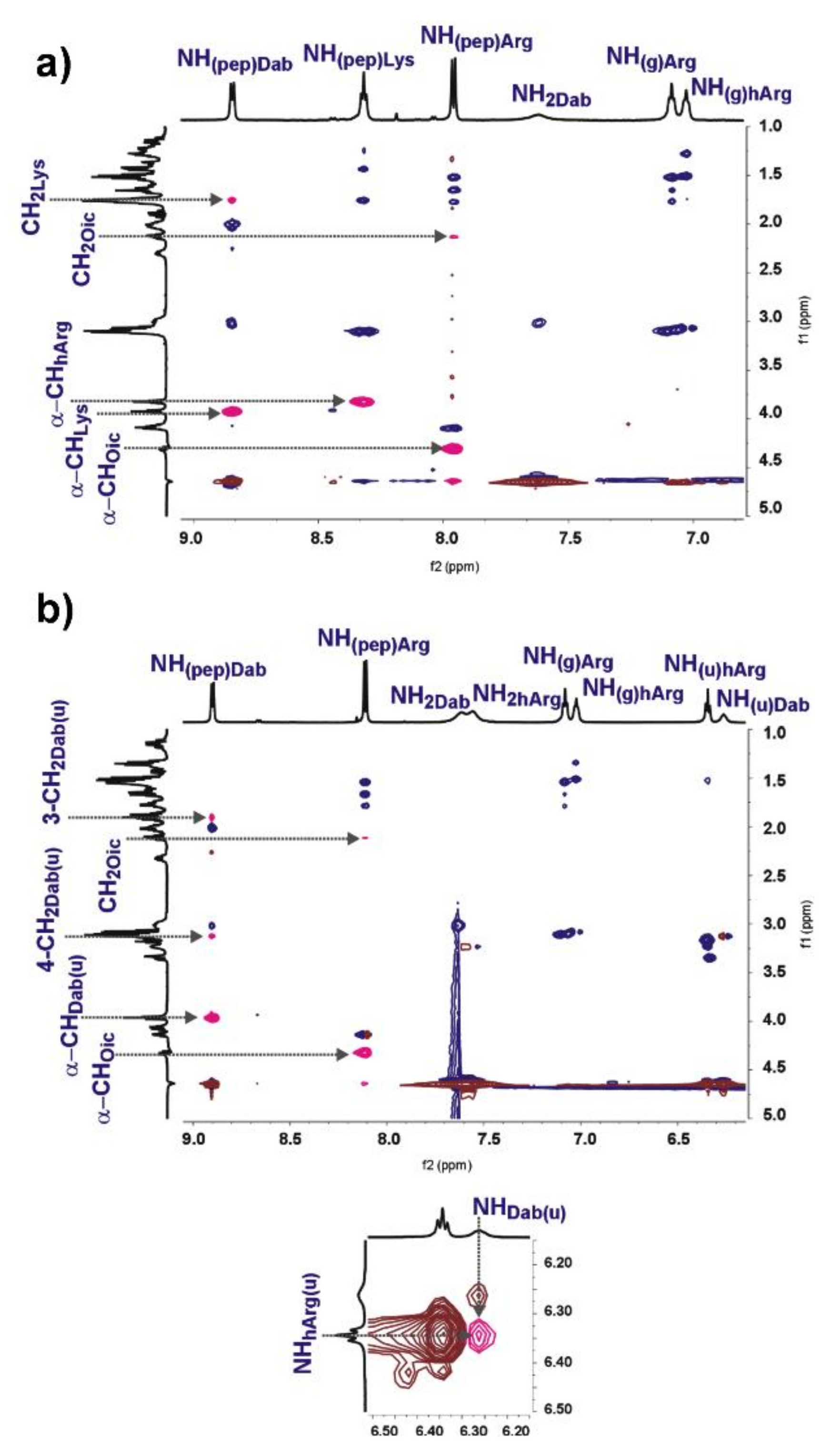
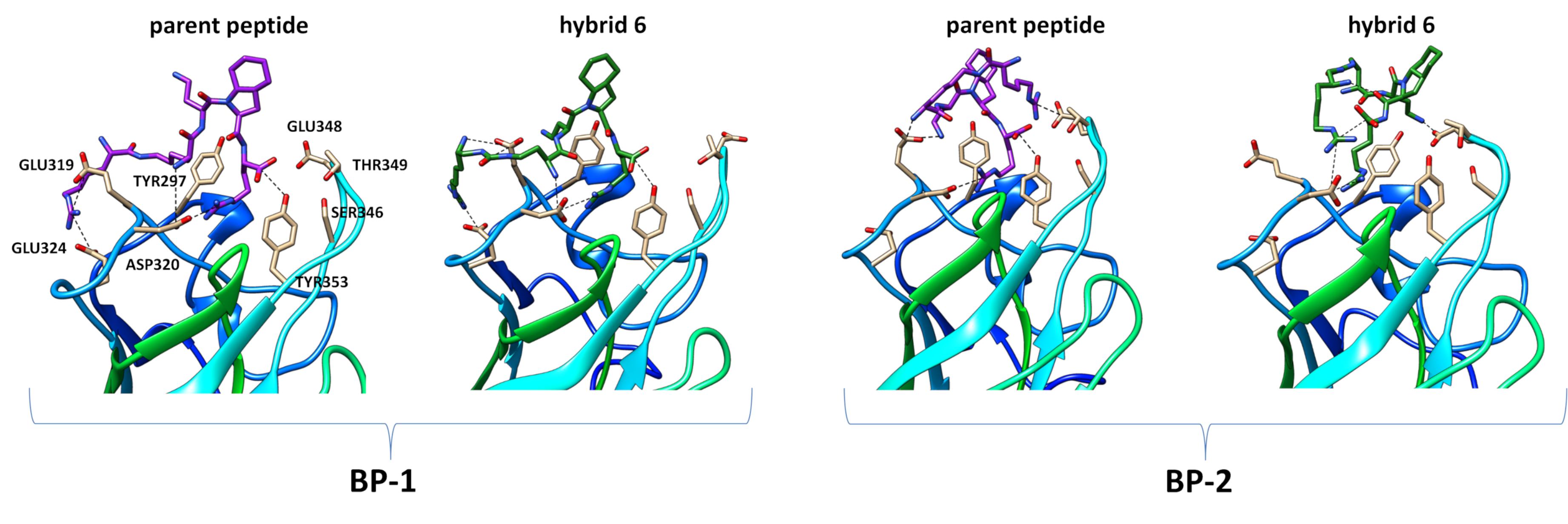
2.4. 2D NMR and Computational Study
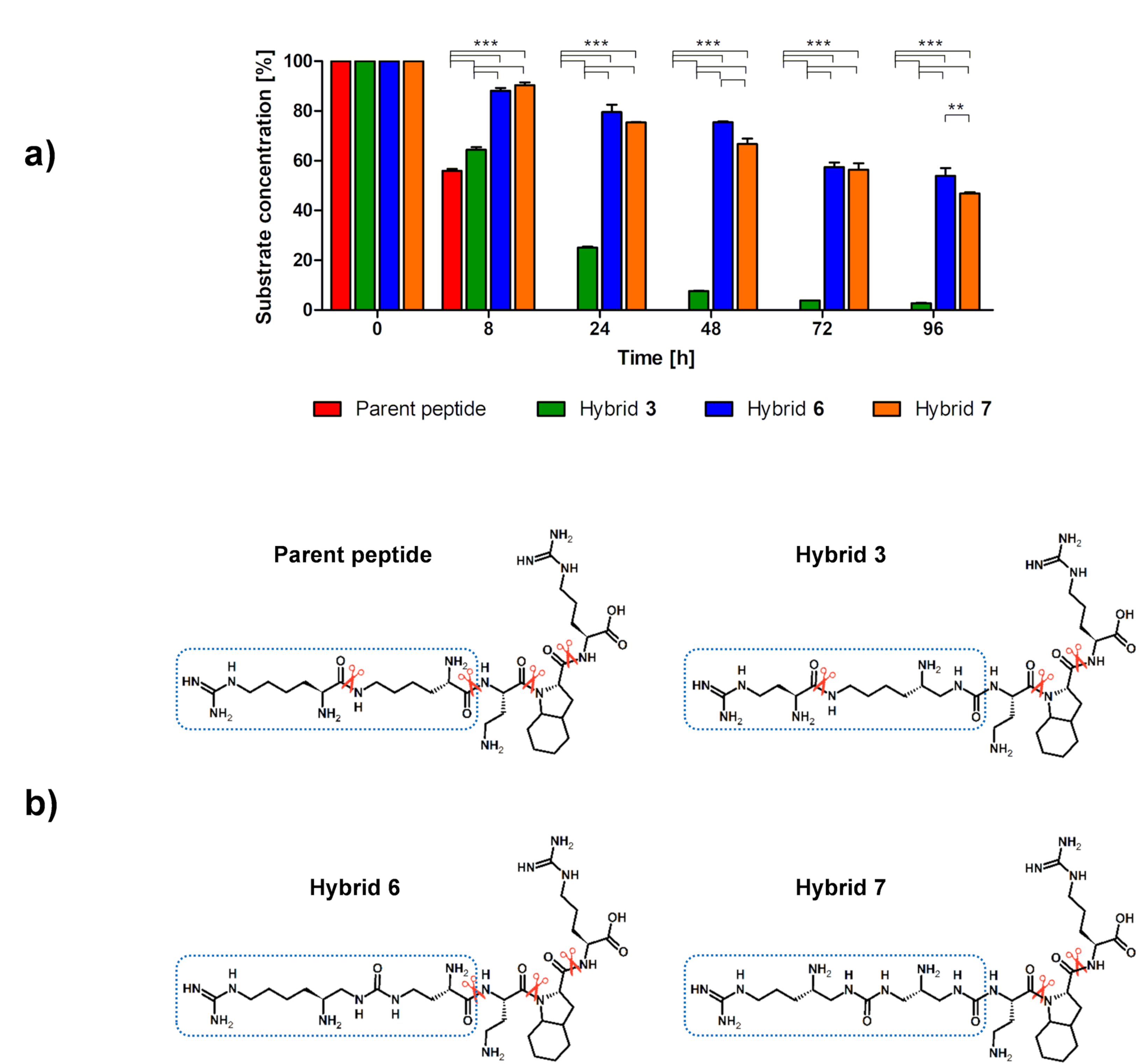
2.5. In Vitro Enzymatic Stability
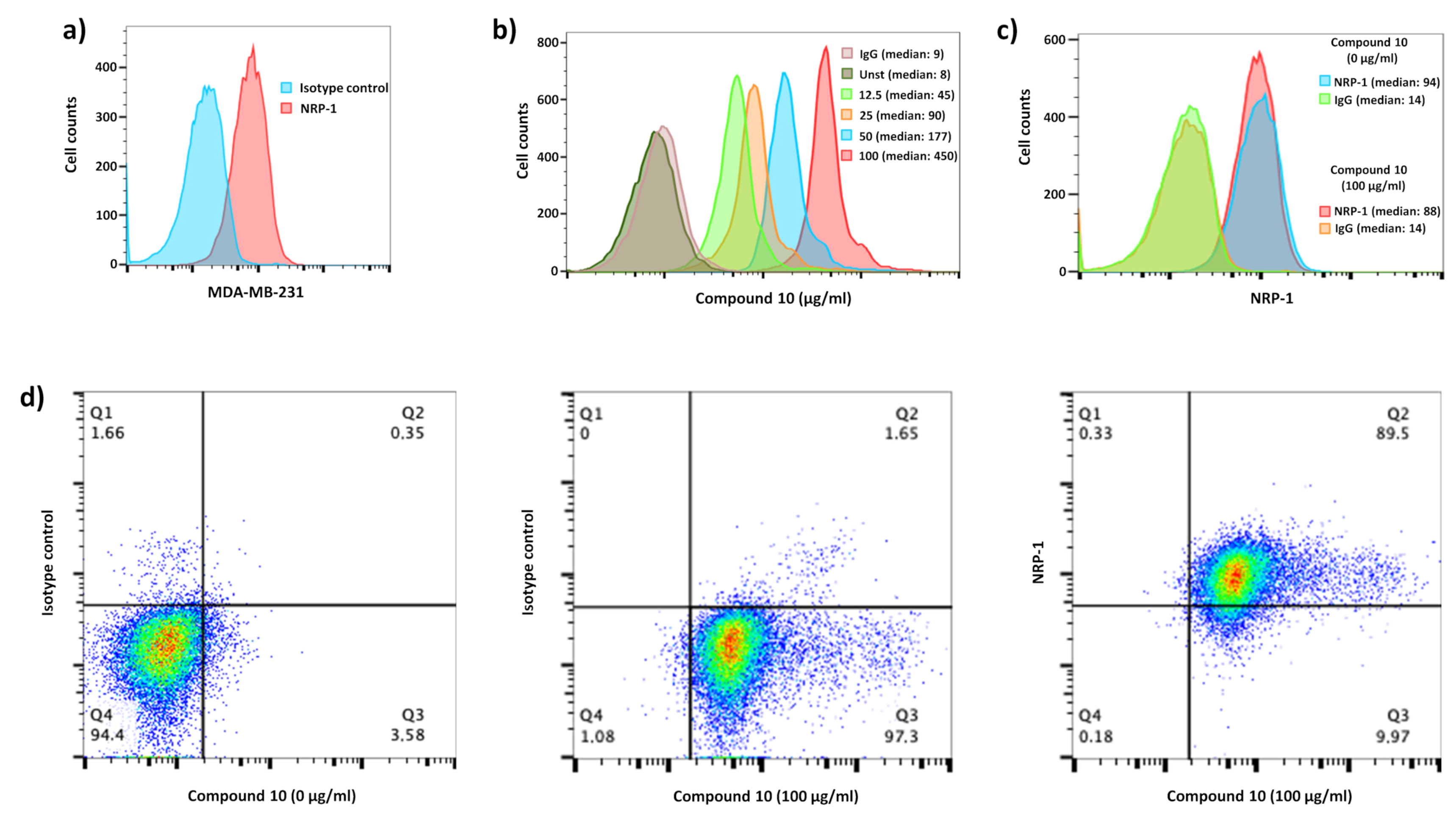
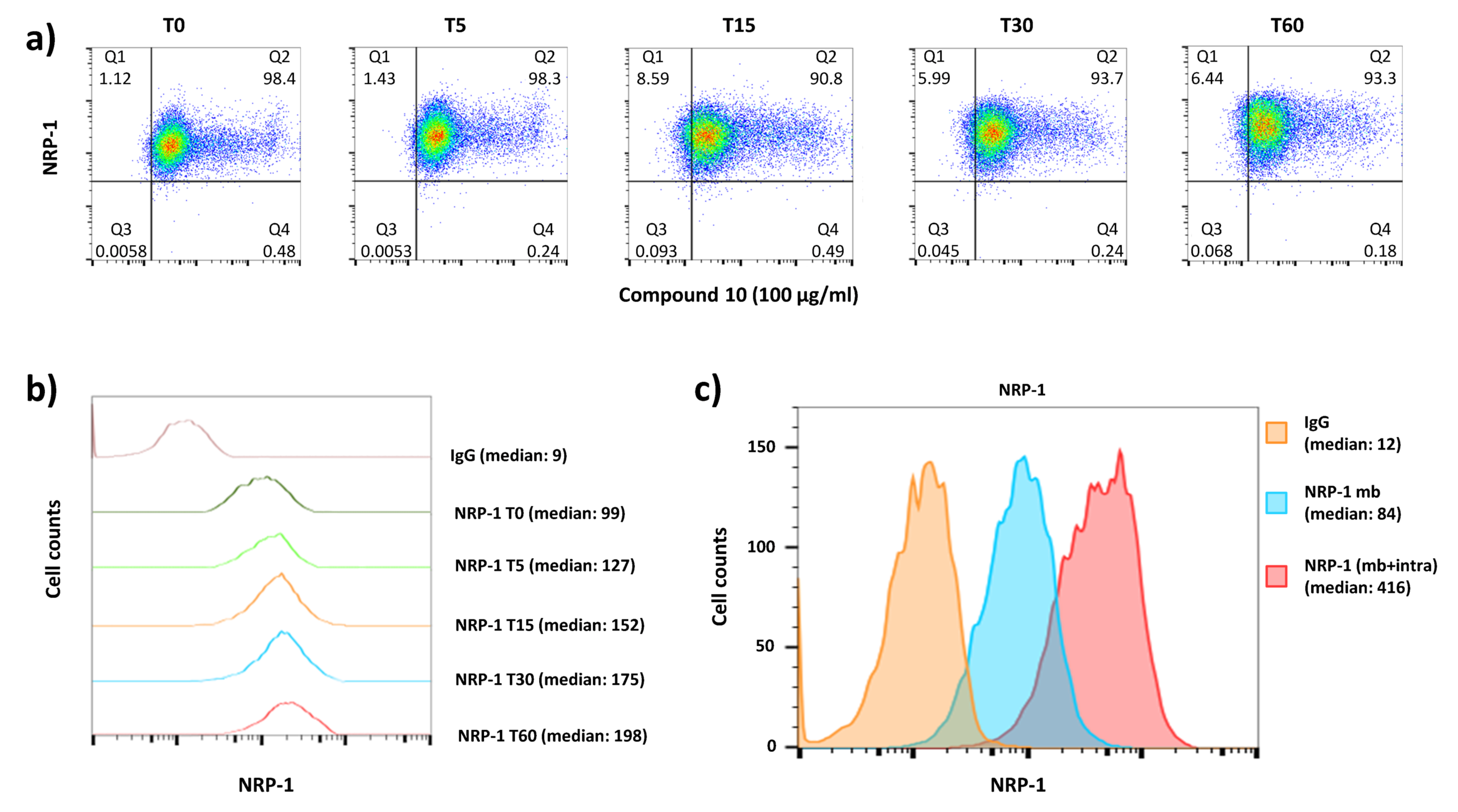
2.6. Binding Assays on Cells
3. Materials and Methods
3.1. Materials
3.2. Succinimidyl Carbamate Building Blocks Synthesis
3.3. Competitive Receptor Binding Assays
3.4. 2D NMR Spectroscopy
3.5. Molecular Dynamic Studies
3.6. Blood Collection and Serum Preparation
3.7. In Vitro Enzymatic Stability Assay
3.8. Cell Culture
3.9. Flow Cytometry Analysis of Membrane and Intracytoplasmic NRP-1 Expression
3.10. Labelled Urea–Peptide Hybrid Binding Assays at 4 °C
3.11. Labelled Urea–Peptide Hybrid Binding Assays at 37 °C
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| anti-Tfr | anti-human transferrin receptor |
| 5/6-FAM | 5/6-carboxyfluorescein |
| Boc | tert-butoxycarbonyl |
| BSA | bovine serum albumin |
| bt | linear dichroism |
| Dab | 2,4-diaminobutyric acid |
| Dap | 2,3-diaminopropionic acid |
| DIPEA | N,N-diisopropylethylamine |
| ELISA | enzyme-linked immunosorbent assay |
| FACS | fluorescence-activated cell sorting |
| FCS | foetal calf serum |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| hArg | homoarginine |
| HCTU | O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| NRP | neuropilin |
| Oic | octahydroindole-2-carboxylic acid |
| PBS | phosphate buffer saline |
| SAR | structure–activity relationship |
| strep-HRP | streptavidin–horseradish peroxidase conjugate |
| SWATH | sequential window acquisition of all theoretical mass spectra |
| TIS | triisopropylsilane |
| TFA | trifluoroacetic acid |
| VEGF | vascular endothelial grow factor |
References
- Ferrara, N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am. J. Physiol. Cell Physiol. 2001, 280, C1358–C1366. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Ellis, L.M. Role of the Vascular Endothelial Growth Factor Pathway in Tumor Growth and Angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Bergantino, F.; Guariniello, S.; Raucci, R.; Colonna, G.; De Luca, A.; Normanno, N.; Costantini, S. Structure–fluctuation–function relationships of seven pro-angiogenic isoforms of VEGFA, important mediators of tumorigenesis. Biochim. Biophys. Acta 2015, 1854, 410–425. [Google Scholar] [CrossRef]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 Is Expressed by Endothelial and Tumor Cells as an Isoform-Specific Receptor for Vascular Endothelial Growth Factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Koch, S. Neuropilin signalling in angiogenesis. Biochem. Soc. Trans. 2012, 40, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Djordjevic, S.; Driscoll, P.C. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov. Today 2013, 18, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, G.; Cohen, T.; Shraga, N.; Lange, T.; Kessler, O.; Herzog, Y. The Neuropilins: Multifunctional Semaphorin and VEGF Receptors that Modulate Axon Guidance and Angiogenesis. Trends Cardiovasc. Med. 2002, 12, 13–19. [Google Scholar] [CrossRef]
- Roy, S.; Bag, A.K.; Singh, R.K.; Talmadge, J.E.; Batra, S.K.; Datta, K. Multifaceted Role of Neuropilins in the Immune System: Potential Targets for Immunotherapy. Front. Immunol. 2017, 8, 1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tordjman, R.; Lepelletier, Y.; Lemarchandel, V.; Cambot, M.; Gaulard, P.; Hermine, O.; Roméo, P.-H. A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat. Immunol. 2002, 3, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Latil, A.; Bièche, I.; Pesche, S.; Valéri, A.; Fournier, G.; Cussenot, O.; Lidereau, R. VEGF overexpression in clinically localized prostate tumors and neuropilin-1 overexpression in metastatic forms. Int. J. Cancer 2000, 89, 167–171. [Google Scholar] [CrossRef]
- Stephenson, J.M.; Banerjee, S.; Saxena, N.K.; Cherian, R.; Banerjee, S.K. Neuropilin-1 is differentially expressed in myoepithelial cells and vascular smooth muscle cells in preneoplastic and neoplastic human breast: A possible marker for the progression of breast cancer. Int. J. Cancer 2002, 101, 409–414. [Google Scholar] [CrossRef]
- Parikh, A.A.; Liu, W.B.; Fan, F.; Stoeltzing, O.; Reinmuth, N.; Bruns, C.J.; Bucana, C.D.; Evans, D.B.; Ellis, L.M. Expression and regulation of the novel vascular endothelial growth factor receptor neuropilin-1 by epidermal growth factor in human pancreatic carcinoma. Cancer 2003, 98, 720–729. [Google Scholar] [CrossRef]
- Parikh, A.A.; Fan, F.; Liu, W.B.; Ahmad, S.A.; Stoeltzing, O.; Reinmuth, N.; Bielenberg, D.; Bucana, C.D.; Klagsbrun, M.; Ellis, L.M. Neuropilin-1 in Human Colon Cancer: Expression, Regulation, and Role in Induction of Angiogenesis. Am. J. Pathol. 2004, 164, 2139–2151. [Google Scholar] [CrossRef]
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Ausprunk, D.H.; Folkman, J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 1977, 14, 53–65. [Google Scholar] [CrossRef]
- Grandclement, C.; Borg, C. Neuropilins: A New Target for Cancer Therapy. Cancers 2011, 3, 1899–1928. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Ware, J.A. Therapeutic angiogenesis in cardiovascular disease. Nat. Rev. Drug Discov. 2003, 2, 863–872. [Google Scholar] [CrossRef]
- Marina, Z.; Sandra, D.; Lucia, M. Development of New Drugs in Angiogenesis. Curr. Drug Targets 2004, 5, 485–493. [Google Scholar] [CrossRef]
- Jarvis, A.; Allerston, C.K.; Jia, H.; Herzog, B.; Garza-Garcia, A.; Winfield, N.; Ellard, K.; Aqil, R.; Lynch, R.; Chapman, C.; et al. Small Molecule Inhibitors of the Neuropilin-1 Vascular Endothelial Growth Factor A (VEGF-A) Interaction. J. Med. Chem. 2010, 53, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Novoa, A.; Pellegrini-Moïse, N.; Bechet, D.; Barberi-Heyob, M.; Chapleur, Y. Sugar-based peptidomimetics as potential inhibitors of the vascular endothelium growth factor binding to neuropilin-1. Biorg. Med. Chem. 2010, 18, 3285–3298. [Google Scholar] [CrossRef] [PubMed]
- Borriello, L.; Montès, M.; Lepelletier, Y.; Leforban, B.; Liu, W.-Q.; Demange, L.; Delhomme, B.; Pavoni, S.; Jarray, R.; Boucher, J.L.; et al. Structure-based discovery of a small non-peptidic Neuropilins antagonist exerting in vitro and in vivo anti-tumor activity on breast cancer model. Cancer Lett. 2014, 349, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Starzec, A.; Miteva, M.A.; Ladam, P.; Villoutreix, B.O.; Perret, G.Y. Discovery of novel inhibitors of vascular endothelial growth factor-A–Neuropilin-1 interaction by structure-based virtual screening. Biorg. Med. Chem. 2014, 22, 4042–4048. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-Q.; Megale, V.; Borriello, L.; Leforban, B.; Montès, M.; Goldwaser, E.; Gresh, N.; Piquemal, J.-P.; Hadj-Slimane, R.; Hermine, O.; et al. Synthesis and structure–activity relationship of non-peptidic antagonists of neuropilin-1 receptor. Bioorg. Med. Chem. Lett. 2014, 24, 4254–4259. [Google Scholar] [CrossRef]
- Liu, W.-Q.; Lepelletier, Y.; Montès, M.; Borriello, L.; Jarray, R.; Grépin, R.; Leforban, B.; Loukaci, A.; Benhida, R.; Hermine, O.; et al. NRPa-308, a new neuropilin-1 antagonist, exerts in vitro anti-angiogenic and anti-proliferative effects and in vivo anti-cancer effects in a mouse xenograft model. Cancer Lett. 2018, 414, 88–98. [Google Scholar] [CrossRef]
- Powell, J.; Mota, F.; Steadman, D.; Soudy, C.; Miyauchi, J.T.; Crosby, S.; Jarvis, A.; Reisinger, T.; Winfield, N.; Evans, G.; et al. Small Molecule Neuropilin-1 Antagonists Combine Antiangiogenic and Antitumor Activity with Immune Modulation through Reduction of Transforming Growth Factor Beta (TGFβ) Production in Regulatory T-Cells. J. Med. Chem. 2018, 61, 4135–4154. [Google Scholar] [CrossRef]
- Brachet, E.; Dumond, A.; Liu, W.-Q.; Fabre, M.; Selkti, M.; Raynaud, F.; Hermine, O.; Benhida, R.; Belmont, P.; Garbay, C.; et al. Synthesis, 3D-structure and stability analyses of NRPa-308, a new promising anti-cancer agent. Bioorg. Med. Chem. Lett. 2019, 29, 126710. [Google Scholar] [CrossRef] [Green Version]
- Binétruy-Tournaire, R.; Demangel, C.; Malavaud, B.; Vassy, R.; Rouyre, S.; Kraemer, M.; Plouët, J.; Derbin, C.; Perret, G.; Mazié, J.C. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000, 19, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- von Wronski, M.A.; Raju, N.; Pillai, R.; Bogdan, N.J.; Marinelli, E.R.; Nanjappan, P.; Ramalingam, K.; Arunachalam, T.; Eaton, S.; Linder, K.E.; et al. Tuftsin Binds Neuropilin-1 through a Sequence Similar to That Encoded by Exon 8 of Vascular Endothelial Growth Factor. J. Biol. Chem. 2006, 281, 5702–5710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starzec, A.; Vassy, R.; Martin, A.; Lecouvey, M.; Di Benedetto, M.; Crépin, M.; Perret, G.Y. Antiangiogenic and antitumor activities of peptide inhibiting the vascular endothelial growth factor binding to neuropilin-1. Life Sci. 2006, 79, 2370–2381. [Google Scholar] [CrossRef] [PubMed]
- Vander Kooi, C.W.; Jusino, M.A.; Perman, B.; Neau, D.B.; Bellamy, H.D.; Leahy, D.J. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starzec, A.; Ladam, P.; Vassy, R.; Badache, S.; Bouchemal, N.; Navaza, A.; du Penhoat, C.H.; Perret, G.Y. Structure–function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF165 binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex. Peptides 2007, 28, 2397–2402. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157. [Google Scholar] [CrossRef] [Green Version]
- Zanuy, D.; Kotla, R.; Nussinov, R.; Teesalu, T.; Sugahara, K.N.; Alemán, C.; Haspel, N. Sequence dependence of C-end rule peptides in binding and activation of neuropilin-1 receptor. J. Struct. Biol. 2013, 182, 78–86. [Google Scholar] [CrossRef]
- Liu, W.-Q.; Borriello, L.; Allain, B.; Pavoni, S.; Lopez, N.; Hermine, O.; Garbay, C.; Raynaud, F.; Lepelletier, Y.; Demange, L. New Peptides Structurally Related to VEGF-A165 Exon-7 and -8 Encoded Domains Antagonize Its Binding to NRP-1 and VEGF-R1. Int. J. Pept. Res. Ther. 2015, 21, 117–124. [Google Scholar] [CrossRef]
- Richard, M.; Chateau, A.; Jelsch, C.; Didierjean, C.; Manival, X.; Charron, C.; Maigret, B.; Barberi-Heyob, M.; Chapleur, Y.; Boura, C.; et al. Carbohydrate-based peptidomimetics targeting neuropilin-1: Synthesis, molecular docking study and in vitro biological activities. Biorg. Med. Chem. 2016, 24, 5315–5325. [Google Scholar] [CrossRef]
- Tymecka, D.; Lipiński, P.F.J.; Fedorczyk, B.; Puszko, A.; Wileńska, B.; Perret, G.Y.; Misicka, A. Structure-activity relationship study of tetrapeptide inhibitors of the Vascular Endothelial Growth Factor A binding to Neuropilin-1. Peptides 2017, 94, 25–32. [Google Scholar] [CrossRef]
- Kamarulzaman, E.E.; Vanderesse, R.; Gazzali, A.M.; Barberi-Heyob, M.; Boura, C.; Frochot, C.; Shawkataly, O.; Aubry, A.; Wahab, H.A. Molecular modelling, synthesis and biological evaluation of peptide inhibitors as anti-angiogenic agent targeting neuropilin-1 for anticancer application. J. Biomol. Struct. Dyn. 2017, 35, 26–45. [Google Scholar] [CrossRef] [Green Version]
- Fedorczyk, B.; Lipiński, P.F.J.; Tymecka, D.; Puszko, A.K.; Wilenska, B.; Perret, G.Y.; Misicka, A. Conformational latitude—Activity relationship of KPPR tetrapeptide analogues toward their ability to inhibit binding of vascular endothelial growth factor 165 to neuropilin-1. J. Pept. Sci. 2017, 23, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Tymecka, D.; Puszko, A.K.; Lipiński, P.F.J.; Fedorczyk, B.; Wilenska, B.; Sura, K.; Perret, G.Y.; Misicka, A. Branched pentapeptides as potent inhibitors of the vascular endothelial growth factor 165 binding to Neuropilin-1: Design, synthesis and biological activity. Eur. J. Med. Chem. 2018, 158, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Fedorczyk, B.; Lipiński, P.F.J.; Puszko, A.K.; Tymecka, D.; Wilenska, B.; Dudka, W.; Perret, G.Y.; Wieczorek, R.; Misicka, A. Triazolopeptides Inhibiting the Interaction between Neuropilin-1 and Vascular Endothelial Growth Factor-165. Molecules 2019, 24, 1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puszko, A.K.; Sosnowski, P.; Tymecka, D.; Raynaud, F.; Hermine, O.; Lepelletier, Y.; Misicka, A. Neuropilin-1 peptide-like ligands with proline mimetics, tested using the improved chemiluminescence affinity detection method. MedChemComm 2019, 10, 332–340. [Google Scholar] [CrossRef]
- Puszko, A.K.; Sosnowski, P.; Pułka-Ziach, K.; Hermine, O.; Hopfgartner, G.; Lepelletier, Y.; Misicka, A. Urea moiety as amide bond mimetic in peptide-like inhibitors of VEGF-A165/NRP-1 complex. Bioorg. Med. Chem. Lett. 2019, 29, 2493–2497. [Google Scholar] [CrossRef]
- Puszko, A.K.; Sosnowski, P.; Raynaud, F.; Hermine, O.; Hopfgartner, G.; Lepelletier, Y.; Misicka, A. Does Cysteine Rule (CysR) Complete the CendR Principle? Increase in Affinity of Peptide Ligands for NRP-1 Through the Presence of N-Terminal Cysteine. Biomolecules 2020, 10, 448. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Bagherzadeh, A.; Hartzoulakis, B.; Jarvis, A.; Löhr, M.; Shaikh, S.; Aqil, R.; Cheng, L.; Tickner, M.; Esposito, D.; et al. Characterization of a Bicyclic Peptide Neuropilin-1 (NP-1) Antagonist (EG3287) Reveals Importance of Vascular Endothelial Growth Factor Exon 8 for NP-1 Binding and Role of NP-1 in KDR Signaling. J. Biol. Chem. 2006, 281, 13493–13502. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Aqil, R.; Cheng, L.; Chapman, C.; Shaikh, S.; Jarvis, A.; Chan, A.W.E.; Hartzoulakis, B.; Evans, I.M.; Frolov, A.; et al. N-Terminal Modification of VEGF-A C Terminus-Derived Peptides Delineates Structural Features Involved in Neuropilin-1 Binding and Functional Activity. ChemBioChem 2014, 15, 1161–1170. [Google Scholar] [CrossRef]
- Grabowska, K.; Puszko, A.K.; Lipiński, P.F.J.; Laskowska, A.K.; Wileńska, B.; Witkowska, E.; Misicka, A. Design, synthesis and in vitro biological evaluation of a small cyclic peptide as inhibitor of vascular endothelial growth factor binding to neuropilin-1. Bioorg. Med. Chem. Lett. 2016, 26, 2843–2846. [Google Scholar] [CrossRef]
- Grabowska, K.; Puszko, A.K.; Lipiński, P.F.J.; Laskowska, A.K.; Wileńska, B.; Witkowska, E.; Perret, G.Y.; Misicka, A. Structure-activity relationship study of a small cyclic peptide H-c[Lys-Pro-Glu]-Arg-OH: A potent inhibitor of Vascular Endothelial Growth Factor interaction with Neuropilin-1. Biorg. Med. Chem. 2017, 25, 597–602. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Biorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Xu, P.; Li, X.; Vander Kooi, C.W. Structural Basis for Selective Vascular Endothelial Growth Factor-A (VEGF-A) Binding to Neuropilin-1. J. Biol. Chem. 2012, 287, 11082–11089. [Google Scholar] [CrossRef] [Green Version]
- Keyt, B.A.; Berleau, L.T.; Nguyen, H.V.; Chen, H.; Heinsohn, H.; Vandlen, R.; Ferrara, N. The Carboxyl-terminal Domain(111165) of Vascular Endothelial Growth Factor Is Critical for Its Mitogenic Potency. J. Biol. Chem. 1996, 271, 7788–7795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soker, S.; Gollamudi-Payne, S.; Fidder, H.; Charmahelli, H.; Klagsbrun, M. Inhibition of Vascular Endothelial Growth Factor (VEGF)-induced Endothelial Cell Proliferation by a Peptide Corresponding to the Exon 7-Encoded Domain of VEGF165. J. Biol. Chem. 1997, 272, 31582–31588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairbrother, W.J.; Champe, M.A.; Christinger, H.W.; Keyt, B.A.; Starovasnik, M.A. Solution structure of the heparin-binding domain of vascular endothelial growth factor. Structure 1998, 6, 637–648. [Google Scholar] [CrossRef]
- Perret, G.Y.; Starzec, A.; Hauet, N.; Vergote, J.; Le Pecheur, M.; Vassy, R.; Léger, G.; Verbeke, K.A.; Bormans, G.; Nicolas, P.; et al. In vitro evaluation and biodistribution of a 99mTc-labeled anti-VEGF peptide targeting neuropilin-1. Nucl. Med. Biol. 2004, 31, 575–581. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Pernot, M.; Vanderesse, R.; Frochot, C.; Guillemin, F.; Barberi-Heyob, M. Stability of peptides and therapeutic success in cancer. Expert Opin. Drug Metab. Toxicol. 2011, 7, 793–802. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, S.; Kawasaki, K.; Okamoto, H.; Umezawa, C.; Okada, Y. Synthesis of Some Pseudo-Peptide Analogs of Thiol Proteinase Inhibitors. Chem. Pharm. Bull. (Tokyo) 1999, 47, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Thomas, N.; Pernot, M.; Vanderesse, R.; Becuwe, P.; Kamarulzaman, E.; Da Silva, D.; François, A.; Frochot, C.; Guillemin, F.; Barberi-Heyob, M. Photodynamic therapy targeting neuropilin-1: Interest of pseudopeptides with improved stability properties. Biochem. Pharmacol. 2010, 80, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Yurek-George, A.; Cecil, A.R.L.; Mo, A.H.K.; Wen, S.; Rogers, H.; Habens, F.; Maeda, S.; Yoshida, M.; Packham, G.; Ganesan, A. The First Biologically Active Synthetic Analogues of FK228, the Depsipeptide Histone Deacetylase Inhibitor. J. Med. Chem. 2007, 50, 5720–5726. [Google Scholar] [CrossRef] [PubMed]
- De Zotti, M.; Biondi, B.; Peggion, C.; De Poli, M.; Fathi, H.; Oancea, S.; Toniolo, C.; Formaggio, F. Partial thioamide scan on the lipopeptaibiotic trichogin GA IV. Effects on folding and bioactivity. Beilstein J. Org. Chem. 2012, 8, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.C.; Nestor, J.J.; Du Vigneaud, V. Synthesis and some pharmacological properties of [1-deamino-9-thioglycine]oxytocin. J. Am. Chem. Soc. 1973, 95, 5677–5679. [Google Scholar] [CrossRef] [PubMed]
- Lankiewicz, L.; Bowers, C.Y.; Reynolds, G.A.; Labroo, V.; Cohen, L.A.; Vonhof, S.; Sirén, A.-L.; Spatola, A.F. Biological activities of thionated thyrotropin-releasing hormone analogs. Biochem. Biophys. Res. Commun. 1992, 184, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Nahrwold, M.; Bogner, T.; Eissler, S.; Verma, S.; Sewald, N. “Clicktophycin-52”: A Bioactive Cryptophycin-52 Triazole Analogue. Org. Lett. 2010, 12, 1064–1067. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.; Arnold, U.; Soellner, M.B.; Raines, R.T. Protein Prosthesis: 1,5-Disubstituted[1,2,3]triazoles as cis-Peptide Bond Surrogates. J. Am. Chem. Soc. 2007, 129, 12670–12671. [Google Scholar] [CrossRef] [Green Version]
- Tischler, M.; Nasu, D.; Empting, M.; Schmelz, S.; Heinz, D.W.; Rottmann, P.; Kolmar, H.; Buntkowsky, G.; Tietze, D.; Avrutina, O. Braces for the Peptide Backbone: Insights into Structure–Activity Relationships of Protease Inhibitor Mimics with Locked Amide Conformations. Angew. Chem. Int. Ed. 2012, 51, 3708–3712. [Google Scholar] [CrossRef]
- Fremaux, J.; Venin, C.; Mauran, L.; Zimmer, R.H.; Guichard, G.; Goudreau, S.R. Peptide-oligourea hybrids analogue of GLP-1 with improved action in vivo. Nat. Commun. 2019, 10, 924. [Google Scholar] [CrossRef] [Green Version]
- Kozikowski, A.P.; Nan, F.; Conti, P.; Zhang, J.; Ramadan, E.; Bzdega, T.; Wroblewska, B.; Neale, J.H.; Pshenichkin, S.; Wroblewski, J.T. Design of Remarkably Simple, Yet Potent Urea-Based Inhibitors of Glutamate Carboxypeptidase II (NAALADase). J. Med. Chem. 2001, 44, 298–301. [Google Scholar] [CrossRef]
- Antunes, S.; Corre, J.-P.; Mikaty, G.; Douat, C.; Goossens, P.L.; Guichard, G. Effect of replacing main-chain ureas with thiourea and guanidinium surrogates on the bactericidal activity of membrane active oligourea foldamers. Biorg. Med. Chem. 2017, 25, 4245–4252. [Google Scholar] [CrossRef] [PubMed]
- Claudon, P.; Violette, A.; Lamour, K.; Decossas, M.; Fournel, S.; Heurtault, B.; Godet, J.; Mély, Y.; Jamart-Grégoire, B.; Averlant-Petit, M.-C.; et al. Consequences of Isostructural Main-Chain Modifications for the Design of Antimicrobial Foldamers: Helical Mimics of Host-Defense Peptides Based on a Heterogeneous Amide/Urea Backbone. Angew. Chem. Int. Ed. 2010, 49, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Cudic, P.; Stawikowski, M. Peptidomimetics: Fmoc Solid-Phase Pseudopeptide Synthesis. In Peptide-Based Drug Design; Otvos, L., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 223–246. [Google Scholar] [CrossRef]
- Tamilarasu, N.; Huq, I.; Rana, T.M. Targeting RNA with peptidomimetic oligomers in human cells. Bioorg. Med. Chem. Lett. 2001, 11, 505–507. [Google Scholar] [CrossRef]
- Douat-Casassus, C.; Pulka, K.; Claudon, P.; Guichard, G. Microwave-Enhanced Solid-Phase Synthesis of N,N′-Linked Aliphatic Oligoureas and Related Hybrids. Org. Lett. 2012, 14, 3130–3133. [Google Scholar] [CrossRef] [PubMed]
- Carpino, L.A.; Han, G.Y. 9-Fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 1972, 37, 3404–3409. [Google Scholar] [CrossRef]
- Hood, C.A.; Fuentes, G.; Patel, H.; Page, K.; Menakuru, M.; Park, J.H. Fast conventional Fmoc solid-phase peptide synthesis with HCTU. J. Pept. Sci. 2008, 14, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Bernatowicz, M.S.; Wu, Y.; Matsueda, G.R. 1H-Pyrazole-1-carboxamidine hydrochloride an attractive reagent for guanylation of amines and its application to peptide synthesis. J. Org. Chem. 1992, 57, 2497–2502. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Mason, J.S.; Overington, J.P. Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol. 2006, 16, 127–136. [Google Scholar] [CrossRef]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational Approaches to Improving Selectivity in Drug Design. J. Med. Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef]
- Rossignol, M.; Gagnon, M.L.; Klagsbrun, M. Genomic Organization of Human Neuropilin-1 and Neuropilin-2 Genes: Identification and Distribution of Splice Variants and Soluble Isoforms. Genomics 2000, 70, 211–222. [Google Scholar] [CrossRef]
- Pellet-Many, C.; Frankel, P.; Jia, H.; Zachary, I. Neuropilins: Structure, function and role in disease. Biochem. J. 2008, 411, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.-F.; Vander Kooi, C.W. Neuropilin Functions as an Essential Cell Surface Receptor. J. Biol. Chem. 2015, 290, 29120–29126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, Y.A.; Li, B.; Christinger, H.W.; Wells, J.A.; Cunningham, B.C.; de Vos, A.M. Vascular endothelial growth factor: Crystal structure and functional mapping of the kinase domain receptor binding site. Proc. Natl. Acad. Sci. USA 1997, 94, 7192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J.; Wright, P.E. Defining Solution Conformations of Small Linear Peptides. Annu. Rev. Biophys. Biophys. Chem. 1991, 20, 519–538. [Google Scholar] [CrossRef]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef]
- Bonner, R.; Hopfgartner, G. SWATH data independent acquisition mass spectrometry for metabolomics. TrAC Trends Anal. Chem. 2019, 120, 115278. [Google Scholar] [CrossRef]
- Raetz, M.; Duchoslav, E.; Bonner, R.; Hopfgartner, G. Hybrid SWATH/MS and HR-SRM/MS acquisition for phospholipidomics using QUAL/QUANT data processing. Anal. Bioanal. Chem. 2019, 411, 5681–5690. [Google Scholar] [CrossRef]
- Cifuentes Girard, M.F.; Ruskic, D.; Böhm, G.; Picenoni, R.; Hopfgartner, G. Automated parallel derivatization of metabolites with SWATH-MS data acquisition for qualitative and quantitative analysis. Anal. Chim. Acta 2020, 1127, 198–206. [Google Scholar] [CrossRef]
- Narazaki, M.; Tosato, G. Ligand-induced internalization selects use of common receptor neuropilin-1 by VEGF165 and semaphorin3A. Blood 2006, 107, 3892–3901. [Google Scholar] [CrossRef] [Green Version]
- Lepelletier, Y.; Camara-Clayette, V.; Jin, H.; Hermant, A.; Coulon, S.; Dussiot, M.; Arcos-Fajardo, M.; Baude, C.; Canionni, D.; Delarue, R.; et al. Prevention of Mantle Lymphoma Tumor Establishment by Routing Transferrin Receptor toward Lysosomal Compartments. Cancer Res. 2007, 67, 1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisel, A.S.; Motulsky, H.J.; Insel, P.A. Propranolol treatment externalizes beta-adrenergic receptors in guinea pig myocardium and prevents further externalization by ischemia. Circ. Res. 1987, 60, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell Jr, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Gfeller, D.; Michielin, O.; Zoete, V. SwissSidechain: A molecular and structural database of non-natural sidechains. Nucleic Acids Res. 2012, 41, D327–D332. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 X(Z)-Dab-Oic-Arg | |||
|---|---|---|---|
| Compound | X(Z) Branching | X(Z) Branching Structure | IC50 [µM] 1 |
| PP | Lys(hArg) |  | 2.3 2 |
| 1 | DabU(hArg) |  | 1.8 3 |
| 2 | OrnU(Arg) |  | 2.1 4 |
| 3 | LysU(gDab) |  | 1.2 5 |
| 4 | Lys(gDabU) |  | 0.49 6 |
| 5 | Orn(ArgU) |  | 0.36 7 |
| 6 | Dab(hArgU) |  | 0.19 8 |
| 7 | DapU(ArgU) |  | 2.1 9 |
| 8 | DabU(gDabU) |  | 2.1 10 |
| 9 | OrnU(gDapU) |  | 1.8 11 |
| Compound | Sequence | IC50 [µM] | |||
|---|---|---|---|---|---|
| NRP-1 | NRP-2 | VEGF-R1 | VEGF-R2 | ||
| 6 | H-Dab(hArgU)Dab-Oic-Arg-OH | 0.19 | 0.48 1 | no affinity | no affinity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puszko, A.K.; Sosnowski, P.; Rignault-Bricard, R.; Hermine, O.; Hopfgartner, G.; Pułka-Ziach, K.; Lepelletier, Y.; Misicka, A. Urea-Peptide Hybrids as VEGF-A165/NRP-1 Complex Inhibitors with Improved Receptor Affinity and Biological Properties. Int. J. Mol. Sci. 2021, 22, 72. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010072
Puszko AK, Sosnowski P, Rignault-Bricard R, Hermine O, Hopfgartner G, Pułka-Ziach K, Lepelletier Y, Misicka A. Urea-Peptide Hybrids as VEGF-A165/NRP-1 Complex Inhibitors with Improved Receptor Affinity and Biological Properties. International Journal of Molecular Sciences. 2021; 22(1):72. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010072
Chicago/Turabian StylePuszko, Anna K., Piotr Sosnowski, Rachel Rignault-Bricard, Olivier Hermine, Gérard Hopfgartner, Karolina Pułka-Ziach, Yves Lepelletier, and Aleksandra Misicka. 2021. "Urea-Peptide Hybrids as VEGF-A165/NRP-1 Complex Inhibitors with Improved Receptor Affinity and Biological Properties" International Journal of Molecular Sciences 22, no. 1: 72. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010072