Mechanisms of Insulin Resistance at the Crossroad of Obesity with Associated Metabolic Abnormalities and Cognitive Dysfunction


Abstract
:1. Introduction
2. Role of Obesity in the Development of IR
3. Role of IR in the Development of Metabolic Dysfunction
4. Role of IR in the Development of Cognitive Dysfunction
5. Reversibility of IR with Lifestyle Interventions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanson, P.; Weickert, M.O.; Barber, T.M. Obesity: Novel and unusual predisposing factors. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820922018. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O. Nutritional modulation of insulin resistance. Scientifica 2012, 2012, 424780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passarello, K.; Kurian, S.; Villanueva, V. Endometrial Cancer: An Overview of Pathophysiology, Management, and Care. Semin. Oncol. Nurs. 2019, 35, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.M.; McCarthy, M.I.; Franks, S.; Wass, J.A. Metabolic syndrome in polycystic ovary syndrome. Endokrynol. Pol. 2007, 58, 34–41. [Google Scholar]
- Barber, T.M.; Dimitriadis, G.K.; Andreou, A.; Franks, S. Polycystic ovary syndrome: Insight into pathogenesis and a common association with insulin resistance. Clin. Med. 2015, 15, s72–s76. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Han, L.; Hu, D. Fasting insulin, insulin resistance and risk of hypertension in the general population: A meta-analysis. Clin. Chim. Acta 2017, 464, 57–63. [Google Scholar] [CrossRef]
- Lam, D.C.; Lam, K.S.; Ip, M.S. Obstructive sleep apnoea, insulin resistance and adipocytokines. Clin. Endocrinol. 2015, 82, 165–177. [Google Scholar] [CrossRef]
- Saboor Aftab, S.A.; Kumar, S.; Barber, T.M. The role of obesity and type 2 diabetes mellitus in the development of male obesity-associated secondary hypogonadism. Clin. Endocrinol. 2013, 78, 330–337. [Google Scholar] [CrossRef]
- Brown, E.; Wilding, J.P.H.; Barber, T.M.; Alam, U.; Cuthbertson, D.J. Weight loss variability with SGLT2 inhibitors and GLP-1 receptor agonists in type 2 diabetes mellitus and obesity: Mechanistic possibilities. Obes. Rev. 2019, 20, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Franz, M.J.; VanWormer, J.J.; Crain, A.L.; Boucher, J.L.; Histon, T.; Caplan, W.; Bowman, J.D.; Pronk, N.P. Weight-loss outcomes: A systematic review and meta-analysis of weight-loss clinical trials with a minimum 1-year follow-up. J. Am. Diet. Assoc. 2007, 107, 1755–1767. [Google Scholar] [CrossRef]
- Lebovitz, H.E. Insulin resistance: Definition and consequences. Exp. Clin. Endocrinol. Diabetes 2001, 109, S135–S148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, T.M.; McCarthy, M.I.; Wass, J.A.; Franks, S. Obesity and polycystic ovary syndrome. Clin. Endocrinol. 2006, 65, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [PubMed] [Green Version]
- Muntoni, S.; Muntoni, S. Insulin resistance: Pathophysiology and rationale for treatment. Ann. Nutr. Metab. 2011, 58, 25–36. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Reaven, G.M. The metabolic syndrome: Requiescat in pace. Clin. Chem. 2005, 51, 931–938. [Google Scholar]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef]
- Amin, M.N.; Hussain, M.S.; Sarwar, M.S.; Rahman Moghal, M.M.; Das, A.; Hossain, M.Z.; Chowdhury, J.A.; Millat, M.S.; Islam, M.S. How the association between obesity and inflammation may lead to insulin resistance and cancer. Diabetes Metab. Syndr. 2019, 13, 1213–1224. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflug. Arch. 2017, 469, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Iossa, S.; Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 2017, 233, R15–R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.K.; Jun, W.; Lee, J. Mechanism of ER Stress and Inflammation for Hepatic Insulin Resistance in Obesity. Ann. Nutr. Metab. 2015, 67, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Nugent, C.A.; Tsompana, M.; Cai, L.; Wang, Y.; Buck, M.J.; Genco, R.J.; Baker, R.D.; Zhu, R.; et al. Gut microbiome may contribute to insulin resistance and systemic inflammation in obese rodents: A meta-analysis. Physiol. Genom. 2018, 50, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Chun, T.H.; Kang, L. Adipose extracellular matrix remodelling in obesity and insulin resistance. Biochem. Pharmacol. 2016, 119, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Moschonis, G.; Androutsos, O.; Hulshof, T.; Dracopoulou, M.; Chrousos, G.P.; Manios, Y. Vitamin D insufficiency is associated with insulin resistance independently of obesity in primary schoolchildren. The healthy growth study. Pediatr. Diabetes 2018, 19, 866–873. [Google Scholar] [CrossRef]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuniga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Pennings, N.; Jaber, J.; Ahiawodzi, P. Ten-year weight gain is associated with elevated fasting insulin levels and precedes glucose elevation. Diabetes Metab. Res. Rev. 2018, 34, e2986. [Google Scholar] [CrossRef]
- Salvetti, A.; Brogi, G.; Di Legge, V.; Bernini, G.P. The inter-relationship between insulin resistance and hypertension. Drugs 1993, 46, 149–159. [Google Scholar] [CrossRef]
- Cwynar, M.; Gasowski, J.; Gryglewska, B.; Gluszewska, A.; Kwater, A.; Krolczyk, J.; Folta, M.; Barton, H.; Grodzicki, T. Insulin Resistance and Renal Sodium Handling Influence Arterial Stiffness in Hypertensive Patients with Prevailing Sodium Intake. Am. J. Hypertens. 2019, 32, 848–857. [Google Scholar] [CrossRef]
- Frontoni, S.; Bracaglia, D.; Gigli, F. Relationship between autonomic dysfunction, insulin resistance and hypertension, in diabetes. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 441–449. [Google Scholar] [CrossRef]
- Klop, B.; Elte, J.W.; Cabezas, M.C. Dyslipidemia in obesity: Mechanisms and potential targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, J.D.; Sparks, C.E.; Adeli, K. Selective hepatic insulin resistance, VLDL overproduction, and hypertriglyceridemia. Arter. Thromb. Vasc. Biol. 2012, 32, 2104–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butkowski, E.G.; Jelinek, H.F. Hyperglycaemia, oxidative stress and inflammatory markers. Redox Rep. 2017, 22, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, C.J.; Sutherland, C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet. Med. 2012, 29, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Liu, J.; Zhu, L.; Tan, F.; Qin, Y.; Huang, H.; Yu, Y. Free fatty acid can induce cardiac dysfunction and alter insulin signaling pathways in the heart. Lipids Health Dis. 2018, 17, 185. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.K.; Zhang, Y.; Cao, C.M.; Hu, X.; Fang, M.; Yao, Y.; Jin, L.; Chen, G.; Jiang, P.; Zhang, S.; et al. Glucose-Sensitive Myokine/Cardiokine MG53 Regulates Systemic Insulin Response and Metabolic Homeostasis. Circulation 2019, 139, 901–914. [Google Scholar] [CrossRef]
- Book, C.B.; Dunaif, A. Selective insulin resistance in the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1999, 84, 3110–3116. [Google Scholar] [CrossRef] [Green Version]
- Sacchi, S.; Marinaro, F.; Tondelli, D.; Lui, J.; Xella, S.; Marsella, T.; Tagliasacchi, D.; Argento, C.; Tirelli, A.; Giulini, S.; et al. Modulation of gonadotrophin induced steroidogenic enzymes in granulosa cells by d-chiroinositol. Reprod. Biol. Endocrinol. 2016, 14, 52. [Google Scholar] [CrossRef] [Green Version]
- Kuscu, N.K.; Koyuncu, F.; Ozbilgin, K.; Inan, S.; Tuglu, I.; Karaer, O. Insulin: Does it induce follicular arrest in the rat ovary? Gynecol. Endocrinol. 2002, 16, 361–364. [Google Scholar] [CrossRef]
- Barber, T.M.; Hanson, P.; Weickert, M.O.; Franks, S. Obesity and Polycystic Ovary Syndrome: Implications for Pathogenesis and Novel Management Strategies. Clin. Med. Insights Reprod. Health 2019, 13, 1179558119874042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef] [PubMed]
- Franks, S.; Stark, J.; Hardy, K. Follicle dynamics and anovulation in polycystic ovary syndrome. Hum. Reprod. Update 2008, 14, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Lee, S.; Tong, M.; Hang, S.; Deochand, C.; de la Monte, S. CSF and Brain Indices of Insulin Resistance, Oxidative Stress and Neuro-Inflammation in Early versus Late Alzheimer’s Disease. J. Alzheimers Dis. Park. 2013, 3, 128. [Google Scholar] [CrossRef]
- Ma, L.; Wang, J.; Li, Y. Insulin resistance and cognitive dysfunction. Clin. Chim. Acta 2015, 444, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar] [CrossRef] [Green Version]
- Fazakerley, D.J.; Naghiloo, S.; Chaudhuri, R.; Koumanov, F.; Burchfield, J.G.; Thomas, K.C.; Krycer, J.R.; Prior, M.J.; Parker, B.L.; Murrow, B.A.; et al. Proteomic Analysis of GLUT4 Storage Vesicles Reveals Tumor Suppressor Candidate 5 (TUSC5) as a Novel Regulator of Insulin Action in Adipocytes. J. Biol. Chem. 2015, 290, 23528–23542. [Google Scholar] [CrossRef] [Green Version]
- Blazquez, E.; Velazquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef] [Green Version]
- Long-Smith, C.M.; Manning, S.; McClean, P.L.; Coakley, M.F.; O’Halloran, D.J.; Holscher, C.; O’Neill, C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromol. Med. 2013, 15, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Neumann, K.F.; Rojo, L.; Navarrete, L.P.; Farias, G.; Reyes, P.; Maccioni, R.B. Insulin resistance and Alzheimer’s disease: Molecular links & clinical implications. Curr. Alzheimer Res. 2008, 5, 438–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, R.; Zhao, Z.; Ji, Z.; Xu, S.; Holscher, C.; Sheng, S. Coexistences of insulin signaling-related proteins and choline acetyltransferase in neurons. Brain Res. 2009, 1249, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Hami, J.; Kheradmand, H.; Haghir, H. Sex differences and laterality of insulin receptor distribution in developing rat hippocampus: An immunohistochemical study. J. Mol. Neurosci. 2014, 54, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Tyagi, E.; Shukla, R.; Nath, C. Insulin receptor signaling in rat hippocampus: A study in STZ (ICV) induced memory deficit model. Eur. Neuropsychopharmacol. 2011, 21, 261–273. [Google Scholar] [CrossRef]
- Santos, G.J.; Ferreira, S.M.; Ortis, F.; Rezende, L.F.; Li, C.; Naji, A.; Carneiro, E.M.; Kaestner, K.H.; Boschero, A.C. Metabolic memory of ss-cells controls insulin secretion and is mediated by CaMKII. Mol. Metab. 2014, 3, 484–489. [Google Scholar] [CrossRef]
- Kar, S.; Chabot, J.G.; Quirion, R. Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II, and [125I]insulin receptor binding sites in developing and adult rat brain. J. Comp. Neurol. 1993, 333, 375–397. [Google Scholar] [CrossRef]
- Spielman, L.J.; Little, J.P.; Klegeris, A. Inflammation and insulin/IGF-1 resistance as the possible link between obesity and neurodegeneration. J. Neuroimmunol. 2014, 273, 8–21. [Google Scholar] [CrossRef]
- Park, S.J.; Chung, Y.H.; Lee, J.H.; Dang, D.K.; Nam, Y.; Jeong, J.H.; Kim, Y.S.; Nabeshima, T.; Shin, E.J.; Kim, H.C. Growth Hormone-Releaser Diet Attenuates Cognitive Dysfunction in Klotho Mutant Mice via Insulin-Like Growth Factor-1 Receptor Activation in a Genetic Aging Model. Endocrinol. Metab. 2014, 29, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.F.; Guo, Z.; Zheng, T.; Jiang, Y.; Yan, Y.; Yin, X.; Chen, Y.; Zhang, B. Intranasal insulin alleviates cognitive deficits and amyloid pathology in young adult APPswe/PS1dE9 mice. Aging Cell 2016, 15, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Brankatschk, M.; Dunst, S.; Nemetschke, L.; Eaton, S. Delivery of circulating lipoproteins to specific neurons in the Drosophila brain regulates systemic insulin signaling. eLife 2014, 3, e02862. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Siu, J.J.; Huang, W.; Askwith, C.; Cao, L. Insulin Modulates Excitatory Synaptic Transmission and Synaptic Plasticity in the Mouse Hippocampus. Neuroscience 2019, 411, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Kamal, A.; Urban, I.J.; Spruijt, B.M.; Erkelens, D.W.; Gispen, W.H. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: Effects of insulin treatment. Brain Res. 1998, 800, 125–135. [Google Scholar] [CrossRef]
- Huang, C.C.; Lee, C.C.; Hsu, K.S. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J. Neurochem. 2004, 89, 217–231. [Google Scholar] [CrossRef]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef]
- Kawano, T.; Iwata, H.; Aoyama, B.; Nishigaki, A.; Yamanaka, D.; Tateiwa, H.; Eguchi, S.; Locatelli, F.M.; Yokoyama, M. The role of hippocampal insulin signaling on postoperative cognitive dysfunction in an aged rat model of abdominal surgery. Life Sci. 2016, 162, 87–94. [Google Scholar] [CrossRef]
- Barbagallo, M.; Dominguez, L.J. Type 2 diabetes mellitus and Alzheimer’s disease. World J. Diabetes 2014, 5, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Song, Y.F.; Yin, J.; Liu, Z.H.; Mo, X.D.; Wang, D.G.; Gao, L.P.; Jing, Y.H. Spatial memory impairment is associated with hippocampal insulin signals in ovariectomized rats. PLoS ONE 2014, 9, e104450. [Google Scholar] [CrossRef]
- Ma, L.; Zhao, Z.; Wang, R.; Zhang, X.; Zhang, J.; Dong, W.; Xu, B.; Zhang, J. Caloric restriction can improve learning ability in C57/BL mice via regulation of the insulin-PI3K/Akt signaling pathway. Neurol. Sci. 2014, 35, 1381–1386. [Google Scholar] [CrossRef]
- Dineley, K.T.; Jahrling, J.B.; Denner, L. Insulin resistance in Alzheimer’s disease. Neurobiol. Dis. 2014, 72, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, R.; Dargahi, L.; Haeri, A.; Moosavi, M.; Mohamed, Z.; Ahmadiani, A. Brain insulin dysregulation: Implication for neurological and neuropsychiatric disorders. Mol. Neurobiol. 2013, 47, 1045–1065. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Taliyan, R. Neuroprotective role of Indirubin-3′-monoxime, a GSKbeta inhibitor in high fat diet induced cognitive impairment in mice. Biochem. Biophys. Res. Commun. 2014, 452, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Rissman, R.A.; Poon, W.W.; Blurton-Jones, M.; Oddo, S.; Torp, R.; Vitek, M.P.; LaFerla, F.M.; Rohn, T.T.; Cotman, C.W. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Investig. 2004, 114, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Aslan, A.K.; Starr, J.M.; Pattie, A.; Deary, I. Cognitive consequences of overweight and obesity in the ninth decade of life? Age Ageing 2015, 44, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Biessels, G.J.; Reagan, L.P. Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef]
- Tokuchi, R.; Hishikawa, N.; Kurata, T.; Sato, K.; Kono, S.; Yamashita, T.; Deguchi, K.; Abe, K. Clinical and demographic predictors of mild cognitive impairment for converting to Alzheimer’s disease and reverting to normal cognition. J. Neurol. Sci. 2014, 346, 288–292. [Google Scholar] [CrossRef] [Green Version]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zheng, W.; Xie, J.W.; Wang, T.; Wang, S.L.; Teng, W.P.; Wang, Z.Y. Insulin deficiency exacerbates cerebral amyloidosis and behavioral deficits in an Alzheimer transgenic mouse model. Mol. Neurodegener. 2010, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Zang, L.; Wu, D.Y.; Li, Y.J.; Zhang, Q.; Wang, H.B.; Tian, G.L.; Mu, Y.M. Pioglitazone improves the ability of learning and memory via activating ERK1/2 signaling pathway in the hippocampus of T2DM rats. Neurosci. Lett. 2017, 651, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Kern, W.; Peters, A.; Fruehwald-Schultes, B.; Deininger, E.; Born, J.; Fehm, H.L. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology 2001, 74, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Borghouts, L.B.; Keizer, H.A. Exercise and insulin sensitivity: A review. Int. J. Sports Med. 2000, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Marson, E.C.; Delevatti, R.S.; Prado, A.K.; Netto, N.; Kruel, L.F. Effects of aerobic, resistance, and combined exercise training on insulin resistance markers in overweight or obese children and adolescents: A systematic review and meta-analysis. Prev. Med. 2016, 93, 211–218. [Google Scholar] [CrossRef]
- Motahari-Tabari, N.; Ahmad Shirvani, M.; Shirzad, E.A.M.; Yousefi-Abdolmaleki, E.; Teimourzadeh, M. The effect of 8 weeks aerobic exercise on insulin resistance in type 2 diabetes: A randomized clinical trial. Glob. J. Health Sci. 2014, 7, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Fujita, H.; Hosono, A.; Shibata, K.; Tsujimura, S.; Oka, K.; Okamoto, N.; Kamiya, M.; Kondo, F.; Wakabayashi, R.; Ichikawa, M.; et al. Physical Activity Earlier in Life Is Inversely Associated with Insulin Resistance Among Adults in Japan. J. Epidemiol. 2019, 29, 57–60. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Zhou, L.; He, T. Potential effect of exercise in ameliorating insulin resistance at transcriptome level. J. Sports Med. Phys. Fit. 2019, 59, 116–125. [Google Scholar] [CrossRef]
- Weickert, M.O.; Mohlig, M.; Koebnick, C.; Holst, J.J.; Namsolleck, P.; Ristow, M.; Osterhoff, M.; Rochlitz, H.; Rudovich, N.; Spranger, J.; et al. Impact of cereal fibre on glucose-regulating factors. Diabetologia 2005, 48, 2343–2353. [Google Scholar] [CrossRef]
- Weickert, M.O.; Mohlig, M.; Schofl, C.; Arafat, A.M.; Otto, B.; Viehoff, H.; Koebnick, C.; Kohl, A.; Spranger, J.; Pfeiffer, A.F. Cereal fiber improves whole-body insulin sensitivity in overweight and obese women. Diabetes Care 2006, 29, 775–780. [Google Scholar] [CrossRef] [Green Version]
- Honsek, C.; Kabisch, S.; Kemper, M.; Gerbracht, C.; Arafat, A.M.; Birkenfeld, A.L.; Dambeck, U.; Osterhoff, M.A.; Weickert, M.O.; Pfeiffer, A.F.H. Fibre supplementation for the prevention of type 2 diabetes and improvement of glucose metabolism: The randomised controlled Optimal Fibre Trial (OptiFiT). Diabetologia 2018, 61, 1295–1305. [Google Scholar] [CrossRef] [Green Version]
- Russell, W.R.; Baka, A.; Bjorck, I.; Delzenne, N.; Gao, D.; Griffiths, H.R.; Hadjilucas, E.; Juvonen, K.; Lahtinen, S.; Lansink, M.; et al. Impact of Diet Composition on Blood Glucose Regulation. Crit. Rev. Food Sci. Nutr. 2016, 56, 541–590. [Google Scholar] [CrossRef]
- Kabisch, S.; Meyer, N.M.T.; Honsek, C.; Gerbracht, C.; Dambeck, U.; Kemper, M.; Osterhoff, M.A.; Birkenfeld, A.L.; Arafat, A.M.; Hjorth, M.F.; et al. Fasting Glucose State Determines Metabolic Response to Supplementation with Insoluble Cereal Fibre: A Secondary Analysis of the Optimal Fibre Trial (OptiFiT). Nutrients 2019, 11, 2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabisch, S.; Meyer, N.M.T.; Honsek, C.; Gerbracht, C.; Dambeck, U.; Kemper, M.; Osterhoff, M.A.; Birkenfeld, A.L.; Arafat, A.M.; Weickert, M.O.; et al. Obesity Does Not Modulate the Glycometabolic Benefit of Insoluble Cereal Fibre in Subjects with Prediabetes-A Stratified Post Hoc Analysis of the Optimal Fibre Trial (OptiFiT). Nutrients 2019, 11, 2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weickert, M.O.; Roden, M.; Isken, F.; Hoffmann, D.; Nowotny, P.; Osterhoff, M.; Blaut, M.; Alpert, C.; Gogebakan, O.; Bumke-Vogt, C.; et al. Effects of supplemented isoenergetic diets differing in cereal fiber and protein content on insulin sensitivity in overweight humans. Am. J. Clin. Nutr. 2011, 94, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Castro-Quezada, I.; Flores-Guillen, E.; Nunez-Ortega, P.E.; Irecta-Najera, C.A.; Sanchez-Chino, X.M.; Mendez-Flores, O.G.; Olivo-Vidal, Z.E.; Garcia-Miranda, R.; Solis-Hernandez, R.; Ochoa-Diaz-Lopez, H. Dietary Carbohydrates and Insulin Resistance in Adolescents from Marginalized Areas of Chiapas, Mexico. Nutrients 2019, 11, 3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, A.N.; Akerman, A.P.; Mann, J. Dietary fibre and whole grains in diabetes management: Systematic review and meta-analyses. PLoS Med. 2020, 17, e1003053. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.M.; Kabisch, S.; Pfeiffer, A.F.H.; Weickert, M.O. The Health Benefits of Dietary Fibre. Nutrients 2020, 12, 3209. [Google Scholar] [CrossRef]
- Isken, F.; Weickert, M.O.; Tschop, M.H.; Nogueiras, R.; Mohlig, M.; Abdelrahman, A.; Klaus, S.; Thorens, B.; Pfeiffer, A.F. Metabolic effects of diets differing in glycaemic index depend on age and endogenous glucose-dependent insulinotrophic polypeptide in mice. Diabetologia 2009, 52, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Weickert, M.O.; Spranger, J.; Holst, J.J.; Otto, B.; Koebnick, C.; Mohlig, M.; Pfeiffer, A.F. Wheat-fibre-induced changes of postprandial peptide YY and ghrelin responses are not associated with acute alterations of satiety. Br. J. Nutr. 2006, 96, 795–798. [Google Scholar] [CrossRef]
- Mohlig, M.; Koebnick, C.; Weickert, M.O.; Lueder, W.; Otto, B.; Steiniger, J.; Twilfert, M.; Meuser, F.; Pfeiffer, A.F.; Zunft, H.J. Arabinoxylan-enriched meal increases serum ghrelin levels in healthy humans. Horm Metab. Res. 2005, 37, 303–308. [Google Scholar] [CrossRef]
- Gruendel, S.; Garcia, A.L.; Otto, B.; Mueller, C.; Steiniger, J.; Weickert, M.O.; Speth, M.; Katz, N.; Koebnick, C. Carob pulp preparation rich in insoluble dietary fiber and polyphenols enhances lipid oxidation and lowers postprandial acylated ghrelin in humans. J. Nutr. 2006, 136, 1533–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, A.L.; Otto, B.; Reich, S.C.; Weickert, M.O.; Steiniger, J.; Machowetz, A.; Rudovich, N.N.; Mohlig, M.; Katz, N.; Speth, M.; et al. Arabinoxylan consumption decreases postprandial serum glucose, serum insulin and plasma total ghrelin response in subjects with impaired glucose tolerance. Eur. J. Clin. Nutr. 2007, 61, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruendel, S.; Garcia, A.L.; Otto, B.; Wagner, K.; Bidlingmaier, M.; Burget, L.; Weickert, M.O.; Dongowski, G.; Speth, M.; Katz, N.; et al. Increased acylated plasma ghrelin, but improved lipid profiles 24-h after consumption of carob pulp preparation rich in dietary fibre and polyphenols. Br. J. Nutr. 2007, 98, 1170–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, A.L.; Steiniger, J.; Reich, S.C.; Weickert, M.O.; Harsch, I.; Machowetz, A.; Mohlig, M.; Spranger, J.; Rudovich, N.N.; Meuser, F.; et al. Arabinoxylan fibre consumption improved glucose metabolism, but did not affect serum adipokines in subjects with impaired glucose tolerance. Horm. Metab. Res. 2006, 38, 761–766. [Google Scholar] [CrossRef]
- Weickert, M.O.; Hattersley, J.G.; Kyrou, I.; Arafat, A.M.; Rudovich, N.; Roden, M.; Nowotny, P.; von Loeffelholz, C.; Matysik, S.; Schmitz, G.; et al. Effects of supplemented isoenergetic diets varying in cereal fiber and protein content on the bile acid metabolic signature and relation to insulin resistance. Nutr. Diabetes 2018, 8, 11. [Google Scholar] [CrossRef]
- Hattersley, J.G.; Pfeiffer, A.F.; Roden, M.; Petzke, K.J.; Hoffmann, D.; Rudovich, N.N.; Randeva, H.S.; Vatish, M.; Osterhoff, M.; Goegebakan, O.; et al. Modulation of amino acid metabolic signatures by supplemented isoenergetic diets differing in protein and cereal fiber content. J. Clin. Endocrinol. Metab. 2014, 99, E2599–E2609. [Google Scholar] [CrossRef] [Green Version]
- Schulze, M.B.; Schulz, M.; Heidemann, C.; Schienkiewitz, A.; Hoffmann, K.; Boeing, H. Fiber and magnesium intake and incidence of type 2 diabetes: A prospective study and meta-analysis. Arch. Intern. Med. 2007, 167, 956–965. [Google Scholar] [CrossRef] [Green Version]
- De Munter, J.S.; Hu, F.B.; Spiegelman, D.; Franz, M.; van Dam, R.M. Whole grain, bran, and germ intake and risk of type 2 diabetes: A prospective cohort study and systematic review. PLoS Med. 2007, 4, e261. [Google Scholar] [CrossRef]
- Weickert, M.O.; Pfeiffer, A.F. Metabolic effects of dietary fiber consumption and prevention of diabetes. J. Nutr. 2008, 138, 439–442. [Google Scholar] [CrossRef]
- Weickert, M.O.; Pfeiffer, A.F.H. Impact of Dietary Fiber Consumption on Insulin Resistance and the Prevention of Type 2 Diabetes. J. Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Weickert, M.O. High fiber intake, dietary protein, and prevention of type 2 diabetes. Expert Rev. Endocrinol. Metab. 2018, 13, 223–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weickert, M.O. What dietary modification best improves insulin sensitivity and why? Clin. Endocrinol. 2012, 77, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Isken, F.; Klaus, S.; Petzke, K.J.; Loddenkemper, C.; Pfeiffer, A.F.; Weickert, M.O. Impairment of fat oxidation under high- vs. low-glycemic index diet occurs before the development of an obese phenotype. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E287–E295. [Google Scholar] [CrossRef] [PubMed]
- Gruendel, S.; Otto, B.; Garcia, A.L.; Wagner, K.; Mueller, C.; Weickert, M.O.; Heldwein, W.; Koebnick, C. Carob pulp preparation rich in insoluble dietary fibre and polyphenols increases plasma glucose and serum insulin responses in combination with a glucose load in humans. Br. J. Nutr. 2007, 98, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Isken, F.; Klaus, S.; Osterhoff, M.; Pfeiffer, A.F.; Weickert, M.O. Effects of long-term soluble vs. insoluble dietary fiber intake on high-fat diet-induced obesity in C57BL/6J mice. J. Nutr. Biochem. 2010, 21, 278–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Track, N.S.; Cawkwell, M.E.; Chin, B.C.; Chiu, S.S.; Haberer, S.A.; Honey, C.R. Guar gum consumption in adolescent and adult rats: Short- and long-term metabolic effects. Can. J. Physiol. Pharmacol. 1985, 63, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Arafat, A.M.; Blaut, M.; Alpert, C.; Becker, N.; Leupelt, V.; Rudovich, N.; Mohlig, M.; Pfeiffer, A.F. Changes in dominant groups of the gut microbiota do not explain cereal-fiber induced improvement of whole-body insulin sensitivity. Nutr. Metab. 2011, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Yannakoulia, M.; Poulimeneas, D.; Mamalaki, E.; Anastasiou, C.A. Dietary modifications for weight loss and weight loss maintenance. Metabolism 2019, 92, 153–162. [Google Scholar] [CrossRef]
- Moran, L.J.; Ko, H.; Misso, M.; Marsh, K.; Noakes, M.; Talbot, M.; Frearson, M.; Thondan, M.; Stepto, N.; Teede, H.J. Dietary composition in the treatment of polycystic ovary syndrome: A systematic review to inform evidence-based guidelines. J. Acad. Nutr. Diet. 2013, 113, 520–545. [Google Scholar] [CrossRef]
- Donga, E.; Romijn, J.A. Sleep characteristics and insulin sensitivity in humans. Handb Clin. Neurol 2014, 124, 107–114. [Google Scholar] [CrossRef]
- De Souza, J.F.T.; Dattilo, M.; de Mello, M.T.; Tufik, S.; Antunes, H.K.M. High-Intensity Interval Training Attenuates Insulin Resistance Induced by Sleep Deprivation in Healthy Males. Front. Physiol. 2017, 8, 992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leproult, R.; Van Cauter, E. Role of sleep and sleep loss in hormonal release and metabolism. Endocr. Dev. 2010, 17, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Taheri, S.; Lin, L.; Austin, D.; Young, T.; Mignot, E. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med. 2004, 1, e62. [Google Scholar] [CrossRef] [PubMed]
- Reutrakul, S.; Van Cauter, E. Sleep influences on obesity, insulin resistance, and risk of type 2 diabetes. Metabolism 2018, 84, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, D.; Balkau, B.; Segrestin, B.; Gottsater, M.; Gabriel, R.; Hatunic, M.; Mari, A.; Dekker, J.M.; Rutters, F.; Group, E.-R.S. Associations between sleep duration and sleep debt with insulin sensitivity and insulin secretion in the EGIR-RISC Study. Diabetes Metab. 2019, 45, 375–381. [Google Scholar] [CrossRef]
- Poroyko, V.A.; Carreras, A.; Khalyfa, A.; Khalyfa, A.A.; Leone, V.; Peris, E.; Almendros, I.; Gileles-Hillel, A.; Qiao, Z.; Hubert, N.; et al. Chronic Sleep Disruption Alters Gut Microbiota, Induces Systemic and Adipose Tissue Inflammation and Insulin Resistance in Mice. Sci. Rep. 2016, 6, 35405. [Google Scholar] [CrossRef] [Green Version]
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 2009, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Barazzoni, R.; Gortan Cappellari, G.; Ragni, M.; Nisoli, E. Insulin resistance in obesity: An overview of fundamental alterations. Eat. Weight Disord. 2018, 23, 149–157. [Google Scholar] [CrossRef]
- Stephen, A.M.; Champ, M.M.; Cloran, S.J.; Fleith, M.; van Lieshout, L.; Mejborn, H.; Burley, V.J. Dietary fibre in Europe: Current state of knowledge on definitions, sources, recommendations, intakes and relationships to health. Nutr. Res. Rev. 2017, 30, 149–190. [Google Scholar] [CrossRef]
- Orzel-Gryglewska, J. Consequences of sleep deprivation. Int. J. Occup. Med. Environ. Health 2010, 23, 95–114. [Google Scholar] [CrossRef]
- McGowan, L.J.; Powell, R.; French, D.P. Older adults’ construal of sedentary behaviour: Implications for reducing sedentary behaviour in older adult populations. J. Health Psychol. 2020, 10, 1359105320909870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McRorie, J.W., Jr.; McKeown, N.M. Understanding the Physics of Functional Fibers in the Gastrointestinal Tract: An Evidence-Based Approach to Resolving Enduring Misconceptions about Insoluble and Soluble Fiber. J. Acad. Nutr. Diet. 2017, 117, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, M.E.; Vaughn, B.V. Poor sleep challenging the health of a Nation. Neurodiagnost. J. 2012, 52, 233–249. [Google Scholar]
- Sun, Y.; Mobasheri, A. Utilizing Crowdsourced Data for Studies of Cycling and Air Pollution Exposure: A Case Study Using Strava Data. Int J. Environ. Res. Public Health 2017, 14, 274. [Google Scholar] [CrossRef]
- Booth, D.A.; Booth, P. Targeting cultural changes supportive of the healthiest lifestyle patterns. A biosocial evidence-base for prevention of obesity. Appetite 2011, 56, 210–221. [Google Scholar] [CrossRef]
- Heaton, P.M. The Covid-19 Vaccine-Development Multiverse. N. Engl. J. Med. 2020, 383, 1986–1988. [Google Scholar] [CrossRef]
- Barber, T.M. COVID-19 and Diabetes Mellitus: Implications for prognosis and clinical management. Expert Rev. Endocrinol. Metab. 2020, 15, 227–236. [Google Scholar] [CrossRef]


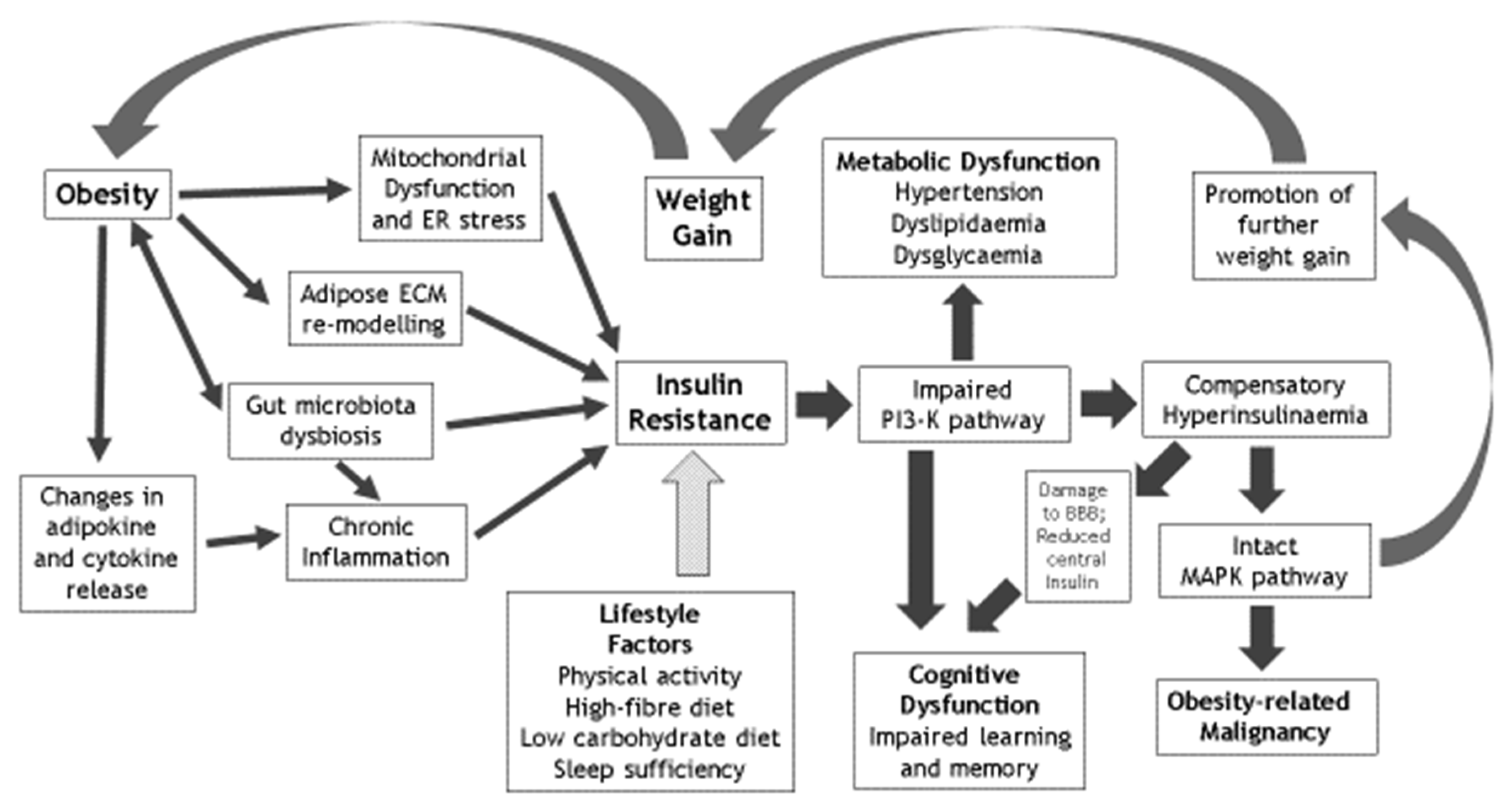{kind=link}
| Factors that Mediate the Effects of Obesity on IR | Metabolic Sequelae of IR | Cognitive Sequelae of IR | Lifestyle Reversibility of IR |
|---|---|---|---|
| Chronic inflammation | Hypertension | Cognitive dysfunction | Physical activity and avoidance of sedentariness |
| Mitochondrial dysfunction | Dyslipidaemia | Learning deficiency | Optimised dietary fibre intake |
| Gut microbiota dysbiosis | Dysglycaemia | Memory impairment | Limited dietary carbohydrate intake |
| Adipose ECM remodelling | Nonalcoholic fatty liver disease & polycystic ovary syndrome | Impaired hippocampal synaptic plasticity | Optimised sleep sufficiency and quality |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barber, T.M.; Kyrou, I.; Randeva, H.S.; Weickert, M.O. Mechanisms of Insulin Resistance at the Crossroad of Obesity with Associated Metabolic Abnormalities and Cognitive Dysfunction. Int. J. Mol. Sci. 2021, 22, 546. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020546
Barber TM, Kyrou I, Randeva HS, Weickert MO. Mechanisms of Insulin Resistance at the Crossroad of Obesity with Associated Metabolic Abnormalities and Cognitive Dysfunction. International Journal of Molecular Sciences. 2021; 22(2):546. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020546
Chicago/Turabian StyleBarber, Thomas M., Ioannis Kyrou, Harpal S. Randeva, and Martin O. Weickert. 2021. "Mechanisms of Insulin Resistance at the Crossroad of Obesity with Associated Metabolic Abnormalities and Cognitive Dysfunction" International Journal of Molecular Sciences 22, no. 2: 546. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020546