Molecular Epidemiology of the Main Druggable Genetic Alterations in Non-Small Cell Lung Cancer

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Molecular Epidemiology of the Main Druggable Genes in LC
2.1. EGFR
2.2. ALK
2.3. ROS1
2.4. KRAS
2.5. BRAF
2.6. MET
2.7. HER2
3. Co-Occurrence of Druggable Genetic Alterations in NSCLC
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Cancer Observatory. Available online: https://gco.iarc.fr (accessed on 30 December 2020).
- Fidler-Benaoudia, M.M.; Torre, L.A.; Bray, F.; Ferlay, J.; Jemal, A. Lung cancer incidence in young women vs. young men: A systematic analysis in 40 countries. Int. J. Cancer 2020, 147, 811–819. [Google Scholar] [CrossRef]
- Jemal, A.; Miller, K.D.; Ma, J.; Siegel, R.L.; Fedewa, S.A.; Islami, F.; Devesa, S.S.; Thun, M.J. Higher lung cancer incidence in young women than young men in the United States. N. Engl. J. Med. 2018, 378, 1999–2009. [Google Scholar] [CrossRef]
- Budroni, M.; Cossu, A.; Paliogiannis, P.; Palmieri, G.; Attene, F.; Cesaraccio, R.; Tanda, F. Epidemiology of malignant pleural mesothelioma in the province of Sassari (Sardinia, Italy). A population-based report. Multidiscip. Respir. Med. 2013, 8, 45. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Cheng, B.; Xiong, S.; Li, C.; Liang, H.; Zhao, Y.; Li, J.; Shi, J.; Ou, L.; Chen, Z.; Liang, P.; et al. An annual review of the remarkable advances in lung cancer clinical research in 2019. J. Thorac. Dis. 2020, 12, 1056–1069. [Google Scholar] [CrossRef]
- Devesa, S.S.; Bray, F.; Vizcaino, A.P.; Parkin, D.M. International lung cancer trends by histologic type: Male:female differences diminishing and adenocarcinoma rates rising. Int. J. Cancer 2005, 117, 294–299. [Google Scholar] [CrossRef] [Green Version]
- Rolfo, C.; Castiglia, M.; Perez, A.; Reclusa, P.; Pauwels, P.; Sober, L.; Passiglia, F.; Russo, A. Liquid biopsy in Non-Small Cell Lung Cancer (NSCLC). In Liquid Biopsy in Cancer Patients, 3rd ed.; Giordano, A., Russo, A., Rolfo, C., Eds.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 103–115. ISBN 978-3-319-55661-1. [Google Scholar]
- Paliogiannis, P.; Attene, F.; Cossu, A.; Defraia, E.; Porcu, G.; Carta, A.; Sotgiu, M.I.; Pazzola, A.; Cordero, L.; Capelli, F.; et al. Impact of tissue type and content of neoplastic cells of samples on the quality of epidermal growth factor receptor mutation analysis among patients with lung adenocarcinoma. Mol. Med. Rep. 2015, 12, 187–191. [Google Scholar] [CrossRef]
- Wheeler, D.A.; Wang, L. From human genome to cancer genome: The first decade. Genome Res. 2013, 23, 1054–1062. [Google Scholar] [CrossRef] [Green Version]
- Arbour, K.C.; Riely, G.J. Systemic therapy for locally advanced and metastatic non-small cell lung cancer: A Review. JAMA 2019, 322, 764–774. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Singh, H.; Kinarivala, N.; Sharma, S. Multi-targeting anticancer agents: Rational approaches, synthetic routes and structure activity relationship. Anticancer Agents Med. Chem. 2019, 19, 842–874. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Bae, N.C.; Chae, M.H.; Lee, M.H.; Kim, K.M.; Lee, E.B.; Kim, C.H.; Park, T.I.; Han, S.B.; Jheon, S.; Jung, T.H.; et al. EGFR, ERBB2, and KRAS mutations in Korean non-small cell lung cancer patients. Cancer Genet. Cytogenet. 2007, 173, 107–113. [Google Scholar] [CrossRef]
- Masood, A.; Kancha, R.K.; Subramanian, J. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in non-small cell lung cancer harboring uncommon EGFR mutations: Focus on afatinib. Semin. Oncol. 2019, 46, 271–283. [Google Scholar] [CrossRef]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007, 11, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Tfayli, A.H.; Fakhri, G.B.; Al Assaad, M.S. Prevalence of the epidermal growth factor receptor mutations in lung adenocarcinoma patients from the Middle East region. Ann. Thorac. Med. 2019, 14, 173–178. [Google Scholar] [CrossRef]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Yuan, J.Q.; Wang, K.F.; Fu, X.H.; Han, X.R.; Threapleton, D.; Yang, Z.Y.; Mao, C.; Tang, J.L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [Green Version]
- Sekine, I.; Yamamoto, N.; Nishio, K.; Saijo, N. Emerging ethnic differences in lung cancer therapy. Br. J. Cancer 2008, 99, 1757–1762. [Google Scholar] [CrossRef] [Green Version]
- Tsao, A.S.; Tang, X.M.; Sabloff, B.; Xiao, L.; Shigematsu, H.; Roth, J. Clinicopathologic characteristics of the EGFR gene mutation in non-small cell lung cancer. J. Thorac. Oncol. 2006, 1, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Benbrahim, Z.; Antonia, T.; Mellas, N. EGFR mutation frequency in Middle East and African non-small cell lung cancer patients: A systematic review and meta-analysis. BMC Cancer 2018, 18, 891. [Google Scholar] [CrossRef]
- Colombino, M.; Paliogiannis, P.; Cossu, A.; Santeufemia, D.A.; Sardinian Lung Cancer (SLC) Study Group; Sini, M.C.; Casula, M.; Palomba, G.; Manca, A.; Pisano, M.; et al. EGFR, KRAS, BRAF, ALK, and cMET genetic alterations in 1440 Sardinian patients with lung adenocarcinoma. BMC Pulm. Med. 2019, 19, 209. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–744. [Google Scholar] [CrossRef]
- Benlloch, S.; Botero, M.L.; Beltran-Alamillo, J.; Mayo, C.; Gimenez-Capitán, A.; de Aguirre, I.; Queralt, C.; Ramirez, J.L.; Ramón y Cajal, S.; Klughammer, B.; et al. Clinical validation of a PCR assay for the detection of EGFR mutations in non-small-cell lung cancer: Retrospective testing of specimens from the EURTAC trial. PLoS ONE 2014, 9, e89518. [Google Scholar] [CrossRef]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Chapman, A.M.; Sun, K.Y.; Ruestow, P.; Cowan, D.M.; Madl, A.K. Lung cancer mutation profile of EGFR, ALK, and KRAS: Meta-analysis and comparison of never and ever smokers. Lung Cancer 2016, 102, 122–134. [Google Scholar] [CrossRef]
- Marchetti, A.; Martella, C.; Felicioni, L.; Barassi, F.; Salvatore, S.; Chella, A.; Camplese, P.P.; Iarussi, T.; Mucilli, F.; Mezzetti, A.; et al. EGFR mutations in non-small-cell lung cancer: Analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J. Clin. Oncol. 2005, 23, 857–865. [Google Scholar] [CrossRef]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Kuwano, H.; Takahashi, T.; Mitsudomi, T. Mutations of the epidermal growth factor receptor gene in lung cancer: Biological and clinical implications. Cancer Res. 2004, 64, 8919–8923. [Google Scholar] [CrossRef] [Green Version]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [Green Version]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Cheng, C.; Wang, R.; Li, Y.; Pan, Y.; Zhang, Y.; Li, H.; Zheng, D.; Zheng, S.; Shen, X.; Sun, Y.; et al. EGFR exon 18 mutations in East Asian patients with lung adenocarcinomas: A comprehensive investigation of prevalence, clinicopathologic characteristics and prognosis. Sci. Rep. 2015, 5, 13959. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Lo, P.C.; Nishino, M.; Dahlberg, S.E.; Lindeman, N.I.; Butaney, M.; Jackman, D.M.; Johnson, B.E.; Jänne, P.A. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J. Thorac. Oncol. 2013, 8, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Xu, Y.; Zhao, J.; Zhong, W.; Zhang, L.; Bi, Y.; Wang, M. Concurrent driver gene mutations as negative predictive factors in epidermal growth factor receptor-positive non-small cell lung cancer. EBioMedicine 2019, 42, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Kawaguchi, T.; Isa, S.; Ando, M.; Tamiya, A.; Kubo, A.; Saka, H.; Takeo, S.; Adachi, H.; Tagawa, T.; et al. Ultra-sensitive detection of the pretreatment EGFR T790M mutation in non-small cell lung cancer patients with an EGFR-activating mutation using droplet digital PCR. Clin. Cancer Res. 2015, 2, 3552–3560. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Molina-Vila, M.A.; Ruan, S.Y.; Su, K.Y.; Liao, W.Y.; Yu, K.; Ho, C.C.; Shih, J.Y.; Yu, C.J.; Yang, J.C.; et al. Coexistence of EGFR T790M mutation and common activating mutations in pretreatment non-small cell lung cancer: A systematic review and meta-analysis. Lung Cancer 2016, 94, 46–53. [Google Scholar] [CrossRef]
- Inamura, K.; Takeuchi, K.; Togashi, Y.; Nomura, K.; Ninomiya, H.; Okui, M.; Satoh, Y.; Okumura, S.; Nakagawa, K.; Soda, M.; et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J. Thorac. Oncol. 2008, 3, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Rosas, G.; Ruiz, R.; Araujo, J.M.; Pinto, J.A.; Mas, L. ALK rearrangements: Biology, detection and opportunities of therapy in non-small cell lung cancer. Crit. Rev. Oncol. Hematol. 2019, 136, 48–55. [Google Scholar] [CrossRef]
- Noh, K.W.; Lee, M.S.; Lee, S.E.; Song, J.Y.; Shin, H.T.; Kim, Y.J.; Oh, D.Y.; Jung, K.; Sung, M.; Kim, M.; et al. Molecular breakdown: A comprehensive view of anaplastic lymphoma kinase (ALK)-rearranged non-small cell lung cancer. J. Pathol. 2017, 243, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- He, Y.; Sun, L.Y.; Gong, R.; Liu, Q.; Long, Y.K.; Liu, F.; Wang, F. The prevalence of EML4-ALK variants in patients with non-small-cell lung cancer: A systematic review and meta-analysis. Biomark. Med. 2019, 13, 1035–1044. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, Y.; Li, Y.; Hu, H.; Wang, L.; Li, H.; Wang, R.; Ye, T.; Luo, X.; Zhang, Y.; et al. ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: A comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung Cancer 2014, 84, 121–126. [Google Scholar] [CrossRef]
- Takahashi, T.; Sonobe, M.; Kobayashi, M.; Yoshizawa, A.; Menju, T.; Nakayama, E.; Mino, N.; Iwakiri, S.; Sato, K.; Miyahara, R.; et al. Clinicopathologic features of non-small-cell lung cancer with EML4-ALK fusion gene. Ann. Surg. Oncol. 2010, 17, 889–897. [Google Scholar] [CrossRef] [Green Version]
- McKeage, M.J.; Tin Tin, S.; Khwaounjoo, P.; Sheath, K.; Dixon-McIver, A.; Ng, D.; Sullivan, R.; Cameron, L.; Shepherd, P.; Laking, G.R.; et al. Screening for Anaplastic Lymphoma Kinase (ALK) gene rearrangements in non-small cell lung cancer (NSCLC) in New Zealand. Intern. Med. J. 2020, 50, 716–725. [Google Scholar] [CrossRef]
- Martelli, M.P.; Sozzi, G.; Hernandez, L.; Pettirossi, V.; Navarro, A.; Conte, D.; Gasparini, P.; Perrone, F.; Modena, P.; Pastorino, U.; et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am. J. Pathol. 2009, 174, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef] [Green Version]
- Hou, H.; Zhang, C.; Qi, X.; Zhou, L.; Liu, D.; Lv, H.; Li, T.; Sun, D.; Zhang, X. Distinctive targetable genotypes of younger patients with lung adenocarcinoma: A cBioPortal for cancer genomics data base analysis. Cancer Biol. Ther. 2020, 21, 26–33. [Google Scholar] [CrossRef]
- Lovly, C.M.; Pao, W. Escaping ALK inhibition: Mechanisms of and strategies to overcome resistance. Sci. Transl. Med. 2012, 4, 120ps2. [Google Scholar] [CrossRef]
- Takeda, M.; Okamoto, I.; Sakai, K.; Kawakami, H.; Nishio, K.; Nakagawa, K. Clinical outcome for EML4-ALK-positive patients with advanced non-small-cell lung cancer treated with first-line platinum-based chemotherapy. Ann. Oncol. 2012, 23, 2931–2936. [Google Scholar] [CrossRef]
- Nagarajan, L.; Louie, E.; Tsujimoto, Y.; Balduzzi, P.C.; Huebner, K.; Croce, C.M. The human c-ros gene (ROS) is located at chromosome region 6q16–6q22. Proc. Natl. Acad. Sci. USA 1986, 83, 6568–6572. [Google Scholar] [CrossRef] [Green Version]
- Acquaviva, J.; Wong, R.; Charest, A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim. Biophys. Acta 2009, 1795, 37–52. [Google Scholar] [CrossRef]
- Cai, W.; Li, X.; Su, C.; Fan, L.; Zheng, L.; Fei, K.; Zhou, C.; Manegold, C.; Schmid-Bindert, G. ROS1 fusions in Chinese patients with non-small-cell lung cancer. Ann. Oncol. 2013, 24, 1822–1827. [Google Scholar] [CrossRef]
- Chin, L.P.; Soo, R.A.; Soong, R.; Ou, S.H. Targeting ROS1 with anaplastic lymphoma kinase inhibitors: A promising therapeutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J. Thorac. Oncol. 2012, 7, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Pal, P.; Khan, Z. ROS1. J. Clin. Pathol. 2017, 70, 1001–1009. [Google Scholar] [CrossRef]
- Charest, A.; Wilker, E.W.; McLaughlin, M.E.; Lane, K.; Gowda, R.; Coven, S.; McMahon, K.; Kovach, S.; Feng, Y.; Yaffe, M.B.; et al. ROS fusion tyrosine kinase activates a SH2 domain-containing phosphatase-2/phosphatidylinositol 3-kinase/mammalian target of rapamycin signaling axis to form glioblastoma in mice. Cancer Res. 2006, 66, 7473–7481. [Google Scholar] [CrossRef] [Green Version]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wu, C.; Ding, W.; Zhang, Z.; Qiu, X.; Mu, D.; Zhang, H.; Xi, Y.; Zhou, J.; Ma, L.; et al. Prevalence of ROS1 fusion in Chinese patients with non-small cell lung cancer. Thorac. Cancer 2019, 10, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Zhan, P.; Zhang, X.; Lv, T.; Song, Y. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2015, 4, 300–309. [Google Scholar]
- Song, Z.; Yu, X.; Zhang, Y. Clinicopathological characteristics and survival of ALK, ROS1 and RET rearrangements in non-adenocarcinoma non-small cell lung cancer patients. Cancer Biol. Ther. 2017, 18, 883–887. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Tseng, D.; Yoda, S.; Dagogo-Jack, I.; Friboulet, L.; Lin, J.J.; Hubbeling, H.G.; Dardaei, L.; Farago, A.F.; Schultz, K.R.; et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-positive non-small-cell lung cancer. JCO Precis. Oncol. 2017, 1, 1–13. [Google Scholar] [CrossRef]
- Shaw, A.T.; Ou, S.H.; Bang, Y.J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, K.; Patell, R.; Rangachari, D.; Costa, D.B. Targeting ROS1 rearrangements in non-small cell lung cancer with crizotinib and other kinase inhibitors. Transl. Cancer Res. 2018, 7, S779–S786. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Paliogiannis, P.; Cossu, A.; Tanda, F.; Palmieri, G.; Palomba, G. KRAS mutational concordance between primary and metastatic colorectal adenocarcinoma. Oncol. Lett. 2014, 8, 1422–1426. [Google Scholar] [CrossRef] [Green Version]
- Polom, K.; Das, K.; Marrelli, D.; Roviello, G.; Pascale, V.; Voglino, C.; Rho, H.; Tan, P.; Roviello, F. KRAS mutation in gastric cancer and prognostication associated with microsatellite instability status. Pathol. Oncol. Res. 2019, 25, 333–340. [Google Scholar] [CrossRef]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted therapies in cholangiocarcinoma: Emerging evidence from clinical trials. Medicina (Kaunas) 2019, 55, 42. [Google Scholar] [CrossRef] [Green Version]
- Sini, M.C.; Doneddu, V.; Paliogiannis, P.; Casula, M.; Colombino, M.; Botti, G.; Ascierto, P.A.; Lissia, A.; Cossu, A.; Palmieri, G. Genetic alterations in main candidate genes during melanoma progression. Oncotarget 2018, 9, 8531–8541. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Freixinos, V.; Lheureux, S.; Mandilaras, V.; Clarke, B.; Dhani, N.C.; Mackay, H.; Butler, M.O.; Wang, L.; Siu, L.L.; Kamel-Reid, S.; et al. Impact of somatic molecular profiling on clinical trial outcomes in rare epithelial gynecologic cancer patients. Gynecol. Oncol. 2019, 153, 304–311. [Google Scholar] [CrossRef]
- Martin, P.; Leighl, N.B.; Tsao, M.S.; Shepherd, F.A. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J. Thorac. Oncol. 2013, 8, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Forbes, S.; Clements, J.; Dawson, E.; Bamford, S.; Webb, T.; Dogan, A.; Flanagan, A.; Teague, J.; Wooster, R.; Futreal, P.A.; et al. COSMIC 2005. Br. J. Cancer 2006, 94, 318–322. [Google Scholar] [CrossRef] [Green Version]
- Mills, N.E.; Fishman, C.L.; Rom, W.N.; Dubin, N.; Jacobson, D.R. Increased prevalence of K-ras oncogene mutations in lung adenocarcinoma. Cancer Res. 1995, 55, 1444–1447. [Google Scholar]
- Jiang, A.G.; Lu, H.Y. k-RAS mutations in non-small cell lung cancer patients treated with TKIs among smokers and non-smokers: A meta-analysis. Contemp. Oncol. (Pozn.) 2016, 20, 124–129. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Ren, Y.; Fang, Z.; Li, C.; Fang, R.; Gao, B.; Han, X.; Tian, W.; Pao, W.; Chen, H.; et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J. Clin. Oncol. 2010, 28, 4616–4620. [Google Scholar] [CrossRef] [Green Version]
- Smits, A.J.; Kummer, J.A.; Hinrichs, J.W.; Herder, G.J.; Scheidel-Jacobse, K.C.; Jiwa, N.M.; Ruijter, T.E.; Nooijen, P.T.; Looijen-Salamon, M.G.; Ligtenberg, M.J.; et al. EGFR and KRAS mutations in lung carcinomas in the Dutch population: Increased EGFR mutation frequency in malignant pleural effusion of lung adenocarcinoma. Cell. Oncol. (Dordr.) 2012, 35, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Fong, K.M.; Zimmerman, P.V.; Smith, P.J. KRAS codon 12 mutations in Australian non-small cell lung cancer. Aust. N. Z. J. Med. 1998, 28, 184–189. [Google Scholar] [CrossRef]
- Riely, G.J.; Marks, J.; Pao, W. KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 2009, 6, 201–205. [Google Scholar] [CrossRef]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linardou, H.; Dahabreh, I.J.; Kanaloupiti, D.; Siannis, F.; Bafaloukos, D.; Kosmidis, P.; Papadimitriou, C.A.; Murray, S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: A systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008, 9, 962–972. [Google Scholar] [CrossRef]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Brägelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Mascaux, C.; Iannino, N.; Martin, B.; Paesmans, M.; Berghmans, T.; Dusart, M.; Lothaire, P.; Meert, A.P.; Noel, S.; Lafitte, J.J.; et al. The role of RAS oncogene in survival of patients with lung cancer: A systematic review of the literature with meta-analysis. Br. J. Cancer 2005, 92, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winton, T.; Livingston, R.; Johnson, D.; Rigas, J.; Johnston, M.; Butts, C.; Cormier, Y.; Cormier, Y.; Goss, G.; Inculet, R.; et al. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N. Engl. J. Med. 2005, 352, 2589–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sithanandam, G.; Kolch, W.; Duh, F.M.; Rapp, U.R. Complete coding sequence of a human B-raf cDNA and detection of B-raf protein kinase with isozyme specific antibodies. Oncogene 1990, 5, 1775–1780. [Google Scholar]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Manca, A.; Paliogiannis, P.; Colombino, M.; Casula, M.; Lissia, A.; Botti, G.; Caracò, C.; Ascierto, P.A.; Sini, M.C.; Palomba, G.; et al. Mutational concordance between primary and metastatic melanoma: A next-generation sequencing approach. J. Transl. Med. 2019, 17, 289. [Google Scholar] [CrossRef] [Green Version]
- Colombino, M.; Lissia, A.; Franco, R.; Botti, G.; Ascierto, P.A.; Manca, A.; Sini, M.C.; Pisano, M.; Paliogiannis, P.; Tanda, F.; et al. Unexpected distribution of cKIT and BRAF mutations among southern Italian patients with sinonasal melanoma. Dermatology 2013, 226, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, M.I.; Junit, S.M.; Ng, K.L.; Jayapalan, J.J.; Karikalan, B.; Hashim, O.H. Papillary thyroid cancer: Genetic alterations and molecular biomarker investigations. Int. J. Med. Sci. 2019, 16, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomba, G.; Doneddu, V.; Cossu, A.; Paliogiannis, P.; Manca, A.; Casula, M.; Colombino, M.; Lanzillo, A.; Defraia, E.; Pazzola, A.; et al. Prognostic impact of KRAS, NRAS, BRAF, and PIK3CA mutations in primary colorectal carcinomas: A population-based study. J. Transl. Med. 2016, 14, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracht, J.W.P.; Karachaliou, N.; Bivona, T.; Lanman, R.B.; Faull, I.; Nagy, R.J.; Drozdowskyj, A.; Berenguer, J.; Fernandez-Bruno, M.; Molina-Vila, M.A.; et al. BRAF mutations classes I, II, and III in NSCLC patients included in the SLLIP trial: The need for a new pre-clinical treatment rationale. Cancers (Basel) 2019, 11, 1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Zhang, H.; Ding, H.; Qian, J.; Lizaso, A.; Lin, J.; Han-Zhang, H.; Xiang, J.; Li, Y.; Zhu, H.; et al. The association between BRAF mutation class and clinical features in BRAF-mutant Chinese non-small cell lung cancer patients. J. Transl. Med. 2019, 17, 298. [Google Scholar] [CrossRef] [Green Version]
- Couraud, S.; Barlesi, F.; Fontaine-Deraluelle, C.; Debieuvre, D.; Merlio, J.P.; Moreau, L.; Beau-Faller, M.; Veillon, R.; Mosser, J.; Al Freijat, F.; et al. Clinical outcomes of non-small-cell lung cancer patients with BRAF mutations: Results from the French Cooperative Thoracic Intergroup biomarkers France study. Eur. J. Cancer 2019, 116, 86–97. [Google Scholar] [CrossRef]
- Leonetti, A.; Facchinetti, F.; Rossi, G.; Minari, R.; Conti, A.; Friboulet, L.; Tiseo, M.; Planchard, D. BRAF in non-small cell lung cancer (NSCLC): Pickaxing another brick in the wall. Cancer Treat. Rev. 2018, 66, 82–94. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Martinez, P.; Yeap, B.Y.; Ambrogio, C.; Ferris, L.A.; Lydon, C.; Nguyen, T.; Jessop, N.A.; Iafrate, A.J.; Johnson, B.E.; et al. Impact of BRAF mutation class on disease characteristics and clinical outcomes in BRAF-mutant lung cancer. Clin. Cancer Res. 2019, 25, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Nguyen, T.; Jessop, N.A.; Iafrate, A.J.; Johnson, B.E.; et al. Author correction: Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 1022. [Google Scholar] [CrossRef] [Green Version]
- Salgia, R. MET in lung cancer: Biomarker selection based on scientific rationale. Mol. Cancer Ther. 2017, 16, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschard, P.; Fournier, T.M.; Lamorte, L.; Naujokas, M.A.; Band, H.; Langdon, W.Y.; Park, M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell. 2001, 8, 995–1004. [Google Scholar] [CrossRef]
- Song, Z.; Wang, H.; Yu, Z.; Lu, P.; Xu, C.; Chen, G.; Zhang, Y. De novo MET amplification in Chinese patients with non-small-cell lung cancer and treatment efficacy with Crizotinib: A multicenter retrospective study. Clin. Lung Cancer 2019, 20, e171–e176. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Cappuzzo, F.; Ou, S.I.; Camidge, D.R. Targeting MET in lung cancer: Will expectations finally be MET? J. Thorac. Oncol. 2017, 12, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.W.; Seo, J.W.; Chung, D.H. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, H.; Okamoto, I.; Okamoto, W.; Tanizaki, J.; Nakagawa, K.; Nishio, K. Targeting MET amplification as a new oncogenic driver. Cancers (Basel) 2014, 6, 1540–1552. [Google Scholar] [CrossRef] [Green Version]
- Baldacci, S.; Kherrouche, Z.; Cockenpot, V.; Stoven, L.; Copin, M.C.; Werkmeister, E.; Marchand, N.; Kyheng, M.; Tulasne, D.; Cortot, A.B. MET amplification increases the metastatic spread of EGFR-mutated NSCLC. Lung Cancer 2018, 125, 57–67. [Google Scholar] [CrossRef]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Landi, L.; Cappuzzo, F. HER2 and lung cancer. Expert. Rev. Anticancer Ther. 2013, 13, 1219–1228. [Google Scholar] [CrossRef]
- Li, X.; Zhao, C.; Su, C.; Ren, S.; Chen, X.; Zhou, C. Epidemiological study of HER-2 mutations among EGFR wild-type lung adenocarcinoma patients in China. BMC Cancer 2016, 16, 828. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigematsu, H.; Takahashi, T.; Nomura, M.; Majmudar, K.; Suzuki, M.; Lee, H.; Nguyen, T.; Jessop, N.A.; Iafrate, A.J.; Johnson, B.E.; et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005, 65, 1642–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Yu, X.; Shi, Z.; Zhao, J.; Zhang, Y. HER2 mutations in Chinese patients with non-small cell lung cancer. Oncotarget 2016, 7, 78152–78158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazières, J.; Peters, S.; Lepage, B.; Cortot, A.B.; Barlesi, F.; Beau-Faller, M.; Besse, B.; Blons, H.; Mansuet-Lupo, A.; Urban, T.; et al. Lung cancer that harbors an HER2 mutation: Epidemiologic characteristics and therapeutic perspectives. J. Clin. Oncol. 2013, 31, 1997–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, S.; Wang, R.; Pan, Y.; Yu, S.; Shen, X.; Li, Y.; Sun, Y.; Chen, H. Clinicopathologic characteristics of patients with HER2 insertions in non-small cell lung cancer. Ann. Surg. Oncol. 2017, 24, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Ross, D.S.; Aisner, D.L.; Chaft, J.E.; Hsu, M.; Kako, S.L.; Kris, M.G.; Varella-Garcia, M.; Arcila, M.E. HER2 Amplification and HER2 mutation are distinct molecular targets in lung cancers. J. Thorac. Oncol. 2016, 11, 414–419. [Google Scholar] [CrossRef]
- Suzuki, M.; Shiraishi, K.; Yoshida, A.; Shimada, Y.; Suzuki, K.; Asamura, H.; Furuta, K.; Kohno, T.; Tsuta, K. HER2 gene mutations in non-small cell lung carcinomas: Concurrence with Her2 gene amplification and Her2 protein expression and phosphorylation. Lung Cancer 2015, 87, 14–22. [Google Scholar] [CrossRef]
- Fan, J.; Dai, X.; Wang, Z.; Huang, B.; Shi, H.; Luo, D.; Zhang, J.; Cai, W.; Nie, X.; Hirsch, F.R. Concomitant EGFR mutation and EML4-ALK rearrangement in lung adenocarcinoma is more frequent in multifocal lesions. Clin. Lung Cancer 2019, 20, e517–e530. [Google Scholar] [CrossRef] [Green Version]
- Galetta, D.; Catino, A.; Misino, A. Concomitant EGFR mutations/ALK rearrangements: Beyond a simple dual target. Transl. Lung Cancer Res. 2016, 5, 143–144. [Google Scholar]
- Ulivi, P.; Chiadini, E.; Dazzi, C.; Dubini, A.; Costantini, M.; Medri, L.; Puccetti, M.; Capelli, L.; Calistri, D.; Verlicchi, A.; et al. Nonsquamous, non-small-cell lung cancer patients who carry a double mutation of EGFR, EML4-ALK or KRAS: Frequency, clinical-pathological characteristics, and response to therapy. Clin. Lung Cancer 2016, 17, 384–390. [Google Scholar] [CrossRef]
- Yang, J.J.; Zhang, X.C.; Su, J.; Xu, C.R.; Zhou, Q.; Tian, H.X.; Xie, Z.; Chen, H.J.; Huang, Y.S.; Jiang, B.Y.; et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: Diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin. Cancer Res. 2014, 20, 1383–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, G.L.; Imbimbo, M.; Corrao, G.; Proto, C.; Signorelli, D.; Vitali, M.; Ganzinelli, M.; Botta, L.; Zilembo, N.; de Braud, F.; et al. Concomitant EML4-ALK rearrangement and EGFR mutation in non-small cell lung cancer patients: A literature review of 100 cases. Oncotarget 2017, 8, 59889–59900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aisner, D.L.; Sholl, L.M.; Berry, L.D.; Rossi, M.R.; Chen, H.; Fujimoto, J.; Moreira, A.L.; Ramalingam, S.S.; Villaruz, L.C.; Otterson, G.A.; et al. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations—The Lung Cancer Mutation Consortium (LCMC2). Clin. Cancer Res. 2018, 24, 1038–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Pan, Y.; Cheng, C.; Zheng, D.; Zhang, Y.; Gao, Z.; Fu, F.; Li, H.; Zheng, S.; Zhuge, L.; et al. EGFR-mutant lung adenocarcinoma harboring co-mutational tumor suppressor genes predicts poor prognosis. J. Cancer Res. Clin. Oncol. 2020, 146, 1781–1789. [Google Scholar] [CrossRef]
- Sun, H.; Liu, S.Y.; Zhou, J.Y.; Xu, J.T.; Zhang, H.K.; Yan, H.H.; Huan, J.J.; Dai, P.P.; Xu, C.R.; Su, J.; et al. Specific TP53 subtype as biomarker for immune checkpoint inhibitors in lung adenocarcinoma. EBioMedicine 2020, 60, 102990. [Google Scholar] [CrossRef]
- Myall, N.J.; Henry, S.; Wood, D.; Neal, J.W.; Han, S.S.; Padda, S.K.; Wakelee, H.A. Natural disease history, outcomes, and co-mutations in a series of patients with BRAF-mutated non-small-cell lung cancer. Clin. Lung Cancer 2019, 20, e208–e217. [Google Scholar] [CrossRef]


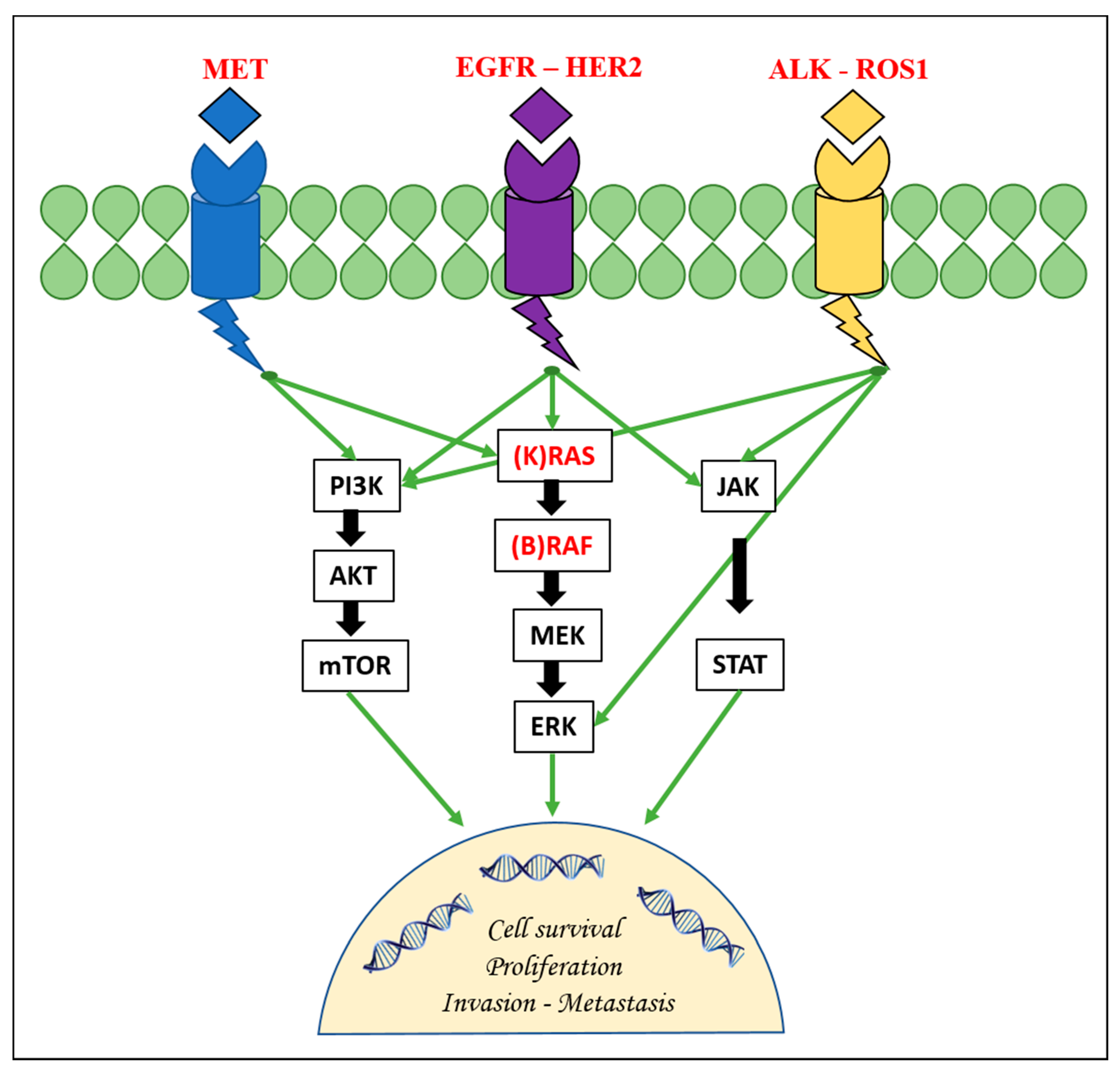{kind=link}
| Gene | Most Common Variants | Prevalence | Age | Gender | Smoking | Histology | Prognostic Significance |
|---|---|---|---|---|---|---|---|
| EGFR | Mutations in exons 19 and 21 | 10–16% in Western populations, 40–50% in Asians. | Younger patients | Females | Never smokers | Adenocarcinoma | Response to specific TKIs, T790M predictor of resistance |
| ALK | EML4-ALK variants | 1–10% of NSCLC | Younger patients | Females? Not clear | Never smokers | Adenocarcinoma | Aggressive tumors, response to specific TKIs |
| ROS1 | CD74-ROS1 variants | 0.9–2.6% of NSCLC | Younger patients | Females | Never smokers | Adenocarcinoma | Less aggressive tumors, response to specific TKIs |
| KRAS | Mutations in codons 12 and 13 | 30–40% of NSCLC, more common in Caucasians | Older ages | Males? Not clear | Smokers | Adenocarcinoma | Poor prognosis or response to TKIs? Not clear |
| BRAF | Mutations in exon 15 | 2–4% of NSCLC | No predilection | V600E in females and others in males | Smokers | Adenocarcinoma | Aggressive tumors and poor prognosis? Not clear. Response to BRAF inhibitors. |
| MET | Mutations in exon 14, amplification | Mutations in 1–10% of NSCLC, amplification in 5–22% | Older ages | Not clear | Smokers | Adenocarcinoma and SCC | Resistance to EGFR-TKIs. Response to MET inhibitors. |
| HER2 | Mutations in exons 18–21, amplification | Mutations in 2–3% and amplifications in 2–5% of adenocarcinomas | Not clear | Mutations in females and amplifications in males | Mutations in never smokers and amplifications in ex-smokers | Adenocarcinoma Rare in SCC | Resistance to EGFR-TKIs? Not clear |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fois, S.S.; Paliogiannis, P.; Zinellu, A.; Fois, A.G.; Cossu, A.; Palmieri, G. Molecular Epidemiology of the Main Druggable Genetic Alterations in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 612. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020612
Fois SS, Paliogiannis P, Zinellu A, Fois AG, Cossu A, Palmieri G. Molecular Epidemiology of the Main Druggable Genetic Alterations in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2021; 22(2):612. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020612
Chicago/Turabian StyleFois, Sara S., Panagiotis Paliogiannis, Angelo Zinellu, Alessandro G. Fois, Antonio Cossu, and Giuseppe Palmieri. 2021. "Molecular Epidemiology of the Main Druggable Genetic Alterations in Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 22, no. 2: 612. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020612