Effects of the Mixing Protocol on the Self-Assembling Process of Water Soluble Porphyrins

 , , and
, , and
Abstract
:1. Introduction
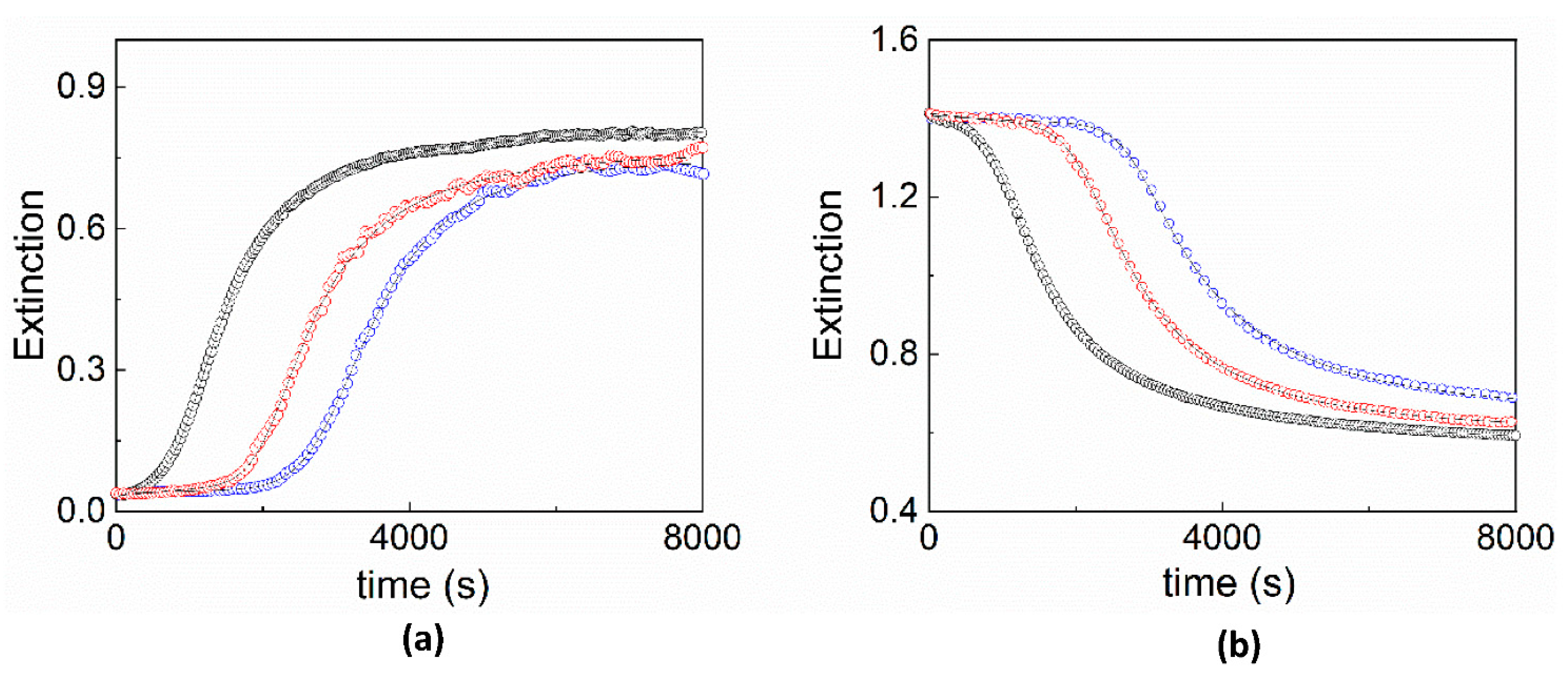
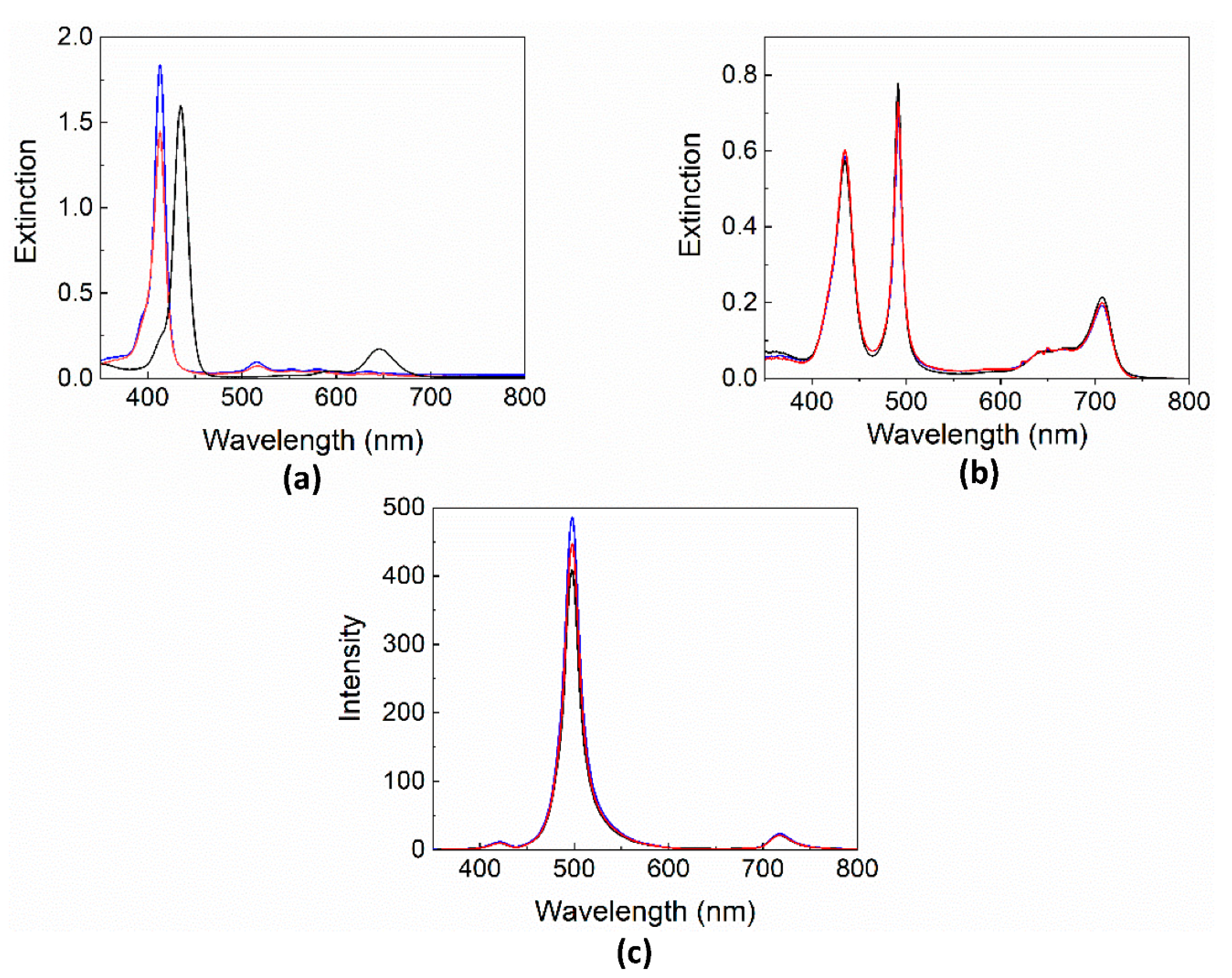
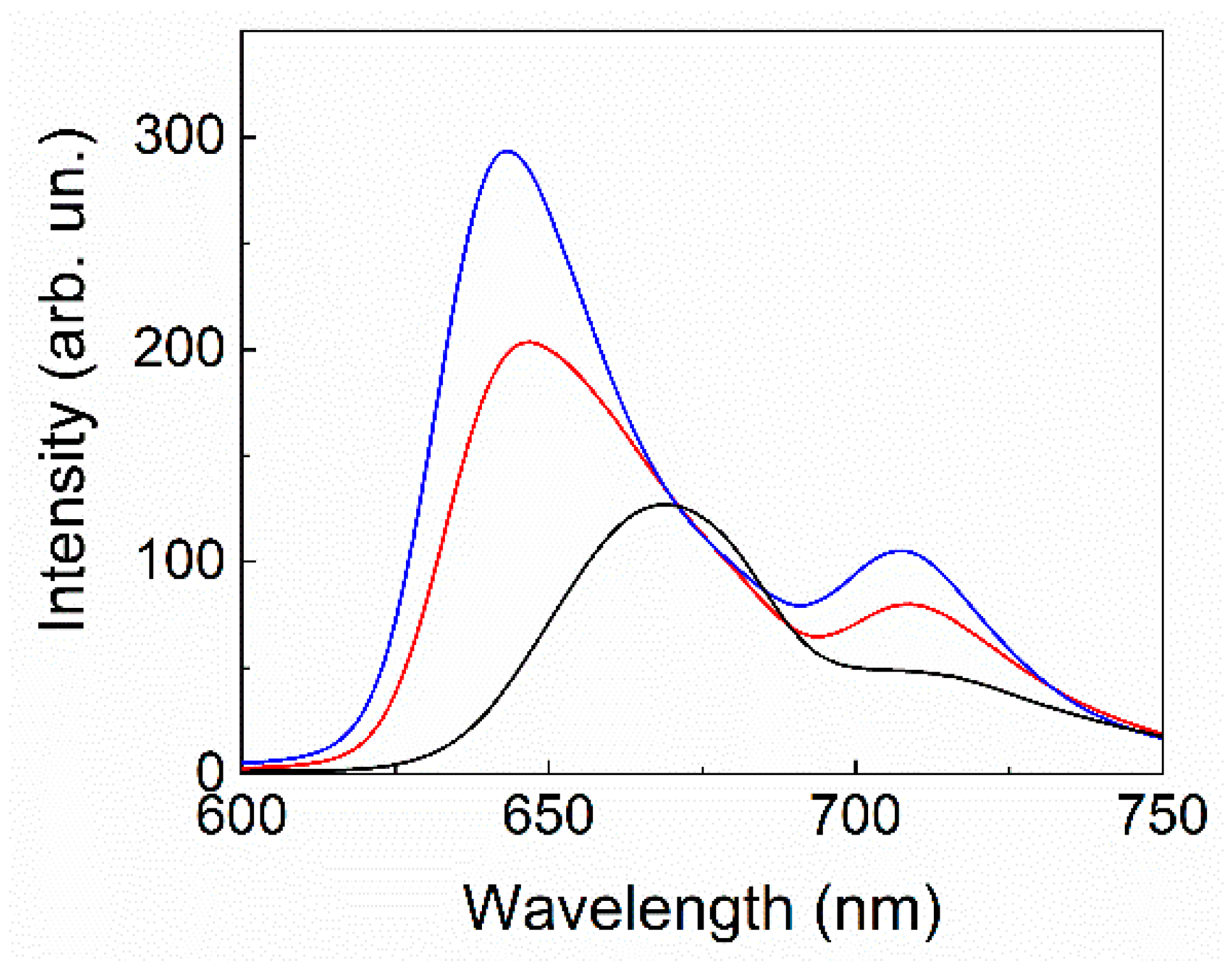
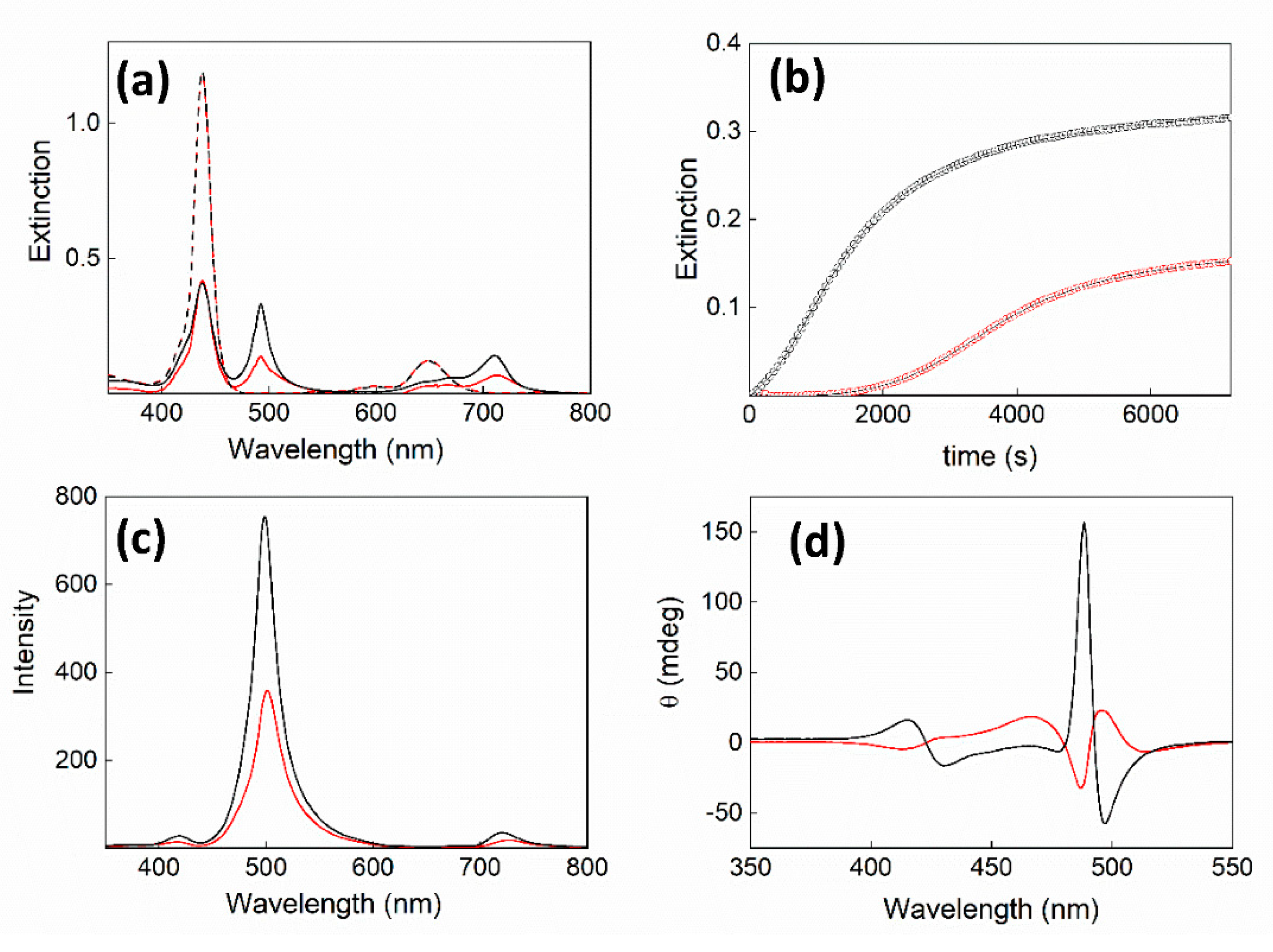
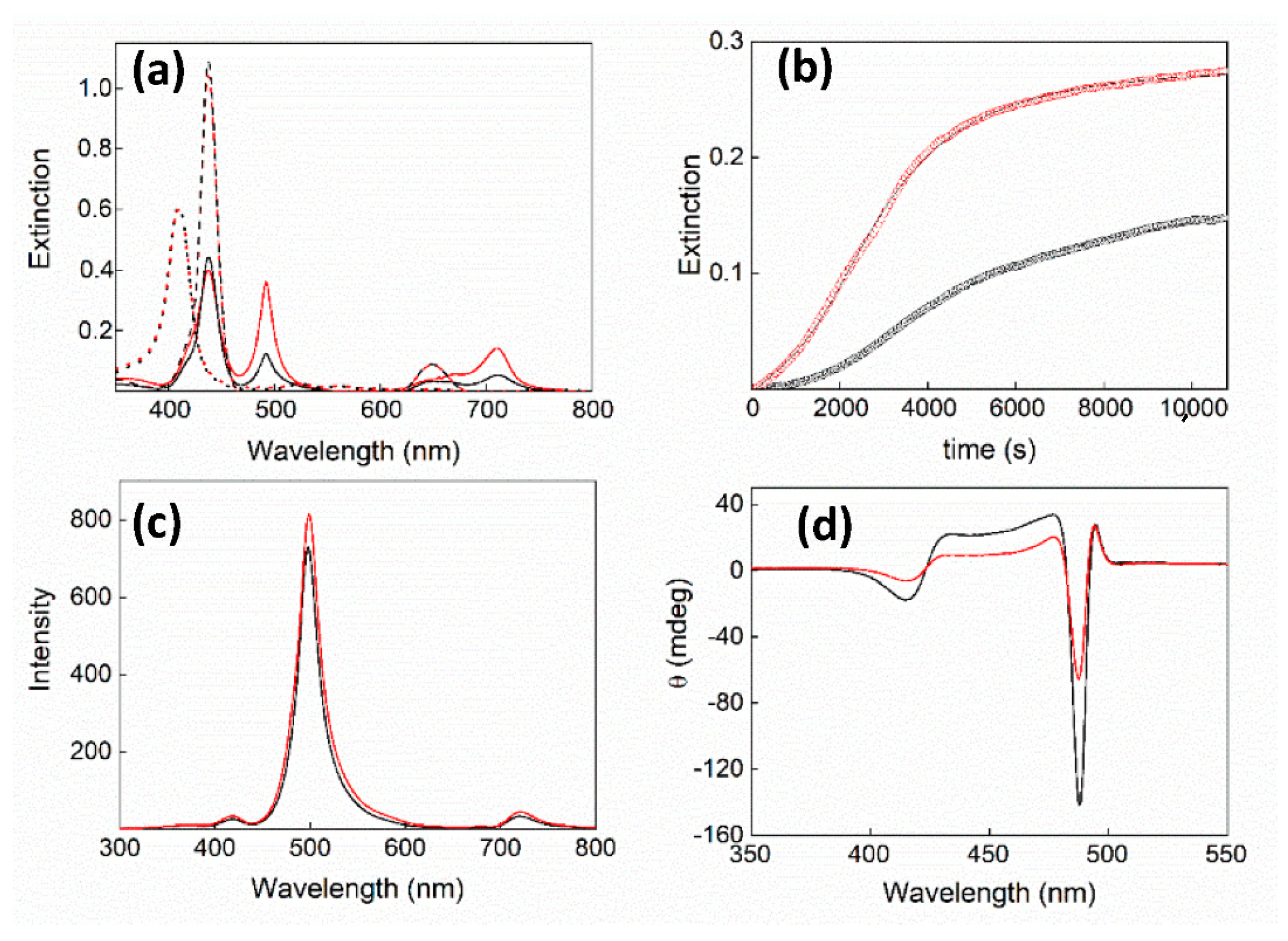
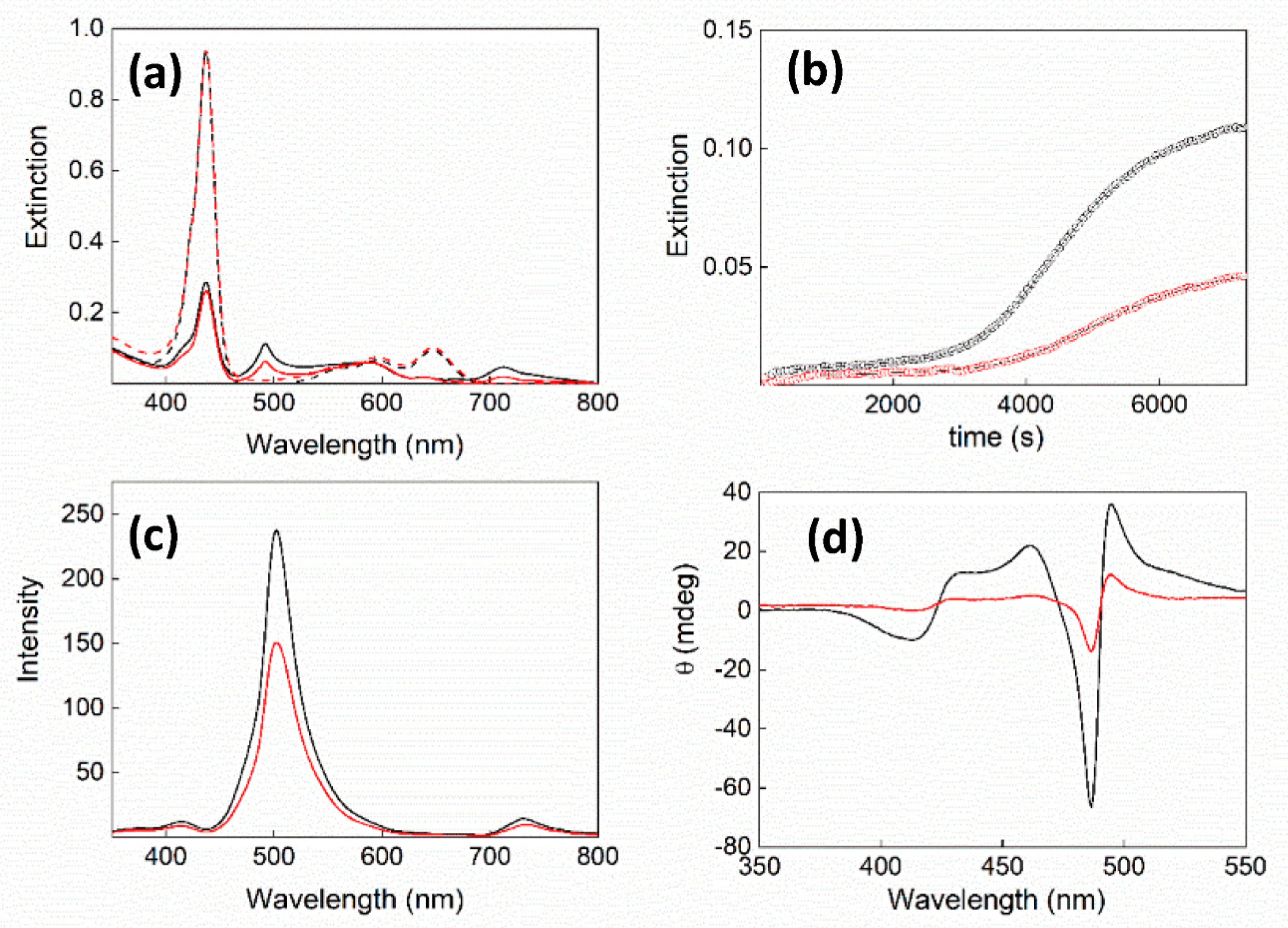
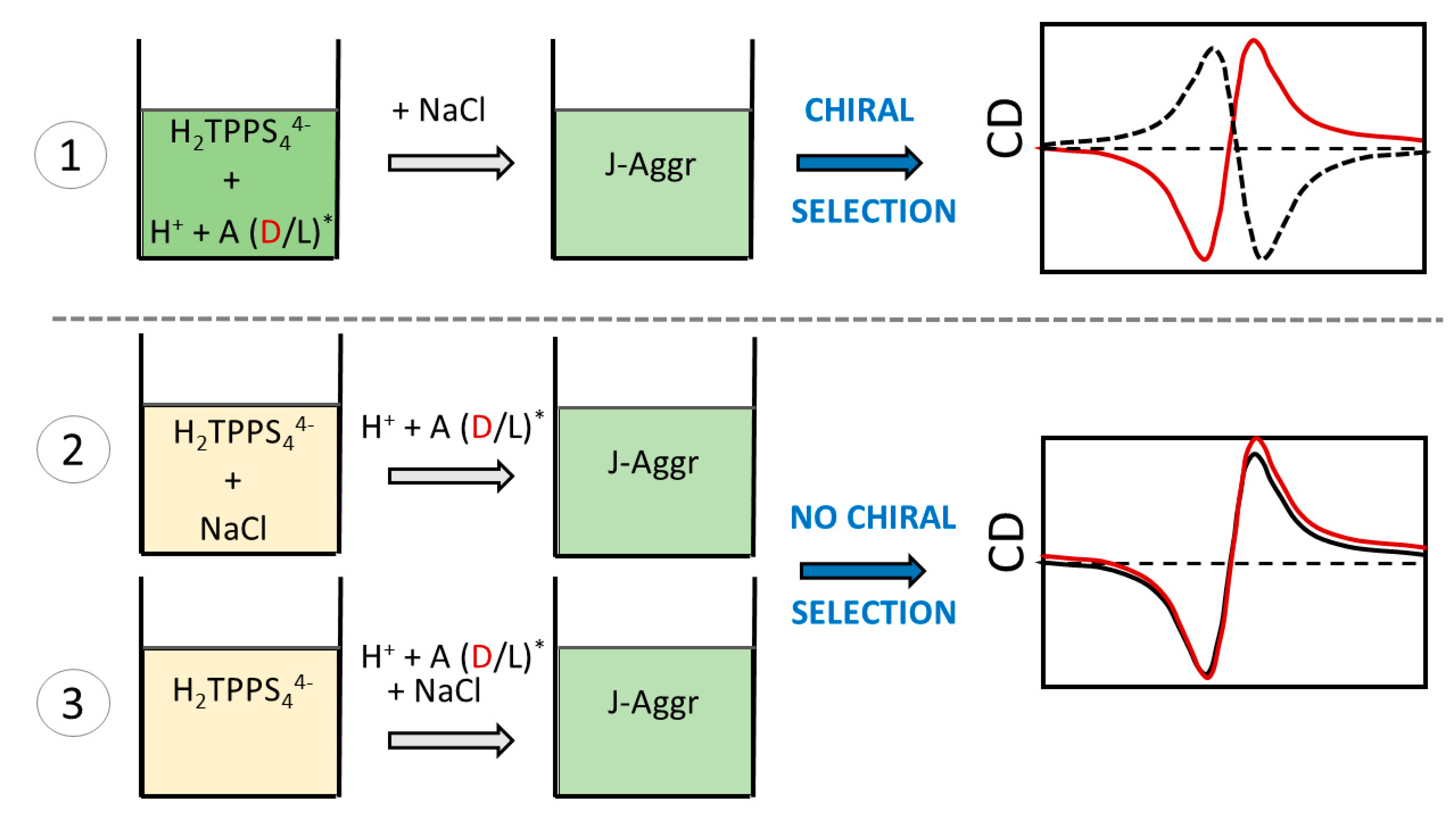
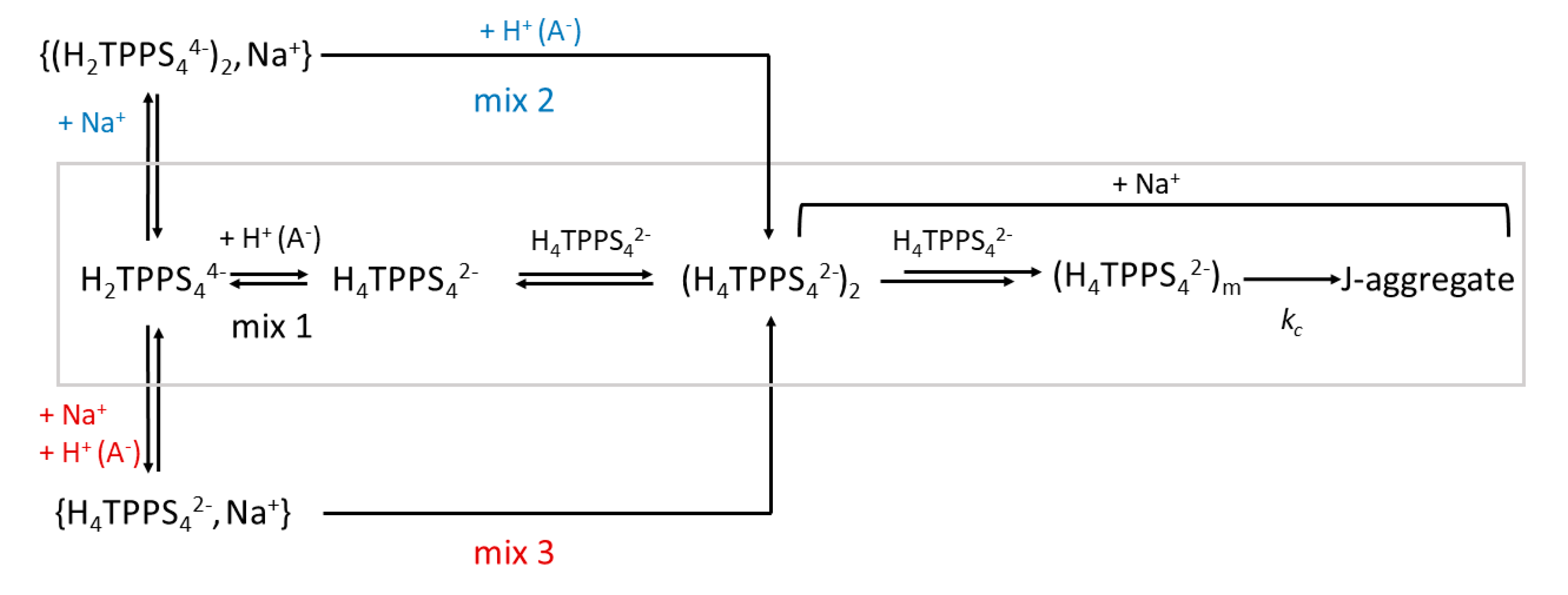
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- White, W.I. 7—Aggregation of Porphyrins and Metalloporphyrins. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; pp. 303–339. [Google Scholar] [CrossRef]
- Kasha, M.; Rawls, H.R.; Ashraf El-Bayoumi, M. The exciton model in molecular spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. [Google Scholar] [CrossRef] [Green Version]
- Maiti, N.C.; Ravikanth, M.; Mazumdar, S.; Periasamy, N. Fluorescence Dynamics of Non covalently Linked Porphyrin Dimers, and Aggregates. J. Phys. Chem. 1995, 99, 17192–17197. [Google Scholar] [CrossRef]
- Kobayashi, T. J-Aggregates; World Scientific Publishing Company: Singapore, 1996; Volume 1. [Google Scholar]
- Koti, A.S.R.; Taneja, J.; Periasamy, N. Control of coherence length and aggregate size in the J-aggregate of porphyrin. Chem. Phys. Lett. 2003, 375, 171–176. [Google Scholar] [CrossRef]
- Schwab, A.D.; Smith, D.E.; Bond-Watts, B.; Johnston, D.E.; Hone, J.; Johnson, A.T.; de Paula, J.C.; Smith, W.F. Photoconductivity of Self-Assembled Porphyrin Nanorods. Nano Lett. 2004, 4, 1261–1265. [Google Scholar] [CrossRef]
- Wang, Z.; Medforth, C.J.; Shelnutt, J.A. Porphyrin Nanotubes by Ionic Self-Assembly. J. Am. Chem. Soc. 2004, 126, 15954–15955. [Google Scholar] [CrossRef] [PubMed]
- Collini, E.; Ferrante, C.; Bozio, R. Strong Enhancement of the Two-Photon Absorption of Tetrakis(4-sulfonatophenyl)porphyrin Diacid in Water upon Aggregation. J. Phys. Chem. B 2005, 109, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Tokunaga, E.; Kobayashi, T. Giant electrooptic response of excitons in porphyrin J-aggregates. Chem. Phys. Lett. 2005, 410, 18–23. [Google Scholar] [CrossRef]
- Collini, E.; Ferrante, C.; Bozio, R.; Lodi, A.; Ponterini, G. Large third-order nonlinear optical response of porphyrin J-aggregates oriented in self-assembled thin films. J. Mater. Chem. 2006, 16, 1573–1578. [Google Scholar] [CrossRef]
- Kirstein, S.; Daehne, S. J-Aggregates of Amphiphilic Cyanine Dyes: Self-Organization of Artificial Light Harvesting Complexes. Int. J. Photoenergy 2006, 1–21. [Google Scholar] [CrossRef]
- Miura, A.; Shibata, Y.; Chosrowjan, H.; Mataga, N.; Tamai, N. Femtosecond fluorescence spectroscopy and near-field spectroscopy of water-soluble tetra(4-sulfonatophenyl)porphyrin and its J-aggregate. J. Photochem. Photobiol. A 2006, 178, 192–200. [Google Scholar] [CrossRef]
- Kobayashi, T. J-Aggregates; World Scientific: Singapore, 2012; Volume 2. [Google Scholar]
- Villari, V.; Mazzaglia, A.; Castriciano, M.A.; de Luca, G.; Romeo, A.; Scolaro, L.M.; Micali, N. Optical enhancement and structural properties of a hybrid organic-inorganic ternary nanocomposite. J. Phys. Chem. C 2011, 115, 5435–5439. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Huber, P.R.; Boyd, P.; Engasser, G.; Francesconi, L.; Gibbs, E.; Fasella, P.; Cerio Venturo, G.; Hinds, L.D. On the Aggregation of Meso-Substituted Water-Soluble Porphyrins. J. Am. Chem. Soc. 1972, 94, 4511–4517. [Google Scholar] [CrossRef] [PubMed]
- Borissevitch, I.E.; Tominaga, T.T.; Imasato, H.; Tabak, M. Fluorescence and optical absorption study of interaction of two water soluble porphyrins with bovine serum albumin. The role of albumin and porphyrin aggregation. J. Lumin. 1996, 69, 65–76. [Google Scholar] [CrossRef]
- Gandini, S.C.M.; Yushmanov, V.E.; Borissevitch, I.E.; Tabak, M. Interaction of the tetra(4-sulfonatophenyl)porphyrin with ionic surfactants: Aggregation and location in micelles. Langmuir 1999, 15, 6233–6243. [Google Scholar] [CrossRef]
- Micali, N.; Mallamace, F.; Romeo, A.; Purrello, R.; Scolaro, L.M. Mesoscopic structure of meso-tetrakis(4-sulfonatophenyl)porphine J-aggregates. J. Phys. Chem. B 2000, 104, 5897–5904. [Google Scholar] [CrossRef]
- Pant, S.; Ohtaka-Saiki, H.; Takezaki, M.; Tominaga, T. Interactions between a tetraanionic porphyrin and the methylviologen dication in methanol studied by fluorescence quenching reaction. J. Mol. Liq. 2001, 90, 121–130. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Romeo, A.; Scolaro, L.M. Aggregation of meso-tetrakis(4-sulfonatophenyl)porphyrin on polyethyleneimine in aqueous solutions and on a glass surface. J. Porphyr. Phthalocyanines 2002, 6, 431–438. [Google Scholar] [CrossRef]
- Koti, A.S.R.; Periasamy, N. Self-Assembly of Template-Directed J-Aggregates of Porphyrin. Chem. Mater. 2003, 15, 369–371. [Google Scholar] [CrossRef]
- Rotomskis, R.; Augulis, R.; Snitka, V.; Valiokas, R.; Liedberg, B. Hierarchical Structure of TPPS4 J-Aggregates on Substrate Revealed by Atomic Force Microscopy. J. Phys. Chem. B 2004, 108, 2833–2838. [Google Scholar] [CrossRef]
- Escudero, C.; El-Hachemi, Z.; Crusats, J.; Ribo, J.M. Zwitterionic vs. porphyrin free-base structures in 4-phenylsulfonic acid meso-substituted porphyrins. J. Porphyr. Phthalocyanines 2005, 9, 852–863. [Google Scholar] [CrossRef]
- Micali, N.; Villari, V.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M. From fractal to nanorod porphyrin J-aggregates. Concentration-induced tuning of the aggregate size. J. Phys. Chem. B 2006, 110, 8289–8295. [Google Scholar] [CrossRef]
- Aggarwal, L.P.F.; Borissevitch, I.E. On the dynamics of the TPPS4 aggregation in aqueous solutions: Successive formation of H and J aggregates. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2006, 63, 227–233. [Google Scholar] [CrossRef]
- Arteaga, O.; Canillas, A.; Crusats, J.; El-Hachemi, Z.; Llorens, J.; Sacristan, E.; Ribo, J.M. Emergence of Supramolecular Chirality by Flows. ChemPhysChem 2010, 11, 3511–3516. [Google Scholar] [CrossRef]
- Romeo, A.; Castriciano, M.A.; Occhiuto, I.; Zagami, R.; Pasternack, R.F.; Scolaro, L.M. Kinetic Control of Chirality in Porphyrin J-Aggregates. J. Am. Chem. Soc. 2014, 136, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Micali, N.; Villari, V.; Romeo, A.; Castriciano, M.A.; Scolaro, L.M. Evidence of the early stage of porphyrin aggregation by enhanced Raman scattering and fluorescence spectroscopy. Phys. Rev. E 2007, 76. [Google Scholar] [CrossRef] [PubMed]
- Castriciano, M.A.; Donato, M.G.; Villari, V.; Micali, N.; Romeo, A.; Scolaro, L.M. Surfactant-like Behavior of Short-Chain Alcohols in Porphyrin Aggregation. J. Phys. Chem. B 2009, 113, 11173–11178. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Carbone, A.; Sacca, A.; Donato, M.G.; Micali, N.; Romeo, A.; De Luca, G.; Scolaro, L.M. Optical and sensing features of TPPS4 J-aggregates embedded in Nafion membranes: Influence of casting solvents. J. Mater. Chem. 2010, 20, 2882–2886. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Leone, N.; Cardiano, P.; Manickam, S.; Scolaro, L.M.; Lo Schiavo, S. A new supramolecular polyhedral oligomeric silsesquioxanes (POSS)-porphyrin nanohybrid: Synthesis and spectroscopic characterization. J. Mater. Chem. C 2013, 1, 4746–4753. [Google Scholar] [CrossRef]
- Zhang, L.; Yuan, J.; Liu, M. Supramolecular Chirality of Achiral TPPS Complexed with Chiral Molecular Films. J. Phys. Chem. B 2003, 107, 12768–12773. [Google Scholar] [CrossRef]
- Purrello, R.; Scolaro, L.M.; Bellacchio, E.; Gurrieri, S.; Romeo, A. Chiral H- and J-Type Aggregates of meso-Tetrakis(4-sulfonatophenyl)porphine on a-Helical Polyglutamic Acid Induced by Cationic Porphyrins. Inorg. Chem. 1998, 37, 3647–3648. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Romeo, A.; De Luca, G.; Villari, V.; Scolaro, L.M.; Micali, N. Scaling the Chirality in Porphyrin J-Nanoaggregates. J. Am. Chem. Soc. 2011, 133, 765–767. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Romeo, A.; Zagami, R.; Micali, N.; Scolaro, L.M. Kinetic effects of tartaric acid on the growth of chiral J-aggregates of tetrakis(4-sulfonatophenyl)porphyrin. Chem. Commun. 2012, 48, 4872–4874. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Liu, M. Aggregation and Induced Chirality of an Anionic meso-Tetraphenylsulfonato Porphyrin (TPPS) on a Layer-by-Layer Assembled DNA/PAH Matrix. J. Phys. Chem. B 2004, 108, 2880–2884. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, M.H. Supramolecular Chirality and Chiral Inversion of Tetraphenylsulfonato Porphyrin Assemblies on Optically Active Polylysine. J. Phys. Chem. B 2009, 113, 14015–14020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, X.; Li, Y.; Ma, R.; An, Y.; Shi, L. Chiral Micelles of Achiral TPPS and Diblock Copolymer Induced by Amino Acids. Macromolecules 2009, 42, 6253–6260. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Escudero, C.; Acosta-Reyes, F.; Casas, M.T.; Altoe, V.; Aloni, S.; Oncins, G.; Sorrenti, A.; Crusats, J.; Campos, J.L.; et al. Structure vs. properties—Chirality, optics and shapes—In amphiphilic porphyrin J-aggregates. J. Mater. Chem. C 2013, 1, 3337–3346. [Google Scholar] [CrossRef] [Green Version]
- Maria, A.C.; Gentili, D.; Romeo, A.; Cavallini, M.; Scolaro, L.M. Spatial control of chirality in supramolecular aggregates. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Randazzo, R.; Gaeta, M.; Gangemi, C.M.A.; Fragalà, M.E.; Purrello, R.; D’Urso, A. Chiral Recognition of L- and D-Amino Acid by Porphyrin Supramolecular Aggregates. Molecules 2018, 24, 84. [Google Scholar] [CrossRef] [Green Version]
- Trapani, M.; Castriciano, M.A.; Romeo, A.; De Luca, G.; Machado, N.; Howes, B.D.; Smulevich, G.; Scolaro, L.M. Nanohybrid Assemblies of Porphyrin and Au-10 Cluster Nanoparticles. Nanomaterials 2019, 9, 1026. [Google Scholar] [CrossRef] [Green Version]
- Trapani, M.; Mazzaglia, A.; Piperno, A.; Cordaro, A.; Zagami, R.; Castriciano, M.A.; Romeo, A.; Monsù Scolaro, L. Novel Nanohybrids Based on Supramolecular Assemblies of Meso-tetrakis-(4-sulfonatophenyl) Porphyrin J-aggregates and Amine-Functionalized Carbon Nanotubes. Nanomaterials 2020, 10, 669. [Google Scholar] [CrossRef] [Green Version]
- Ribo, J.M.; Crusats, J.; Sagues, F.; Claret, J.; Rubires, R. Chiral sign induction by vortices during the formation of mesophases in stirred solutions. Science 2001, 292, 2063–2066. [Google Scholar] [CrossRef] [PubMed]
- Escudero, C.; Crusat, J.; Diez-Perez, I.; El-Hachemi, Z.; Ribo, J.M. Folding and hydrodynamic forces in J-aggregates of 5-phenyl-10,15,20-tris-(4-sulfo-phenyl)porphyrin. Angew. Chem. Int. Ed. 2006, 45, 8032–8035. [Google Scholar] [CrossRef]
- Arteaga, O.; Canillas, A.; Purrello, R.; Ribo, J.M. Evidence of induced chirality in stirred solutions of supramolecular nanofibers. Opt. Lett. 2009, 34, 2177–2179. [Google Scholar] [CrossRef] [PubMed]
- El-Hachemi, Z.; Balaban, T.S.; Campos, J.L.; Cespedes, S.; Crusats, J.; Escudero, C.; Kamma-Lorger, C.S.; Llorens, J.; Malfois, M.; Mitchell, G.R.; et al. Effect of Hydrodynamic Forces on meso-(4-Sulfonatophenyl)-Substituted Porphyrin J-Aggregate Nanoparticles: Elasticity, Plasticity and Breaking. Chem. Eur. J. 2016, 22, 9740–9749. [Google Scholar] [CrossRef]
- Crusats, J.; El-Hachemi, Z.; Ribo, J.M. Hydrodynamic effects on chiral induction. Chem. Soc. Rev. 2010, 39, 569–577. [Google Scholar] [CrossRef]
- D’Urso, A.; Randazzo, R.; Lo Faro, L.; Purrello, R. Vortexes and Nanoscale Chirality. Angew. Chem. Int. Ed. Engl. 2010, 49, 108–112. [Google Scholar] [CrossRef]
- Auwärter, W.; Écija, D.; Klappenberger, F.; Barth, J.V. Porphyrins at interfaces. Nat. Chem. 2015, 7, 105–120. [Google Scholar] [CrossRef]
- Micali, N.; Engelkamp, H.; van Rhee, P.G.; Christianen, P.C.M.; Scolaro, L.M.; Maan, J.C. Selection of supramolecular chirality by application of rotational and magnetic forces. Nat. Chem. 2012, 4, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.S.; Li, Y.K.; Yan, F.S.; Liu, C.; Sang, Y.T.; Tian, F.; Feng, Q.; Duan, P.F.; Zhang, L.; Shi, X.H.; et al. Control over the emerging chirality in supramolecular gels and solutions by chiral microvortices in milliseconds. Nat. Commun. 2018, 9, 2599. [Google Scholar] [CrossRef] [PubMed]
- Romeo, A.; Castriciano, M.A.; Zagami, R.; Pollicino, G.; Monsu Scolaro, L.; Pasternack, R.F. Effect of zinc cations on the kinetics for supramolecular assembling and the chirality of porphyrin J-aggregates. Chem. Sci. 2017, 8, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Occhiuto, I.G.; Castriciano, M.A.; Trapani, M.; Zagami, R.; Romeo, A.; Pasternack, R.F.; Monsù Scolaro, L. Controlling J-Aggregates Formation and Chirality Induction through Demetallation of a Zinc(II) Water Soluble Porphyrin. Int. J. Mol. Sci. 2020, 21, 4001. [Google Scholar] [CrossRef] [PubMed]
- Castriciano, M.; Romeo, A.; Villari, V.; Micali, N.; Scolaro, L.M. Structural rearrangements in 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin J-aggregates under strongly acidic conditions. J. Phys. Chem. B 2003, 107, 8765–8771. [Google Scholar] [CrossRef]
- Romeo, A.; Castriciano, M.A.; Scolaro, L.M. Spectroscopic and kinetic investigations on porphyrin J-aggregates induced by polyamines. J. Porphyrins Phthalocyanines 2010, 14, 713–721. [Google Scholar] [CrossRef]
- Scolaro, L.M.; Romeo, A.; Castriciano, M.A.; Micali, N. Unusual optical properties of porphyrin fractal J-aggregates. Chem. Commun. 2005, 3018–3020. [Google Scholar] [CrossRef] [PubMed]
- De Napoli, M.; Nardis, S.; Paolesse, R.; Vicente, M.G.H.; Lauceri, R.; Purrello, R. Hierarchical Porphyrin Self-Assembly in Aqueous Solution. J. Am. Chem. Soc. 2004, 126, 5934–5935. [Google Scholar] [CrossRef]
- Lauceri, R.; De Napoli, M.; Mammana, A.; Nardis, S.; Romeo, A.; Purrello, R. Hierarchical self-assembly of water-soluble porphyrins. Synth. Met. 2004, 147, 49–55. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Photochemistry of Polypyridine and Porphyrin Complexes; Academic Press: London, UK, 1992; p. 428. [Google Scholar]
- Ohno, O.; Kaizu, Y.; Kobayashi, H. J-Aggregate Formation of a Water-Soluble Porphyrin in Acidic Aqueous-Media. J. Chem. Phys. 1993, 99, 4128–4139. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Gibbs, E.J.; Collings, P.J.; dePaula, J.C.; Turzo, L.C.; Terracina, A. A nonconventional approach to supramolecular formation dynamics. The kinetics of assembly of DNA-bound porphyrins. J. Am. Chem. Soc. 1998, 120, 5873–5878. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Fleming, C.; Herring, S.; Collings, P.J.; dePaula, J.; DeCastro, G.; Gibbs, E.J. Aggregation kinetics of extended porphyrin and cyanine dye assemblies. Biophys. J. 2000, 79, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Maiti, N.C.; Mazumdar, S.; Periasamy, N. J- and H-aggregates of porphyrin-surfactant complexes: Time-resolved fluorescence and other spectroscopic studies. J. Phys. Chem. B 1998, 102, 1528–1538. [Google Scholar] [CrossRef]
- Zagami, R.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M. Spectroscopic investigations on chiral J-aggregates induced by tartaric acid in alcoholic solution. J. Porphyr. Phthalocyanines 2017, 21, 327–333. [Google Scholar] [CrossRef]
- Short, J.M.; Berriman, J.A.; Kübel, C.; El-Hachemi, Z.; Naubron, J.-V.; Balaban, T.S. Electron Cryo-Microscopy of TPPS4⋅2HCl Tubes Reveals a Helical Organisation Explaining the Origin of their Chirality. ChemPhysChem 2013, 14, 3209–3214. [Google Scholar] [CrossRef] [Green Version]
- Pasternack, R.F.; Collings, P.J. Resonance Light-Scattering—A New Technique for Studying Chromophore Aggregation. Science 1995, 269, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Purrello, R. Supramolecular chemistry: Lasting chiral memory. Nat. Mater. 2003, 2, 216–217. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 105 × k0/s−1 | 104 × kc/s−1 | m | n | |
|---|---|---|---|---|
| Mix 1 | 7.61 ± 0.24 | 10.30 ± 0.01 | 3.2 ± 0.1 | 3.5 ± 0.1 |
| Mix 2 | 1.93 ± 0.21 | 5.27 ± 0.01 | 4.1 ± 0.1 | 7.8 ± 0.2 |
| Mix 3 | 0.90 ± 0.18 | 3.85 ± 0.01 | 3.0 ± 0.1 | 7.6 ± 0.2 |
| PF-HCl a | 0.78 ± 0.03 | 1.95 ± 0.01 | 3.7 ± 0.1 | 7.4. ± 0.2 |
| PF-HCl b [27] | 22.01 ± 0.04 | 8.91 ± 0.02 | 2.4 ± 0.1 | 4.8 ± 0.1 |
| 105 × k0/s−1 | 104 × kc/s−1 | m | n | |
|---|---|---|---|---|
| L | ||||
| Mix 1 | 20.2 ± 0.1 | 10.2 ± 0.1 | 3.1 ± 0.1 | 1.8 ± 0.1 |
| Mix 2 | 26.1 ± 0.2 | 3.9 ± 0.1 | 10.8 ± 0.9 | 4.0 ± 0.2 |
| Mix 3 | 1.9 ± 0.1 | 2.9 ± 0.1 | 2.8 ± 0.1 | 6.8 ± 0.1 |
| D | ||||
| Mix 1 | 0.4 ± 0.1 | 3.8 ± 0.5 | 2.5 ± 0.1 | 3.6 ± 0.1 |
| Mix 2 | 12.3 ± 1.1 | 5.1 ± 0.1 | 2.6 ± 0.1 | 2.6 ± 0.2 |
| Mix 3 | 1.7 ± 0.2 | 2.9 ± 0.01 | 2.8 ± 0.4 | 6.6 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castriciano, M.A.; Cardillo, S.; Zagami, R.; Trapani, M.; Romeo, A.; Scolaro, L.M. Effects of the Mixing Protocol on the Self-Assembling Process of Water Soluble Porphyrins. Int. J. Mol. Sci. 2021, 22, 797. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020797
Castriciano MA, Cardillo S, Zagami R, Trapani M, Romeo A, Scolaro LM. Effects of the Mixing Protocol on the Self-Assembling Process of Water Soluble Porphyrins. International Journal of Molecular Sciences. 2021; 22(2):797. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020797
Chicago/Turabian StyleCastriciano, Maria Angela, Sergio Cardillo, Roberto Zagami, Mariachiara Trapani, Andrea Romeo, and Luigi Monsù Scolaro. 2021. "Effects of the Mixing Protocol on the Self-Assembling Process of Water Soluble Porphyrins" International Journal of Molecular Sciences 22, no. 2: 797. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020797