Clinical and Molecular Insights in Erythropoiesis Regulation of Signal Transduction Pathways in Myelodysplastic Syndromes and β-Thalassemia

 , , ,
, , ,
Abstract
:1. Introduction
2. Targeting Transforming Growth Factor (TGF)-β Signaling to Improve Erythropoiesis
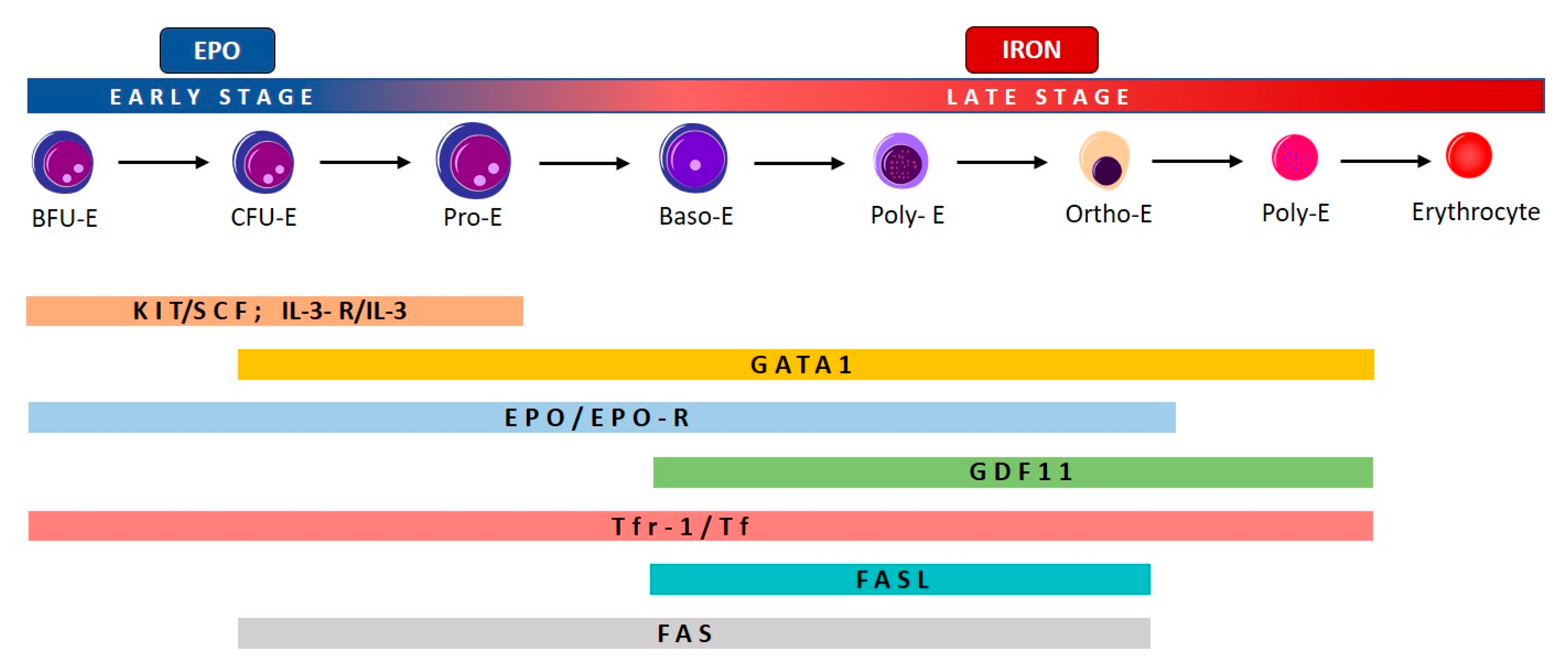
2.1. Effective and Ineffective Erythropoiesis
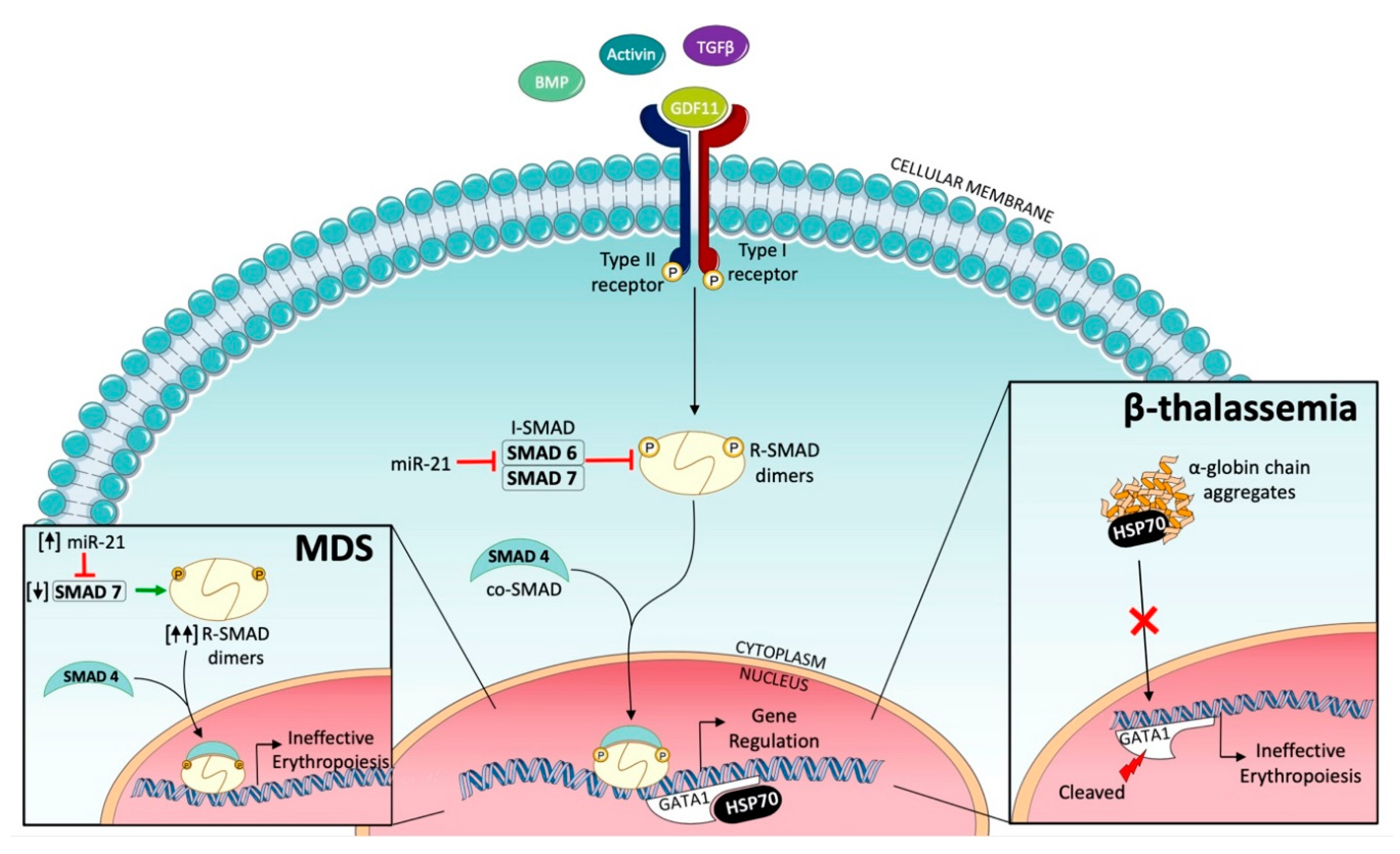
2.2. Ineffective Erythropoiesis in Myelodysplastic Syndromes (MDS)
2.3. Ineffective Erythropoiesis in β-Thalassemia
3. Targeting SMAD2/3 Signaling to Correct Ineffective Erythropoiesis
3.1. Activin Receptor Ligand Traps
3.2. Activin Receptor Ligand Traps: Preclinical Studies
3.3. Activin Receptor Ligand Traps: Clinical Evaluation
3.4. Clinical Studies in MDS
3.5. Clinical Studies in β-Thalassemia
4. Targeting HIF: Clinical Development of Roxadustat, a New Erythropoiesis Stimulating Agent
5. Targeting Inositide-Dependent Signal Transduction Pathways
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE-011 | Sotatercept |
| ACE-536 | Luspatercept |
| ActRIIA | Activin receptor type IIA |
| ActRIIA-Fc | Fc domain of human IgG1 |
| ActRIIB | Activin receptor type IIB |
| ActRIIB-Fc | Fc domain of human IgG1 |
| ALK1-7 | Activin-like receptors |
| Bcl-x | B-cell lymphoma-extra large |
| BFU-E | Erythroid burst-forming units |
| BMPs | Bone morphogenetic proteins |
| CD34+ | Cluster of differentiation 34+ |
| CFU-E | Erythroid colony-forming units |
| CKD | Chronic kidney disease |
| DAG | Dyacylglicerol |
| DCYTB | Duodenal Cytochrome B |
| DMT1 | Divalent Metal Transporter 1 |
| DOAJ | Directory of open access journals |
| EPC | EPO-producing cells |
| EPO | Erythropoietin |
| EPO-R | Erythropoietin receptor |
| ESAs | Erythropoiesis-stimulating agents |
| FASL | FAS ligand |
| GDF11 | Growth/differentiating factor 11 |
| HI-E | Hematological erythroid improvement |
| HIF | Hypoxia inducible factor |
| HIF-PHD enzymes | Hypoxia inducible factor prolyl hydrolase domain enzymes |
| HSC | Hematopoietic stem cells |
| HSP70 | Heat shock protein 70 |
| IL-3 | Interleukin 3 |
| IP3 | Inositol-1,4,5-trisphospate |
| I-SMAD | Inhibitory small mother against decapentaplegic |
| JAK 2 | Janus Kinase 2 |
| LD | Linear dichroism |
| MAD | Mothers against decapentaplegic |
| MDS | Myelodysplastic syndromes |
| MDS-RS | Myelodysplastic syndromes with ring sideroblasts |
| NTDT | Non-transfusion-dependent thalassemia |
| PI | Phosphoinositides |
| PIP2 | Phosphatidylinositol (4,5) bisphosphate |
| PIP3 | PtdIns(3,4,5)P3 |
| PI3K | Phosphatidilinositol 3-kinases |
| PLCs | Phospholipases C |
| p-SMAD | Phosphorylated small mother against decapentaplegic |
| ROS | Radical oxygen species |
| R-SMAD | Regulatory small mother against decapentaplegic |
| SCF | Stem cell factor |
| SF3B1 | Splicing factor 3B subunit 1 |
| SMAD | Small mother against decapentaplegic |
| STAT5 | Signal Transducer and Activator of Transcription 5 |
| TDT | Transfusion-dependent thalassemia |
| TEA | Treatment-emergent adverse events |
| TGF | Transforming growth factor |
| TNF | Tumor necrosis factor |
| WHO | World Health Organization |
References
- Zivot, A.; Lipton, J.M.; Narla, A.; Blanc, L. Erythropoiesis: Insights into pathophysiology and treatments in 2017. Mol. Med. 2018, 24, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valent, P.; Büsche, G.; Theurl, I.; Uras, I.Z.; Germing, U.; Stauder, R.; Sotlar, K.; Füreder, W.; Bettelheim, P.; Pfeilstöcker, M.; et al. Normal and pathological erythropoiesis in adults: From gene regulation to targeted treatment concepts. Haematologica 2018, 103, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Blank, U.; Karlsson, S. TGF-β signaling in the control of hematopoietic stem cells. Blood 2015, 125, 3542–3550. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Pham, E.; Macdougall, I.C. Erythropoietins: A common mechanism of action. Exp. Hematol. 2008, 36, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef] [Green Version]
- Zermati, Y.; Fichelson, S.; Valensi, F.; Freyssinier, J.M.; Rouyer-Fessard, P.; Cramer, E.; Guichard, J.; Varet, B.; Hermine, O. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp. Hematol. 2000, 28, 885–894. [Google Scholar] [CrossRef]
- Xie, Y.; Bai, H.; Liu, Y.; Hoyle, D.L.; Cheng, T.; Wang, Z.Z. Cooperative Effect of Erythropoietin and TGF-β Inhibition on Erythroid Development in Human Pluripotent Stem Cells. J. Cell. Biochem. 2015, 116, 2735–2743. [Google Scholar] [CrossRef]
- Nimer, S.D. Myelodysplastic syndromes. Blood 2008, 111, 4841–4851. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Malcovati, L.; Cazzola, M. Recent advances in the understanding of myelodysplastic syndromes with ring sideroblasts. Br. J. Haematol. 2016, 174, 847–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Li, X.; Chang, C.; Xu, F.; He, Q.; Guo, J.; Tao, Y.; Liu, Y.; Liu, L.; Shi, W. SF3B1-mutated myelodysplastic syndrome with ring sideroblasts harbors more severe iron overload and corresponding over-erythropoiesis. Leuk. Res. 2016, 44, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Suragani, R.N.; Aluri, S.; Shah, N.; Bhagat, T.D.; Alexander, M.J.; Komrokji, R.; Kumar, R. Biological basis for efficacy of activin receptor ligand traps in myelodysplastic syndromes. J. Clin. Investig. 2020, 130, 582–589. [Google Scholar] [CrossRef]
- Zhou, L.; Nguyen, A.N.; Sohal, D.; Ying Ma, J.; Pahanish, P.; Gundabolu, K.; Hayman, J.; Chubak, A.; Mo, Y.; Bhagat, T.D.; et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood 2008, 112, 3434–3443. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; McMahon, C.; Bhagat, T.; Alencar, C.; Yu, Y.; Fazzari, M.; Sohal, D.; Heuck, C.; Gundabolu, K.; Ng, C.; et al. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase. Cancer Res. 2011, 71, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Blank, U.; Karlsson, G.; Moody, J.L.; Utsugisawa, T.; Magnusson, M.; Singbrant, S.; Larsson, J.; Karlsson, S. Smad7 promotes self-renewal of hematopoietic stem cells. Blood 2006, 108, 4246–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, V.; Valcárcel, D.; Platzbecker, U.; Komrokji, R.S.; Cleverly, A.L.; Lahn, M.M.; Janssen, J.; Zhao, Y.; Chiang, A.; Giagounidis, A.; et al. Phase II Study of the ALK5 Inhibitor Galunisertib in Very Low-, Low-, and Intermediate-Risk Myelodysplastic Syndromes. Clin. Cancer Res. 2019, 25, 6976–6985. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, T.D.; Zhou, L.; Sokol, L.; Kessel, R.; Caceres, G.; Gundabolu, K.; Tamari, R.; Gordon, S.; Mantzaris, I.; Jodlowski, T.; et al. miR-21 mediates hematopoietic suppression in MDS by activating TGF-β signaling. Blood 2013, 121, 2875–2881. [Google Scholar] [CrossRef] [Green Version]
- Velu, C.S.; Chaubey, A.; Phelan, J.D.; Horman, S.R.; Wunderlich, M.; Guzman, M.L.; Jegga, A.G.; Zeleznik-Le, N.J.; Chen, J.; Mulloy, J.C.; et al. Therapeutic antagonists of microRNAs deplete leukemia-initiating cell activity. J. Clin. Investig. 2014, 124, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Rivella, S. Ineffective erythropoiesis and thalassemias. Curr. Opin. Hematol. 2009, 16, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Pilo, F.; Angelucci, E. Luspatercept to treat β-thalassemia. Drugs Today 2020, 56, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 2016, 172, 512–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mies, A.; Hermine, O.; Platzbecker, U. Activin Receptor II Ligand Traps and Their Therapeutic Potential in Myelodysplastic Syndromes with Ring Sideroblasts. Curr. Hematol. Malig. Rep. 2016, 11, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S. Activin Receptor II Ligand Traps: New Treatment Paradigm for Low-Risk MDS. Curr. Hematol. Malig. Rep. 2019, 14, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Martinez-Hackert, E. Transforming Growth Factor-β Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J. Biol. Chem. 2016, 291, 10792–10804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suragani, R.N.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef]
- Sako, D.; Grinberg, A.V.; Liu, J.; Davies, M.V.; Castonguay, R.; Maniatis, S.; Andreucci, A.J.; Pobre, E.G.; Tomkinson, K.N.; Monnell, T.E.; et al. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J. Biol. Chem. 2010, 285, 21037–21048. [Google Scholar] [CrossRef] [Green Version]
- Suragani, R.N.; Cawley, S.M.; Li, R.; Wallner, S.; Alexander, M.J.; Mulivor, A.W.; Gardenghi, S.; Rivella, S.; Grinberg, A.V.; Pearsall, R.S.; et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine β-thalassemia. Blood 2014, 123, 3864–3872. [Google Scholar] [CrossRef] [Green Version]
- Dussiot, M.; Maciel, T.T.; Fricot, A.; Chartier, C.; Negre, O.; Veiga, J.; Grapton, D.; Paubelle, E.; Payen, E.; Beuzard, Y.; et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia. Nat. Med. 2014, 20, 398–407. [Google Scholar] [CrossRef]
- Ruckle, J.; Jacobs, M.; Kramer, W.; Pearsall, A.E.; Kumar, R.; Underwood, K.W.; Seehra, J.; Yang, Y.; Condon, C.H.; Sherman, M.L. Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J. Bone Miner. Res. 2009, 24, 744–752. [Google Scholar] [CrossRef]
- Sherman, M.L.; Borgstein, N.G.; Mook, L.; Wilson, D.; Yang, Y.; Chen, N.; Kumar, R.; Kim, K.; Laadem, A. Multiple-dose, safety, pharmacokinetic, and pharmacodynamic study of sotatercept (ActRIIA-IgG1), a novel erythropoietic agent, in healthy postmenopausal women. J. Clin. Pharmacol. 2013, 53, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Attie, K.M.; Allison, M.J.; McClure, T.; Boyd, I.E.; Wilson, D.M.; Pearsall, A.E.; Sherman, M.L. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am. J. Hematol. 2014, 89, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, E. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823. [Google Scholar] [CrossRef]
- Raftopoulos, H.; Laadem, A.; Hesketh, P.J.; Goldschmidt, J.; Gabrail, N.; Osborne, C.; Ali, M.; Sherman, M.L.; Wang, D.; Glaspy, J.A.; et al. Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia in patients with metastatic breast cancer or advanced or metastatic solid tumors treated with platinum-based chemotherapeutic regimens: Results from two phase 2 studies. Support. Care Cancer 2016, 24, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Brancaleoni, V.; Nava, I.; Delbini, P.; Duca, L.; Motta, I. Activin Receptor-Ligand Trap for the Treatment of β-thalassemia: A Serendipitous Discovery. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020075. [Google Scholar] [PubMed]
- Komrokji, R.; Garcia-Manero, G.; Ades, L.; Prebet, T.; Steensma, D.P.; Jurcic, J.G.; Sekeres, M.A.; Berdeja, J.; Savona, M.R.; Beyne-Rauzy, O.; et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: A phase 2, dose-ranging trial. Lancet Haematol. 2018, 5, e63–e72. [Google Scholar] [CrossRef]
- Platzbecker, U.; Germing, U.; Götze, K.S.; Kiewe, P.; Mayer, K.; Chromik, J.; Radsak, M.; Wolff, T.; Zhang, X.; Laadem, A.; et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): A multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017, 18, 1338–1347. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Piga, A.; Perrotta, S.; Gamberini, M.R.; Voskaridou, E.; Melpignano, A.; Filosa, A.; Caruso, V.; Pietrangelo, A.; Longo, F.; Tartaglione, I.; et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with β-thalassemia. Blood 2019, 133, 1279–1289. [Google Scholar] [CrossRef] [Green Version]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231. [Google Scholar] [CrossRef]
- Mittelman, M.; Gardyn, J.; Carmel, M.; Malovani, H.; Barak, Y.; Nir, U. Analysis of the erythropoietin receptor gene in patients with myeloproliferative and myelodysplastic syndromes. Leuk. Res. 1996, 20, 459–466. [Google Scholar] [CrossRef]
- Backx, B.; Broeders, L.; Hoefsloot, L.H.; Wognum, B.; Löwenberg, B. Erythropoiesis in myelodysplastic syndrome: Expression of receptors for erythropoietin and kit ligand. Leukemia 1996, 10, 466–472. [Google Scholar]
- Hoefsloot, L.H.; van Amelsvoort, M.P.; Broeders, L.C.; van der Plas, D.C.; van Lom, K.; Hoogerbrugge, H.; Touw, I.P.; Löwenberg, B. Erythropoietin-induced activation of STAT5 is impaired in the myelodysplastic syndrome. Blood 1997, 89, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.S.; Percy, M.J. The HIF pathway and erythrocytosis. Annu. Rev. Pathol. 2011, 6, 165–192. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodial. Int. 2017, 21 (Suppl. 1), S110–S124. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.; Saad, M. A New Approach to the Management of Anemia in CKD Patients: A Review on Roxadustat. Adv. Ther. 2017, 34, 848–853. [Google Scholar] [CrossRef] [Green Version]
- Besarab, A.; Provenzano, R.; Hertel, J.; Zabaneh, R.; Klaus, S.J.; Lee, T.; Leong, R.; Hemmerich, S.; Yu, K.H.; Neff, T.B. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol. Dial. Transplant. 2015, 30, 1665–1673. [Google Scholar] [CrossRef]
- Provenzano, R.; Besarab, A.; Wright, S.; Dua, S.; Zeig, S.; Nguyen, P.; Poole, L.; Saikali, K.G.; Saha, G.; Hemmerich, S.; et al. Roxadustat (FG-4592) Versus Epoetin Alfa for Anemia in Patients Receiving Maintenance Hemodialysis: A Phase 2, Randomized, 6- to 19-Week, Open-Label, Active-Comparator, Dose-Ranging, Safety and Exploratory Efficacy Study. Am. J. Kidney Dis. 2016, 67, 912–924. [Google Scholar] [CrossRef] [Green Version]
- Besarab, A.; Chernyavskaya, E.; Motylev, I.; Shutov, E.; Kumbar, L.M.; Gurevich, K.; Chan, D.T.; Leong, R.; Poole, L.; Zhong, M.; et al. Roxadustat (FG-4592): Correction of Anemia in Incident Dialysis Patients. J. Am. Soc. Nephrol. 2016, 27, 1225–1233. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef]
- Manzoli, F.A.; Maraldi, N.M.; Cocco, L.; Capitani, S.; Facchini, A. Chromatin phospholipids in normal and chronic lymphocytic leukemia lymphocytes. Cancer Res. 1977, 37, 843–849. [Google Scholar] [PubMed]
- Obeng, E.O.; Rusciano, I.; Marvi, M.V.; Fazio, A.; Ratti, S.; Follo, M.Y.; Xian, J.; Manzoli, L.; Billi, A.M.; Mongiorgi, S.; et al. Phosphoinositide-Dependent Signaling in Cancer: A Focus on Phospholipase C Isozymes. Int. J. Mol. Sci. 2020, 21, 2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follo, M.Y.; Faenza, I.; Piazzi, M.; Blalock, W.L.; Manzoli, L.; McCubrey, J.A.; Cocco, L. Nuclear PI-PLCbeta1: An appraisal on targets and pathology. Adv. Biol. Regul. 2014, 54, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Manzoli, L.; Mongiorgi, S.; Clissa, C.; Finelli, C.; Billi, A.M.; Poli, A.; Quaranta, M.; Cocco, L.; Follo, M.Y. Strategic Role of Nuclear Inositide Signalling in Myelodysplastic Syndromes Therapy. Mini Rev. Med. Chem. 2014, 14, 873–883. [Google Scholar] [CrossRef]
- Mongiorgi, S.; Finelli, C.; Yang, Y.R.; Clissa, C.; McCubrey, J.A.; Billi, A.M.; Manzoli, L.; Suh, P.G.; Cocco, L.; Follo, M.Y. Inositide-dependent signaling pathways as new therapeutic targets in myelodysplastic syndromes. Expert Opin. Ther. Targets 2016, 20, 677–687. [Google Scholar] [CrossRef]
- Xian, J.; Obeng, E.O.; Ratti, S.; Rusciano, I.; Marvi, M.V.; Fazio, A.; De Stefano, A.; Mongiorgi, S.; Cappellini, A.; Ramazzotti, G.; et al. Nuclear Inositides and Inositide-Dependent Signaling Pathways in Myelodysplastic Syndromes. Cells 2020, 9, 697. [Google Scholar] [CrossRef] [Green Version]
- Mongiorgi, S.; Follo, M.Y.; Yang, Y.R.; Ratti, S.; Manzoli, L.; McCubrey, J.A.; Billi, A.M.; Suh, P.G.; Cocco, L. Selective Activation of Nuclear PI-PLCbeta1 During Normal and Therapy-Related Differentiation. Curr. Pharm. Des. 2016, 22, 2345–2348. [Google Scholar] [CrossRef]
- Follo, M.Y.; Mongiorgi, S.; Clissa, C.; Paolini, S.; Martinelli, G.; Martelli, A.M.; Fioravanti, G.; Manzoli, L.; Finelli, C.; Cocco, L. Activation of nuclear inositide signalling pathways during erythropoietin therapy in low-risk MDS patients. Leukemia 2012, 26, 2474–2482. [Google Scholar] [CrossRef] [Green Version]
- Poli, A.; Ratti, S.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Lonetti, A.; Cappellini, A.; Catozzi, A.; Barraco, M.; Suh, P.G.; et al. Nuclear translocation of PKC-alpha is associated with cell cycle arrest and erythroid differentiation in myelodysplastic syndromes (MDSs). FASEB J. 2018, 32, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Hacein-Bey-Abina, S.; Estienne, M.; Bessoles, S.; Echchakir, H.; Pederzoli-Ribeil, M.; Chiron, A.; Aldaz-Carroll, L.; Leducq, V.; Zhang, Y.Y.; Souyri, M.; et al. Erythropoietin is a major regulator of thrombopoiesis in thrombopoietin-dependent and-independent contexts. Exp. Hematol. 2020, 88, 15–27. [Google Scholar] [CrossRef]
- Bellio, M.; Caux, M.; Vauclard, A.; Chicanne, G.; Gratacap, M.P.; Terrisse, A.D.; Severin, S.; Payrastre, B. Phosphatidylinositol 3 monophosphate metabolizing enzymes in blood platelet production and in thrombosis. Adv. Biol. Regul. 2020, 75, 100664. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Kiladjian, J.J.; Platzbecker, U. Luspatercept for the treatment of anemia in myelodysplastic syndromes and primary myelofibrosis. Blood 2019, 133, 790–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bewersdorf, J.P.; Zeidan, A.M. Transforming growth factor (TGF)-β pathway as a therapeutic target in lower risk myelodysplastic syndromes. Leukemia 2019, 33, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.; Navada, S.C.; Silverman, L.R. Myelo-deception: Luspatercept & TGF-Beta ligand traps in myeloid diseases & anemia. Leuk. Res. 2020, 97, 106430. [Google Scholar]



{kind=link}
{kind=link}
| Drug | Patients | Primary Endpoint | Outcomes | Ref | |
|---|---|---|---|---|---|
| MDS | Sotatercept | Total = 74 | Hematological response (HI-E) | 49% | [36] |
| Luspatercept | Total = 58 | Hematological response (HI-E) | 63% | [37] | |
| Luspatercept | Total = 229 | Transfusion independence for ≥8 weeks during weeks 1–24 | [38] | ||
| * | ||||
| β-thalassemia | Luspatercept | Total = 64 | [39] | ||
| Mean hemoglobin increase ≥1.5 g/dL for 14 days | 58% | |||
| Transfusion burden reduction | 81% | |||
| Luspatercept | Total = 336 | Reduction in the transfusion burden during 13–24 weeks | [40] | ||
| * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parisi, S.; Finelli, C.; Fazio, A.; De Stefano, A.; Mongiorgi, S.; Ratti, S.; Cappellini, A.; Billi, A.M.; Cocco, L.; Follo, M.Y.; et al. Clinical and Molecular Insights in Erythropoiesis Regulation of Signal Transduction Pathways in Myelodysplastic Syndromes and β-Thalassemia. Int. J. Mol. Sci. 2021, 22, 827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020827
Parisi S, Finelli C, Fazio A, De Stefano A, Mongiorgi S, Ratti S, Cappellini A, Billi AM, Cocco L, Follo MY, et al. Clinical and Molecular Insights in Erythropoiesis Regulation of Signal Transduction Pathways in Myelodysplastic Syndromes and β-Thalassemia. International Journal of Molecular Sciences. 2021; 22(2):827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020827
Chicago/Turabian StyleParisi, Sarah, Carlo Finelli, Antonietta Fazio, Alessia De Stefano, Sara Mongiorgi, Stefano Ratti, Alessandra Cappellini, Anna Maria Billi, Lucio Cocco, Matilde Y. Follo, and et al. 2021. "Clinical and Molecular Insights in Erythropoiesis Regulation of Signal Transduction Pathways in Myelodysplastic Syndromes and β-Thalassemia" International Journal of Molecular Sciences 22, no. 2: 827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020827