
Mechanisms Applied by Protein Inhibitors to Inhibit Cysteine Proteases



1
Department of Biochemistry and Molecular and Structural Biology, Jozef Stefan Institute, Jamova cesta 39, 1000 Ljubljana, Slovenia
2
Centre of Excellence for Integrated Approaches in Chemistry and Biology of Proteins (CIPKeBiP), Jamova cesta 39, 1000 Ljubljana, Slovenia
3
Faculty of Chemistry, University of Ljubljana, Večna pot 113, 1000 Ljubljana, Slovenia
4
Institute of Regenerative Medicine, I.M. Sechenov First Moscow State Medical University, Bol’shaya Pirogovskaya Ulitsa, 19c1, 119146 Moscow, Russia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(3), 997; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030997
Submission received: 13 December 2020
/
Revised: 13 January 2021
/
Accepted: 16 January 2021
/
Published: 20 January 2021
(This article belongs to the Special Issue Biocatalysis: Mechanisms of Proteolytic Enzymes)
Abstract
:Protein inhibitors of proteases are an important tool of nature to regulate and control proteolysis in living organisms under physiological and pathological conditions. In this review, we analyzed the mechanisms of inhibition of cysteine proteases on the basis of structural information and compiled kinetic data. The gathered structural data indicate that the protein fold is not a major obstacle for the evolution of a protease inhibitor. It appears that nature can convert almost any starting fold into an inhibitor of a protease. In addition, there appears to be no general rule governing the inhibitory mechanism. The structural data make it clear that the “lock and key” mechanism is a historical concept with limited validity. However, the analysis suggests that the shape of the active site cleft of proteases imposes some restraints. When the S1 binding site is shaped as a pocket buried in the structure of protease, inhibitors can apply substrate-like binding mechanisms. In contrast, when the S1 binding site is in part exposed to solvent, the substrate-like inhibition cannot be employed. It appears that all proteases, with the exception of papain-like proteases, belong to the first group of proteases. Finally, we show a number of examples and provide hints on how to engineer protein inhibitors.
1. Introduction
Previously, we reviewed cysteine protease protein inhibitors and their role in regulation of proteolysis [1]. In the review, we classified the inhibitors according to their physiological roles using quantitative criteria of enzyme kinetics, the delay time of inhibition, and the stability time of inhibition, established by Joseph Bieth in the 1980s [2,3]. Delay time, d(t), is the time needed to achieve ≈99% of inhibition (d(t) = ln 2/Io × kass), and roughly equals seven half-lives of the reaction. In this equation, Io represents the physiological concentration of the inhibitor and kass is an approximation of the association rate constant. The stability time of reversible inhibitors is defined as the minimal time in which the EI (E, enzyme; I, inhibitor) complex remains undissociated (t(s) = ln 2/kdiss), where kdiss represents the dissociation rate constant. This suggest that inhibitors were of physiological relevance when the delay time was below 1 s and, for reversible inhibitors, when the stability time was above 10 min [2]. Two types of inhibitors were introduced, emergency and regulatory [1,4]. Emergency inhibitors rapidly trap a protease and maintain it in a stable complex preventing any undesired activity. Regulatory inhibitors, by comparison, modulate the protease activity under physiological conditions. They can be further divided into threshold, buffer, delay, and pro-inhibitor sub-types. The threshold-type inhibitors prevent undesired protease activation. The buffer-type inhibitors reversibly and rapidly bind proteases, and when their physiological substrate appears, they also rapidly release them and thereby prevent undesired and potentially harmful proteolysis in the absence of their substrate. The delay-type inhibitors irreversibly (or pseudo-irreversibly) and slowly bind their target, thereby enabling proteolysis for a limited amount of time, whereas pro-inhibitors require initial processing by a protease to become inhibitory. Detailed kinetic studies in which Kass and Kdiss are measured are seldomly performed. To gain insight into the relative differences between various protease inhibitor interactions, we rely predominantly on their ratio, Ki. This link between biochemical principles of inhibition and physiology does not require an update, however, the determination of a number of new structures of cysteine protease inhibitors indicates that canonical mechanisms of inhibition should be updated [5]. Because no review, including ours, completely covers a broad topic such as protein inhibitors of cysteine proteases, we want to remind readers that other related reviews have been undertaken that describe various aspects of cysteine protease inhibition [6,7,8,9,10,11,12,13,14].
Cysteine proteases use the reactive site cysteine as the catalytic nucleophile and the histidine to perform peptide bond hydrolysis. In MEROPS [15], an online database that provides an insight into peptidases, there are 16 clans of cysteine peptidases and some that are unclassified, of which four among them include proteases with mixed catalytic types. They are further divided into 97 families of structurally and sequentially related peptidases, of which 18 families belong to the four clans of mixed catalytic types. Activity of many of these is regulated by protein inhibitors, which are either endogenous or originate from the invading organisms [15]. The MEROPS list of clans and families of protein inhibitors of proteases contains 27 clans and about four times as many structurally and sequentially related families. Their classification has little relation to the type of protease they target; inhibitors such as macrocypins, thyropins, and serpins can simultaneously bind two different families of cysteine proteases with their two distinct reactive sites. Table 1, Table 2, Table 3, Table 4 and Table 5 present the available structures of the complexes between protein inhibitors and cysteine proteases, including their family classification, Protein Data Bank (PDB) codes [16], and publication references. Table A1 shows the binding and kinetic constants Ki for their interaction with target proteases to provide an experimental basis for their classification. Due to the differences in Ki values, the same inhibitor can belong to several types in respect to the protease they inhibit. For example, cystatins differentiate among exo- and endo-peptidases, and the inhibitory fragment of the p41 form of the invariant chain associated with the major histocompatibility class II molecule (the p41 fragment) can be, in respect to the target, emergency and buffer inhibitors, and also the delay type and pro-inhibitor (a detailed explanation and references are provided below). For our review of the mechanisms of inhibition of cysteine proteases, we selected crystal structures of diverse types of inhibitors, which either target large groups of related proteases, such as papain-like proteases and caspases, or others including calpastatin and securin, with unique mechanism(s) of inhibition.
2. Inhibitors of Papain-Like Cysteine Proteases
Papain-like cysteine proteases are the largest family (C1 according to MEROPS) among the cysteine proteases, and likely the most studied. The subgroup of cysteine cathepsins is involved in a myriad of physiological functions from protein turnover to processing of antigens, hormones, and bone remodeling [11]. Moreover, the crystal structure of papain was among the first enzyme structures determined [17], and Schechter and Berger introduced the nomenclature of substrate binding sites and positioning of the substrate when studying papain interaction with a polyalanine peptide [18]. Papain-like cysteine proteases are inhibited by several groups of protein inhibitors that are involved in the regulation of physiological and pathogenic conditions. In this section, we present cystatins as the largest group of inhibitors [7,10], followed by falstatins, chagasins, thyropins, clitocypins and macrocypins, and staphostatins inhibiting a papain-related protease staphopain from family C47. Serpins are an important group of inhibitors of serine and cysteine proteases, including papain-like cysteine proteases. To the best of our knowledge, the structure of their complex with a representative of cysteine proteases is still lacking. Thus, we include a brief overview of their mechanism in Section 3.4.
2.1. Cystatins
Cystatins were the first discovered and are the best studied endogenous inhibitors of cysteine cathepsins [19]. Their major function appears to be protection of the organism from undesired endogenous proteases; however, they also protect against invading microorganisms and parasites, which apply cysteine proteases to invade the host. Cystatins are divided into three families: the stefins, the cystatins, and the kininogens. Stefins and cystatins are single-domain proteins, whereas kininogens contain three cystatin domain repeats. The cystatin fold was revealed by the crystal structure of chicken cystatin ([20], PDB code 1CEW), which provided the basis for the elephant trunk model of their interaction with papain-like cysteine proteases. The model was later confirmed by the crystal structure of stefin B in complex with papain ([21], PDB code 1STF), shown in Figure 1a. Cystatins block the reactive site with the N-terminal trunk and a loop. The positions of the N-terminal trunk and the loop in the structure are stabilized by a β-sheet, which is at the concave side stabilized by an α-helix. The second loop of cystatins interacts with the active site cleft and contributes to the binding [22], however, it is not directly involved in blocking the access to the cysteine histidine pair of reactive site residues. They bind to their targets in a two-step mechanism, with the loops providing the initial binding and the N-terminus locking the complex and strengthening the interaction [23], indicating that the inhibitor undergoes a conformational change on binding. The structure of the complex showed that cystatins do not interact with the reactive site of the target protease in a substrate-like manner; hence, cystatins are not slowly degrading substrates, in contrast to at the time most studied inhibitors of serine proteases, such as bovine pancreatic trypsin inhibitor (BPTI) ([24], PDB code 2TGP). Cystatins reversibly bind papain-like cysteine endopeptidases in the nM to fM range [22,25,26,27,28], whereas cysteine exopeptidases are inhibited in the mM to nM range [29,30,31,32]. Nevertheless, they bind to cathepsins B and H, as demonstrated by the crystal structure of their complexes ([33], PDB code 3K9M; [34], PDB code 1NB5). Due to the span in the binding constants, stefins and cystatins are emergency and buffer inhibitors. Table 1 shows complexes for the cystatin family.
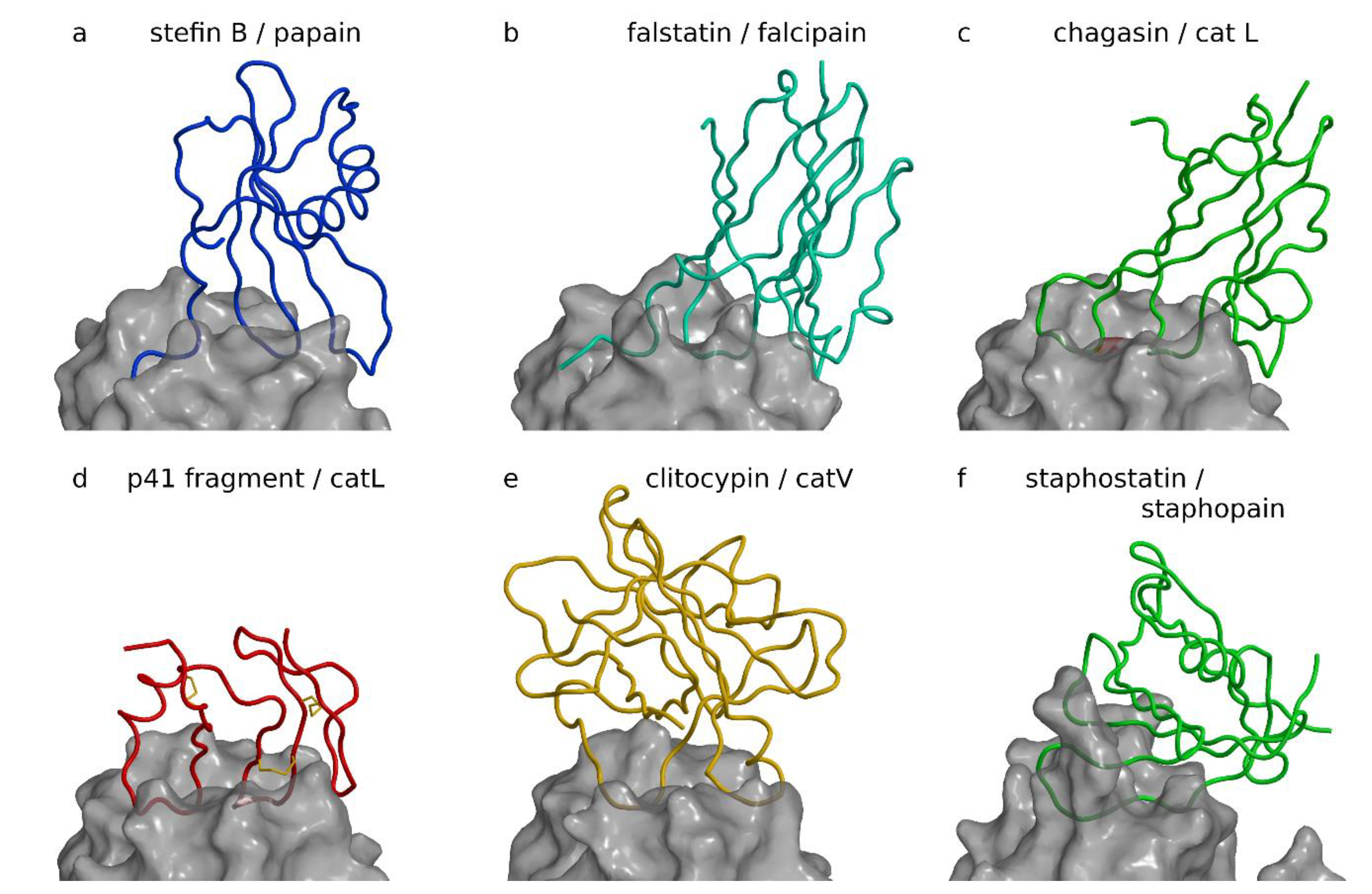
Figure 1.
Inhibitors of papain-like and related proteases. Complexes are shown with the same view across the active site cleft and the same scale after superimposition of proteases to cathepsin L in the p41 fragment complex. Figure was prepared using MAIN [35] and rendered with Raster3d [36]. (a) Stefin B papain complex ([21], PDB code 1STF). The stefin B chain is shown as a blue coil on the semitransparent background of the white surface of papain. (b) Inhibitor of cysteine protease (ICP) (falstatin) falcipain complex ([37], PDB code 3PNR). ICP, also known as falstatin from Plasmodium berghei, is shown as a cyan coil on the semitransparent background of the white surface of falcipain-2. (c) Chagasin cathepsin L complex ([38], PDB code 2NQD). The chagasin chain is shown as a green coil on the semitransparent background of the white surface of cathepsin L. (d) p41 fragment cathepsin L complex ([39], PDB code 1ICF). p41 fragment chain shown as a red coil on the semitransparent background of the white surface of cathepsin L. The three disulfide bonds of the p41 fragment are shown as yellow sticks. (e) Clitocypin cathepsin V complex ([40], PDB code 3H6S). The clitocypin chain is shown as a yellow coil on the semitransparent background of the white surface of cathepsin V. (f) Staphostatin staphopain complex ([41], PDB code 1PXV). The staphostatin chain is shown as a green coil on the semitransparent background of the white surface of staphopain.
Figure 1.
Inhibitors of papain-like and related proteases. Complexes are shown with the same view across the active site cleft and the same scale after superimposition of proteases to cathepsin L in the p41 fragment complex. Figure was prepared using MAIN [35] and rendered with Raster3d [36]. (a) Stefin B papain complex ([21], PDB code 1STF). The stefin B chain is shown as a blue coil on the semitransparent background of the white surface of papain. (b) Inhibitor of cysteine protease (ICP) (falstatin) falcipain complex ([37], PDB code 3PNR). ICP, also known as falstatin from Plasmodium berghei, is shown as a cyan coil on the semitransparent background of the white surface of falcipain-2. (c) Chagasin cathepsin L complex ([38], PDB code 2NQD). The chagasin chain is shown as a green coil on the semitransparent background of the white surface of cathepsin L. (d) p41 fragment cathepsin L complex ([39], PDB code 1ICF). p41 fragment chain shown as a red coil on the semitransparent background of the white surface of cathepsin L. The three disulfide bonds of the p41 fragment are shown as yellow sticks. (e) Clitocypin cathepsin V complex ([40], PDB code 3H6S). The clitocypin chain is shown as a yellow coil on the semitransparent background of the white surface of cathepsin V. (f) Staphostatin staphopain complex ([41], PDB code 1PXV). The staphostatin chain is shown as a green coil on the semitransparent background of the white surface of staphopain.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: cystatins.
Table 1.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: cystatins.
| Protein Inhibitor | Cysteine Protease | ||||||
|---|---|---|---|---|---|---|---|
| MEROPS ID | Name | Organism | MEROPS ID | Name | Organism | PDB ID | Reference |
| I25 | Cystatin family | ||||||
| I25.001 | stefin A | Homo sapiens | C01.060 | cathepsin B | Homo sapiens | 3K9M | [33] |
| I25.001 | stefin A | Homo sapiens | C01.040 | cathepsin H | Sus scrofa | 1NB3/1NB5 | [34] |
| I25.001 | stefin A | Homo sapiens | C01.032 | cathepsin L | Homo sapiens | 3KSE | |
| I25.001 | stefin A | Homo sapiens | C01.009 | cathepsin V | Homo sapiens | 3KFQ | |
| I25.003 | stefin B | Homo sapiens | C01.001 | papain | Carica papaya | 1STF | [21] |
| I25.006 | cystatin E/M (cystatin 6) | Homo sapiens | C13.004 | legumain, animal-type | Homo sapiens | 4N6N | [42] |
| I25.011 | ovocystatin | Gallus gallus | C01.046 | falcipain-2 | Plasmodium falciparum 3D7 | 1YVB | [43] |
| I25.014 | tarocystatin | Colocasia esculenta | C01.001 | papain | Carica papaya | 3IMA | |
| I25.014 | CTD of tarocystatin | Colocasia esculenta | C01.001 | papain | Carica papaya | 3LFY | |
2.2. Falstatins
Falstatins, which are inhibitors of cysteine proteases (ICPs) from Plasmodium spp., as demonstrated by the crystal structure of a falcipain 2 complex ([37], PDB code 3PNR, Table 2), seemingly use the same interaction pattern to inhibit their target cysteine protease falcipain as cystatins—the N-terminal peptide interacts with the protease surface in a similar manner to stefin B, followed by a two-loop arrangement filling the active site (Figure 1b). This is the extent of the similarity, however, because the Plasmodium spp. ICPs are based on a β-sandwich related to the immunoglobulin fold, and the apparent N-terminal trunk is a partially disordered loop, which thereby lacks structure, whereas the part in contact with the target protease is ordered. Hence, ICPs from Plasmodium spp. utilize a three-loop arrangement to block the active site of papain-like proteases. The visible interaction loop is the second loop and longer than the first interaction loop in cystatins because it must span the space between the two β-sheets in the fold. ICPs from Plasmodium spp. bind non-selectively, and bind to papain-like and related cysteine proteases in the pM to nM range [44]. Falstatin does not inhibit cysteine proteases with exopeptidase activity (cathepsins B and C), and proteases of other catalytic classes, such as serine proteases (trypsin and chymotrypsin), aspartic proteases (pepsin and renin), and metalloproteases (collagenase and matrix metalloprotease-2). Falstatins are also supposed to inhibit calpain-1 in the sub-nM range, and caspases-3 and -8 in the nM range [44], yet these results were later disputed by Hansen et al. [37].
Table 2.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: falstatin, chagasins, thyropin, clitocypin, and staphostatins.
Table 2.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: falstatin, chagasins, thyropin, clitocypin, and staphostatins.
| Protein Inhibitor | Cysteine Protease | ||||||
|---|---|---|---|---|---|---|---|
| MEROPS ID | Name | Organism | MEROPS ID | Name | Organism | PDB ID | References |
| I71 | Falstatin family | ||||||
| I71.001 | falstatin (PbICP-C) | Plasmodium berghei | C01.046 | falcipain-2 | Plasmodium falciparum 3D7 | 3PNR | [37] |
| I42 | Chagasin family | ||||||
| I42.001 | chagasin | Trypanosoma brucei | C01.001 | papain | Carica papaya | 2CIO | [45] |
| I42.001 | chagasin | Trypanosoma cruzi | C01.001 | papain | Carica papaya | 3E1Z | [46] |
| I42.001 | chagasin | Trypanosoma cruzi | C01.046 | falcipain-2 | Plasmodium falciparum 3D7 | 2OUL | [32] |
| I42.001 | chagasin | Trypanosoma cruzi | C01.060 | cathepsin B | Homo sapiens | 3CBJ/3CBK | [47] |
| I42.001 | chagasin | Trypanosoma cruzi | C01.032 | cathepsin L | Homo sapiens | 2NQD | [38] |
| I31 | Thyropin family | ||||||
| I31.002 | MHC II invariant chain p41 form | Homo sapiens | C01.032 | cathepsin L | Homo sapiens | 1ICF | [39] |
| I48 | Clitocypin family | ||||||
| I48.001 | clitocypin | Clitocybe nebularis | C01.009 | cathepsin V | Homo sapiens | 3H6S | [40,48,49] |
| I57 | Staphostatin family | ||||||
| I57.001 | staphostatin B | Staphylococcus aureus | C47.002 | staphopain B | Staphylococcus aureus | 1PXV | [41] |
| I57.001 | staphostatin B | Staphylococcus aureus | C47.002 | staphopain B | Staphylococcus aureus | 1Y4H | [50] |
2.3. Chagasins
Chagasins are endogenous inhibitors of papain-like cysteine proteases from parasites such as Trypanosoma cruzi and Leishmainia mexicana. They have a similar fold as that of ICPs from Plasmodium spp. In Figure 1c, the structure of the complex of chagasin from Trypanosoma cruzi with cathepsin L is shown ([38], PDB code 2NQD, Table 2). The N-terminal trunk has been replaced by a loop, hence their interaction with the active site is based on a three-loop arrangement. Several complexes of chagasin with other cysteine cathepsins ([47], PDB code 3CBK, Table 2), papain ([46], PDB code 3E1Z; [45], PDB code 2CIO, Table 2), and falcipain-2 ([32], PDB code 2OUL, Table 2) have been reported. Chagasin is a nonspecific inhibitor of papain-like proteases, with Ki values in the pM to 100 nM range [32,38]. Several structures of complexes of chagasin with other cysteine proteases, cathepsin B ([47], PDB code 3CBK, Table 2), papain ([46], PDB code 3E1Z, Table 2), and falcipain 2 ([32], PDB code 2OUL, Table 2) have been reported. Endogenous physiological inhibition of cruzipain by chagasin is reversible and tight-binding with a Ki value in the pM range [51].
2.4. p41 Fragment
The p41 fragment sequence is embedded in the much larger invariant chain associated with the major histocompatibility complex (MHC) class II-associated p41 invariant chain fragment. The p41 fragment is homologous to sequential repeats, called thryoglobulin type-1 domains, due to their numerous occurrences in thyrogobulin [52,53]. The inhibitors with this sequential repeat are called thyropins [54]. The crystal structure of the p41 fragment in a complex with cathepsin L ([39], PDB code 1ICF, Table 2) revealed its fold, which is stabilized by three disulfide bonds (Figure 1d). The CWCV sequence, the signature of the fold, is at its core, and with which two disulfide bonds stabilize the three stranded β-sheet. The third disulfide bond attaches the helix to the body of the fold. The p41 fragment is the smallest of the cysteine protease inhibitors composed of only 64 amino acid residues. Similarly to inhibitors of cysteine proteases (ICPs) and chagasin, it uses a three-loop arrangement to bind to the active site. The first and the second loops of the p41 fragment occlude the reactive site, whereas the third forms additional contacts at the primed side of the active site cleft. The p41 fragment is more selective than cystatins. It inhibits the endopeptidases cathepsins L and V in the pM range, cathepsins K and F in the nM range, and cathepsin S in the µM range, but does not inhibit exopeptidases [55,56]. On the basis of these values and its concentration, we can consider the p41 fragment an emergency inhibitor (cathepsins L and V) or a buffer-type inhibitor (cathepsin S) [56]. It may even be possible that its inhibitory role is activated after processing of the invariant chain, hence the p41 fragment may also be a pro-inhibitor or a delay-type inhibitor. In addition to thyroglobulin, several other proteins contain this structural motif, such as saxiphilin, which binds saxitoxin, a toxin from bullfrogs [57]. However, the inhibitory function has been confirmed for few of these, including equistatin inhibiting cysteine cathepsins [58]; testicans-2 and -3, which inhibit matrix metalloproteases and serine proteases’ saxiphilin, which inhibits papain and cathepsin L; and the inhibitor from salmon egg, which inhibits papain and cysteine cathepsins [58,59,60,61,62,63,64]. In other proteins, such as nidogen, insulin growth factor-binding proteins, and the human carcinoma marker protein GA733 (also called TROP2 [65]), the inhibitory role has not been demonstrated, although it has been suggested that their thyroglobulin type-1 domains may serve as a buffer for the activity of endosomal proteases during thyroglobulin processing [66,67,68]. Hence, the conclusion that the thyroglobulin type-1 repeat is a structural motif occasionally employed as an inhibitor of proteases still applies [69].
2.5. Clitocypins and Macrocypins
Clitocypins and macrocypins from mushroom species (basidiomycetes) Clitocybe nebularis and Macrolepiota procera, respectively, are primarily inhibiting papain-like cysteine proteases, although inhibition of legumain and serine proteases has also been observed [49]. This property is due to their β-trefoil fold, the hallmark of Kunitz-type inhibitors, which are the classical serine protease inhibitors ([40], PDB code 3H6S, Table 2). The β-trefoil fold has a remarkably simple core composed of a sixfold β-barrel and six exposed loops stabilized by short β-antiparallel strand arrangements (reviewed in [48]). The crystal structure of the complex between clitocypin and human cathepsin V (Figure 1e) shows that clitocypin (and similar macrocypins) binds in the active site of papain-like proteases with two broad loops occluding the catalytic site residues from both sides of the active site cleft. Two broad loops are a common denominator in all complexes presented in Figure 1 and indicate convergence in the mechanism of inhibiting papain-like proteases. An exception is the staphostatin/staphopain complex shown in Figure 1f, in which the protease staphopain is not papain-like, but a papain-fold-related protease. Clitocypin and macrocypins inhibit papain-like proteases in the 10 pm to 100 nM range [49,70].
2.6. Staphostatins A and B
Staphostatins A and B are endogenous inhibitors of the secreted cysteine proteases from Staphylococcus aureus, staphopains A and B, which are remotely related to cysteine cathepsins. Each staphostatin specifically inhibits its target staphopain. Staphostatins have a β-barrel fold, which is similar to that of lipopains and different from that of cystatins [71]. The crystal structure of staphostatin B in complex with the staphopain B catalytic site mutant C243A ([41], PDB code 1PXV; [50], PDB code 1Y4H, Table 2) shows that staphostatin B binds in the active site cleft of staphopain in a substrate-like manner with the sequence IGTS mimicking the P2 to P2′ substrate residues (Figure 1f), which explains why staphostatins are slowly degraded substrates. Filipek et al. [50] further showed that the G98 residue is crucial for inhibitory activity because its mutations in other residues converted staphostatin B to a significantly better substrate of staphopain B. The extended conformation of the G98-T99 section is stabilized by a hydrogen-bonding ladder with the antiparallel positioned S93-S92-T91 section in the central β-sheet, likely keeping the G98-T99 peptide bond at a distance to prevent its hydrolyses. Hence, rather than a loop, an antiparallel β-sheet hydrogen bonding ladder stabilizes the bound conformation.
2.7. Serpins
Serpins obtained their name from their ability to inhibit serine proteases, however, they are cross-class inhibitors that also inhibit cysteine proteases such as cathepsins, calpains, and caspases. With over 1500 representatives in Archaea, Prokarya, and Eukarya, in addition to a number of viruses, serpins are the largest and most widely dispersed family of peptidase inhibitors [72], and include 37 human serpins [73]. Squamous cell carcinoma antigen 1 (SCCA1 also called serpin B3) is an epithelial-derived serpin that inhibits the endopeptidases cathepsins K, L, and S [74,75]. Heparin was also found to enhance the interaction with the target proteases, similar to the interaction of several serpins with plasma serine proteases [76]. In addition to SCCA1, cathepsin L was shown to be inhibited by the cross-class inhibitor endopin 2C [77], which preferentially inhibits cathepsin L over papain and elastase.
Among cathepsins, cathepsin L is specifically targeted with another cross-class serpin, hurpin (serpin B13) [78]. Another cross class inhibitor SRP-6 was shown to inhibit cathepsins K and L, and calpain-2 [79]. Although serpins are primarily endogenous inhibitors, they are also employed as part of a defense against pathogens. SCCA1 was shown to inhibit staphopains [80] and falcipain-2 [81], whereas SCCA 2 was shown to inhibit mite allergen cysteine protease Der p1 [82]. Inhibition of caspases by serpins is described below.
3. Inhibitors of Caspases
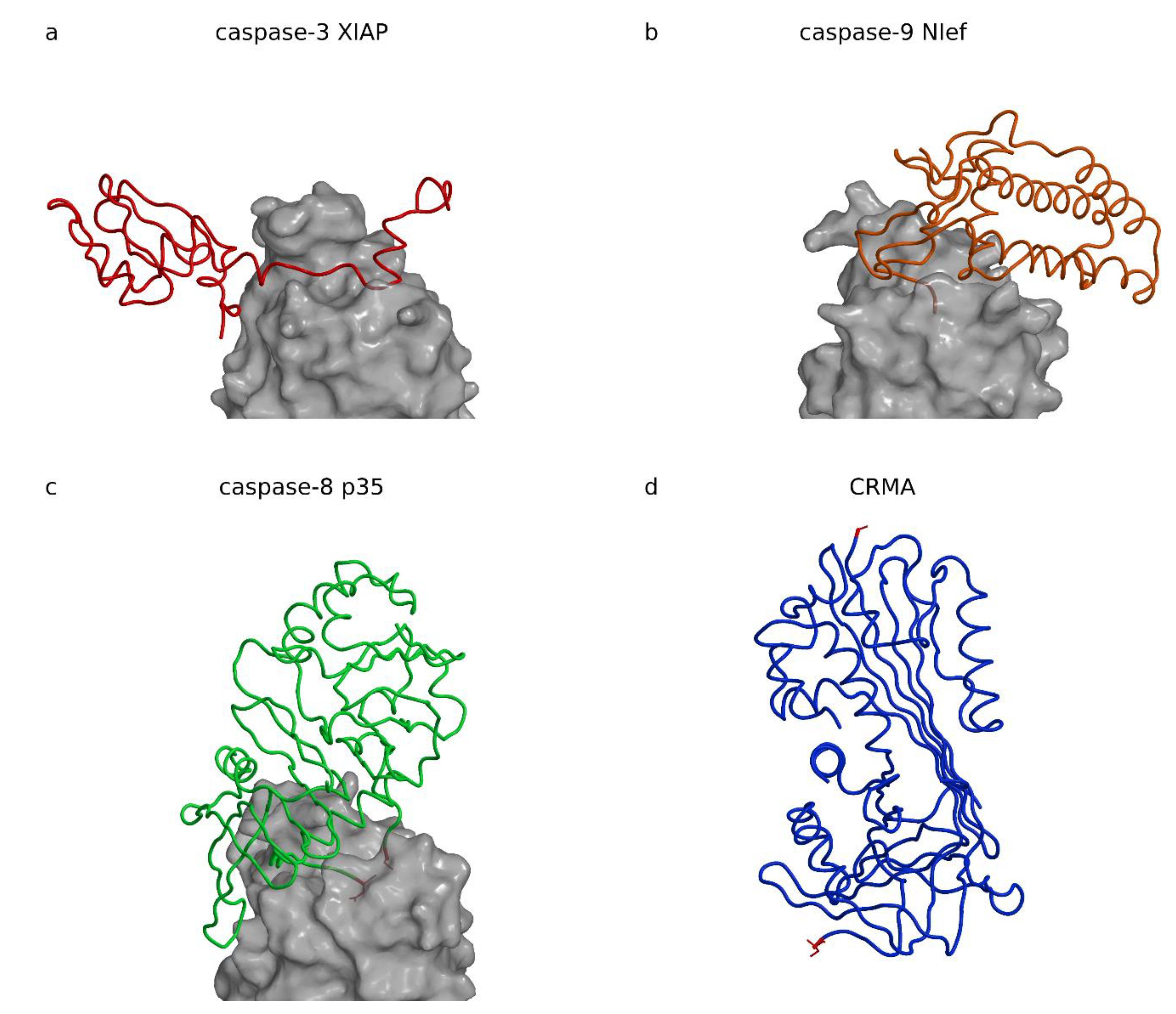
Cell apoptosis is an important mechanism during embryogenesis and organism growth to make place for new cells and tissues. Later in life, apoptosis is required for removal of defected, infected, and malicious cells, and is crucial for organism survival. Caspases take a central role in the apoptosis initiation and execution phases. They are cysteine proteases belonging to their own C14 family (MEROPS: [15]). Because removal of infected cells by apoptosis is also a defense mechanism against infections, it is no coincidence that caspase inhibitors have been found in cell invaders such as viruses (baculovirus inhibitor of apoptosis proteins (IAPs) and protein p35, cowpox virus serpin cytokine response modifier A (CrmA)) and bacteria (Escherichia coli effector protein Nlef). Figure 2 shows structures of four caspase inhibitors, three in complexes made with X-linked inhibitor of apoptosis (XIAP), Nlef, and p35, and the serpin CrmA. As Figure 2 demonstrates, the folds of these four inhibitors have no common structural motif, which suggests their common role or at least an evolutionary relationship. In addition, the view in which all three caspases are superimposed shows that XIAP, Nlef, and p35 are positioned at positions most widely spread from left to right in respect to the active site of their target caspases.
3.1. XIAP
XIAP belongs to the protein family of inhibitor of apoptosis proteins (IAPs) present in viruses [87,88] and eukaryotes. IAPs activity is embedded in the baculoviral IAP repeat (BIR) domains, the homologues of which are present throughout all eucaryotic kingdoms [89]. They are involved in regulation of the activity of executioner and initiator caspases-3, -7, and -9. The crystal structures of XIAP (BIR domain 2) in complex with caspase-7 ([90]; PDB code 1I51, Table 3); [91]; PDB code 1I4O, Table 3) or caspase-3 ([83]; PDB code 1I3O, Table 3) revealed that XIAP binds along the active site cleft of the caspase (Figure 3a).
Table 3.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: IAPs, p35, and NIef.
Table 3.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: IAPs, p35, and NIef.
| Protein Inhibitor | Cysteine Protease | ||||||
|---|---|---|---|---|---|---|---|
| MEROPS ID | Name | Organism | MEROPS ID | Name | Organism | PDB ID | Reference |
| I32 | IAP family | ||||||
| I32.002 | cIAP-BIR3 | Homo sapiens | C14.010 | caspase-9 (Nter pept) | Homo sapiens | 3D9T | [92] |
| I32.004 | XIAP | Homo sapiens | C14.004 | caspase-7 | Homo sapiens | 1I4O | [93] |
| I32.004 | XIAP | Homo sapiens | C14.004 | caspase-7 | Homo sapiens | 1I51 | [90] |
| I32.004 | XIAP-BIR2 | Homo sapiens | C14.004 | caspase-7 | Homo sapiens | 1KMC | |
| I32.004 | XIAP-BIR2 | Homo sapiens | C14.003 | caspase-3 | Homo sapiens | 1I3O | [83] |
| I32.004 | XIAP-BIR3 | Homo sapiens | C14.010 | caspase-9 | Homo sapiens | 1NW9 | [94] |
| I32.009 | DIAP1 | Drosophila melanogaster | C14.019 | caspase Dronc (pept) | Drosophila melanogaster | 1Q4Q | [95] |
| I32.009 | DIAP1-BIR1 | Drosophila melanogaster | C14.015 | drICE | Drosophila melanogaster | 3SIP | [96] |
| I50 | Baculovirus p35 family | ||||||
| I50A | p35 | Autographa californica nucleopolyhedrovirus | C14.009 | caspase-8 | Homo sapiens | 1I4E | [85] |
| I50A | p35 | Autographa californica nucleopolyhedrovirus | C14.009 | caspase-8 | Homo sapiens | 2FUN | [97] |
| Family I94 | |||||||
| I94.001 | NIeF | Escherichia coli | C14.010 | caspase-9 | Homo sapiens | 3V3K | [84] |
XIAP has a short helical region the authors called a “hook” followed by a long linker, which runs along the active site cleft of the caspase and the terminal BIR2 domain with the “sinker” interacting with the S4 pocket.
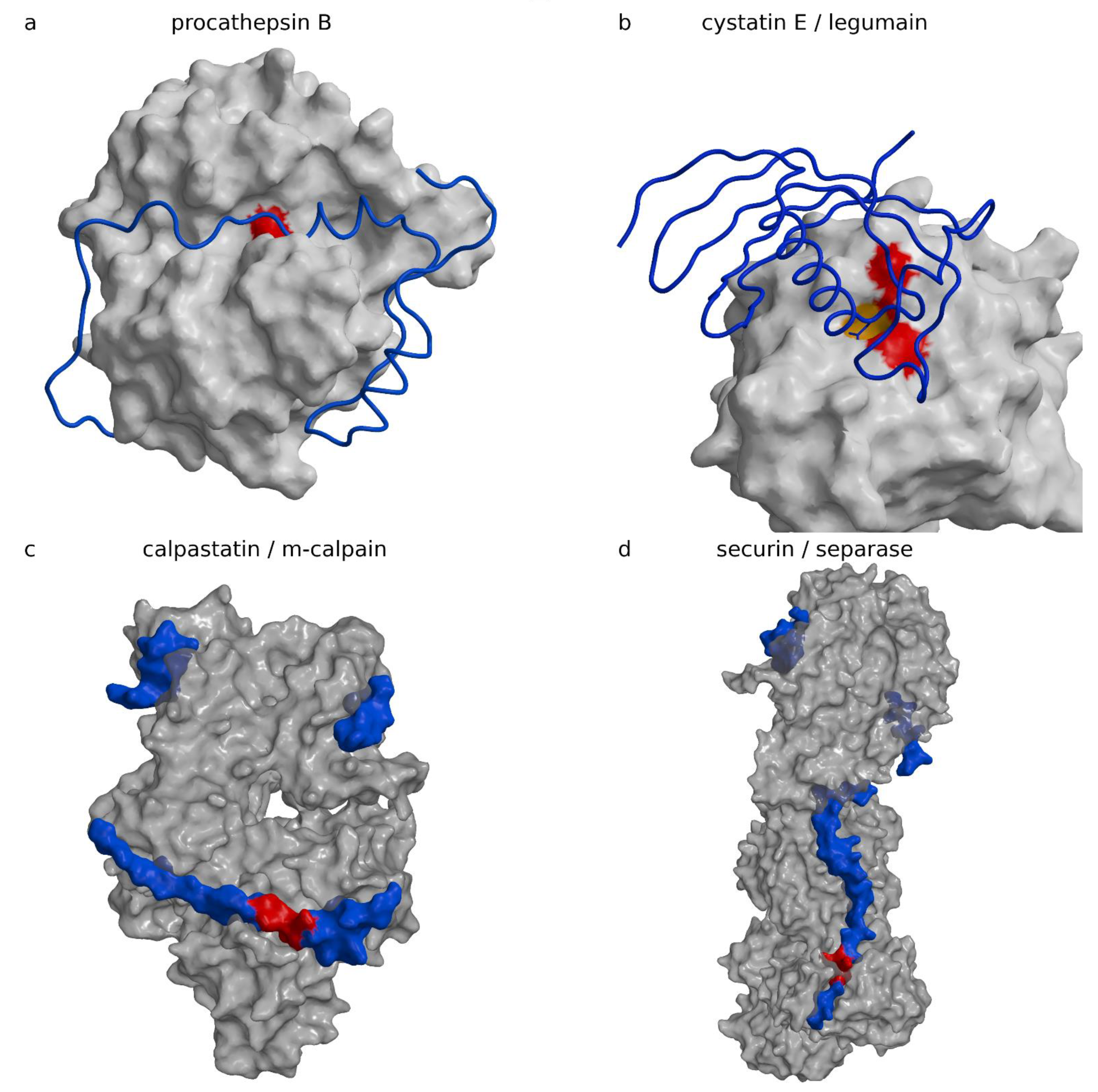
The “hook” and the “sinker” with the BIR2 domain attach to the caspase surface and stretch the linker. The linker runs along the active site cleft in the direction opposite to the substrate binding, thereby precluding its cleavage. The concept is reminiscent of cysteine cathepsin inhibition by their propeptides in their zymogen form (Figure 3a shows procathepsin B; [98], PDB code 3PBH, Table 4), in particular, the smallest one of cathepsin X ([101], PDB code 1DEU, Table 4). The similarity is dual: (i) the propeptides of cysteine cathepsins run along the active site cleft in the direction opposite to the substrate, and (ii) the propeptides form smaller and larger domains, which all begin with a helix positioned at the prime side of the active site cleft approximately above the reactive site cysteine histidine pair. Because prodomains of cysteine cathepsins and BIR2 lie on opposite sides of the active site cleft, the similarity reflects a convergent solution. The fold of the BIR domain is, interestingly, not important for inhibition, as noted earlier [1]. An important contribution of the XIAP BIR2 domain to a two-site interaction inhibition of caspases-3 and -7 has been proposed by Scott et al. [102], in which the weak interaction of the linker sequence that inhibits activity must be stabilized by the binding of the BIR2 domain surface groove that binds caspase-7 at a site exposed only during the maturation cleavage. IAP proteins inhibit caspases by several distinct mechanisms. For example, the BIR3 domain of XIAP inhibits caspase-9 by blocking the dimerization of the catalytically-inactive monomers that is required for activity ([94], PDB code 1NW9).
Table 4.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: proenzymes.
Table 4.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: proenzymes.
| Zymogens | ||||
|---|---|---|---|---|
| MEROPS ID | Name | Organism | PDB ID | References |
| C10.001 | exotoxin B (streptopain) | Streptococcus pyogenes | 1DKI | [103] |
| C01.060 | procathepsin B | Homo sapiens | 3PBH | [98,104] |
| C25.003 | gingipain RgpB | Porphyromonas gingivalis W83 | 4IEF | [105] |
| C01.032 | procathepsin L | Homo sapiens | 1CJL/1CS8 | [106] |
| C01.001 | propapain | Carica papaya | 3TNX | [107] |
| C01.003 | procaricain | Carica papaya | 1PCI | [108] |
| C01.013 | procathepsin X | Homo sapiens | 1DEU | [101] |
| C01.060 | procathepsin B | Rattus norvegicus | 1MIR | [109] |
| C01.036 | procathepsin K | Homo sapiens | 7PCK | [110] |
| C01.036 | procathepsin K | Homo sapiens | 1BY8 | [111] |
| C01.034 | procathepsin S | Homo sapiens | 2C0Y | [112] |
| C01.040 | procathepsin H | Homo sapiens | 6CZK/6CZS | [113] |
3.2. Escherichia coli Effector Protein Nlef
Escherichia coli effector protein Nlef inhibits caspases-4, -8, and -9 [84,114,115,116,117,118]. The crystal structure of the complex between Nlef and human caspase-9 indicates two Nlef segments interacting with the active site cleft of caspase-9, the protein C-terminal sequences L196, Q197, C198, and G199; and the H145, H146, and S157 (Figure 2b) ([84], PDB code 3V3K, Table 3). The Nlef can be considered a substrate analog, only falling short of one residue to be cleaved. Instead, its C-terminal residue lacks the side chain and deploys the C-terminal carboxylic group of G199 to mimic the aspartate at the P1 position. This concept is reminiscent of the cathepsin C exclusion domain-binding mechanism, which provides the N-terminal carboxylic group of the aspartic residue to block access in the active site beyond the S2 site, and thereby restricts activity of cathepsin C to a di-amino-peptidase [119]. The similarity is even more striking in the case of a metalloprotease inhibitor from Erwinia chrisantemi, which fills half of the active site with its N-terminus and thereby blocks access to substrates [120]. Interestingly, it was shown that Nlef is only one among many Escherichia coli effector proteins causing delay and inhibition of apoptosis. Its role appears minor due to its low expression profile, however, when over expressed it can severely impact apoptosis [84].
3.3. Baculovirus Protein P35
Baculovirus protein p35 is a broad-spectrum caspase inhibitor. It has a flexible reactive loop with the caspase recognition sequence DQMD ([121]; PDB code 1P35). The crystal structure of the p35 in complex with caspase-8 ([85], PDB code 1I4E, Table 3) shows how the cleaved loop remains trapped in its covalent attachment to the enzyme with D87 forming the thioester bond to the caspase-8 C360 reactive site cysteine and D84 bound to the S4 binding site. The covalent interaction explains why the crystal structure of the complex shows loose packing of the p35 chains entering the active site cleft (Figure 2c). These two chains belong to two different N-termini sequentially far apart: the D87 at the N-terminus resulting from the p35 cleavage and the repositioned residue C2 at the p35 N-terminus. Later, the sulfhydril group of C2 was shown to be crucial for preventing hydrolysis of the caspase C360p35 D87 thioester bond by trapping it in the exchange with the p35 thioester bond of C2D87, as demonstrated by Lu et al. ([97], PDB code 2FUN, Table 3), who found that the N-terminal fragment of p35 appeared as a circular peptide after dissociation from the complex.
3.4. Cowpox Virus CrmA
The cowpox virus CrmA structure was chosen to represent serpins as inhibitors of caspases. Its crystal structure was determined in the cleaved form (Figure 2d), with the P1 and P1′ residues A359 and S359A, respectively, more than 60 Å apart ([86], PDB code 1F0C; [122], PDB code 1M93). At conditions preventing hydrolysis, it was shown that serpins’ reactive site loop binds in the active site cleft of trypsin in extended conformation ([123], PDB code 1K9O), which is in a strong contrast with the cleaved form structure. The mechanism of inhibition of cysteine proteases was not demonstrated with a crystal structure of a complex of the cleaved form, however, the typical serpin insertion of the reactive site sequence in the central β-sheet suggests the trypsin-like mechanism of inhibition ([124], PDB code 1EZX), and formation of a thiol ester with the catalytic cysteine that in part unfolds the target protease [125,126,127]. CrmA is a minimal serpin. It targets caspases-1 with Ki in the pM range [128,129] and caspase-8 in the sub-nM range [130,131,132], but poorly inhibits executioner caspases-3, -6, and -7 [131,132], and probably caspase-10 [130]. Similar to a number of other serpins, such as serpin B9 [133] and myxoma virus serpin serp2 [130], it also inhibits serine protease granzyme B [130]. Although serpins eventually separate from their target protease, they cannot bind back to it. Serpins are suicide substrates irreversibly changed upon reaction. In fact, the pathway, called the suicide substrate branched pathway mechanism, is even more complicated because it involves one two-way and four one-way processes, all of which end in separation of serpin from its target (reviewed in [12]). Therefore, in such cases Ki does not apply. For simple comparison of inhibition rates of serpins, we advise the use the kinetic constant kass of the first step only.
4. Some Other Types of Inhibitors
4.1. Propeptides of Papain-Like Cysteine Proteases
Propeptides of papain-like cysteine proteases are in their essence inhibitors attached to the framework of the mature protease structure [134,135]. They are not entirely specific to their cognate enzyme and may inhibit other enzymes in the family [136]. All propeptides share the same architecture. They fold around the L-domain of the mature enzyme, as shown for the propeptide of cathepsin B (Figure 3a) ([98], PDB code 3PBH, Table 4). Exceptionally, the papain-like enzyme is shown in an orientation in which the active site cleft runs from left to right (standard view is from bottom to the top), which brings the so-called R- and L-domains to the bottom and top of the image. They wrap around the R-domain of the mature part of the enzyme. They build an N-terminal, predominantly a helical domain of various sizes, which binds to the surface of the L-domain of the enzyme on the prime side (left in the figure) and enters the active site cleft with an α-helix ending above the pair of catalytic cysteine and histidine residues (colored red). Then, the chain continues in the direction opposite to substrate binding along the active site and turns down where it joins with the enzyme’s N-terminus. Several structures of proenzymes of papain-like proteases (Table 4) have been determined (procathepsins B ([109], PDB code 1MIR; [98], PDB code 3PBH), L ([106], PDB code 1CJL, 1CS8), H ([113], PDB code 6CZK/6CZS), K ([110], PDB code 7PCK; [111], PDB code 1BY8), and S ([112], PDB code 2C0Y), and propapain ([107], PDB code 3TNX)); among them, the propeptide of procathepsin X appears the shortest ([101], PDB code 1DEU). Table 4 shows the zymogens.
Its N-terminal domain is composed of a short peptide only, for which the reactive site attachment is strengthened by a disulfide formed between the reactive site C31 and propeptide C10P. In contrast, the propeptide of cathepsin L has the largest N-terminal domain, composed of 96 residues. The role of N-terminal domains appears to be the same; they anchor the propetide in the primed side of the structure to enable its stretched binding along the active site cleft to the enzyme’s N-terminus.
4.2. Cystatin E and Macrocypins
Cystatin E and macrocypins, such as macrocypins 1 and 3, can, in addition to papain-like proteases, also inhibit legumain (known also as asparagine endopeptidase or AEP) and macrocypin 4, and even the serine protease trypsin [49]. Among these, only the crystal structure of the complex between cystatin E and legumain has been determined (Figure 3b) ([42], PDB code 4N6N). In the complex, cystain E makes contact with legumain with two loops. The first, called the reactive center loop, encompasses residues from G37 to I41, which bind across the reactive site in a substrate-like manner with the N39 side chain binding in the pocket S1, which specifically recognizes asparagine residues and, under acidic conditions, also accepts aspartic residues. The second loop from D72 to Q96, called the exosite loop, includes residues from R74 to D81, which make contact in the region of the primed substrate binding sites. Because the reactive site loop binds as a substrate, Dall et al. [42] investigated the possible cleavage and observed that cystatin was indeed cleaved after N39 and that, over time, the ratio between the cleaved and uncleaved cystatin remained constant. On the basis of the subsequent analysis, which included chemical modification of the reactive site C189 with S-methyl methanethiosulfonate, the authors arrived at the conclusion that, at neutral pH, legumain behaves as a ligase and C189 is not involved in this reaction. They assigned the catalytic activity to a different catalytic center, which they assigned in an unorthodox manner to succinate 147, a chemical modification of D147, clearly recognizable in the electron density map. In a follow-up theoretical work, they confirmed the initial idea that cysteine is not involved in the ligase reaction, however, the catalytic center was assigned to H148 [137], and succinate 147 carbonyl was used to stabilize the side chain of H148. The ligase activity of cysteine proteases as a consequence of the pH of the media may be a common phenomenon. It was also observed for papain at pH above 9 [138] and cathepsin C at neutral pH [139,140].
4.3. Calpastatin
Calpastatin is a highly selective inhibitor of calpains, which are Ca2+-dependent multidomain cysteine proteases with the catalytic domain that shares some resemblance to the papain fold. Calpains are involved in a number of processes including cell migration, cell death, insulin secretion to synaptic function, and muscle homeostasis [141,142,143,144,145], whereas under pathological conditions they have been linked to cell death by necrosis induced by stroke [91], neuronal injury and perhaps Alzheimer’s disease [91,146], heart disease [146], cataract formation [91,146], type 2 diabetes [91,146,147], cancer, and limb-girdle muscular dystrophy type 2A [91,146,147].
The crystal structure of the complex between m-calpain and the first repeat of calpastatin truncated to the residues from 119 to 238 ([99], PDB code 3DF0, Table 5) reveals that calpastatin is a polypetide that adopts a three-dimensional structure in the presence of its target, calcium-activated m-calpain (Figure 3c). Parts of the chain remained unstructured even after binding to calpain. In Figure 3c, calpastatin is presented as a surface model because the chain trace of the calpastatin coil appears too small to be resolved. The calpastatin chain binds in the active site cleft in the direction of the substrate with L175 filling the specificity pocket S1 (please note that we do not follow the authors numbering from the publication, but instead follow the numbering of residues in the PDB file).
Table 5.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: calpastatins, securing, and designed ankyrin repeat proteins (DARPins).
Table 5.
List of the available structures of the complexes between protein inhibitors and cysteine proteases: calpastatins, securing, and designed ankyrin repeat proteins (DARPins).
| Protein Inhibitor | Cysteine Protease | ||||||
|---|---|---|---|---|---|---|---|
| MEROPS ID | Name | Organism | MEROPS ID | Name | Organism | PDB ID | Reference |
| I27 | Calpastatin family | ||||||
| I27.001 | calpastatin | Rattus norvegicus | C02.002 | calpain-2 | Rattus norvegicus | 3DF0 | [99] |
| I27.001 | calpastatin | Rattus norvegicus | C02.002 | calpain-2 | Rattus norvegicus | 3BOW | [148] |
| securin | Saccharomyces cerevisiae S288C | C50.001 | separin | Saccharomyces cerevisiae S288C | 5U1S/5U1T | [100] | |
| Interactor of FizzY protein | Caenorhabditis elegans | C50.004 | separase | Caenorhabditis elegans | 5MZ6 | [149] | |
| DARPin | synthetic construct | C14.006 | Caspase-2 | Homo sapiens | 2P2C | [150] | |
| DARPin-3.4 | synthetic construct | C14.003 | Caspase-3 | Homo sapiens | 2XZD | [151] | |
| DARPin C7_16 | synthetic construct | C14.004 | Caspase-7 | Homo sapiens | 4JB8 | [152] | |
| DARPin D7.18 | synthetic construct | C14.004 | Caspase-7 | Homo sapiens | 4LSZ | [153] | |
| DARPin 8h6 | synthetic construct | C01.060 | Cathepsin B | Homo sapiens | 5MBM | [154] | |
| DARPin 81 | synthetic construct | C01.060 | Cathepsin B | Homo sapiens | 5MBL | [154] | |
However, the chain then turns away from the catalytic residues, forming a cross-over of the reactive site with the IKEGT sequence called “loop out”, colored red in Figure 3c, and only thereafter following the active site cleft with I182 and a pair of proline residues that lie at the N-terminus of a helix. The helix just beyond the active site cleft in the primed side is reminiscent of cathepsin propeptide structures; however, their chains run in the opposite directions so that the helix N-terminus of calpastatin, rather than the C-terminus, is positioned near the active site. The exact sequence and length of the loop out region appears crucial for inhibition because the deletion mutant of K178 abolishes any inhibition.
4.4. Securin
Securin inhibits separase, a protease involved in separation of sister chromatids during chromosome segregation during somatic cell division at mitosis and meiosis [155,156,157,158,159,160,161]. The crystal structure ([100], PDB code 5ULS, 5ULT) and the electron microscopy (EM) structure of intermediate resolution ([149], PDB code 5MZ6) of the complex were determined almost simultaneously (Table 5). Due to having a substantially more complete model and more accurate insight, we have shown the crystal structure of Saccharomyces cerevisiae securin (Figure 3d). Separases are large four-domain proteins with chains longer than 1600 residues. The C-terminal domain is catalytic. There was a disagreement between Luo et al. [100] and Boland et al. [149] regarding whether separase is a caspase-like enzyme. FatCat [162] found that 146 residues could be aligned between the human caspase-3 and the catalytic domain of S. cervisiae separase with an root mean square deviation (RMSD) of 3.1 Å and 8% sequence identities. Hence, folds are superimposable. In addition, upon visual inspection, a high similarity of the folds is clearly recognizable. However, a number of secondary structure elements do not share deviations within the 3 Å of RMSD. Moreover, separase substrate specificity differs from the specificity of caspases. In contrast to caspases, separase cleave substrates after an arginine at P1, which is, however, characteristic of metacaspases, and glutamate at P4 [156,161,163]. In addition, loops surrounding the active site cleft provide a different means of entry to the substrate and potential inhibitors. Hence, in addition to the difference in molecular size and the number of domains, separases and caspases belong to two different protease families according to MEROPS classification [15]. The structures of the complex reveal that securin—which starts in the structure with M257 and runs to E361, and is unstructured in the naked form—binds along and around all four domains of separase, including the whole active site of the enzyme. No own secondary structure motifs stabilize its conformation, which relies completely on binding to discontinuous grooves of its target. Consequently, a substantial part of the securin structure in the complex remains unstructured. When preparing Figure 3d, we decided to show securin in a similar fashion to that of calpastatin in Figure 3c, that is, as a surface rather than a coil, due to its small size in comparison to the large separase structure. The region P263 to R265 is colored red to indicate the position of the reactive site. Although the securin chain binds in the active site cleft in the direction of a substrate, it is not a substrate analogue. S3 is filled with I261, however, the S1 pocket is instead filled with arginine covered with a proline residue P263, conformational rigidity of which likely makes securin non-cleavable, and then turns away from the catalytic pair C2110–H2083 and bypasses it. Securin regulates separase activity; however, it interacts with all domains, including those carrying no proteolytic activity, and hence its regulatory function is far more complex than the simple regulation of separase proteolytic activity.
5. Mechanisms of Inhibition
The brief analysis presented here suggests that the protein fold does not present a major obstacle for the evolution of a protease inhibitor. Numerous folds are adopted to inhibit proteases. Simple and small folds exist, such as cysteine cathepsins’ propeptide domains based on the α-helical folds, cystatins and p41 fragments using a β-sheet combined with an α-helix, as well as ICPs from Plasmodium, staphostatin, chagasin, clitocypins and macrocypins, and the exclusion domain of cathepsin C [119] using β-barrels of various strand numbers and architectures. More complex folds also exist that contain several motifs that combine α-helical and β-sheet elements, such as in serpin CrmA, p35, and XIAP. Some of the inhibitors are embedded in larger protein chains, such as the p41 fragment, and some appear as multiple repeats including kininogens [10], equistatin [58], and calpastatins ([99]). In addition, peptide-like inhibitors exist without a folding pattern, such as calpastatin and securin, which appear to fold only in contact with their target protease. Our understanding of protein inhibitor protease interactions follows the understanding of the substrate enzyme interaction models. The first model to explain the match between a substrate and an enzyme, introduced by Fischer, used the “lock and key” analogy [164]. In the 1950s, this model was enriched by Koshland with the ”induced fit” theory [165]. More recently “conformational selection” was introduced to describe the dynamic of binding events [166]. From the behavior of partially or completely unfolded inhibitors, it appears that their dynamic surpasses the extent of dynamics in the conformational selection model because some protease inhibitors appear to lack discernable conformations in the pre-bound state.
Our survey of the structures of the majority of cysteine protease protein inhibitors in complexes with cysteine proteases shows that the diversity of inhibition mechanisms appears to be unlimited. It appears as if nature has found numerous means of successively overcome almost any starting fold. Thus, it is to be expected that the folds first observed in protein inhibitors, such as cystatins and serpins, may be used in proteins exhibiting other physiological roles and functions, such as monellin, a sweet-tasting protein with the cystatin fold (reviewed in [167]), or the non-histone architectural protein myeloid and erythroid nuclear termination stage-specific protein (MENT), which participates in DNA and chromatin condensation [168]. The challenge is whether we can do the same. Does science provide enough insight, understanding, and tools to enable us to design protein inhibitors of proteases for medical, agricultural, and industrial uses?
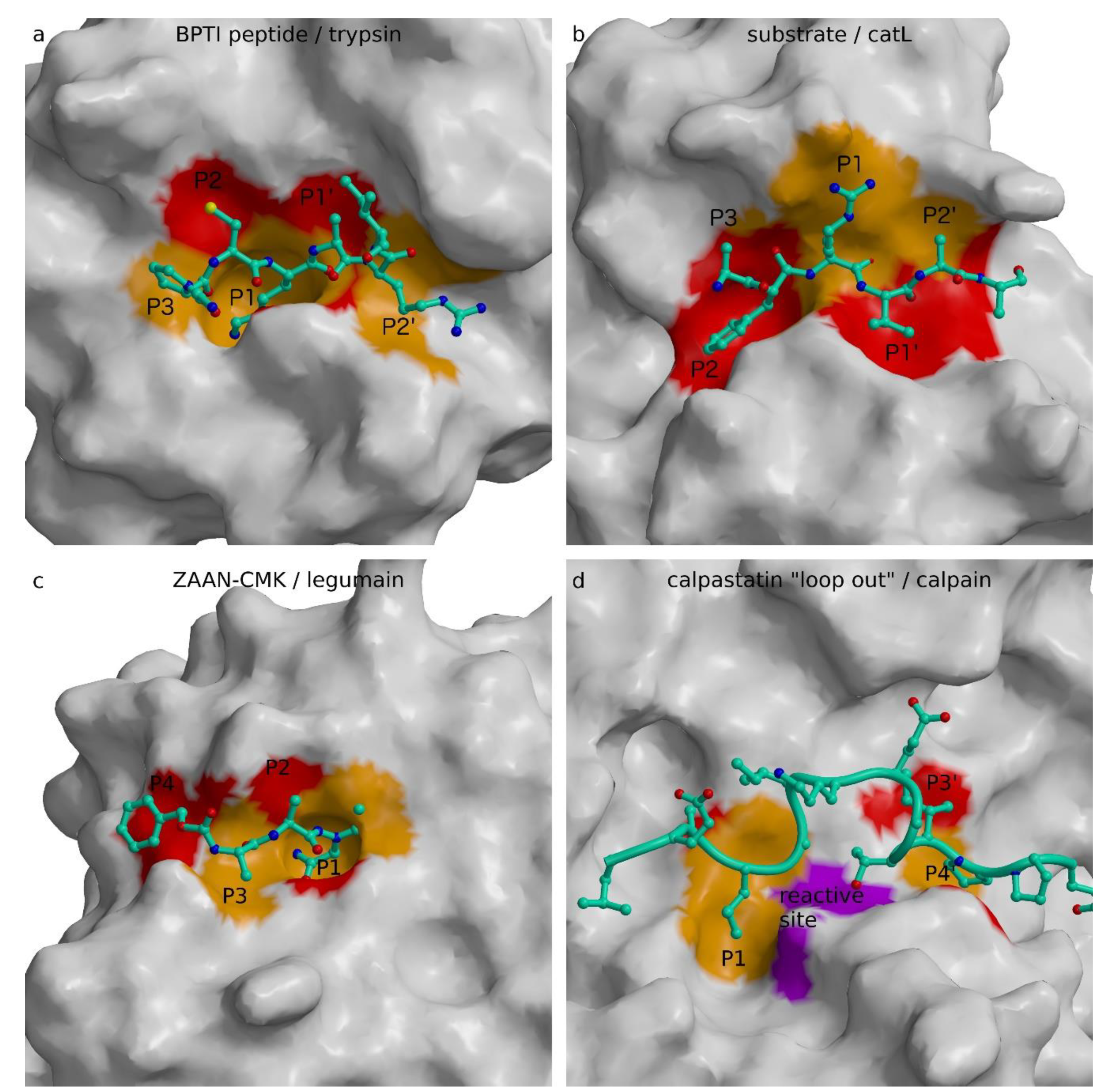
To provide insight, we revisited the review of Bode and Huber of the interactions of natural protein protease inhibitors [5]. This seminal work suggested that the era of the substrate-like canonical serine protease inhibitors of different folds, and the same active site binding geometry and product-like inhibition of carboxypeptidase inhibitor, ended with complexes of stefin B with papain [21] and hirudin with thrombin ([169], PDB code 3HTC), which revealed non-substrate-like interactions. In addition, protein inhibitors of cysteine proteases can be divided into two groups: those mimicking a protease substrate and those that do not. To demonstrate the requirements imposed by the structure of the active site cleft, we prepared Figure 4 with the canonical region of the BPTI in the complex with trypsin to compare it with the modeled substrate in the active site of cathepsin L, with the substrate analogue, inhibitor Z-Ala-Ala-Asn-chloromethyketone (ZAAN-CMK), in the complex with legumain, and the loop out construct of calpastatin. The canonical conformation of BPTI and the substrate model of cathepsin L, as well as ZAAN-CMK, all bind in an extended conformation along the active site cleft in the direction of the N- to C-peptidyl termini running from left to right. The up and down directions of the side chains clearly demonstrate the opposite patterns. Using the same alternate red and orange coloring for the surface of substrate-binding sites from S3 to S2′, one can expose a reverse color pattern between the active sites of trypsin (Figure 4a) and legumain (Figure 4c) on the one side and cathepsin L (Figure 4b) on the other. Whereas the trypsin and legumain upper parts of the surfaces are red and lower orange, the opposite coloring pattern is present in the cathepsin L surface. The orange S1 pocket pointing inwards in trypsin is the structural feature that dominates its arginine/lysine specificity, similar to the S1 pocket in legumain with asparagine/aspartate specificity, whereas the S1 in cathepsin L has no pocket—it is merely a surface to which substrate side chains can attach from a side. Hence, papain-like proteases provide significantly less structural restraints and enable broader selectivity of residues at P1. There is one important consequence of the shape and position of the S1 binding site, namely, the presence of the S1 pocket that requires the substrate P1 residue side chain to point away from the substrate surface—that is, towards the inside of a protein substrate or towards the solvent in the case of structural restraints free of peptide chains. This suggests that cathepsin L-like substrates must exhibit considerable flexibility to be able to adopt their binding geometry to the active site of cathepsin L–like protease. As a consequence, inhibitors of papain-like cysteine proteases cannot mimic a “canonical” substrate-like geometry because it embeds flexibility of the putative “canonical” region. Hence, their inhibitors also cannot mimic a substrate without being cleaved. We are thereby compelled to suggest that protein inhibitors targeting papain-like proteases in a substrate-like manner do not exist. Serpins are no exception to this rule, because they do not bind their target papain-like protease in a stable canonical conformation, but are essentially suicide substrates that, rather than remaining bound, use a flexible reactive site loop to pull out a part of catalytic site before the reaction can be completed, and remain loosely attached to the enzyme outside the immediate active site region, as indicated by the structure of the trypsin anti-trypsin complex [124]. Serpins are a cross-class type of protease inhibitors because they can pull out the catalytic residue using the ester bond-formed nucleophilic residues, such as serine and cysteine. Because ester bonds attached to the enzyme cannot be formed with the solvent molecule, which plays the role of a nucleophile in aspartic/glutamic and metalloproteases, we are therefore unlikely to find a serpin inhibiting these two classes of proteases.
Overall, the division of proteases according to those with the S1 binding site shaped as a pocket, and those with the S1 binding site loosely formed at the surface, enables identification of the families of proteases that can be targeted with protein inhibitors that bind in the active site cleft in a substrate-like manner. Among the proteases inspected here, only papain-like cysteine proteases do not possess an S1 binding pocket. This explains why protein inhibitors apply combinations of loops or chain-loop constructs to occlude the reactive site, while binding into the active site cleft of their targets. However, when the concept of spanning the active site by a peptidyl chain is applied, as in the case of propeptides, the chain can only run in the direction opposite to the substrate binding.
For inhibition of the families of proteases that possess S1 shaped as a specific binding pocket, there appear to be fewer restraints in the concept. It appears that the S1 binding pocket implies smaller dynamics of a peptidyl substrate, and thus inhibitors can implement a single chain that spans the active site and still bypass the catalytic site. Among these are solutions such as “loop out” in calpastatin (Figure 4d) and specific uncleavable sequences with residues at P1, such as glycine in staphostatin B and proline in securin. The mechanism of cystatin inhibition of legumain is the closest to the “canonical” conformation of serine proteases because it applies the substrate-like binding, however, in combination with a loop. In concept, this is similar to the p35 inhibition mechanism of caspases. P35 uses residue D87, which remains bound to the enzyme after cleavage. This concept is, in turn, similar to the Nlef inhibition mechanism. Nlef binds with its last four C-terminal residues in the non-primed part of the caspase active site cleft and provides the negatively charged C-terminal G199 to bind in the S1 pocket.
6. Concluding Remarks
In the age of protein engineering, we would like to go beyond repurposing natural design. We would like to apply our knowledge and understanding to the design of protein inhibitors that regulate, mark, or block the activity of proteases in biological systems. We found no inhibitors in clinical trials or in use as a drug (WHO International Clinical Trials Registry Platform ICTRP: https://www.who.int/ictrp/en/; ClinicalTrials.gov: https://clinicaltrials.gov/ct2/results?cond=COVID-19). Of interest, however, Novartis developed a small molecule inhibitor, LCL-161, a second mitochondria-derived activator of caspase (SMAC) mimetic, which binds to XIAP and loosens the binding of XIAP to caspase-9, thereby promoting cell apoptosis [171]. LCL-161 is in clinical phase II for the treatment of breast cancer. We are still at the beginning of the “de novo” design of proteins; however, we are capable of adopting existing concepts found in nature. For substrate-like inhibitors that bind to the non-primed part of the active site cleft, the simplest approach appears to be to tap the substrate specificity with high throughput screens such as [172], and build these sequences into the inhibitor of interest. Serpins, baculovirus p35, macrocypins, Nlef and likely others, including BPTI, could be used to engineer the desired specificity. Serpins appear to be an ideal scaffold because their flexibility and unique mechanism likely address every protease class that contains a reactive site nucleophile capable of forming ester intermediates. For example, Whisstock and his team engineered α1-antitrypsin to inhibit cysteine cathepsins L, V, and K [173]. Moreover, the scaffold of stefins has also been used to develop a targeted drug delivery system [174] and to generate binders for proteins not related to cysteine proteases [175,176]. The binding loop of cystatins was engineered in plant cyclotide, a 35-residues-long cyclic peptide cross-linked with three disulfide bonds called McoTI-II, and reached μM binding [177]. However, to engineer tight binding inhibitors of papain-like proteases (in addition to serpins) on the basis of non-substrate-like approaches, generic tools should be utilized, including antibodies or structural repeats such as designed ankyrin repeat proteins (DARPins), which were engineered to inhibit cathepsin B in the pM range. These were found to have a substantially higher affinity than those shown for any of endogenous inhibitors [154]. One of the most studied protein serine protease inhibitors, BPTI, under the name aprotinin, has been in and out of and again in use in surgery to slow fibrinolysis (blood clot degradation) during complex surgical procedures. Recently, reexamination of its potential in the treatment of pancreatitis was encouraged by analysis of previous studies, which argued that previous clinical studies lacked an adequate biochemical background [178]. It appears that the technology and knowledge of protein inhibitor engineering has not yet reached a level comparable to that of small molecule design. One obstacle is the specificity of binding. Protein inhibitors do not inhibit a single protease molecule, but usually bind to a group of related enzymes with different affinities. Nevertheless, we believe that the potential exists. Most mechanistic studies mentioned in this review targeted the main interaction regions within the vicinity of the reactive sites. Only a few studies, such as the study of interactions between the p41 fragment and cysteine cathepsins [49], systematically addressed the binding affinity of an inhibitor against a group of related enzymes using site-directed mutagenesis. We hope that the analysis and understanding of interactions between protein inhibitors and their protease targets presented here may encourage and assist in the application of protein inhibitors in medical, agricultural, and industrial applications.
Author Contributions
D.T., A.U., B.T. and L.T. wrote the manuscript, D.T. made the figures, L.T. and A.U. prepared the tables. All authors have read and agreed to the published version of the manuscript.
Funding
This review was funded by Slovenian Research Agency, grant number P1-0048 (awarded to D.T.) and grant number P1-0140 (awarded to B.T.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Table A1.
Compiled kinetic constants Ki for the comparison of inhibitors potency presented as ranges of values reported by the researchers in the publications. (a) Ranges of binding constants for cathepsin inhibitors Ki (nM). (b) Range of binding constants for caspase inhibitors Ki (nM). (c) Ranges of binding constants for papain and similar protease inhibitors Ki (nM). (d) Ranges of binding constants for the small group of protease inhibitors Ki (nM).
Table A1.
Compiled kinetic constants Ki for the comparison of inhibitors potency presented as ranges of values reported by the researchers in the publications. (a) Ranges of binding constants for cathepsin inhibitors Ki (nM). (b) Range of binding constants for caspase inhibitors Ki (nM). (c) Ranges of binding constants for papain and similar protease inhibitors Ki (nM). (d) Ranges of binding constants for the small group of protease inhibitors Ki (nM).
| (a) | ||||||||||||
| Inhibitors of Cathepsins Ki (nM) | Cathepsin B | Cathepsin L | Cathepsin H | Cathepsin K | Cathepsin S | Cathepsin V | Cathepsin C | Cathepsin X | Cathepsin F | References | ||
| Stefin A | 1.79->10,000 | 0.0034–1.3 | 0.069–2400 | 0.053–0.27 | 1.1–5340 | 1.7 | [34,56,174,179,180] | |||||
| Stefin B | 73 | 0.23–13 | 0.14–930 | >250 | [11,34,179,181] | |||||||
| Cystatin | 101 | 11.5 | 0.63 | 25.4 | [32] | |||||||
| Cystatin A | 8.2 | 1.3 | 0.31 | 0.05 | 33 | [6,30] | ||||||
| Cystatin B | 16–73 | 0.23 | 0.58 | 0.07 | 0.23 | [6,30,46] | ||||||
| Cystatin C | 0.25–18,000 | <0.02 | <0.005–1.8 | 0.008 | 0.5–3.5 | 12 | [6,23,31,34,38,179,182,183,184] | |||||
| Cystatin Chicken | 0.07–4 | 0.019 | 0.064 | 0.35 | 15 | [6,27,31,98,179] | ||||||
| Cystatin D | >1000 | 5.8–25 | 7.5–18 | 0.24 | [11,30,38,182,183] | |||||||
| Cystatin E | 32 | [182] | ||||||||||
| Cystatin E/M | 31–32 | [38,183] | ||||||||||
| Cystatin F | >1000 | 0.31 | [38,183] | |||||||||
| Cystatin SN | 19 | [30] | ||||||||||
| Kininogens HMW | 400 | 0.019–0.109 | 1.1 | [6,11,27,183] | ||||||||
| Kininogens LMW | 600 | 0.017–0.048 | 0.72–1.2 | >130 | >1000 | [6,30,179,185] | ||||||
| Kininogen segment 1 | >100 | [6] | ||||||||||
| Kininogen segment 2 | 0.14 | [6] | ||||||||||
| Kininogen segment 3 | 0.005 | [6] | ||||||||||
| Falstatin | 0.032 | 0.052 | 0.025 | [44] | ||||||||
| Chagasin | 0.35–100 | 0.039–0.35 | 15 | 2 | [32,37,38,46,47,51] | |||||||
| p41 fragment | >1000 | 0.00149–0.01 | 5.3 | 0.09 | 0.27–208 | 0.0072 | >1000 | 0.51 | [11,55,186] | |||
| Clitocypin | >1000 | 0.02–0.03 | 0.02–0.03 | 2.2–3.2 | 0.08–1.9 | [11,40,49,55,70,186] | ||||||
| Macrocypin | 515 | 3.81 | 370 | 4.5 | 47.1 | 8.5–12.6 | [40,49] | |||||
| Macrocypin 1 | 490 | 0.64 | 100 | 170 | 23.1 | 0.69–12.5 | [40,49] | |||||
| Macrocypin 3 | >1000 | 0.31 | 24 | 17.5 | 5.1 | 0.45–3.43 | [40,49] | |||||
| Macrocypin 4 | 125 | 2.76 | 32 | 21.8 | 6.3 | 1.44–10.2 | [40,49] | |||||
| Serpins | 35.85–71.06 | [81] | ||||||||||
| Equistatin | 1.4 | 0.051 | [58] | |||||||||
| Leupeptin | 0.37 | 0.52 | 3.2 | 0.64 | [32] | |||||||
| Saxiphilin | 1.67 | 0.02 | [58] | |||||||||
| Cysteine proteinase inhibitor | 15.8–281 | 0.062–3.607 | [64,185,187] | |||||||||
| Propeptide Cathepsin K | >600 | 3.6 | 5.5 | 6.3 | [136] | |||||||
| Propeptide Cathepsin L | >600 | 0.12 | 0.27 | 65 | [136] | |||||||
| Propeptide Cathepsin S | >600 | 0.46 | 7 | 7.6 | [136] | |||||||
| Propeptide Cathepsin B | 0.4–64 | [134] | ||||||||||
| Propeptide Cruzipain | 2.05 | 5.18 | 0.032 | [188] | ||||||||
| (b) | ||||||||||||
| Inhibitors of Caspases Ki (nM) | Caspase-1 | Caspase-2 | Caspase-3 | Caspase-4 | Caspase-6 | Caspase-5 | Caspase-7 | Caspase-8 | Caspase-9 | Caspase-10 | Pro-Caspase 7 | References |
| XIAP | <0.4->1000 | <0.05->1000 | [102] | |||||||||
| XIAP BIR2 BIR3 | 3 | 1 | [189,190] | |||||||||
| XIAP BIR3 | >1000 | >1000 | 13 | [189,190] | ||||||||
| XIAP BIR1 | >1000 | >1000 | [189] | |||||||||
| XIAP BIR1 BIR2 | 3 | 1 | [189] | |||||||||
| XIAP BIR2 | <0.4->1000 | <0.05->1000 | [83,91,102,189] | |||||||||
| MLBIR | 3200 | [190] | ||||||||||
| MLBIR-Q | <<10–4500 | [190] | ||||||||||
| MLXBIR3 | <<10–960 | [190] | ||||||||||
| p35 | 9 | 0.11–34,000 | 0.38 | 1.8 | 0.48–400 | 7 | [85,114,191] | |||||
| Serpin Darpin AR_F8 | 0.29 | [150] | ||||||||||
| Serpin DARPin D3.4 | 3.49–16.8 | [151] | ||||||||||
| Serpin DARPin D3.8 | 6.7 | [151] | ||||||||||
| Serpin DARPins D7.18 | 144 | 295 | [153] | |||||||||
| Serpin DARPins D7.43 | 24.7 | 32.2 | [153] | |||||||||
| DARPin | 33–597 | [152] | ||||||||||
| Serpin SERP2 | 0.1–0.2 | 0.5–7.4 | 1.3–200 | [130] | ||||||||
| CrmA | 0.01 | >10,000 | 500–1600 | 1.1 | 110–1300 | <0.1 | >10,000 | <0.34 | <2.3 | 17 | [129,130,131,132] | |
| Falstatin | 376 | 80 | [44] | |||||||||
| (c) | ||||||||||||
| Inhibitors of Proteases Ki (nM) | Papain | Calpain | Calpain-1 | Falcipain | Falcipain-2 | Falcipain-3 | Ficin | Cruzi-pain | Legu-main | Actini-din | References | |
| Stefin A | 0.019–87.20 | 0.021–0.072 | [174,179,180,192] | |||||||||
| Stefin B | 0.12–110 | 0.06 | [11,34,179,181,192] | |||||||||
| Cystatin | 6.5 | 100 | [32] | |||||||||
| Cystatin A | 0.019 | >10,000 | [6,7,30] | |||||||||
| Cystatin B | 0.034–0.12 | >10,000 | [30,46] | |||||||||
| Cystatin C | 0.00001–11 | >10,000 | 0.0013–29 | 0.0048–0.014 | 19-≥40,000 | [6,7,23,38,179,182,184,192,193] | ||||||
| Stefin A | 0.019 | 0.0072 | [11] | |||||||||
| Stefin B | 0.12 | 0.06 | [11] | |||||||||
| Cystatin Chicken | 0.00006–0.005 | >10,000 | 0.00005 | 0.001–0.0028 | [6,27,31,179,192] | |||||||
| Cystatin C | 0.00001–1.7 | 23–29 | 0.014 | 1100–40,000 | [11,23] | |||||||
| Cystatin D | 0.9–1.2 | [7,11,38,182] | ||||||||||
| Cystatin E | 0.39 | [182] | ||||||||||
| Cystatin E/M | 0.39–0.46 | [38,183] | ||||||||||
| Cystatin F | 1.1 | [38,183] | ||||||||||
| Cystatin S | 108 | [7,30] | ||||||||||
| Cystatin SA | 0.32 | [7,30] | ||||||||||
| Cystatin SN | 0.016 | [7,30] | ||||||||||
| Kininogens HMW | 0.02 | [6,185] | ||||||||||
| Kininogens LMW | 0.015–0.017 | 1 | 0.041 | [6,30,179,185,192] | ||||||||
| Kininogen segment 1 | >100 | >100 | [6] | |||||||||
| Kininogen segment 2 | 0.083 | 1 | [6] | |||||||||
| Kininogen segment 3 | 0.03 | >100 | [6] | |||||||||
| Falstatin | 53.5 | 0.196 | 0.021–0.045 | 0.223 | [44] | |||||||
| Serpin B3 | 220.48–293.49 | 277.27–338.11 | [81] | |||||||||
| Chagasins | 0.013–0.036 | 1.7 | 4.8 | 0.062 | 0.0067–0.095 | [32,37,38,46,47,51] | ||||||
| p41 fragment | 1.4 | 0.058 | [55,186] | |||||||||
| Clitocypin | 2.5–6.2 | 7.1–21.5 | [40,49,70] | |||||||||
| Macrocypin | 5.04 | 110 | [40,49] | |||||||||
| Macrocypin 1 | 0.95 | 3.38 | [40,49] | |||||||||
| Macrocypin 3 | 0.12 | 9.17 | [40,49] | |||||||||
| Macrocypin 4 | 0.19 | 2.86->1000 | [40,49] | |||||||||
| 205-residue N-terminal prodomain | [105] | |||||||||||
| Equistatin | 0.57->1000 | [58] | ||||||||||
| Leupeptin | 0.2 | 0.3 | [32] | |||||||||
| Phosphonoformate | [194] | |||||||||||
| Saxiphilin | 1.72 | [62] | ||||||||||
| Cysteine proteinase inhibitor | 0.034–0.35 | 8.7–37.1 | [37,64,185,187] | |||||||||
| Propeptide Cathepsin K | >600 | [136] | ||||||||||
| Propeptide Cathepsin L | >600 | [136] | ||||||||||
| Propeptide Cathepsin S | [136] | |||||||||||
| Propeptide Cruzipain | 0.018–0.2637 | [188] | ||||||||||
| Propeptide Cathepsin B | 2800–5600 | [134] | ||||||||||
| (d) | ||||||||||||
| Inhibitors of Proteases Ki (nM) | Vivapain-3 | Berghepain-2 | Vinckepain-2 | Knowlepain-2 | Gingipain | FosA | Leishmainia mexicana CPB2.8∆CTE | Brucipain | Thrombin | References | ||
| Falstatin | 0.065 | 0.048 | 0.15 | 0.078 | [44] | |||||||
| 205-residue N-terminal prodomain | 6.2 | [105] | ||||||||||
| Phosphonoformate | 400 | [194] | ||||||||||
| ICP | 0.071–0.495 | [37,64,187] | ||||||||||
| Propeptide cruzipain | 0.0163 | [188] | ||||||||||
| Hirudin | 0.00001–0.01 | [169] | ||||||||||
References
- Turk, B.; Turk, D.; Salvesen, G.S. Regulating cysteine protease activity: Essential role of protease inhibitors as guardians and regulators. Med. Chem. Rev. 2005, 2, 283–297. [Google Scholar] [CrossRef]
- Bieth, J.G. In vivo significance of kinetic constants of protein proteinase inhibitors. Biochem. Med. 1984, 32, 387–397. [Google Scholar] [CrossRef]
- Bieth, J.G. Theoretical and practical aspects of proteinase inhibition kinetics. Methods Enzymol. 1995, 248, 59–84. [Google Scholar] [CrossRef]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Huber, R. Natural protein proteinase inhibitors and their interaction with proteinases. JBIC J. Biol. Inorg. Chem. 1992, 204, 433–451. [Google Scholar] [CrossRef]
- Otto, H.-H.; Schirmeister, T. Cysteine Proteases and Their Inhibitors. Chem. Rev. 1997, 97, 133–172. [Google Scholar] [CrossRef]
- Grzonka, Z.; Jankowska, E.; Kasprzykowski, F.; Kasprzykowska, R.; Lankiewicz, L.; Wiczk, W.; Wieczerzak, E.; Ciarkowski, J.; Drabik, P.; Janowski, R.; et al. Structural studies of cysteine proteases and their inhibitors. Acta Biochim. Pol. 2001, 48, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Dubin, G. Proteinaceous cysteine protease inhibitors. Cell. Mol. Life Sci. 2005, 62, 653–669. [Google Scholar] [CrossRef]
- Callus, B.A.; Vaux, D.L. Caspase inhibitors: Viral, cellular and chemical. Cell Death Differ. 2006, 14, 73–78. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef] [Green Version]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gettins, P.G.W.; Olson, S.T. Inhibitory serpins. New insights into their folding, polymerization, regulation and clearance. Biochem. J. 2016, 473, 2273–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, J.; Pan, G.; Poncz, M.; Wei, J.; Ran, M.; Zhou, Z.-Y. Serpin functions in host-pathogen interactions. PeerJ 2018, 6, e4557. [Google Scholar] [CrossRef] [Green Version]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drenth, J.; Jansonius, J.N.; Koekoek, R.; Swen, H.M.; Wolthers, B.G. Structure of Papain. Nat. Cell Biol. 1968, 218, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Fossum, K.; Whitaker, J.R. Ficin and papain inhibitor from chicken egg white. Arch. Biochem. Biophys. 1968, 125, 367–375. [Google Scholar] [CrossRef]
- Bode, W.; Engh, R.; Musil, D.; Thiele, U.; Huber, R.; Karshikov, A.; Brzin, J.; Kos, J.; Turk, V. The 2.0 A X-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. EMBO J. 1988, 7, 2593–2599. [Google Scholar] [CrossRef]
- Stubbs, M.; Laber, B.; Bode, W.; Huber, R.; Jerala, R.; Lenarčič, B.; Turk, V. The refined 2.4 A X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: A novel type of proteinase inhibitor interaction. EMBO J. 1990, 9, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Björk, I.; Alriksson, E.; Ylinenjärvi, K.; Bjoerk, I.; Ylinenjaervi, K. Kinetics of binding of chicken cystatin to papain. Biochemistry 1989, 28, 1568–1573. [Google Scholar] [CrossRef] [PubMed]
- Björk, I.; Brieditis, I.; Abrahamson, M. Probing the functional role of the N-terminal region of cystatins by equilibrium and kinetic studies of the binding of Gly-11 variants of recombinant human cystatin C to target proteinases. Biochem. J. 1995, 306, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquart, M.; Walter, J.; Deisenhofer, J.; Bode, W.; Huber, R. The geometry of the reactive site and of the peptide groups in trypsin, trypsinogen and its complexes with inhibitors. Acta Crystallogr. Sect. B Struct. Sci. 1983, 39, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Barrett, A.J. Cystatin, the egg white inhibitor of cysteine proteinases. Methods Enzymol. 1981, 771–778. [Google Scholar] [CrossRef]
- Barrett, A.J.; Davies, M.; Grubb, A. The place of human γ-trace (cystatin C) amongst the cysteine proteinase inhibitors. Biochem. Biophys. Res. Commun. 1984, 120, 631–636. [Google Scholar] [CrossRef]
- Barrett, A.J. The cystatins: A diverse superfamily of cysteine peptidase inhibitors. Biomed. Biochim. Acta 1986, 45, 1363. [Google Scholar]
- Lindahl, P.; Alriksson, E.; Jörnvall, H.; Björk, I.; Bjoerk, I.; Joernvall, H. Interaction of the cysteine proteinase inhibitor chicken cystatin with papain. Biochemistry 1988, 27, 5074–5082. [Google Scholar] [CrossRef]
- Keilova, H.; Tomášek, V. Effect of papain inhibitor from chicken egg white on cathepsin B1. Biochim. Biophys. Acta (BBA) Enzym. 1974, 334, 179–186. [Google Scholar] [CrossRef]
- Abrahamson, M. Cystatins. Methods Enzymol. 1994, 244, 685–700. [Google Scholar] [CrossRef]
- Björk, I.; Pol, E.; Raub-Segall, E.; Abrahamson, M.; Rowan, A.D.; Mort, J.S. Differential changes in the association and dissociation rate constants for binding of cystatins to target proteinases occurring on N-terminal truncation of the inhibitors indicate that the interaction mechanism varies with different enzymes. Biochem. J. 1994, 299, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.X.; Pandey, K.C.; Scharfstein, J.; Whisstock, J.C.; Huang, R.K.; Jacobelli, J.; Fletterick, R.J.; Rosenthal, P.J.; Abrahamson, M.; Brinen, L.S.; et al. The Structure of Chagasin in Complex with a Cysteine Protease Clarifies the Binding Mode and Evolution of an Inhibitor Family. Structure 2007, 15, 535–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renko, M.; Požgan, U.; Majera, D.; Turk, D. Stefin A displaces the occluding loop of cathepsin B only by as much as required to bind to the active site cleft. FEBS J. 2010, 277, 4338–4345. [Google Scholar] [CrossRef]
- Jenko, S.; Dolenc, I.; Gunčar, G.; Doberšek, A.; Podobnik, M.; Turk, D. Crystal Structure of Stefin A in Complex with Cathepsin H: N-terminal Residues of Inhibitors can Adapt to the Active Sites of Endo- and Exopeptidases. J. Mol. Biol. 2003, 326, 875–885. [Google Scholar] [CrossRef]
- Turk, D. MAINsoftware for density averaging, model building, structure refinement and validation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1342–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, E.A.; Bacon, D.J. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 1997, 277, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Hansen, G.; Heitmann, A.; Witt, T.; Li, H.; Jiang, H.; Shen, X.; Heussler, V.T.; Rennenberg, A.; Hilgenfeld, R. Structural Basis for the Regulation of Cysteine-Protease Activity by a New Class of Protease Inhibitors in Plasmodium. Structure 2011, 19, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Ljunggren, A.; Redzynia, I.; Alvarez-Fernandez, M.; Abrahamson, M.; Mort, J.S.; Krupa, J.C.; Jaskolski, M.; Bujacz, G.D. Crystal Structure of the Parasite Protease Inhibitor Chagasin in Complex with a Host Target Cysteine Protease. J. Mol. Biol. 2007, 371, 137–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunčar, G.; Pungercic, G.; Klemenčič, I.; Turk, V.; Turk, D. Crystal structure of MHC class II-associated p41 Ii fragment bound to cathepsin L reveals the structural basis for differentiation between cathepsins L and S. EMBO J. 1999, 18, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Renko, M.; Sabotič, J.; Mihelič, M.; Brzin, J.; Kos, J.; Turk, D. Versatile Loops in Mycocypins Inhibit Three Protease Families. J. Biol. Chem. 2010, 285, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Filipek, R.; Rzychon, M.; Oleksy, A.; Gruca, M.; Dubin, A.; Potempa, J.; Bochtler, M. The Staphostatin-Staphopain Complex: A forward binding inhibitor in complex with its target cysteine protease. J. Biol. Chem. 2003, 278, 40959–40966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall, E.; Fegg, J.C.; Briza, P.; Brandstetter, H. Structure and Mechanism of an Aspartimide-Dependent Peptide Ligase in Human Legumain. Angew. Chem. Int. Ed. 2015, 54, 2917–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.X.; Pandey, K.C.; Somoza, J.R.; Sijwali, P.S.; Kortemme, T.; Brinen, L.S.; Fletterick, R.J.; Rosenthal, P.J.; McKerrow, J.H. Structural basis for unique mechanisms of folding and hemoglobin binding by a malarial protease. Proc. Natl. Acad. Sci. USA 2006, 103, 11503–11508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, K.C.; Singh, N.; Arastu-Kapur, S.; Bogyo, M.; Rosenthal, P.J. Falstatin, a Cysteine Protease Inhibitor of Plasmodium falciparum, Facilitates Erythrocyte Invasion. PLoS Pathog. 2006, 2, e117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alphey, M.S.; Hunter, W.N. High-resolution complex of papain with remnants of a cysteine protease inhibitor derived fromTrypanosoma brucei. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 504–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redzynia, I.; Ljunggren, A.; Bujacz, A.; Abrahamson, M.; Jaskolski, M.; Bujacz, G. Crystal structure of the parasite inhibitor chagasin in complex with papain allows identification of structural requirements for broad reactivity and specificity determinants for target proteases. FEBS J. 2009, 276, 793–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redzynia, I.; Ljunggren, A.; Abrahamson, M.; Mort, J.S.; Krupa, J.C.; Jaskolski, M.; Bujacz, G. Displacement of the Occluding Loop by the Parasite Protein, Chagasin, Results in Efficient Inhibition of Human Cathepsin B. J. Biol. Chem. 2008, 283, 22815–22825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renko, M.; Sabotič, J.; Turk, D. β-Trefoil inhibitors—From the work of Kunitz onward. Biol. Chem. 2012, 393, 1043–1054. [Google Scholar] [CrossRef]
- Sabotič, J.; Popovič, T.; Puizdar, V.; Brzin, J. Macrocypins, a family of cysteine protease inhibitors from the basidiomycete Macrolepiota procera. FEBS J. 2009, 276, 4334–4345. [Google Scholar] [CrossRef]
- Filipek, R.; Potempa, J.; Bochtler, M. A Comparison of Staphostatin B with Standard Mechanism Serine Protease Inhibitors. J. Biol. Chem. 2005, 280, 14669–14674. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, A.C.; Abrahamson, M.; Lima, A.P.; Vannier-Santos, M.A.; Scharfstein, J. Identification, characterization and localization of chagasin, a tight-binding cysteine protease inhibitor in Trypanosoma cruzi. J. Cell Sci. 2001, 114, 3933–3942. [Google Scholar] [PubMed]
- Molina, F.; Bouanani, M.; Pau, B.; Granier, C. Characterization of the Type-1 Repeat from Thyroglobulin, a Cysteine-Rich Module Found in Proteins from Different Families. JBIC J. Biol. Inorg. Chem. 1996, 240, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Coscia, F.; Taler-Verčič, A.; Chang, V.T.; Sinn, L.; O’Reilly, F.J.; Izoré, T.; Renko, M.; Berger, I.; Rappsilber, J.; Turk, D.; et al. The structure of human thyroglobulin. Nat. Cell Biol. 2020, 578, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Lenarcic, B.; Bevec, T. Thyropins--new structurally related proteinase inhibitors. Biol. Chem. 1998, 379, 3933–3942. [Google Scholar]
- Bevec, T.; Stoka, V.; Pungercic, G.; Dolenc, I.; Turk, V. Major histocompatibility complex class II-associated p41 invariant chain fragment is a strong inhibitor of lysosomal cathepsin L. J. Exp. Med. 1996, 183, 1331–1338. [Google Scholar] [CrossRef]
- Mihelič, M.; Teuscher, C.; Turk, V.; Turk, D. Mouse stefins A1 and A2 (Stfa1andStfa2) differentiate between papain-like endo- and exopeptidases. FEBS Lett. 2006, 580, 4195–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, T.-J.; Lolicato, M.; Thomas-Tran, R.; Du Bois, J.; Minor, D.L. Structure of the saxiphilin:saxitoxin (STX) complex reveals a convergent molecular recognition strategy for paralytic toxins. Sci. Adv. 2019, 5, eaax2650. [Google Scholar] [CrossRef] [Green Version]
- Lenarčič, B.; Ritonja, A.; Štrukelj, B.; Turk, B.; Turk, V. Equistatin, a New Inhibitor of Cysteine Proteinases fromActinia equina, Is Structurally Related to Thyroglobulin Type-1 Domain. J. Biol. Chem. 1997, 272, 13899–13903. [Google Scholar] [CrossRef] [Green Version]
- Vannahme, C.; Schübel, S.; Herud, M.; Gösling, S.; Hülsmann, H.; Paulsson, M.; Hartmann, U.; Maurer, P. Molecular Cloning of Testican-2: Defining a novel calcium-binding proteoglycan family expressed in brain. J. Neurochem. 2002, 73, 12–20. [Google Scholar] [CrossRef]
- Nakada, M.; Yamada, A.; Takino, T.; Miyamori, H.; Takahashi, T.; Sato, H.; Sato, H.; Nakada, M.; Takahashi, T.; Yamashita, J. Suppression of membrane-type I matrix metalloproteinase (MMP)-mediated MMP-2 activation and tumor invasion by testican 3 and its splicing variant gene product, N-Tes. Cancer Res. 2001, 61, 8896–8902. [Google Scholar]
- Lenarcic, B.; Turk, V. Thyroglobulin Type-1 Domains in Equistatin Inhibit Both Papain-like Cysteine Proteinases and Cathepsin D. J. Biol. Chem. 1999, 274, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenarčič, B.; Krishnan, G.; Borukhovich, R.; Ruck, B.; Turk, V.; Moczydlowski, E. Saxiphilin, a Saxitoxin-binding Protein with Two Thyroglobulin Type 1 Domains, Is an Inhibitor of Papain-like Cysteine Proteinases. J. Biol. Chem. 2000, 275, 15572–15577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Konagaya, S. Increase in catheptic activity and appearance of phagocytes in the white muscle of Chum salmon during spawning migration. Biomed. Biochim. Acta 1991, 50, 565–567. [Google Scholar] [PubMed]
- Yamashita, M.; Konagaya, S. A Novel Cysteine Protease Inhibitor of the Egg of Chum Salmon, Containing a Cysteine-rich Thyroglobulin-like Motif. J. Biol. Chem. 1996, 271, 1282–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szala, S.; Froehlich, M.; Scollon, M.; Kasai, Y.; Steplewski, Z.; Koprowski, H.; Linnenbach, A.J. Molecular cloning of cDNA for the carcinoma-associated antigen GA733-2. Proc. Natl. Acad. Sci. USA 1990, 87, 3542–3546. [Google Scholar] [CrossRef] [Green Version]
- Nakae, H.; Sugano, M.; Ishimori, Y.; Endo, T.; Obinata, T. Ascidian entactin/nidogen. Implication of evolution by shuffling two kinds of cysteine-rich motifs. JBIC J. Biol. Inorg. Chem. 1993, 213, 11–19. [Google Scholar] [CrossRef]
- Headey, S.J.; Keizer, D.W.; Yao, S.; Brasier, G.; Kantharidis, P.; Bach, L.A.; Norton, R.S. C-Terminal Domain of Insulin-Like Growth Factor (IGF) Binding Protein-6: Structure and Interaction with IGF-II. Mol. Endocrinol. 2004, 18, 2740–2750. [Google Scholar] [CrossRef] [Green Version]
- Novinec, M.; Kordis, D.; Turk, V.; Lenarčič, B. Diversity and Evolution of the Thyroglobulin Type-1 Domain Superfamily. Mol. Biol. Evol. 2005, 23, 744–755. [Google Scholar] [CrossRef] [Green Version]
- Mihelič, M.; Turk, D. Two decades of thyroglobulin type-1 domain research. Biol. Chem. 2007, 388, 1123–1130. [Google Scholar] [CrossRef]
- Sabotič, J.; Galeša, K.; Popovic, T.; Leonardi, A.; Brzin, J. Comparison of natural and recombinant clitocypins, the fungal cysteine protease inhibitors. Protein Expr. Purif. 2007, 53, 104–111. [Google Scholar] [CrossRef]
- Rzychon, M.; Sabat, A.; Kosowska, K.; Potempa, J.; Dubin, A. Staphostatins: An expanding new group of proteinase inhibitors with a unique specificity for the regulation of staphopains, Staphylococcus spp. cysteine proteinases. Mol. Microbiol. 2003, 49, 1051–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, R.H.; Zhang, Q.; McGowan, S.; Buckle, A.M.; Silverman, G.A.; Wong, W.; Rosado, C.J.; Langendorf, C.G.; Pike, R.N.; Bird, P.I.; et al. An overview of the serpin superfamily. Genome Biol. 2006, 7, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, G.A.; Bird, P.I.; Carrell, R.W.; Church, F.C.; Coughlin, P.B.; Gettins, P.G.W.; Irving, J.A.; Lomas, D.A.; Luke, C.J.; Moyer, R.W.; et al. The Serpins Are an Expanding Superfamily of Structurally Similar but Functionally Diverse Proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 2001, 276, 33293–33296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, A.; Yamamoto, T.; Nakamura, Y.; Takahashi, T.; Hibino, T. Squamous cell carcinoma antigen is a potent inhibitor of cysteine proteinase cathepsin L. FEBS Lett. 1995, 359, 78–80. [Google Scholar] [CrossRef] [Green Version]
- Schick, C.; Pemberton, P.A.; Shi, G.-P.; Kamachi, Y.; Çataltepe, S.; Bartuski, A.J.; Gornstein, E.R.; Brömme, D.; Chapman, A.H.A.; Silverman, G.A. Cross-Class Inhibition of the Cysteine Proteinases Cathepsins K, L, and S by the Serpin Squamous Cell Carcinoma Antigen 1: A Kinetic Analysis. Biochemistry 1998, 37, 5258–5266. [Google Scholar] [CrossRef]
- Higgins, W.J.; Fox, D.M.; Kowalski, P.S.; Nielsen, J.E.; Worrall, D.M. Heparin Enhances Serpin Inhibition of the Cysteine Protease Cathepsin L. J. Biol. Chem. 2010, 285, 3722–3729. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.-R.; Stoka, V.; Turk, A.V.; Hook, V.Y.H. The Novel Bovine Serpin Endopin 2C Demonstrates Selective Inhibition of the Cysteine Protease Cathepsin L Compared to the Serine Protease Elastase, in Cross-Class Inhibition. Biochemistry 2005, 44, 7757–7767. [Google Scholar] [CrossRef]
- Walz, M.; Kellermann, S.; Bylaite, M.; Andree, B.; Ruther, U.; Paus, R.; Kloepper, J.E.; Reifenberger, J.; Ruzicka, T. Expression of the human Cathepsin L inhibitor hurpin in mice: Skin alterations and increased carcinogenesis. Exp. Dermatol. 2007, 16, 715–723. [Google Scholar] [CrossRef]
- Luke, C.J.; Pak, S.C.; Askew, Y.S.; Naviglia, T.L.; Askew, D.J.; Nobar, S.M.; Vetica, A.C.; Long, O.S.; Watkins, S.C.; Stolz, D.B.; et al. An Intracellular Serpin Regulates Necrosis by Inhibiting the Induction and Sequelae of Lysosomal Injury. Cell 2007, 130, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Kantyka, T.; Plaza, K.; Koziel, J.; Florczyk, D.; Stennicke, H.R.; Thogersen, I.B.; Enghild, J.J.; Silverman, G.A.; Pak, S.C.; Potempa, J. Inhibition of Staphylococcus aureus cysteine proteases by human serpin potentially limits staphylococcal virulence. Biol. Chem. 2011, 392, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Alam, B.; Biswas, S. Inhibition of Plasmodium falciparum cysteine protease falcipain-2 by a human cross-class inhibitor serpinB3: A mechanistic insight. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2019, 1867, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Sakata, Y.; Arima, K.; Takai, T.; Sakurai, W.; Masumoto, K.; Yuyama, N.; Suminami, Y.; Kishi, F.; Yamashita, T.; Kato, T.; et al. The Squamous Cell Carcinoma Antigen 2 Inhibits the Cysteine Proteinase Activity of a Major Mite Allergen, Der p 1. J. Biol. Chem. 2004, 279, 5081–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, S.J.; Renatus, M.; Schwarzenbacher, R.; Zhou, Q.; Sun, C.; Fesik, S.W.; Liddington, R.C.; Salvesen, G.S. Structural Basis for the Inhibition of Caspase-3 by XIAP. Cell 2001, 104, 791–800. [Google Scholar] [CrossRef]
- Blasche, S.; Mörtl, M.; Steuber, H.; Siszler, G.; Nisa, S.; Schwarz, F.; Lavrik, I.; Gronewold, T.M.A.; Maskos, K.; Donnenberg, M.S.; et al. The E. coli Effector Protein NleF Is a Caspase Inhibitor. PLoS ONE 2013, 8, e58937. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Cirilli, M.; Huang, Y.; Rich, R.L.; Myszka, D.G.; Wu, H. Covalent inhibition revealed by the crystal structure of the caspase-8/p35 complex. Nat. Cell Biol. 2001, 410, 494–497. [Google Scholar] [CrossRef]
- Renatus, M.; Zhou, Q.; Stennicke, H.R.; Snipas, S.J.; Turk, D.; Bankston, L.A.; Liddington, R.C.; Salvesen, G.S. Crystal structure of the apoptotic suppressor CrmA in its cleaved form. Structure 2000, 8, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Crook, N.E.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 1993, 67, 2168–2174. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, M.J.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J. Virol. 1994, 68, 2521–2528. [Google Scholar] [CrossRef] [Green Version]
- Uren, A.G.; Coulson, E.J.; Vaux, D.L. Conservation of baculovirus inhibitor of apoptosis repeat proteins (BIRPs) in viruses, nematodes, vertebrates and yeasts. Trends Biochem. Sci. 1998, 23, 159–162. [Google Scholar] [CrossRef]
- Chai, J.; Shiozaki, E.; Srinivasula, S.M.; Wu, Q.; Dataa, P.; Alnemri, E.S.; Shi, Y. Structural Basis of Caspase-7 Inhibition by XIAP. Cell 2001, 104, 769–780. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, K.K. The calpain family and human disease. Trends Mol. Med. 2001, 7, 355–362. [Google Scholar] [CrossRef]
- Kulathila, R.; Vash, B.; Sage, D.; Cornell-Kennon, S.; Wright, K.; Koehn, J.; Stams, T.; Clark, K.; Price, A.C. The structure of the BIR3 domain of cIAP1 in complex with the N-terminal peptides of SMAC and caspase-9. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 65, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Park, Y.C.; Rich, R.L.; Segal, D.; Myszka, D.G.; Wu, H. Structural Basis of Caspase Inhibition by XIAP. Cell 2001, 104, 781–790. [Google Scholar] [CrossRef] [Green Version]
- Shiozaki, E.N.; Chai, J.; Rigotti, D.J.; Riedl, S.J.; Li, P.; Srinivasula, S.M.; Alnemri, E.S.; Fairman, R.; Shi, Y. Mechanism of XIAP-Mediated Inhibition of Caspase-9. Mol. Cell 2003, 11, 519–527. [Google Scholar] [CrossRef]
- Chai, J.; Yan, N.; Huh, J.R.; Wu, J.-W.; Li, W.; Hay, B.A.; Shi, Y. Molecular mechanism of Reaper-Grim-Hid-mediated suppression of DIAP1-dependent Dronc ubiquitination. Nat. Struct. Mol. Biol. 2003, 10, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Shi, Y. Structural mechanisms of DIAP1 auto-inhibition and DIAP1-mediated inhibition of drICE. Nat. Commun. 2011, 2, 408. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Min, T.; Eliezer, D.; Wu, H. Native Chemical Ligation in Covalent Caspase Inhibition by p35. Chem. Biol. 2006, 13, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Podobnik, M.; Kuhelj, R.; Turk, V.; Turk, D. Crystal structure of the wild-type human procathepsin B at 2.5 Å resolution reveals the native active site of a papain-like cysteine protease zymogen. J. Mol. Biol. 1997, 271, 774–788. [Google Scholar] [CrossRef]
- Moldoveanu, T.; Gehring, K.; Green, D.R. Concerted multi-pronged attack by calpastatin to occlude the catalytic cleft of heterodimeric calpains. Nat. Cell Biol. 2008, 456, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Tong, S.L.L. Molecular mechanism for the regulation of yeast separase by securin. Nat. Cell Biol. 2017, 542, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Sivaraman, J.; Nägler, D.K.; Zhang, R.; Ménard, R.; Cygler, M. Crystal structure of human procathepsin X: A cysteine protease with the proregion covalently linked to the active site cysteine. J. Mol. Biol. 2000, 295, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.L.; Denault, J.-B.; Riedl, S.J.; Shin, H.; Renatus, M.; Salvesen, G.S. XIAP inhibits caspase-3 and -7 using two binding sites: Evolutionarily conserved mechanism of IAPs. EMBO J. 2005, 24, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, T.F.; Cooney, J.C.; Baker, H.M.; McSweeney, S.; Liu, M.; Gubba, S.; Musser, J.M.; Baker, E.N. Crystal structure of the zymogen form of the group A Streptococcus virulence factor SpeB: An integrin-binding cysteine protease. Proc. Natl. Acad. Sci. USA 2000, 97, 2235–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, D.; Podobnik, M.; Kuhelj, R.; Dolinar, M.; Turk, V. Crystal structures of human procathepsin B at 3.2 and 3.3 Å resolution reveal an interaction motif between a papain-like cysteine protease and its propeptide. FEBS Lett. 1996, 384, 211–214. [Google Scholar] [CrossRef] [Green Version]
- De Diego, I.; Veillard, F.T.; Guevara, T.; Potempa, B.; Sztukowska, M.; Potempa, J.; Gomis-Rüth, F.X. Porphyromonas gingivalis Virulence Factor Gingipain RgpB Shows a Unique Zymogenic Mechanism for Cysteine Peptidases. J. Biol. Chem. 2013, 288, 14287–14296. [Google Scholar] [CrossRef] [Green Version]
- Coulombe, R.; Grochulski, P.; Sivaraman, J.; Ménard, R.; Mort, J.S.; Cygler, M. Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J. 1996, 15, 5492–5503. [Google Scholar] [CrossRef]
- Roy, S.; Choudhury, D.; Aich, P.; Dattagupta, J.K.; Biswas, S. The structure of a thermostable mutant of pro-papain reveals its activation mechanism. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1591–1603. [Google Scholar] [CrossRef]
- Groves, M.R.; Taylor, M.A.; Scott, M.; Cummings, N.J.; Pickersgill, R.W.; Jenkins, J.A. The prosequence of procaricain forms an α-helical domain that prevents access to the substrate-binding cleft. Structure 1996, 4, 1193–1203. [Google Scholar] [CrossRef] [Green Version]
- Cygler, M.; Sivaraman, J.; Grochulski, P.; Coulombe, R.; Storer, A.C.; Mort, J.S. Structure of rat procathepsin B: Model for inhibition of cysteine protease activity by the proregion. Structure 1996, 4, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Sivaraman, J.; Lalumière, M.; Ménard, R.; Cygler, M. Crystal structure of wild-type human procathepsin K. Protein Science 1999. [Google Scholar] [CrossRef]
- LaLonde, J.M.; Zhao, B.; Janson, C.A.; D’Alessio, K.J.; McQueney, M.S.; Orsini, M.J.; Debouck, A.C.M.; Smith, W.W. The Crystal Structure of Human Procathepsin K. Biochemistry 1999, 38, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Kaulmann, G.; Palm, G.J.; Schilling, K.; Hilgenfeld, R.; Wiederanders, B. The crystal structure of a Cys25 → Ala mutant of human procathepsin S elucidates enzyme-prosequence interactions. Protein Sci. 2006, 15, 2619–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Purtha, W.; Cortesio, C.; Rui, H.; Gu, Y.; Chen, H.; Sickmier, E.A.; Manzanillo, P.; Huang, X. Crystal structures of human procathepsin H. PLoS ONE 2018, 13, e0200374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Krebs, J.F.; Snipas, S.J.; Price, A.; Alnemri, E.S.; Tomaselli, A.K.J.; Salvesen, G.S. Interaction of the Baculovirus Anti-apoptotic Protein p35 with Caspases. Specificity, Kinetics, and Characterization of the Caspase/p35 Complex. Biochemistry 1998, 37, 10757–10765. [Google Scholar] [CrossRef] [PubMed]
- Vucic, D.; Kaiser, W.J.; Miller, L.K. Inhibitor of Apoptosis Proteins Physically Interact with and Block Apoptosis Induced by Drosophila Proteins HID and GRIM. Mol. Cell. Biol. 1998, 18, 3300–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Echtenkamp, F.; Deng, W.; Wickham, M.E.; Vazquez, A.; Puente, J.L.; Thanabalasuriar, A.; Gruenheid, S.; Finlay, B.B.; Hardwidge, P.R. Characterization of the NleF effector protein from attaching and effacing bacterial pathogens. FEMS Microbiol. Lett. 2008, 281, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.X.; Divangahi, M.; Remold, H.G. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Turk, D.; Janjić, V.; Štern, I.; Podobnik, M.; Lamba, D.; Dahl, S.W.; Lauritzen, C.; Pedersen, J.; Turk, V.; Turk, B. Structure of human dipeptidyl peptidase I (cathepsin C): Exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001, 20, 6570–6582. [Google Scholar] [CrossRef]
- Baumann, U.; Bauer, M.; Létoffé, S.; Delepelaire, P.; Wandersman, C. Crystal Structure of a Complex BetweenSerratia marcescens Metallo-protease and an Inhibitor from Erwinia chrysanthemi. J. Mol. Biol. 1995, 248, 653–661. [Google Scholar] [CrossRef]
- Fisher, A.J.; Cruz, W.D.; Zoog, S.J.; Schneider, C.L.; Friesen, P.D. Crystal structure of baculovirus P35: Role of a novel reactive site loop in apoptotic caspase inhibition. EMBO J. 1999, 18, 2031–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonovic, M.; Gettins, P.G.W.; Volz, K. Crystal structure of viral serpin crmA provides insights into its mechanism of cysteine proteinase inhibition. Protein Sci. 2000, 9, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Cech, A.L.; Belmares, R.; Bergstrom, R.C.; Tong, Y.; Corey, D.R.; Kanost, M.R.; Goldsmith, E.J. The structure of a Michaelis serpin-protease complex. Nat. Genet. 2001, 8, 979–983. [Google Scholar] [CrossRef]
- Huntington, J.A.; Read, R.J.; Carrell, R.W. Structure of a serpin–protease complex shows inhibition by deformation. Nat. Cell Biol. 2000, 407, 923–926. [Google Scholar] [CrossRef]
- Tesch, L.D.; Raghavendra, M.P.; Bedsted-Faarvang, T.; Gettins, P.G.; Olson, S.T. Specificity and reactive loop length requirements for crmA inhibition of serine proteases. Protein Sci. 2005, 14, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Swanson, R.; Raghavendra, M.P.; Zhang, W.; Froelich, C.; Gettins, P.G.W.; Olson, S.T. Serine and Cysteine Proteases Are Translocated to Similar Extents upon Formation of Covalent Complexes with Serpins: Fluorescence perturbation and fluores-cence resonance energy transfer mapping of the protease binding site in CrmA complexes with granzyme. J. Biol. Chem. 2007, 282, 2305–2313. [Google Scholar] [CrossRef] [Green Version]
- Best, S.M. Viral Subversion of Apoptotic Enzymes: Escape from Death Row. Annu. Rev. Microbiol. 2008, 62, 171–192. [Google Scholar] [CrossRef] [Green Version]
- Ray, C.A.; Black, R.A.; Kronheim, S.R.; Greenstreet, T.A.; Sleath, P.R.; Salvesen, G.S.; Pickup, D.J. Viral inhibition of inflammation: Cowpox virus encodes an inhibitor of the interleukin-1β converting enzyme. Cell 1992, 69, 597–604. [Google Scholar] [CrossRef]
- Komiyama, T.; Ray, C.A.; Pickup, D.J.; Howard, A.D.; Thornberry, N.A.; Peterson, E.P.; Salvesen, G. Inhibition of interleu-kin-1β converting enzyme by the cowpox virus serpin CrmA. An example of cross-class inhibition. J. Biol. Chem. 1994, 269, 19331–19337. [Google Scholar] [CrossRef]
- MacNeill, A.L.; Turner, P.C.; Moyer, R.W. Mutation of the Myxoma virus SERP2 P1-site to prevent proteinase inhibition causes apoptosis in cultured RK-13 cells and attenuates disease in rabbits, but mutation to alter specificity causes apoptosis without reducing virulence. Virology 2006, 356, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Snipas, S.; Orth, K.; Muzio, M.; Dixit, V.M.; Salvesen, G.S. Target Protease Specificity of the Viral Serpin CrmA. J. Biol. Chem. 1997, 272, 7797–7800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Calvo, M.; Peterson, E.P.; Leiting, B.; Ruel, R.; Nicholson, D.W.; Thornberry, N.A. Inhibition of Human Caspases by Peptide-based and Macromolecular Inhibitors. J. Biol. Chem. 1998, 273, 32608–32613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Whisstock, J.C.; Harriott, P.; Walker, B.; Novak, A.; Thompson, P.E.; Smith, A.I.; Bird, P.I. Importance of the P4′ Residue in Human Granzyme B Inhibitors and Substrates Revealed by Scanning Mutagenesis of the Proteinase Inhibitor 9 Reactive Center Loop. J. Biol. Chem. 2001, 276, 15177–15184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, T.; De Miguel, E.; Mort, J.S.; Storer, A.C. Potent slow-binding inhibition of cathepsin B by its propeptide. Biochemistry 1992, 31, 12571–12576. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Briggs, G.S.; Baker, K.C.; Cummings, N.J.; Pratt, K.A.; Freedman, R.B.; Goodenough, P.W. Expression of the pro-regions of papain and papaya proteinase IV in Escherichia coli and their inhibition of mature cysteine proteinases. Biochem. Soc. Trans. 1995, 23, 80S. [Google Scholar] [CrossRef]
- Guay, J.; Falgueyret, J.-P.; Ducret, A.; Percival, M.D.; Mancini, J.A. Potency and selectivity of inhibition of cathepsin K, L and S by their respective propeptides. JBIC J. Biol. Inorg. Chem. 2000, 267, 6311–6318. [Google Scholar] [CrossRef] [Green Version]
- Elsässer, B.; Zauner, F.B.; Messner, J.; Soh, W.T.; Dall, E.; Brandstetter, H. Distinct Roles of Catalytic Cysteine and Histidine in the Protease and Ligase Mechanisms of Human Legumain As Revealed by DFT-Based QM/MM Simulations. ACS Catal. 2017, 7, 5585–5593. [Google Scholar] [CrossRef]
- Mitin, Y.V.; Zapevalova, N.P.; Gorbunova, E.Y. Peptide synthesis catalyzed by papain at alkaline pH values. Int. J. Pept. Protein Res. 1984. [Google Scholar] [CrossRef]
- Metrione, R.M.; Neves, A.G.; Fruton, J.S. Purification and Properties of Dipeptidyl Transferase (Cathepsin C)*. Biochemistry 1966, 5, 1597–1604. [Google Scholar] [CrossRef]
- Turk, B.; Turk, D.; Dolenc, I.; Turk, V. Dipeptidyl-Peptidase I. In Handbook of Proteolytic Enzymes; Elsevier BV: Amsterdam, The Netherlands, 2013; pp. 1968–1974. [Google Scholar]
- Wang, K.K.W. Calpain and caspase: Can you tell the difference? Trends Neurosci. 2000, 23, 20–26. [Google Scholar] [CrossRef]
- Franco, S.J.; Huttenlocher, A. Regulating cell migration: Calpains make the cut. J. Cell Sci. 2005, 118, 3829–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, F.; Biswas, S.; Singh, J.; Dennison, S.; Phoenix, D.A. Calpains and Their Multiple Roles in Diabetes Mellitus. Ann. N. Y. Acad. Sci. 2006, 1084, 452–480. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Beckmann, J.S.; Spencer, M.J. Molecular and cellular basis of calpainopathy (limb girdle muscular dystrophy type 2A). Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2007, 1772, 128–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, M.C.; Wang, K.K. Calpain in the CNS: From Synaptic Function to Neurotoxicity. Sci. Signal. 2008, 1, re1. [Google Scholar] [CrossRef]
- Sáez, M.E.; Ramirez-Lorca, R.; Morón, F.J.; Ruiz, A. The therapeutic potential of the calpain family: New aspects. Drug Discov. Today 2006, 11, 917–923. [Google Scholar] [CrossRef]
- Suzuki, K.; Hata, S.; Kawabata, Y.; Sorimachi, H. Structure, Activation, and Biology of Calpain. Diabetes 2004, 53, S12–S18. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.A.; Campbell, R.L.; Davies, P.L. Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nat. Cell Biol. 2008, 456, 409–412. [Google Scholar] [CrossRef]
- Boland, A.; Martin, T.G.; Zhang, Z.; Yang, J.; Bai, X.-C.; Chang, L.; Scheres, S.H.W.; Barford, D. Cryo-EM structure of a metazoan separase–securin complex at near-atomic resolution. Nat. Struct. Mol. Biol. 2017, 24, 414–418. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, A.; Roschitzki-Voser, H.; Amstutz, P.; Briand, C.; Gulotti-Georgieva, M.; Prenosil, E.; Binz, H.K.; Capitani, G.; Baici, A.; Simon, M.; et al. Inhibition of Caspase-2 by a Designed Ankyrin Repeat Protein: Specificity, Structure, and Inhibition Mechanism. Structure 2007, 15, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, T.; Barandun, J.; Flütsch, A.; Briand, C.; Mittl, P.R.E.; Grütter, M.G. Specific Inhibition of Caspase-3 by a Competitive DARPin: Molecular Mimicry between Native and Designed Inhibitors. Structure 2013, 21, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Seeger, M.A.; Zbinden, R.; Flütsch, A.; Gutte, P.G.M.; Engeler, S.; Roschitzki-Voser, H.; Grütter, M.G. Design, construction, and characterization of a second-generation DARPin library with reduced hydrophobicity. Protein Sci. 2013, 22, 1239–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flütsch, A.; Ackermann, R.; Schroeder, T.; Lukarska, M.; Hausammann, G.J.; Weinert, C.; Briand, C.; Grütter, M.G. Combined inhibition of caspase 3 and caspase 7 by two highly selective DARPins slows down cellular demise. Biochem. J. 2014, 461, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L.; Renko, M.; Završnik, J.; Turk, D.; Seeger, M.A.; Vasiljeva, O.; Grütter, M.G.; Turk, V.; Turk, B. Non-invasive in vivo imaging of tumour-associated cathepsin B by a highly selective inhibitory DARPin. Theranostics 2017, 7, 2806–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlmann, F.; Wernic, D.; Poupart, M.-A.; Koonin, E.V.; Nasmyth, K.A. Cleavage of Cohesin by the CD Clan Protease Separin Triggers Anaphase in Yeast. Cell 2000, 103, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Hauf, S.; Waizenegger, I.C.; Peters, J.-M. Cohesin Cleavage by Separase Required for Anaphase and Cytokinesis in Human Cells. Science 2001, 293, 1320–1323. [Google Scholar] [CrossRef]
- Nasmyth, K. Segregating Sister Genomes: The Molecular Biology of Chromosome Separation. Science 2002, 297, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.M. The anaphase-promoting complex: Proteolysis in mitosis and beyond. Mol. Cell 2002, 9, 931–943. [Google Scholar] [CrossRef]
- Uhlmann, F. Separase regulation during mitosis. Biochem. Soc. Symp. 2003, 70, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Yanagida, M. Cell cycle mechanisms of sister chromatid separation; Roles of Cut1/separin and Cut2/securin. Genes Cells 2000, 5, 1–8. [Google Scholar] [CrossRef]
- Sullivan, M.; Hornig, N.C.D.; Porstmann, T.; Uhlmann, F. Studies on Substrate Recognition by the Budding Yeast Separase. J. Biol. Chem. 2004, 279, 1191–1196. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Jaroszewski, L.; Iyer, M.; Sedova, M.; Godzik, A. FATCAT 2.0: Towards a better understanding of the structural diversity of proteins. Nucleic Acids Res. 2020, 48, W60–W64. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F.; Lottspeich, F.; Nasmyth, K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nat. Cell Biol. 1999, 400, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme. Eur. J. Inorg. Chem. 1894, 27, 2985–2993. [Google Scholar] [CrossRef] [Green Version]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Esposito, V.; Temussi, P.A. Cystatins: A versatile family. Biomol. Concepts 2011, 2, 95–102. [Google Scholar] [CrossRef]
- McGowan, S.; Buckle, A.M.; Irving, J.A.; Ong, P.C.; Bashtannyk-Puhalovich, T.A.; Kan, W.-T.; Henderson, K.N.; Bulynko, Y.A.; Popova, E.Y.; Smith, A.I.; et al. X-ray crystal structure of MENT: Evidence for functional loop–sheet polymers in chromatin condensation. EMBO J. 2006, 25, 3144–3155. [Google Scholar] [CrossRef]
- Rydel, T.J.; Ravichandran, K.G.; Tulinsky, A.; Bode, W.; Huber, R.; Roitsch, C.; Fenton, J.W. The structure of a complex of recombinant hirudin and human alpha-thrombin. Science 1990, 249, 277–280. [Google Scholar] [CrossRef]
- Dall, E.; Brandstetter, H. Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 10940–10945. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein–protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Targ. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Kasperkiewicz, P.; Poreba, M.; Snipas, S.J.; Parker, H.; Winterbourn, C.C.; Salvesen, G.S.; Drag, M. Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl. Acad. Sci. USA 2014, 111, 2518–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, J.A.; Pike, R.N.; Dai, W.; Brömme, D.; Worrall, D.M.; Silverman, G.A.; Coetzer, T.H.T.; Dennison, C.; Bottomley, S.P.; Whisstock, J.C. Evidence That Serpin Architecture Intrinsically Supports Papain-like Cysteine Protease Inhibition: Engineering α1-Antitrypsin To Inhibit Cathepsin Proteases. Biochemistry 2002, 41, 4998–5004. [Google Scholar] [CrossRef] [PubMed]
- Bratovš, A.; Kramer, L.; Mikhaylov, G.; Vasiljeva, O.; Turk, B. Stefin A-functionalized liposomes as a system for cathepsins S and L-targeted drug delivery. Biochimie 2019, 166, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Stadler, L.K.J.; Busby, M.; Song, Q.; Buxton, A.T.; Wagner, S.D.; Davis, J.J.; Ferrigno, P.K. Structure-function studies of an engineered scaffold protein derived from stefin A. I: Development of the SQM variant. Protein Eng. Des. Sel. 2010, 23, 403–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, L.K.J.; Hoffmann, T.; Tomlinson, D.C.; Song, Q.; Lee, T.; Busby, M.; Nyathi, Y.; Gendra, E.; Tiede, C.; Flanagan, K.; et al. Structure−function studies of an engineered scaffold protein derived from Stefin A. II: Development and applications of the SQT variant. Protein Eng. Des. Sel. 2011, 24, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Mishra, M.; Singh, V.; Tellis, M.B.; Joshi, R.S.; Singh, S. Repurposing the McoTI-II Rigid Molecular Scaffold in to Inhibitor of ‘Papain Superfamily’ Cysteine Proteases. Pharmaceuticals 2021, 14, 7. [Google Scholar] [CrossRef]
- Smith, M.; Kocher, H.M.; Hunt, B.J. Aprotinin in severe acute pancreatitis. Int. J. Clin. Pr. 2009, 64, 84–92. [Google Scholar] [CrossRef]
- Klemenčič, I.; Carmona, A.K.; Cezari, M.H.S.; Juliano, M.A.; Juliano, L.; Guncar, G.; Turk, D.; Križaj, I.; Turk, V.; Turk, B. Biochemical characterization of human cathepsin X revealed that the enzyme is an exopeptidase, acting as carboxymonopeptidase or carboxydipeptidase. JBIC J. Biol. Inorg. Chem. 2000, 267, 5404–5412. [Google Scholar] [CrossRef]
- Turk, B.; Ritonja, A.; Björk, I.; Stoka, V.; Dolenc, I.; Turk, V. Identification of bovine stefin A, a novel protein inhibitor of cysteine proteinases. FEBS Lett. 1995, 360, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Jerala, R.; Kroon-Zitko, L.; Popovic, T.; Turk, V. Elongation on the Amino-terminal Part of Stefin B Decreases Inhibition of Cathepsin H. JBIC J. Biol. Inorg. Chem. 1994, 224, 797–802. [Google Scholar] [CrossRef]
- Ni, J.; Abrahamson, M.; Zhang, M.; Fernandez, M.A.; Grubb, A.; Su, J.; Yu, G.-L.; Li, Y.; Parmelee, D.; Xing, L.; et al. Cystatin E is a Novel Human Cysteine Proteinase Inhibitor with Structural Resemblance to Family 2 Cystatins. J. Biol. Chem. 1997, 272, 10853–10858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.; Fernandez, M.A.; Danielsson, L.; Chillakuru, R.A.; Zhang, J.; Grubb, A.; Su, J.; Gentz, R.; Abrahamson, M. Cystatin F Is a Glycosylated Human Low Molecular Weight Cysteine Proteinase Inhibitor. J. Biol. Chem. 1998, 273, 24797–24804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimerman, N.; Prebanda, M.T.; Turk, B.; Popovič, T.; Dolenc, I.; Turk, V. Interaction of cystatin C variants with papain and human cathepsins B, H and L. J. Enzym. Inhib. 1999, 14, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Müller-Esterl, W.; Fritz, H.; Machleidt, W.; Ritonja, A.; Brzin, J.; Kotnik, M.; Turk, V.; Kellermann, J.; Lottspeich, F. Human plasma kininogens are identical with α-cysteine proteinase inhibitors. Evidence from immunological, enzymological and se-quence data. FEBS Lett. 1985, 182, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Mihelič, M.; Doberšek, A.; Gunčar, G.; Turk, D. Inhibitory Fragment from the p41 Form of Invariant Chain Can Regulate Activity of Cysteine Cathepsins in Antigen Presentation. J. Biol. Chem. 2008, 283, 14453–14460. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, S.; Westrop, G.; Scharfstein, J.; Mottram, J.; Coombs, G. Functional conservation of a natural cysteine peptidase inhibitor in protozoan and bacterial pathogens 1. FEBS Lett. 2003, 542, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Reis, F.C.G.; Costa, T.F.R.; Sulea, T.; Mezzetti, A.; Scharfstein, J.; Brömme, D.; Ménard, R.; Lima, A.P.C.A. The propeptide of cruzipain—A potent selective inhibitor of the trypanosomal enzymes cruzipain and brucipain, and of the human enzyme cathepsin F. FEBS J. 2007, 274, 1224–1234. [Google Scholar] [CrossRef]
- Takahashi, R.; Deveraux, Q.; Tamm, I.; Welsh, K.; Assa-Munt, N.; Salvesen, G.S.; Reed, J.C. A Single BIR Domain of XIAP Sufficient for Inhibiting Caspases. J. Biol. Chem. 1998, 273, 7787–7790. [Google Scholar] [CrossRef] [Green Version]
- Vucic, M.; Franklin, M.C.; Wallweber, H.J.A.; Das, K.; Eckelman, B.P.; Shin, H.; Elliott, L.O.; Kadkhodayan, S.; Deshayes, K.; Salvesen, G.S.; et al. Engineering ML-IAP to produce an extraordinarily potent caspase 9 inhibitor: Implications for Smac-dependent anti-apoptotic activity of ML-IAP. Biochem. J. 2004, 385, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Rich, R.L.; Steegborn, C.; Min, T.; Huang, Y.; Myszka, D.G.; Wu, H. Mutational Analyses of the p35-Caspase Interaction: A bowstring kinetic model of caspase inhibition by p35. J. Biol. Chem. 2002, 278, 5455–5461. [Google Scholar] [CrossRef] [Green Version]
- Stoka, V.; Nycander, M.; Lenarčič, B.; Labriola, C.; Cazzulo, J.J.; Björk, I.; Turk, V. Inhibition of cruzipain, the major cysteine proteinase of the protozoan parasite, Trypanosoma cruzi, by proteinase inhibitors of the cystatin superfamily. FEBS Lett. 1995, 370, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, P.; Abrahamson, M.; Björk, I. Interaction of recombinant human cystatin C with the cysteine proteinases papain and actinidin. Biochem. J. 1992, 281, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigsby, R.E.; Rife, C.L.; Fillgrove, K.L.; Newcomer, M.E.; Armstrong, R.N. Phosphonoformate: A Minimal Transition State Analogue Inhibitor of the Fosfomycin Resistance Protein, FosA. Biochemistry 2004, 43, 13666–13673. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Inhibitors of caspases. Complexes are shown with the same view and scale aligned after superimposition of the caspases to the caspase-3 in the complex with X-linked inhibitor of apoptosis (XIAP). Figure was prepared using MAIN [35] and rendered with Raster3d [36]. (a) Human XIAP caspase-3 complex ([83], PDB code 1I3O). The XIAP chain is shown as a red coil on the semitransparent background of the white surface of caspase-3. (b) Escherichia coli Nlef caspase-9 complex ([84], PDB code 3V3K). The Nlef chain is shown as an orange coil on the semitransparent background of the white surface of caspase-9. (c) Baculovirus p35 caspase-8 complex ([85], PDB code 1I4E). The p35 chain is shown as a blue coil on the semitransparent background of the white surface of caspase-8. (d) The CrmA chain ([86], PDB code 1F0C) is shown as a blue coil with the cleaved residues A359 and S359A shown as stick model in red.
Figure 2.
Inhibitors of caspases. Complexes are shown with the same view and scale aligned after superimposition of the caspases to the caspase-3 in the complex with X-linked inhibitor of apoptosis (XIAP). Figure was prepared using MAIN [35] and rendered with Raster3d [36]. (a) Human XIAP caspase-3 complex ([83], PDB code 1I3O). The XIAP chain is shown as a red coil on the semitransparent background of the white surface of caspase-3. (b) Escherichia coli Nlef caspase-9 complex ([84], PDB code 3V3K). The Nlef chain is shown as an orange coil on the semitransparent background of the white surface of caspase-9. (c) Baculovirus p35 caspase-8 complex ([85], PDB code 1I4E). The p35 chain is shown as a blue coil on the semitransparent background of the white surface of caspase-8. (d) The CrmA chain ([86], PDB code 1F0C) is shown as a blue coil with the cleaved residues A359 and S359A shown as stick model in red.

Figure 3.
Other inhibitors. Complexes of inhibitors are shown in views and scales adjusted to each complex and size. Figure was prepared using MAIN [35] and rendered with Raster3d [36]. (a) Procathepsin B ([98], PDB code 3PBH). The chain of cathepsin B propeptide is shown as a blue coil on the white surface of mature enzyme part of the structure. The surface part corresponding to the catalytic pair of C29 H199 residues is colored red. (b) Cystain E legumain complex ([42], PDB code 4N6N). Cystatin E is shown as a blue ribbon, with the P1 residue N39 side chain bound in to the legumain S1 site shown as a red stick model. Legumain is shown as a white surface with the S1 binding pocket colored orange and the part corresponding to the reactive site residues C189 H148 colored red. (c) Calpastatin m-calpain complex ([99], PDB code 3DF0). Calpastatin is shown as a blue surface with the loop out region indicating the position above the reactive site of calpain shown in red. Calpain-m is shown as a semitransparent white surface. (d) Securin separase complex ([100], PDB code 5ULS, 5ULT). Securin is shown as a blue surface with the region from 262 to 265 bound above the reactive site of separase shown in red. Securin is shown as a semitransparent white surface.
Figure 3.
Other inhibitors. Complexes of inhibitors are shown in views and scales adjusted to each complex and size. Figure was prepared using MAIN [35] and rendered with Raster3d [36]. (a) Procathepsin B ([98], PDB code 3PBH). The chain of cathepsin B propeptide is shown as a blue coil on the white surface of mature enzyme part of the structure. The surface part corresponding to the catalytic pair of C29 H199 residues is colored red. (b) Cystain E legumain complex ([42], PDB code 4N6N). Cystatin E is shown as a blue ribbon, with the P1 residue N39 side chain bound in to the legumain S1 site shown as a red stick model. Legumain is shown as a white surface with the S1 binding pocket colored orange and the part corresponding to the reactive site residues C189 H148 colored red. (c) Calpastatin m-calpain complex ([99], PDB code 3DF0). Calpastatin is shown as a blue surface with the loop out region indicating the position above the reactive site of calpain shown in red. Calpain-m is shown as a semitransparent white surface. (d) Securin separase complex ([100], PDB code 5ULS, 5ULT). Securin is shown as a blue surface with the region from 262 to 265 bound above the reactive site of separase shown in red. Securin is shown as a semitransparent white surface.

Figure 4.
Substrate and substrate-like binding. Peptidyl substrates with their positions marked are shown as ball (nitrogen: blue, oxygens: red, carbons: cyan) and stick (cyan) models on the background of the protease surface. The surface is white with the exception of the substrate binding sites S3, S1, and S2′, respectively corresponding to substrate positions P3, P1, and P2′ colored orange, and S4, S2, and S1′ respectively corresponding to substrate positions P4, P2, and P1′ colored red. The figure was prepared using MAIN [35] and rendered with Raster3d [36]. (a) Canonical conformation of BPTI peptide bound to trypsin ([24], PDB code 2TGP). BPTI peptide is shown as a ball and stick model on the background of trypsin structure shown in white, with the exception of the substrate-binding sites surface from S3 to S3′ colored alternatively red and orange. (b) Substrate model bound to cathepsin L. The cathepsin L model was used from a previous study [11]. (c) Peptidyl inhibitor bound to legumain ([170], PDB code 4AWB). Z-Ala-Ala-Asn (ZAAN) binds to the non-prime region of the active site cleft. (d) Calpastatin loop out region bound to calpain-m ([99], PDB code 3DF0). The 172–185 region of calpastatin is shown as a coil for the main chain trace, and a ball and stick model for side chains on the background of the protease surface. The calpain surface was generated with the residues from S241 to V253, I260, and Q261 excluded to enable the view in the active site cleft. The surface of the reactive site residues C105S and H262 is purple.
Figure 4.
Substrate and substrate-like binding. Peptidyl substrates with their positions marked are shown as ball (nitrogen: blue, oxygens: red, carbons: cyan) and stick (cyan) models on the background of the protease surface. The surface is white with the exception of the substrate binding sites S3, S1, and S2′, respectively corresponding to substrate positions P3, P1, and P2′ colored orange, and S4, S2, and S1′ respectively corresponding to substrate positions P4, P2, and P1′ colored red. The figure was prepared using MAIN [35] and rendered with Raster3d [36]. (a) Canonical conformation of BPTI peptide bound to trypsin ([24], PDB code 2TGP). BPTI peptide is shown as a ball and stick model on the background of trypsin structure shown in white, with the exception of the substrate-binding sites surface from S3 to S3′ colored alternatively red and orange. (b) Substrate model bound to cathepsin L. The cathepsin L model was used from a previous study [11]. (c) Peptidyl inhibitor bound to legumain ([170], PDB code 4AWB). Z-Ala-Ala-Asn (ZAAN) binds to the non-prime region of the active site cleft. (d) Calpastatin loop out region bound to calpain-m ([99], PDB code 3DF0). The 172–185 region of calpastatin is shown as a coil for the main chain trace, and a ball and stick model for side chains on the background of the protease surface. The calpain surface was generated with the residues from S241 to V253, I260, and Q261 excluded to enable the view in the active site cleft. The surface of the reactive site residues C105S and H262 is purple.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tušar, L.; Usenik, A.; Turk, B.; Turk, D. Mechanisms Applied by Protein Inhibitors to Inhibit Cysteine Proteases. Int. J. Mol. Sci. 2021, 22, 997. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030997
AMA Style
Tušar L, Usenik A, Turk B, Turk D. Mechanisms Applied by Protein Inhibitors to Inhibit Cysteine Proteases. International Journal of Molecular Sciences. 2021; 22(3):997. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030997
Chicago/Turabian StyleTušar, Livija, Aleksandra Usenik, Boris Turk, and Dušan Turk. 2021. "Mechanisms Applied by Protein Inhibitors to Inhibit Cysteine Proteases" International Journal of Molecular Sciences 22, no. 3: 997. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030997
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.