
The Role of mTOR Signaling as a Therapeutic Target in Cancer


1
Laboratory of Receptor Cell Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Miklukho-Maklaya Str., 16/10, 117997 Moscow, Russia
2
Institute of Biochemistry and Signal Transduction, University Medical Center Hamburg-Eppendorf, Martinistraße 52, 20246 Hamburg, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(4), 1743; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041743
Submission received: 10 January 2021
/
Revised: 30 January 2021
/
Accepted: 3 February 2021
/
Published: 9 February 2021
(This article belongs to the Special Issue mTOR Signaling Network in Cell Biology and Human Disease)
Abstract
:The aim of this review was to summarize current available information about the role of phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling in cancer as a potential target for new therapy options. The mTOR and PI3K/AKT/mTORC1 (mTOR complex 1) signaling are critical for the regulation of many fundamental cell processes including protein synthesis, cell growth, metabolism, survival, catabolism, and autophagy, and deregulated mTOR signaling is implicated in cancer, metabolic dysregulation, and the aging process. In this review, we summarize the information about the structure and function of the mTOR pathway and discuss the mechanisms of its deregulation in human cancers including genetic alterations of PI3K/AKT/mTOR pathway components. We also present recent data regarding the PI3K/AKT/mTOR inhibitors in clinical studies and the treatment of cancer, as well the attendant problems of resistance and adverse effects.
1. Mechanistic/Mammalian Target of Rapamycin (mTOR)
The mechanistic/mammalian target of rapamycin (mTOR) is a 289-kDa serine/threonine protein kinase of the phosphatidylinositol-3-kinase (PI3K) family-related protein kinases (PIKKs) that, in humans, is encoded by the MTOR gene [1,2,3]. Mammals express one mTOR protein that serves as a core component of two multi-subunit complexes, mTOR complex 1 (mTORC1) and mTORC2 [4]. The complexes perform separate functions in the cell: mTORC1 primarily controls cell growth, while mTORC2 participates in the control of cell survival and proliferation [5].
mTORC1 is composed of three core components: mTOR itself, mammalian lethal with SEC13 protein 8 (mLST8) (also known as G protein β-subunit-like protein GβL [6]), regulatory-associated protein of mTOR (RAPTOR) [7,8], and two non-core components: proline-rich AKT1 substrate 1 (PRAS40) [9] and DEP domain-containing mTOR-interacting protein (DEPTOR) [10] (Figure 1). RAPTOR is fundamental for mTORC1 assembly, stability, correct subcellular localization, and substrate recruitment [7,11,12]. PRAS40 blocks mTORC1 activity until growth factor receptor signaling unlocks PRAS40-mediated mTORC1 inhibition [13].
Like mTORC1, mTORC2 is formed by mTOR and mLST8 [14]. mTORC2 also contains two essential specific components, rapamycin insensitive companion of mTOR (RICTOR) [15] and stress-activated map kinase-interacting protein 1 (mSIN1) [16]; additionally, mTORC2 is associated with the facultative subunit protein observed with RICTOR (Protor)-1/2 [17]. RICTOR is necessary for mTORC2 assembly, stability, and substrate interactions [18]. mSIN1 acts as a negative regulator of mTORC2 kinase activity [16]. Protor-1 may play a role in enabling mTORC2 to efficiently phosphorylate serum and glucocorticoid-activated kinase 1 (SGK1) [19].
Rapamycin allosterically inhibits mTORC1 activity, while mTORC2 demonstrates short-term rapamycin insensitivity [4,20]. The mechanism of rapamycin action remained elusive for more than 20 years after the time of its isolation in 1972. In the 1990s, it was demonstrated that rapamycin acts by binding the peptidyl-prolyl cis-trans isomerase FK506-binding protein 12 (FKBP12) to form a complex that broadly inhibits cell growth and proliferation [21,22]. Biochemical experiments in mammalian cells have revealed that the rapamycin–FKBP12 complex specifically targets and inhibits mTOR [1,2,3]. The rapamycin–FKBP12 complex binds to the FKBP12-rapamycin-binding (FRB) domain on mTOR and inhibits the kinase by directly blocking substrate recruitment and by further restricting active-site access [23].
The structure of mTORC2 obtained by cryogenic electron microscopy showed that RICTOR and mSIN1 together generate steric hindrance, inhibiting the FKBP12–rapamycin complex binding site on mTOR and thereby rendering mTORC2 insensitive to acute inhibition by rapamycin [24,25]. Nonetheless, prolonged rapamycin treatment can inhibit mTORC2 signaling by sequestering the cellular pool of mTOR into rapamycin-bound complexes that cannot nucleate new mTORC2 [26,27].
2. Activation of mTOR
2.1. PI3K/AKT/mTOR Signalling Pathway
Growth factors, such as insulin or insulin-like growth factor-1 (IGF-1), activate mTORC1 primarily through the stimulation of a pathway involving class I PI3K and its downstream effector AKT [28]. The growth factor binds to extracellular regions of receptor tyrosine kinases (RTKs), and the receptor is activated by ligand-induced receptor dimerization and/or oligomerization [29]. The activation of RTKs results in kinase activation and the autophosphorylation of tyrosine residues in the receptor C-terminal tail (Figure 2). The autophosphorylation of RTKs also recruits and activates a variety of downstream signaling proteins that contain Src homology-2 (SH2) or phosphotyrosine-binding (PTB) domains. These domains bind to specific phosphotyrosine residues within the receptor and engage downstream mediators that propagate critical cellular signaling pathways [30]. PI3K is recruited to the membrane by binding to the phosphotyrosine consensus residues of growth factor receptors or adaptors through one of the two SH2 domains in its adaptor subunit. PI3K generates at the plasma membrane phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) from phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) [31]. A direct antagonist of PI3K is the tumor suppressor phosphatase and tensin homolog (PTEN). PTEN dephosphorylates PtdIns(3,4,5)P3 into PtdIns(4,5)P2 to reverse the activity of PI3K, thereby functioning as an important negative control of incoming signals [32,33]. PtdIns(3,4,5)P3 is a second messenger that recruits phosphoinositide-dependent kinase 1 (PDK1) and AKT to the plasma membrane, and AKT is then phosphorylated by PDK1 at Thr308 [34,35]. To become fully active in response to growth factors, AKT must be further phosphorylated on S473 [36]. While PDK1 catalyzes phosphorylation of T308 [37], mTORC2 phosphorylates AKT at S473 [38,39]. The phosphorylation of S473 on its own is not sufficient to promote AKT activity, but it boosts it by up to ten-fold by promoting a conformational change that stabilizes the active conformation of the kinase domain [40]. AKT phosphorylates several intracellular proteins, including forkhead box O transcription factors (FoxO), BCL2-associated agonist of cell death (BAD) and glycogen synthase kinase 3 (GSK3), to promote cell cycle entry and cell survival. AKT also directly phosphorylates five residues (S939, S981, S1130, S1132, and T1462) on tuberous sclerosis complex 2 (TSC2) within the TSC complex that results in the dissociation of TSC from the lysosomal surface and the activation of Rheb (Ras homolog enriched in the brain) and mTORC1 [41,42,43].
TSC is a heterotrimeric complex that comprises TSC1, TSC2, and TBC1 domain family member 7 (TBC1D7) [44,45]. TSC functions as a GTPase activating protein (GAP) for the small GTPase Rheb [46,47]. TSC stimulates the conversion from the active Rheb-GTP state to the inactive GDP-bound state, and the TSC blockade results in the activation of Rheb. The GTP-bound form of Rheb directly binds the catalytic domain of mTOR and, therefore, activates mTORC1 [9,48]. The activation of mTORC1 occurs via recruitment to the surface of the lysosomes [49], a major hub for the degradation and recycling of macromolecules.
Additionally, AKT activation by growth factors can activate mTORC1 in a TSC-independent manner. AKT phosphorylates mTORC1 inhibitory subunit PRAS40 at T246 that causes PRAS40/RAPTOR dissociation and mTORC1 activation [9,13,50]. TSC can also be inhibited by the TSC2 phosphorylation by the MAP (mitogen-activated protein) kinase extracellular signal-regulated kinase (ERK) [51] and p90 ribosomal S6 kinase (RSK) [52], two downstream substrates of the Ras receptor tyrosine kinase signaling pathway.
Activated mTOR further phosphorylates downstream substrates, thus implicating a variety of cellular processes. mTORC1 directly regulates protein synthesis; plays a central role in lipid and nucleotide synthesis and energetic homeostasis, ribosome biogenesis, nucleotide metabolism, and cell cycle progression; and also negatively regulates catabolic processes such as autophagy, therefore controlling the balance between anabolism and catabolism in response to environmental conditions [53]. mTORC2 mainly regulates cell proliferation, survival, cytoskeletal remodeling, and cell migration [54].
mTORC1 regulates protein synthesis through the phosphorylation of two key molecules, S6K1 (p70 S6 kinase) and 4E-BP1 (eukaryotic initiation factor 4E-binding protein), that promote translation and protein synthesis [55]. Unphosphorylated 4E-BP1 suppresses the initiation of translation by binding and sequestering translation initiation factor 4E (eIF4E). Upon phosphorylation by mTORC1, 4E-BP1 dissociates from eIF4E, allowing for 5′cap-dependent mRNA translation to occur [56,57,58].
S6K1 and mTORC1 upregulate the transcription of rRNA by enhancing the activity of RNA polymerase I and RNA polymerase III through the phosphorylation of the upstream binding factor (UBF) [59], transcription initiation factor 1A (TIF-1A) [60], and MAF1 regulatory factors [61,62]. S6K1 also enhances protein synthesis by activating eIF4B [63], a positive regulator of cap-dependent translation, and by degrading the eIF4A inhibitor PDCD4 (programmed cell death protein 4) [64].
The activation of PI3K-mTOR signaling is normally controlled at numerous levels. Activated RTKs are dephosphorylated by protein tyrosine phosphatases. To control the extent of mTORC1 activation and restore TSC regulation after this stimulus, the mTORC1 substrate S6K1 then directly phosphorylates insulin receptor substrate 1 (IRS-1) as part of a negative feedback loop, blocking the further insulin-mediated activation of the PI3K–AKT pathway [65,66]. S6K1 can also phosphorylate the mTORC2 component mSIN1 on the T86 and T398 sites, which results in the suppression of the mTORC2-mediated activation of AKT [67]. Phosphatidylinositol second messengers at the membrane are dephosphorylated by lipid phosphatases, including PTEN, inositol polyphosphate 4-phosphatase type II (INPP4B), synaptojanin, and Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase (SHIP1/2) [68,69]. PTEN is a dual-specificity phosphatase that selectively removes phosphate from the 3′-hydroxyl group of PtdIns(3,4,5)P3 to produce PtdIns(4,5)P2. Synaptojanins and SHIPs dephosphorylate the 5′-hydroxyl group of PtdIns(3,4,5)P3 to produce PtdIns(3,4)P2. INPP4B removes a phosphate group from the 4′ position of PtdIns(3,4)P2 to produce PtdIns(3)P. The phosphorylation and activation of AKT is also countered by multiple phosphatases including protein phosphatase 2 (PP2A), pleckstrin homology (PH) domain and leucine rich repeat protein phosphatases 1 (PHLPP1), and PHLPP2 [70]. AKT phosphorylation is also inhibited by carboxyl-terminal modulator protein (CTMP), which binds to the AKT carboxyl-terminal regulatory domain [71].
2.2. Regulation of mTORC1 by Other Factors
The activity of mTORC1 is also regulated by amino acids, energy status, phosphatidic acid, and oxidative stress (reviewed in [72,73]).
Unlike growth factors, amino acids activate mTORC1 through a PI3K-, AKT-, and TSC-independent mechanism [74,75]. The amino acid-dependent activation of mTORC1 requires the Rag subfamily of Ras small GTPases [76,77]. Amino acids stimulate the loading of RagA or RagB with GTP, which enables RagA/B to interact with the RAPTOR component of mTORC1 [77]. This interaction results in the translocation of mTORC1 to the lysosomal surface, where the Rag GTPases dock on a multi-subunit complex called Ragulator, and lysosomal Rheb stimulates mTORC1 kinase activity [49].
Another important regulator of mTORC1 signaling is the AMP-activated protein kinase (AMPK), the principal energy sensor in most eukaryotic cells [78]. In contrast to growth factors, the depletion of cellular energy inhibits mTORC1 activity and cell growth. During periods of energy or oxygen deprivation, AMP levels rise and promote the activation of AMPK [78,79]. Activated AMPK directly inhibits mTORC1 by phosphorylating RAPTOR [80], and indirectly inhibits mTORC1 by phosphorylating TSC2 on T1227 and S1345, as well as enhancing the ability of TSC2 to inhibit mTOR signaling [81].
An additional regulatory mechanism of mTORC1 signaling involves the lipid second messenger phosphatidic acid (PA) [72]. The potential role of PA in the regulation of mTORC1 signaling was revealed in a study that demonstrated that PA can directly bind to the FRB domain of mTOR and stimulate the activation of the mTOR substrate S6K1 and the phosphorylation of 4E-BP1 in HEK293 cells [82]. It was further demonstrated that phospholipase D (PLD), an enzyme that promotes the hydrolysis of phosphatidylcholine to yield choline and PA [83,84], is a critical regulator in the activation of mTOR signaling by a variety of stimuli [85,86,87]. The elevated expression of either PLD1 or PLD2 activates mTORC1 signaling in various types of cells (reviewed in [72]).
3. Deregulation of the PI3K/AKT/mTOR Pathway in Cancer
According to the published data, the PI3K/AKT/mTOR pathway is activated in approximately 70% of ovarian or breast cancers [88,89]. For instance, the aberrant activation of the PI3K/AKT/mTOR pathway has been found in 90% of lung adenocarcinomas (ADCs) and 40% of squamous cell carcinomas (SCCs) [90].
In normal physiology, regulatory mechanisms tightly control the activity and homeostasis of the PI3K/AKT/mTOR pathway, but it can be constitutively activated in various cancers. These mechanisms include the amplification or mutation of genes encoding PI3K subunits, AKT, and other pathway members; the activation of receptor tyrosine kinases, mutation, or overexpression of growth factor receptors, e.g., epithelial growth factor receptor (EGFR) or human epidermal growth factor receptor 2 (HER2); the inactivating mutations in the genes encoding key tumor suppressors PTEN or INPP4B; the inactivating mutations in the genes encoding mTOR regulators such as TSC1 and TSC2; and the activating mutations in MTOR itself [91,92,93].
3.1. PI3K in Human Cancer
PI3Ks belong to a family of lipid kinases that phosphorylate the 3-OH group of phosphoinositides [94]. Based on their primary structures and in vitro lipid substrate specificity, PI3Ks are classified into three classes: class I PI3Ks, class II PI3Ks, and class III PI3Ks [28]. PI3K-related kinases, which are sometimes referred to as class IV PI3Ks, are protein serine/threonine kinases with a structure similar to the catalytic subunits of PI3Ks. Examples of PI3K-related kinases are mTOR and the DNA-dependent protein kinase (DNA-PK).
Class I PI3Ks are heterodimeric molecules composed of regulatory and catalytic subunits. Class I PI3Ks are further divided into two subclasses—IA and IB—based on their regulatory subunit and upstream activator.
Class IA PI3Ks, activated by receptor tyrosine kinases and RAS [95], consist of a p110 catalytic subunit (p110α, p110β, or p110δ) and one of five p85-like regulatory subunits (p85α, p55α, p50α, p85β, or p55γ). Class IB PI3Ks, activated by G protein-coupled receptors, consist of the catalytic subunit p110γ and one of two regulatory subunits (p101 or p87) [96]. While p110α and p110β are ubiquitously expressed, p110δ and p110γ expression is largely restricted to leukocytes [97].
Class ΙA PI3Ks are widely present in carcinogenic processes. The PIK3CA gene, encoding the p110α subunit, is frequently mutated or amplified in the most common human cancers, including those of the breast, colon, gastric, endometrial, cervical, prostate, and lung, as well as glioblastoma [98,99,100,101,102,103]. Most of these mutations occur at two hot-spots—E545K and H1047R—located in the helical domain and the kinase domain of p110α, respectively [103].
Catalytic subunit p110α normally binds to p85, which stabilizes it and controls its enzymatic activity [104]. p110α with the E545K mutation gains the ability to associate with IRS-1 independently of the p85 regulatory subunit, resulting in the constitutive activation of the PI3K pathway and increased cell proliferation, survival, and motility [105]. Another highly recurrent PIK3CA mutation, H1047R, increases protein activity through alteration of its catalytic site, resulting in the upregulation of PI3K signaling. However, these two mutants have slightly different phenotypic impact and expression of p110α, as E545K produces a more severe metastatic phenotype than that induced by expressing p110α H1047R in a breast cancer cell line [106].
The PIK3CA gene was found to be mutated, on average, in 15% of human cancers, and cancers of the liver, breast, and colon harbor the most PIK3CA mutations, with average mutational frequencies of 36%, 26%, and 25%, respectively [99]. A detailed analysis of the COSMIC (Catalogue Of Somatic Mutations in Cancer, UK) database revealed that PIK3CA is indeed most frequently mutated in breast cancer (28.83%), especially in estrogen receptor (ER)-positive carcinomas (38.88%), as well as endometrium (27.39%) and urinary tract (20.2%) cancers (Table 1). By contrast, elevated PIK3CA expression has been found in about 32% of lung cancers (Table 1).
3.2. AKT in Cancers
AKT is an evolutionarily conserved serine/threonine kinase that belongs to the AGC kinase family. Three highly conserved AKT isoforms have been identified: AKT1 (i.e., AKT), AKT2, and AKT3. The alteration of AKT activity is associated with several human diseases, including cancer and diabetes [107].
AKT is composed of three conservative structure domains: an N-terminal PH domain, a central kinase domain, and a C-terminal regulatory domain. A single amino acid substitution, E17K, in the lipid-binding PH domain of AKT-1 is a recurrent somatic cell mutation that occurs in breast cancer, meningioma, colorectal, endometrial, and ovarian cancers, and the mutation results in constitutive AKT1 activation [108,109]. This mutation dramatically increases the affinity of E17K AKT to PtdIns(4,5)P2 [110], activates AKT1 by means of pathological localization to the plasma membrane, and stimulates downstream signaling [111,112].
According to the COSMIC database, the frequency of AKT1 mutations is about 4% in breast cancer and 15% in meningiomas, and elevated expression is more common for endometrium (8.14%) and lung (8.73%) cancers (Table 2).
The overexpression of phosphorylated AKT (p-AKT) is also considered to be an indicator of poor prognosis in many malignancies. A meta-analysis conducted to evaluate the association of p-AKT overexpression with breast cancer prognosis showed that high p-AKT expression was significantly associated with a higher risk of death and disease recurrence [113]. In meta-analyses, p-AKT overexpression was also associated with worse survival in NSCLC (non-small cell lung carcinoma) [114,115], lymph node metastasis and poor prognosis in patients with gastric cancer [116], and poor overall survival and progression-free survival in patients with epithelial ovarian cancer [117].
3.3. Alterations of Receptor Tyrosine Kinases
The RTK-mediated activation of the PI3K/AKT/mTOR pathway is crucially important for its oncogenic activity and is clearly linked to the RTK signaling. Examples include PI3K activation by EGFR in lung cancers harboring somatic-activating mutations in EGFR [118] and HER2 mutations in breast cancers with HER2 amplification [119]. EGFR mutations have also been reported in colorectal cancers [120] and glioblastomas [121].
EGFR/HER1/ErbB1 and HER2/ErbB2 are the members of the ErbB receptor family. Activated receptors bind various signaling proteins and stimulate activation of many signaling pathways, including the PI3K/AKT/mTOR, Ras/RAF/MEK/ERK, phospholipase C (PLC)-γ1, and SRC pathways (reviewed in [122]). It has been shown that many cancer cells are characterized by EGFR hyperactivation, gene amplification leading to receptor overexpression, or mutants with dysregulated signaling [123]. In the EGFR gene, mutations occur within exons 18–21, which encode a portion of the EGFR kinase domain. For instance, approximately 80–90% of patients with EGFR-mutated, non–small cell lung cancer have either deletions in exon 19 or substitutions of leucine for arginine (L858R) in exon 21 of the EGFR gene [124].
An analysis of the COSMIC database displayed that the incidence of EGFR mutations significantly varies among the different types of cancers. The highest frequency is detected in lung cancers (26.58%) and especially in adenocarcinoma (30.61%), where AKT is also overexpressed (Table 3). In contrast to lung cancer, the EGFR mutations are rare in breast cancer [125,126], with a frequency of no more than 5–6% (Table 3). However, the overexpression of EGFR is observed in 15–30% of breast carcinomas and is associated with a large tumor size and poor clinical outcomes [126].
3.4. PTEN
The PTEN protein consists of two major domains, the N-terminal phosphatase catalytic domain (residues 7–185) and a C-terminal domain (residues 186–351) [127]. The N-terminal phosphatase domain of PTEN contains a consensus PtdIns(4,5)P2-binding motif, and the C-terminal part contains the lipid-binding C2 domain. The C2 domain is believed to be required for the correct positioning of PTEN at the plasma membrane, the site of the lipid substrates of PTEN [128].
PTEN antagonizes the PI3K by dephosphorylating PtdIns(3,4,5)P3 to PtdIns(4,5)P2; therefore, PtdIns(3,4,5)P3 levels are strictly regulated by PTEN in normal cells. PTEN is frequently inactivated in human cancers through several mechanisms, including mutation, a loss of heterozygosity, methylation, the aberrant expression of regulatory microRNA, and protein instability [129,130]. Elevated PtdIns(3,4,5)P3 levels, through the loss of PTEN function, cause the constitutive activation of AKT and downstream cascades such as mTOR signaling [41]. This leads to cell survival, growth, proliferation, and decreased apoptosis [131,132,133].
PTEN has been shown to be lost or inactivated by multiple mechanisms in a wide spectrum of human cancer types: a loss of heterozygosity of PTEN was found in 60–80% of patients with glioblastoma and in 45% of endometrioid carcinoma of the ovary; a loss of PTEN protein expression was found in 20–40% of patients with colorectal cancer; and a decreased PTEN expression was found in 30% of patients with head and neck cancer, PTEN mutations were found in up to 40% of patients with glioblastoma and in 15–50% of patients with prostate cancer [134]. Mutations of the human PTEN gene are also frequently observed in breast cancer, glioblastoma, endometrial cancer, malignant melanoma, and prostate cancer [135]. Deletions of PTEN have also been shown in lung cancer. The loss of PTEN expression assessed by immunohistochemistry has been demonstrated in up to 24% of 125 resected early stage NSCLC specimens [136]. Another series of early stage NSCLC specimens revealed that PTEN protein expression was reduced or lost in 74% of tumors [137]. Elevated levels of miR-21 in NSCLC, relative to adjacent non-tumor tissues, were correlated with decreased PTEN mRNA levels and advanced tumor stages [138].
An analysis of the COSMIC database revealed PTEN mutations in 55.71% of endometrioid carcinomas, in 39.68% of endometrium cancers, in 13.36% of central nervous system cancers, and in 8.4% of skin cancers (Table 4).
3.5. mTOR and Cancer Development
Receptor tyrosine kinase hyperactivation or overexpression, PTEN loss of function, and mutations in PIK3CA and AKT are not the only mechanisms that can lead to mTOR activation. mTOR can become activated via additional molecular mechanisms that include gene amplification and mutation (reviewed in [139]). It was reported that two different point mutations, S2215Y and R2505P, identified in the COSMIC database, confer the constitutive activation of mTOR signaling in cell cultures even under nutrient starvation conditions. S2215Y was identified in large intestine adenocarcinoma, whereas R2505P was identified in renal cell carcinoma [140]. In cell cultures, the association of the mTOR P2229R mutation with the activation of both the mTORC1 and mTORC2 pathways, increased cell proliferation, and cell survival was also demonstrated [141]. Grabiner et al. identified 33 MTOR mutations that occur in multiple cancer types and confer pathway hyperactivation [142]. It was also found that resistant colonies emerged after the exposure of the MCF-7 breast cancer cell line to high concentrations of a first generation mTORC1 inhibitor, rapamycin, or a second generation mTOR ATP competitive inhibitor (AZD8055) for three months. Deep sequencing revealed that the AZD8055-resistant clones harbored an mTOR mutation located in the kinase domain at the M2327I position, while two rapamycin-resistant clones contained mutations located in the FKBP12-rapamycin binding domain at positions A2034V and F2108L [143].
An analysis of the COSMIC database revealed an overexpression of MTOR in ovary (9.77%), urinary tract (8.33%), and skin (8.25%) cancers and a downregulation in tumors of the central nervous system (13.06%). The highest incidence of MTOR mutations is in meninges (18.63%), endometrium cancer (10.43%), and, especially, endometrioid carcinoma (12.63%) (Table 5).
Many clinical reports have attempted to investigate the roles and the potential prognostic value of mTOR and p-mTOR in a variety of cancers. A meta-analysis of data from 915 patients with esophageal squamous cell carcinoma (ESCC) was conducted to evaluate the prognostic and clinicopathological significance of mTOR/p-mTOR expression. The pooled analysis identified that positive mTOR/p-mTOR expression was significantly correlated with the worse conditions of differentiation degree, depth of tumor invasion, and lymph node metastasis but had no relationship to gender. Moreover, mTOR/p-mTOR expression was also significantly associated with worse overall survival, disease-free survival, and cancer-specific survival of patients with ESCC [144].
The prognostic impact of the PI3K/AKT/mTOR signaling pathway in advanced esophageal squamous cell carcinoma was also assessed in 145 tumor and 145 non-tumor samples of patients from China. The PI3K/AKT/mTOR signaling pathway was shown to be significantly upregulated and PTEN was largely downregulated in tumor tissue. The expression level of PTEN, mTOR, p-mTOR, and S6K1 was closely related to the presence of lymph node metastases. The expression of PTEN, mTOR, and S6K1 were also correlated to the TNM stage and overall survival [145].
4. Targeting PI3K/AKT/mTOR
The PI3K/AKT/mTOR signaling pathway plays an important role in cell growth, proliferation, and survival, and it is one of the most commonly deregulated pathways found in human cancers, which makes components of this pathway attractive targets for anticancer therapy.
4.1. PI3K Inhibitors
PI3K inhibitors can be subdivided into pan-PI3K inhibitors, isoform-selective PI3K inhibitors, and dual PI3K/mTOR inhibitors [146]. Dozens of PI3K inhibitors have been developed as potential chemotherapeutic drugs (reviewed in [146,147]). The first PI3K inhibitors wortmannin and LY294002 did not reach clinical trials due to the problems with stability, solubility, and toxicity [148], and the development of the PI3K inhibitor as antitumor agent has been a hotspot area since 2006 when ZSTK474, a new pan-class I PI3K inhibitor with less toxic effects, was first presented [149].
Pan-PI3K inhibitors act on each of the four catalytic isoforms of class I PI3K and have, as expected, broad inhibition potential in a number of tumors. However, this broad activity may lead to a higher risks of side effects and toxicities [150]. Until the end of 2020, two pan-PI3K inhibitors, i.e., copanlisib and duvelisib, and two isoform-selective PI3K inhibitors, i.e., idelalisib and alpelisib, were approved for cancer treatment by the Food and Drug Administration (FDA) (Figure 2).
Copanlisib (Aliqopa), an inhibitor of PI3K, predominantly against the isoforms PI3Kα and PI3Kδ, was approved by the FDA in September 2017 for the treatment of adult patients with relapsed follicular lymphoma who have received at least two prior systemic therapies (https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-relapsed-follicular-lymphoma; 14 September 2017).
Another pan-PI3K inhibitor, duvelisib (Copiktra), is an oral dual inhibitor of PI3Kγ and PI3Kδ. It was approved in September 2018 for the treatment of adult patients with relapsed or refractory chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). It was recommended mainly after at least two prior systemic therapies (https://www.fda.gov/drugs/resources-information-approved-drugs/duvelisib-copiktra-verastem-inc-adult-patients-relapsed-or-refractory-chronic-lymphocytic-leukemia; 24 September 2018).
Isoform-selective inhibitors are emerging as next-generation of PI3K inhibitors with improved, precise targeting and reduced toxicity. This class of inhibitors has been developed to target cancers that are connected to one of the PI3K isoforms. The selective inhibition of specific PI3K isoforms may allow for the administration of therapeutic doses of drugs and have fewer toxicities compared to pan-PI3K inhibitors. On the other hand, isoform-specific PI3K inhibitors have the narrowest profile and may require careful patient selection based on potential biomarkers of sensitivity and resistance [151]. It was also shown that the acquired amplification and mutation of PIK3CA cause resistance to selective PI3K inhibitors [152]. It was proposed that PTEN loss can lead to clinical PI3Kα inhibitor resistance [153].
Idelalisib (Zydelig), a PI3Kδ inhibitor, was approved in July 2014. Zydelig is currently approved by the FDA for the treatment of relapsed chronic lymphocytic leukemia in combination with rituximab and for the treatment of relapsed follicular B-cell non-Hodgkin’s lymphoma or relapsed small lymphocytic lymphoma in patients who have received at least two prior systemic therapies (https://www.fda.gov/drugs/drug-safety-and-availability/fda-alerts-healthcare-professionals-about-clinical-trials-zydelig-idelalisib-combination-other; 14 March 2016).
Alpelisib (Piqray) is a PI3Kα-specific inhibitor, approved by the FDA in May 2019 for use in combination with endocrine therapy fulvestrant for the treatment of hormone receptor (HR)-positive, HER2-negative, PIK3CA-mutated, advanced, or metastatic breast cancer (https://www.fda.gov/news-events/press-announcements/fda-approves-first-pi3k-inhibitor-breast-cancer; 24 May 2019).
4.2. AKT Inhibitors
AKT could be a promising target in PI3K/AKT/mTOR pathway-activated tumors as one of the key effector nodes. However, the number of AKT inhibitors that have been explored in clinical trials is less than that of PI3K inhibitors. Most AKT inhibitors in clinical development inhibit AKT 1, 2, and 3 and are therefore termed as pan-AKT inhibitors. There are two distinct classes of AKT inhibitors currently in clinical development: ATP-competitive and allosteric inhibitors. Allosteric inhibitors can prevent the localization of AKT with its PH domain to the plasma membrane, thereby blocking AKT phosphorylation and activation [154]. ATP-competitive inhibitors bind to the active conformation in which the PH domain has swung out from the kinase domain and expose the ATP-binding pocket [155,156]. Allosteric inhibitors are generally better than ATP-competitive inhibitors because they show less toxicity, reduced side-effects, and greater specificity [157].
MK2206, TAS-117, uprosertib (GSK2141795), afuresertib (GSK2110183), capivasertib (AZD5363), and ipatasertib have shown preliminary activity in phase I trials, and they are being tested in phase II trials in a range of tumors as monotherapy or in combination with the MEK inhibitor selumetinib (+MK2206), the MEK inhibitor trametinib (+GSK2141795), docetaxel (+ipatasertib), and paclitaxel (+AZD5363 or ipatasertib). Phase III studies of ipatasertib and capivasertib in combination with various drugs are also ongoing or will begin soon (Table 6).
4.3. mTOR Inhibitors
mTORC1 functions as a downstream effector for many frequently mutated oncogenic pathways including the PI3K/AKT and Ras/RAF/MEK/ERK (MAPK) pathways, and mTOR signaling is hyperactive in a range of 40–90% in different tumor entities [158], which makes mTOR an attractive target for cancer therapy.
4.3.1. The First Generation: Allosteric mTOR Inhibitors
Rapamycin and its analogs (rapalogs) are the first generation of mTOR inhibitors, which selectively inhibit the activity of mTORC1 by binding to FKBP-12 and forming a ternary complex with mTOR. Rapamycin is a macrolide, that is produced by the microorganism Streptomyces hygroscopicus and has shown antifungal properties [159]. Shortly after its discovery, immunosuppressive properties were detected, which later led to the establishment of rapamycin as an immunosuppressant [160,161]. In the 1980s, rapamycin was also found to have anticancer activity [162].
Rapamycin is an allosteric inhibitor of mTOR, and it inhibits some of the functions of mTORC1, such as the phosphorylation of the protein kinase S6K1. The clinical use of rapamycin is limited due to its poor water solubility and stability. Thus, several pharmaceutical companies have developed rapamycin analogs with improved pharmacokinetic properties (Figure 2) (Table 7).
Rapalogs differ in their chemical properties in terms of drug solubility and metabolism. For example, temsirolimus, a prodrug of rapamycin, and ridaforolimus (MK-8669) are water soluble and may be administered intravenously, whereas rapamycin and everolimus display low solubility and are therefore only available for oral formulations [163]. Rapalogs have been undergoing clinical trials for various malignancies and have already been approved by the FDA for the treatment of specific types of cancers (Table 7).
Though rapamycin inhibits mTOR with a high specificity, its effectiveness is dose-dependent in different contexts. Two mTOR complexes, mTORC1 and mTORC2, have different sensitivities to rapamycin; different doses of rapamycin are needed to suppress mTOR in different cell lines, as well as the phosphorylation of different mTOR substrates, and these properties of rapamycin dosage can be largely attributed to the competition between rapamycin and phosphatidic acid for mTOR (reviewed in [164]).
While rapamycin inhibits S6K1, it does not fully inhibit 4E-BP1 phosphorylation, thus making it ineffective in blocking cap-dependent translation in most cell types [165]. Phosphorylated 4E-BP1 inhibits pro-oncogenic eIF4E. eIF4E-mediated translation is upregulated in tumors, and blocking this pathway may be crucial to preventing tumor growth in specific cancers [166,167,168]. On the other hand, the inhibition of mTORC1 may lead to the feedback activation of IGF-IR and AKT, which compromise the anti-cancer effect of rapalogs [169]. Rapalogs have proved more cytostatic than cytotoxic, perhaps because they also only partially block 4E-BP-dependent translation and fail to inhibit the pro-survival pathways regulated by mTORC2–AKT [170,171].
The combination of rapamycin with 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) was reported to make rapamycin cytotoxic rather than cytostatic at doses that are clinically tolerated [172]. AICAR is a compound that activates AMPK, and a reciprocal regulation of PLD by AMPK and AMPK by PLD was demonstrated, i.e., the suppression of AMPK activity led to an increase in PLD activity and the suppression of PLD activity resulted in elevated AMPK activity [173]. The authors reported that the suppression of PLD activity by AICAR can improve the efficacy of rapamycin for both mTORC1 and mTORC2, and tolerable doses of rapamycin in combination with AICAR suppress both 4E-BP1 and AKT phosphorylation, as well as inducing apoptosis in cancer cells [172].
4.3.2. The Second Generation: ATP-Competitive mTOR Inhibitors
To more completely inhibit mTOR and target both mTORC1 and mTORC2, a number of ATP-competitive mTOR inhibitors have been developed. A second generation of mTOR inhibitors are small molecular ATP analogues that compete with ATP to occupy the kinase active site of mTOR. Differently from rapalogs, these molecules—also called selective mTOR kinase inhibitors (TORKIs)—ensure a complete block of both mTORC1 and 2, thus preventing AKT phosphorylation due to mTORC2 and avoiding the resistance observed in rapalogs.
In the majority of in vitro studies, compared with rapalogs, the ATP-competitive inhibitors have shown significantly higher inhibitory effects. Large scale trials have not yet been conducted to show greater and convincing efficacy than the currently available best treatment options; hence, TORKIs are still not approved by the FDA.
4.3.3. The Third Generation: RapaLink
Third generation mTOR inhibitors were synthesized and investigated to address the treatment resistance issues found in the use of the rapalogs and TORKIs. The new compounds are called Rapalink because they are made by the conjugation of rapamycin and ATP-competitive mTOR inhibitors, and they can bind both to FRB and the mTOR kinase domain [143]. This dual binding may serve to increase affinity and stability, both of which likely contribute to efficacy. Rapalink-1 has shown increased and durable inhibitory action compared to the first and second-generation inhibitors in glioblastoma and follicular lymphoma in vitro and in vivo [174,175,176]. A resistance of cancer cell cultures to rapalogs and TORKIs was shown to be overcome by the use of Rapalink [143].
The effectiveness of Rapalink-1 was also demonstrated in reducing prostate cancer tumor growth using an in vitro organoids assay and ex vivo tumor slice drug assays. The exposure of LAPC9 to Rapalink-1 was found to block mTORC1/2 signaling and reduce the fraction of CD44+ in vitro. Mice treated with Rapalink-1 showed a significantly delayed tumor growth, and cells recovered from the tumors of treated animals showed a marked decrease of CD44 expression [177]. The in vitro and in vivo therapeutic efficacy of Rapalink-1 against renal cell carcinoma (RCC) was evaluated and compared to temsirolimus. Rapalink-1 showed significantly greater effects against proliferation, migration, invasion, and colony formation in RCC cells. RNA sequencing showed that Rapalink-1 suppressed not only the mTOR signaling pathway but also a part of the MAPK signaling pathway, the ErbB signaling pathway, and ATP-binding cassette (ABC) transporters [178]. No large-sample clinical data have been reported for Rapalink.
4.4. Dual PI3K/mTOR Inhibitors
Though the inhibition of mTORC1 and mTORC2 can downregulate AKT S473 phosphorylation, mTOR inhibitors may paradoxically enhance the PI3K/PDK1 axis. Thus, an inhibitor targeting both PI3K and mTOR may have better anti-cancer activity compared to targeting mTOR alone [179,180].
PI3K and mTOR both belong to the PI3K-related kinase (PIKK) superfamily, and the catalytic isoform of the p110 subunit and mTOR have structural similarities; as a consequence, certain inhibitory compounds target both kinases. Dual PI3K/mTOR inhibitors are active against PI3K isoforms and both mTORC1 and mTORC2, thus targeting three most critical nodes of the same pathway. AKT activation would also be blocked by these inhibitors since PI3K blocking would diminish the production of PtdIns(3,4,5)P3, which acts as a docking site for AKT and PDK1. The inhibition of mTORC2 would also block the feedback activation of AKT. There are some promising trials with dual PI3K/mTOR inhibitors but only in phase 1 or 2 (Table 8). Therefore, more studies are needed to determine if the dual PI3K/mTOR inhibitors would be more effective than mTOR inhibitors.
Another possibility may be the dual inhibition with mTOR and AKT inhibitors to prevent the feedback activation of PI3K/AKT signaling after mTOR inhibition. Indeed, our own studies with a combinatorial treatment of hepatocellular carcinoma and cholangiocarcinoma cells with RAD001 and MK2206 showed synergistic effects on tumor growth in vitro and in xenotransplantation mouse models in vivo [181,182].
5. Summary and Conclusions
The discovery of mTOR is a fundamental breakthrough in the understanding of cell growth, metabolism, and diseases. Studies to determine the regulators and effectors of mTOR signaling have revealed multiple networks that interact together to integrate growth factor, nutrient, and nucleotide signaling. The understanding of the critical role of the mTOR pathway in tumorigenesis has driven to the development of a growing list of PI3K/AKT/mTOR inhibitors, though major clinical success has not been achieved.
The long-term use of the same inhibitor in tumors can lead to drug resistance, which is a main challenge in cancer therapy. Resistance can be developed by several mechanisms including the incomplete inhibition of mTORC1 functions, resistance mutations in MTOR, the compensatory activation of different pathways because of the mTORC1 inhibition, and the suppression of negative feedback loops (reviewed in [183]). More research to understand the molecular basis of mTOR networks and potential resistance mechanisms in mTOR-targeted cancer therapy are necessary to rationally apply mTOR inhibitors for the effective treatment of cancer.
A second problem comprises the clinical adverse effects associated with PI3K/AKT/mTOR inhibition that include hyperglycemia, hyperlipidemia, bone marrow suppression, pneumonitis, stomatitis, and hepatotoxicity [184]. Severe toxicities associated with PI3K/AKT/mTOR inhibitors may limit the clinical application and approval of these agents. It is necessary to analyze the mechanism leading to the toxicities of PI3K/AKT/mTOR inhibitors that may help to develop optimal prevention and treatment strategies.
High-throughput molecular profiling, including next-generation sequencing, can give insights into the mechanism(s) of intrinsic sensitivity/resistance and the mechanisms of acquired resistance to PI3K/AKT/mTOR inhibitors. Work must be continued to validate predictive biomarkers, which may help to identify the patients who will most likely benefit from treatment with mTOR inhibitors and allow for the better determination of rational combinations of anti-cancer agents.
Author Contributions
N.V.P. and M.J. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AICAR | 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside |
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| BAD | BCL2-associated agonist of cell death |
| CTMP | carboxyl-terminal modulator protein |
| DEPTOR | DEP domain-containing mTOR-interacting protein |
| DNA-PK | DNA-dependent protein kinase |
| eIF4E | eukaryotic translation initiation factor 4E |
| ERK | extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| FKBP12 | FK506-binding protein 12 |
| FoxO | forkhead box O transcription factors |
| FRB | FKBP12-rapamycin-binding |
| GAP | GTPase activating protein |
| GβL | G protein β-subunit-like protein |
| GSK3 | glycogen synthase kinase 3 |
| IGF-1 | insulin-like growth factor-1 |
| INPP4B | inositol polyphosphate 4-phosphatase type II |
| IRS-1 | insulin receptor substrate 1 |
| MAP kinase | mitogen-activated protein kinase |
| mLST8 | mammalian lethal with SEC13 protein 8 |
| mSIN1 | stress-activated map kinase-interacting protein 1 |
| mTOR | mechanistic/mammalian target of rapamycin |
| PDCD4 | programmed cell death protein 4 |
| PDK1 | phosphoinositide-dependent kinase 1 |
| PI3K | phosphatidylinositol-3-kinase |
| PIKK | phosphatidylinositol 3-kinase-related kinase |
| PI(4,5)P2 | phosphatidylinositol 4,5 bisphosphate |
| PI(3,4,5)P3 | phosphatidylinositol 3,4,5 trisphosphate |
| PLD | phospholipase D |
| PRAS40 | proline-rich AKT1 substrate 1 |
| Protor | protein observed with RICTOR |
| PTEN | phosphatase and tensin homolog |
| RAPTOR | regulatory-associated protein of mTOR |
| Rheb | Ras homolog enriched in brain |
| RICTOR | rapamycin insensitive companion of mTOR |
| RSK | ribosomal S6 kinase |
| RTK | receptor tyrosine kinase |
| S6K1 | p70 S6 kinase, ribosomal protein S6 kinase beta-1 |
| SGK1 | serum and glucocorticoid-activated kinase 1 |
| SHIP | Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase |
| TBC1D7 | TBC1 domain family member 7 |
| TIF-1A | transcription initiation factor 1A |
| TSC | tuberous sclerosis complex |
| UBF | upstream binding factor |
References
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A Mammalian Protein Targeted by G1-Arresting Rapamycin-Receptor Complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A Mammalian Protein That Binds to FKBP12 in a Rapamycin-Dependent Fashion and Is Homologous to Yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a Protein Target of the FKBP12-Rapamycin Complex in Mammalian Cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR Complexes, Only One of Which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.P.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GβL, a Positive Regulator of the Rapamycin-Sensitive Pathway Required for the Nutrient-Sensitive Interaction between Raptor and MTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. MTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex That Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the MTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an MTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The Mammalian Target of Rapamycin (MTOR) Partner, Raptor, Binds the MTOR Substrates P70 S6 Kinase and 4E-BP1 through Their TOR Signaling (TOS) Motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [Green Version]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS Motif-Mediated Raptor Binding Regulates 4E-BP1 Multisite Phosphorylation and Function. Curr. Biol. CB 2003, 13, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Haar, E.V.; Lee, S.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin Signalling to MTOR Mediated by the Akt/PKB Substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Scaiola, A.; Mangia, F.; Imseng, S.; Boehringer, D.; Berneiser, K.; Shimobayashi, M.; Stuttfeld, E.; Hall, M.N.; Ban, N.; Maier, T. The 3.2-Å Resolution Structure of Human MTORC2. Sci. Adv. 2020, 6, eabc1251. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of MTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway That Regulates the Cytoskeleton. Curr. Biol. CB 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.-L. Identification of Sin1 as an Essential TORC2 Component Required for Complex Formation and Kinase Activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.M.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a Novel Rictor-Binding Component of MTOR Complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-H.; Sarbassov, D.D. The MTOR (Mammalian Target of Rapamycin) Kinase Maintains Integrity of MTOR Complex 2. J. Biol. Chem. 2011, 286, 40386–40394. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Sommer, E.M.; Sakamoto, K.; Wullschleger, S.; Alessi, D.R. Protor-1 Is Required for Efficient MTORC2-Mediated Activation of SGK1 in the Kidney. Biochem. J. 2011, 436, 169–179. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR Complex 2 Controls the Actin Cytoskeleton and Is Rapamycin Insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Bierer, B.E.; Mattila, P.S.; Standaert, R.F.; Herzenberg, L.A.; Burakoff, S.J.; Crabtree, G.; Schreiber, S.L. Two Distinct Signal Transmission Pathways in T Lymphocytes Are Inhibited by Complexes Formed between an Immunophilin and Either FK506 or Rapamycin. Proc. Natl. Acad. Sci. USA 1990, 87, 9231–9235. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-FKBP Specifically Blocks Growth-Dependent Activation of and Signaling by the 70 Kd S6 Protein Kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. MTOR Kinase Structure, Mechanism and Regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, M.; Tian, Y.; Li, J.; Qi, Y.; Zhao, D.; Wu, Z.; Huang, M.; Wong, C.C.L.; Wang, H.-W.; et al. Cryo-EM Structure of Human MTOR Complex 2. Cell Res. 2018, 28, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Stuttfeld, E.; Aylett, C.H.; Imseng, S.; Boehringer, D.; Scaiola, A.; Sauer, E.; Hall, M.N.; Maier, T.; Ban, N. Architecture of the Human MTORC2 Core Complex. eLife 2018, 7, e33101. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-Induced Insulin Resistance Is Mediated by MTORC2 Loss and Uncoupled from Longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits MTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Fruman, D.A.; Meyers, R.E.; Cantley, L.C. Phosphoinositide Kinases. Annu. Rev. Biochem. 1998, 67, 481–507. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Pawson, T.; Gish, G.D.; Nash, P. SH2 Domains, Interaction Modules and Cellular Wiring. Trends Cell Biol. 2001, 11, 504–511. [Google Scholar] [CrossRef]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Maehama, T.; Dixon, J.E. PTEN: A Tumour Suppressor That Functions as a Phospholipid Phosphatase. Trends Cell Biol. 1999, 9, 125–128. [Google Scholar] [CrossRef]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative Regulation of PKB/Akt-Dependent Cell Survival by the Tumor Suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Cantley, L.C. Regulation of MTORC1 by PI3K Signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.; Komander, D.; van Aalten, D.M.F.; Alessi, D.R. PDK1, the Master Regulator of AGC Kinase Signal Transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-Phosphoinositide-Dependent Protein Kinase Which Phosphorylates and Activates Protein Kinase Balpha. Curr. Biol. CB 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Stephens, L.; Anderson, K.; Stokoe, D.; Erdjument-Bromage, H.; Painter, G.F.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; McCormick, F.; Tempst, P.; et al. Protein Kinase B Kinases That Mediate Phosphatidylinositol 3,4,5-Trisphosphate-Dependent Activation of Protein Kinase B. Science 1998, 279, 710–714. [Google Scholar] [CrossRef]
- Hresko, R.C.; Mueckler, M. MTOR.RICTOR Is the Ser473 Kinase for Akt/Protein Kinase B in 3T3-L1 Adipocytes. J. Biol. Chem. 2005, 280, 40406–40416. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The Nuts and Bolts of AGC Protein Kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 Is Phosphorylated and Inhibited by Akt and Suppresses MTOR Signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of MTORC1 at the Lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt Regulates Growth by Directly Phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 Is a Third Subunit of the TSC1-TSC2 Complex Upstream of MTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [Green Version]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between Hamartin and Tuberin, the TSC1 and TSC2 Gene Products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase Is a Direct Target of TSC2 GAP Activity and Regulates MTOR Signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control MTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb Binds and Regulates the MTOR Kinase. Curr. Biol. CB 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets MTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C. PRAS40 Regulates MTORC1 Kinase Activity by Functioning as a Direct Inhibitor of Substrate Binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and Functional Inactivation of TSC2 by Erk Implications for Tuberous Sclerosis and Cancer Pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-Promoting Phorbol Esters and Activated Ras Inactivate the Tuberous Sclerosis Tumor Suppressor Complex via P90 Ribosomal S6 Kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Fu, W.; Hall, M.N. Regulation of MTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 Phosphorylation of the Translational Regulators P70 S6 Kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar]
- Brunn, G.J.; Hudson, C.C.; Sekulić, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C.; Abraham, R.T. Phosphorylation of the Translational Repressor PHAS-I by the Mammalian Target of Rapamycin. Science 1997, 277, 99–101. [Google Scholar] [CrossRef]
- Gingras, A.-C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 Phosphorylation: A Novel Two-Step Mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar]
- Hara, K.; Yonezawa, K.; Kozlowski, M.T.; Sugimoto, T.; Andrabi, K.; Weng, Q.P.; Kasuga, M.; Nishimoto, I.; Avruch, J. Regulation of EIF-4E BP1 Phosphorylation by MTOR. J. Biol. Chem. 1997, 272, 26457–26463. [Google Scholar] [CrossRef] [Green Version]
- Hannan, K.; Brandenburger, Y.; Jenkins, A.; Sharkey, K.; Cavanaugh, A.; Rothblum, L.; Moss, T.; Poortinga, G.; Mcarthur, G.; Pearson, R.; et al. MTOR-Dependent Regulation of Ribosomal Gene Transcription Requires S6K1 and Is Mediated by Phosphorylation of the Carboxy-Terminal Activation Domain of the Nucleolar Transcription Factor UBF†. Mol. Cell. Biol. 2004, 23, 8862–8877. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. MTOR-Dependent Activation of the Transcription Factor TIF-IA Links RRNA Synthesis to Nutrient Availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Michels, A.A.; Robitaille, A.M.; Buczynski-Ruchonnet, D.; Hodroj, W.; Reina, J.H.; Hall, M.N.; Hernandez, N. MTORC1 Directly Phosphorylates and Regulates Human MAF1. Mol. Cell. Biol. 2010, 30, 3749–3757. [Google Scholar] [CrossRef] [Green Version]
- Shor, B.; Wu, J.; Shakey, Q.; Toral-Barza, L.; Shi, C.; Follettie, M.; Yu, K. Requirement of the MTOR Kinase for the Regulation of Maf1 Phosphorylation and Control of RNA Polymerase III-Dependent Transcription in Cancer Cells. J. Biol. Chem. 2010, 285, 15380–15392. [Google Scholar] [CrossRef] [Green Version]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. MTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and BetaTRCP-Mediated Degradation of PDCD4 Promotes Protein Translation and Cell Growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 Tumor Suppressor Controls Insulin–PI3K Signaling via Regulation of IRS Proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Shah, O.J.; Wang, Z.; Hunter, T. Inappropriate Activation of the TSC/Rheb/MTOR/S6K Cassette Induces IRS1/2 Depletion, Insulin Resistance, and Cell Survival Deficiencies. Curr. Biol. 2004, 14, 1650–1656. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 Phosphorylation Impairs MTORC2 Complex Integrity and Inhibits Downstream Akt Signalling to Suppress Tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef]
- Bunney, T.D.; Katan, M. Phosphoinositide Signalling in Cancer: Beyond PI3K and PTEN. Nat. Rev. Cancer 2010, 10, 342–352. [Google Scholar] [CrossRef]
- Woscholski, R.; Finan, P.M.; Radley, E.; Totty, N.F.; Sterling, A.E.; Hsuan, J.J.; Waterfield, M.D.; Parker, P.J. Synaptojanin Is the Major Constitutively Active Phosphatidylinositol-3,4,5-Trisphosphate 5-Phosphatase in Rodent Brain. J. Biol. Chem. 1997, 272, 9625–9628. [Google Scholar] [CrossRef] [Green Version]
- Brognard, J.; Sierecki, E.; Gao, T.; Newton, A.C. PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms. Mol. Cell 2007, 25, 917–931. [Google Scholar] [CrossRef]
- Maira, S.M.; Galetic, I.; Brazil, D.P.; Kaech, S.; Ingley, E.; Thelen, M.; Hemmings, B.A. Carboxyl-Terminal Modulator Protein (CTMP), a Negative Regulator of PKB/Akt and v-Akt at the Plasma Membrane. Science 2001, 294, 374–380. [Google Scholar] [CrossRef]
- Foster, D.A. Regulation of MTOR by Phosphatidic Acid? Cancer Res. 2007, 67, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Bond, P. Regulation of MTORC1 by Growth Factors, Energy Status, Amino Acids and Mechanical Stimuli at a Glance. J. Int. Soc. Sports Nutr. 2016, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gulati, P.; Thomas, G. Nutrient Sensing in the MTOR/S6K1 Signalling Pathway. Biochem. Soc. Trans. 2007, 35, 236–238. [Google Scholar] [CrossRef]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino Acids Mediate MTOR/Raptor Signaling through Activation of Class 3 Phosphatidylinositol 3OH-Kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in Nutrient Response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to MTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK—A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-Activated/SNF1 Protein Kinases: Conserved Guardians of Cellular Energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic Acid-Mediated Mitogenic Activation of MTOR Signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef]
- Saito, M.; Kanfer, J. Phosphatidohydrolase Activity in a Solubilized Preparation from Rat Brain Particulate Fraction. Arch. Biochem. Biophys. 1975, 169, 318–323. [Google Scholar] [CrossRef]
- Singer, W.D.; Brown, H.A. Sternweis, P.C. regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu. Rev. Biochem. 1997, 66, 475–509. [Google Scholar] [CrossRef]
- Ballou, L.M.; Jiang, Y.-P.; Du, G.; Frohman, M.A.; Lin, R.Z. Ca(2+)- and Phospholipase D-Dependent and -Independent Pathways Activate MTOR Signaling. FEBS Lett. 2003, 550, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Park, I.-H.; Wu, A.-L.; Du, G.; Huang, P.; Frohman, M.A.; Walker, S.J.; Brown, H.A.; Chen, J. PLD1 Regulates MTOR Signaling and Mediates Cdc42 Activation of S6K1. Curr. Biol. CB 2003, 13, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Kam, Y.; Exton, J.H. Role of Phospholipase D1 in the Regulation of MTOR Activity by Lysophosphatidic Acid. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 311–319. [Google Scholar] [CrossRef]
- du Rusquec, P.; Blonz, C.; Frenel, J.S.; Campone, M. Targeting the PI3K/Akt/MTOR Pathway in Estrogen-Receptor Positive HER2 Negative Advanced Breast Cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920940939. [Google Scholar] [CrossRef]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/MTOR Signaling Pathway as a Therapeutic Target for Ovarian Cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef]
- Dobashi, Y.; Watanabe, Y.; Miwa, C.; Suzuki, S.; Koyama, S. Mammalian Target of Rapamycin: A Central Node of Complex Signaling Cascades. Int. J. Clin. Exp. Pathol. 2011, 4, 476–495. [Google Scholar]
- Fedele, C.G.; Ooms, L.M.; Ho, M.; Vieusseux, J.; O’Toole, S.A.; Millar, E.K.; Lopez-Knowles, E.; Sriratana, A.; Gurung, R.; Baglietto, L.; et al. Inositol Polyphosphate 4-Phosphatase II Regulates PI3K/Akt Signaling and Is Lost in Human Basal-like Breast Cancers. Proc. Natl. Acad. Sci. USA 2010, 107, 22231–22236. [Google Scholar] [CrossRef] [Green Version]
- Saal, L.H.; Gruvberger-Saal, S.K.; Persson, C.; Lövgren, K.; Jumppanen, M.; Staaf, J.; Jönsson, G.; Pires, M.M.; Maurer, M.; Holm, K.; et al. Recurrent Gross Mutations of the PTEN Tumor Suppressor Gene in Breast Cancers with Deficient DSB Repair. Nat. Genet. 2008, 40, 102–107. [Google Scholar] [CrossRef]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An Integrative Genomic and Proteomic Analysis of PIK3CA, PTEN, and AKT Mutations in Breast Cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [Green Version]
- Toker, A.; Cantley, L.C. Signalling through the Lipid Products of Phosphoinositide-3-OH Kinase. Nature 1997, 387, 673–676. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH Kinase as a Direct Target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The Evolution of Phosphatidylinositol 3-Kinases as Regulators of Growth and Metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Vanhaesebroeck, B. PI3K in Lymphocyte Development, Differentiation and Activation. Nat. Rev. Immunol. 2003, 3, 317–330. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA Oncogene in Human Cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Lee, H.W.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. PIK3CA Gene Is Frequently Mutated in Breast Carcinomas and Hepatocellular Carcinomas. Oncogene 2005, 24, 1477–1480. [Google Scholar] [CrossRef] [Green Version]
- Levine, D.A.; Bogomolniy, F.; Yee, C.J.; Lash, A.; Barakat, R.R.; Borgen, P.I.; Boyd, J. Frequent Mutation of the PIK3CA Gene in Ovarian and Breast Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 2875–2878. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.Y.; Wei, S.J.; Lin, Y.C.; Lung, J.C.; Chang, T.C.; Whang-Peng, J.; Liu, J.M.; Yang, D.M.; Yang, W.K.; Shen, C.Y. PIK3CA as an Oncogene in Cervical Cancer. Oncogene 2000, 19, 2739–2744. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [Green Version]
- Vadas, O.; Burke, J.E.; Zhang, X.; Berndt, A.; Williams, R.L. Structural Basis for Activation and Inhibition of Class I Phosphoinositide 3-Kinases. Sci. Signal. 2011, 4, re2. [Google Scholar] [CrossRef]
- Hao, Y.; Wang, C.; Cao, B.; Hirsch, B.M.; Song, J.; Markowitz, S.D.; Ewing, R.M.; Sedwick, D.; Liu, L.; Zheng, W.; et al. Gain of Interaction with IRS1 by P110α-Helical Domain Mutants Is Crucial for Their Oncogenic Functions. Cancer Cell 2013, 23, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Pang, H.; Flinn, R.; Patsialou, A.; Wyckoff, J.; Roussos, E.T.; Wu, H.; Pozzuto, M.; Goswami, S.; Condeelis, J.S.; Bresnick, A.R.; et al. Differential Enhancement of Breast Cancer Cell Motility and Metastasis by Helical and Kinase Domain Mutations of Class IA Phosphoinositide 3-Kinase. Cancer Res. 2009, 69, 8868–8876. [Google Scholar] [CrossRef] [Green Version]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, Regulation and Function of PKB/AKT—a Major Therapeutic Target. Biochim. Biophys. Acta BBA—Proteins Proteom. 2004, 1697, 3–16. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Felicioni, L.; Buttitta, F.; Lamba, S.; Cardone, L.; Rodolfo, M.; Scarpa, A.; Leenstra, S.; Frattini, M.; Barbareschi, M.; et al. AKT1(E17K) in Human Solid Tumours. Oncogene 2008, 27, 5648–5650. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Huang, L.; Dong, Y.; Tao, C.; Zhang, R.; Shao, H.; Shen, H. Effect of AKT1 (p. E17K) Hotspot Mutation on Malignant Tumorigenesis and Prognosis. Front. Cell Dev. Biol. 2020, 8, 996. [Google Scholar] [CrossRef]
- Landgraf, K.E.; Pilling, C.; Falke, J.J. Molecular Mechanism of an Oncogenic Mutation That Alters Membrane Targeting: Glu17Lys Modifies the PIP Lipid Specificity of the AKT1 PH Domain. Biochemistry 2008, 47, 12260–12269. [Google Scholar] [CrossRef] [Green Version]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A Transforming Mutation in the Pleckstrin Homology Domain of AKT1 in Cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef]
- Mancini, M.L.; Lien, E.C.; Toker, A. Oncogenic AKT1(E17K) Mutation Induces Mammary Hyperplasia but Prevents HER2-Driven Tumorigenesis. Oncotarget 2016, 7, 17301–17313. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-Y.; Di, M.-Y.; Yuan, J.-Q.; Shen, W.-X.; Zheng, D.-Y.; Chen, J.-Z.; Mao, C.; Tang, J.-L. The Prognostic Value of Phosphorylated Akt in Breast Cancer: A Systematic Review. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.-X.; Zhang, K.; Qiu, X.-S.; Zhou, M.; Li, W.-M. The Prognostic Value of Phosphorylated AKT Expression in Non-Small Cell Lung Cancer: A Meta-Analysis. PLoS ONE 2013, 8, e81451. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, J.; Zhai, X.; Fu, Z.; Tang, Z.; Liu, L.; Chen, M.; Zhu, Y. Prognostic Value of Phospho-Akt in Patients with Non-Small Cell Lung Carcinoma: A Meta-Analysis. Int. J. Cancer 2014, 135, 1417–1424. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Zhang, C.; Han, W.; Gao, X.-J.; Ma, J.; Hu, Y.-W.; Gu, X.; Ding, H.-Z.; Zhu, L.-X.; Liu, Q. P-Akt as a Potential Poor Prognostic Factor for Gastric Cancer: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 59878–59888. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Xu, L.; Tang, H.; Yang, Q.; Yi, X.; Fang, Y.; Zhu, Y.; Wang, Z. The Role of the PTEN/PI3K/Akt Pathway on Prognosis in Epithelial Ovarian Cancer: A Meta-Analysis. Oncologist 2014, 19, 528–535. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective Use of PI3K and MEK Inhibitors to Treat Mutant Kras G12D and PIK3CA H1047R Murine Lung Cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, P.J.A.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.L.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-Kinase Hyperactivation Results in Lapatinib Resistance That Is Reversed by the MTOR/Phosphatidylinositol 3-Kinase Inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [Google Scholar] [CrossRef] [Green Version]
- McKay, J.A.; Murray, L.J.; Curran, S.; Ross, V.G.; Clark, C.; Murray, G.I.; Cassidy, J.; McLeod, H.L. Evaluation of the Epidermal Growth Factor Receptor (EGFR) in Colorectal Tumours and Lymph Node Metastases. Eur. J. Cancer Oxf. Engl. 2002, 38, 2258–2264. [Google Scholar] [CrossRef]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal Growth Factor Receptor in Glioma: Signal Transduction, Neuropathology, Imaging, and Radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Pines, G. The ERBB Network: At Last, Cancer Therapy Meets Systems Biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Engelman, J.A. ERBB Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [Green Version]
- Castellanos, E.; Feld, E.; Horn, L. Driven by Mutations: The Predictive Value of Mutation Subtype in EGFR-Mutated Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 612–623. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, R.; Gerald, W.L.; Li, A.R.; Pan, Q.; Lal, P.; Ladanyi, M.; Chen, B. EGFR Gene Amplification in Breast Cancer: Correlation with Epidermal Growth Factor Receptor MRNA and Protein Expression and HER-2 Status and Absence of EGFR-Activating Mutations. Mod. Pathol. 2005, 18, 1027–1033. [Google Scholar] [CrossRef]
- Hsu, J.L.; Hung, M.-C. The Role of HER2, EGFR, and Other Receptor Tyrosine Kinases in Breast Cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Worby, C.A.; Dixon, J.E. PTEN. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal Structure of the PTEN Tumor Suppressor: Implications for Its Phosphoinositide Phosphatase Activity and Membrane Association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Sansal, I.; Sellers, W.R. The Biology and Clinical Relevance of the PTEN Tumor Suppressor Pathway. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 2954–2963. [Google Scholar] [CrossRef]
- Tamguney, T.; Stokoe, D. New Insights into PTEN. J. Cell Sci. 2007, 120, 4071–4079. [Google Scholar] [CrossRef] [Green Version]
- Cristofano, A.D.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten Is Essential for Embryonic Development and Tumour Suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Furnari, F.B.; Lin, H.; Huang, H.-J.S.; Cavenee, W.K. Growth Suppression of Glioma Cells by PTEN Requires a Functional Phosphatase Catalytic Domain. Proc. Natl. Acad. Sci. USA 1997, 94, 12479–12484. [Google Scholar] [CrossRef] [Green Version]
- Weng, L.-P.; Smith, W.M.; Dahia, P.L.M.; Ziebold, U.; Gil, E.; Lees, J.A.; Eng, C. PTEN Suppresses Breast Cancer Cell Growth by Phosphatase Activity-Dependent G1 Arrest Followed by Cell Death. Cancer Res. 1999, 59, 5808–5814. [Google Scholar]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; McGowan, E.M.; Nassif, N.T. PTEN/PTENP1: ‘Regulating the Regulator of RTK-Dependent PI3K/Akt Signalling’, New Targets for Cancer Therapy. Mol. Cancer 2018, 17, 1–14. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the Human PTEN Gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Soria, J.-C.; Lee, H.-Y.; Lee, J.I.; Wang, L.; Issa, J.-P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN Expression in Non-Small Cell Lung Cancer Could Be Related to Promoter Methylation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 1178–1184. [Google Scholar]
- Marsit, C.J.; Zheng, S.; Aldape, K.; Hinds, P.W.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. PTEN Expression in Non-Small-Cell Lung Cancer: Evaluating Its Relation to Tumor Characteristics, Allelic Loss, and Epigenetic Alteration. Hum. Pathol. 2005, 36, 768–776. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G. MicroRNA-21 (MiR-21) Represses Tumor Suppressor PTEN and Promotes Growth and Invasion in Non-Small Cell Lung Cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef]
- Murugan, A.K. MTOR: Role in Cancer, Metastasis and Drug Resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef]
- Sato, T.; Nakashima, A.; Guo, L.; Coffman, K.; Tamanoi, F. Single Amino-Acid Changes That Confer Constitutive Activation of MTOR Are Discovered in Human Cancer. Oncogene 2010, 29, 2746–2752. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Park, G.; Huuhtanen, J.; Lundgren, S.; Khajuria, R.K.; Hurtado, A.M.; Muñoz-Calleja, C.; Cardeñoso, L.; Gómez-García de Soria, V.; Chen-Liang, T.H.; et al. Somatic MTOR Mutation in Clonally Expanded T Lymphocytes Associated with Chronic Graft versus Host Disease. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A Diverse Array of Cancer-Associated MTOR Mutations Are Hyperactivating and Can Predict Rapamycin Sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming MTOR Resistance Mutations with a New-Generation MTOR Inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, Z.; Huang, J.; Cheng, S.; Du, H.; Che, G.; Peng, Y. Clinicopathological and Prognostic Significance of MTOR and Phosphorylated MTOR Expression in Patients with Esophageal Squamous Cell Carcinoma: A Systematic Review and Meta-Analysis. BMC Cancer 2016, 16, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Du, Z.; Zhu, Y.; Song, Y.; Pang, L.; Chen, Z. The Expression and Prognostic Impact of the PI3K/AKT/MTOR Signaling Pathway in Advanced Esophageal Squamous Cell Carcinoma. Technol. Cancer Res. Treat. 2018, 17, 1533033818758772. [Google Scholar] [CrossRef]
- Zhao, W.; Qiu, Y.; Kong, D. Class I Phosphatidylinositol 3-Kinase Inhibitors for Cancer Therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Workman, P.; Clarke, P.A.; Raynaud, F.I.; Montfort, R.L.M. van Drugging the PI3 Kinome: From Chemical Tools to Drugs in the Clinic. Cancer Res. 2010, 70, 2146–2157. [Google Scholar] [CrossRef] [Green Version]
- Yaguchi, S.; Fukui, Y.; Koshimizu, I.; Yoshimi, H.; Matsuno, T.; Gouda, H.; Hirono, S.; Yamazaki, K.; Yamori, T. Antitumor Activity of ZSTK474, a New Phosphatidylinositol 3-Kinase Inhibitor. JNCI J. Natl. Cancer Inst. 2006, 98, 545–556. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K Pathway in Cancer: Are We Making Headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Janku, F. Phosphoinositide 3-Kinase (PI3K) Pathway Inhibitors in Solid Tumors: From Laboratory to Patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Huw, L.-Y.; O’Brien, C.; Pandita, A.; Mohan, S.; Spoerke, J.M.; Lu, S.; Wang, Y.; Hampton, G.M.; Wilson, T.R.; Lackner, M.R. Acquired PIK3CA Amplification Causes Resistance to Selective Phosphoinositide 3-Kinase Inhibitors in Breast Cancer. Oncogenesis 2013, 2, e83. [Google Scholar] [CrossRef] [Green Version]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent Loss of PTEN Leads to Clinical Resistance to a PI(3)Kα Inhibitor. Nature 2015, 518, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.-I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.A.; Brandhuber, B.J. Crystal Structure of Human AKT1 with an Allosteric Inhibitor Reveals a New Mode of Kinase Inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the Potential of AKT Inhibitors as Anti-Cancer Treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Huck, B.R.; Mochalkin, I. Recent Progress towards Clinically Relevant ATP-Competitive Akt Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2838–2848. [Google Scholar] [CrossRef]
- Lu, S.; Li, S.; Zhang, J. Harnessing Allostery: A Novel Approach to Drug Discovery. Med. Res. Rev. 2014, 34, 1242–1285. [Google Scholar] [CrossRef]
- Menon, S.; Manning, B.D. Common Corruption of the MTOR Signaling Network in Human Tumors. Oncogene 2008, 27, S43–S51. [Google Scholar] [CrossRef] [Green Version]
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a New Antifungal Antibiotic. I. Taxonomy of the Producing Streptomycete and Isolation of the Active Principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef]
- Calne, R.Y.; Collier, D.S.; Lim, S.; Pollard, S.G.; Samaan, A.; White, D.J.; Thiru, S. Rapamycin for Immunosuppression in Organ Allografting. Lancet Lond. Engl. 1989, 2, 227. [Google Scholar] [CrossRef]
- Thomson, A.W.; Woo, J. Immunosuppressive Properties of FK-506 and Rapamycin. Lancet Lond. Engl. 1989, 2, 443–444. [Google Scholar] [CrossRef]
- Douros, J.; Suffness, M. New Antitumor Substances of Natural Origin. Cancer Treat. Rev. 1981, 8, 63–87. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Faivre, S.; Serova, M.; Raymond, E. MTORC1 Inhibitors: Is Temsirolimus in Renal Cancer Telling Us How They Really Work? Br. J. Cancer 2008, 99, 1197–1203. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Frias, M.A.; Chatterjee, A.; Yellen, P.; Foster, D.A. The Enigma of Rapamycin Dosage. Mol. Cancer Ther. 2016, 15, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Choo, A.Y.; Yoon, S.-O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin Differentially Inhibits S6Ks and 4E-BP1 to Mediate Cell-Type-Specific Repression of MRNA Translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Zhang, X.; Lu, T.; Zhang, X.; Wang, X.; Meng, B.; Zhang, H.; Wang, P.; Vose, J.M.; Chan, W.C.; et al. Inhibition of 4EBP Phosphorylation Mediates the Cytotoxic Effect of Mechanistic Target of Rapamycin Kinase Inhibitors in Aggressive B-Cell Lymphomas. Haematologica 2017, 102, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Costa, M.; Zollo, O.; Davis, C.; Feldman, M.E.; Testa, J.R.; Meyuhas, O.; Shokat, K.M.; Ruggero, D. Genetic Dissection of the Oncogenic MTOR Pathway Reveals Druggable Addiction to Translational Control via 4EBP-EIF4E. Cancer Cell 2010, 17, 249–261. [Google Scholar] [CrossRef] [Green Version]
- Mallya, S.; Fitch, B.A.; Lee, J.S.; So, L.; Janes, M.R.; Fruman, D.A. Resistance to MTOR Kinase Inhibitors in Lymphoma Cells Lacking 4EBP1. PLoS ONE 2014, 9, e88865. [Google Scholar] [CrossRef]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. MTORC2 Promotes Type I Insulin-like Growth Factor Receptor and Insulin Receptor Activation through the Tyrosine Kinase Activity of MTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The Translational Landscape of MTOR Signalling Steers Cancer Initiation and Metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Rojo, F.; Calvo, E.; Burris, H.; Judson, I.; Hazell, K.; Martinelli, E.; Ramon y Cajal, S.; Jones, S.; Vidal, L.; et al. Dose- and Schedule-Dependent Inhibition of the Mammalian Target of Rapamycin Pathway with Everolimus: A Phase I Tumor Pharmacodynamic Study in Patients with Advanced Solid Tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 1603–1610. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-Carboxamide-1-β-4-Ribofuranoside (AICAR) Enhances the Efficacy of Rapamycin in Human Cancer Cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frias, M.A.; Foster, D.A. Reciprocal Regulation of AMP-Activated Protein Kinase and Phospholipase D. J. Biol. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.-Q.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to MTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [Green Version]
- Oricchio, E.; Katanayeva, N.; Donaldson, M.C.; Sungalee, S.; Pasion, J.P.; Béguelin, W.; Battistello, E.; Sanghvi, V.R.; Jiang, M.; Jiang, Y.; et al. Genetic and Epigenetic Inactivation of SESTRIN1 Controls MTORC1 and Response to EZH2 Inhibition in Follicular Lymphoma. Sci. Transl. Med. 2017, 9, eaak9969. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Toscano, A.; Nickel, A.-C.; Li, G.; Kamp, M.A.; Muhammad, S.; Leprivier, G.; Fritsche, E.; Barker, R.A.; Sabel, M.; Steiger, H.-J.; et al. Rapalink-1 Targets Glioblastoma Stem Cells and Acts Synergistically with Tumor Treating Fields to Reduce Resistance against Temozolomide. Cancers 2020, 12, 3859. [Google Scholar] [CrossRef]
- La Manna, F.; De Menna, M.; Patel, N.; Karkampouna, S.; De Filippo, M.; Klima, I.; Kloen, P.; Beimers, L.; Thalmann, G.N.; Pelger, R.C.M.; et al. Dual-MTOR Inhibitor Rapalink-1 Reduces Prostate Cancer Patient-Derived Xenograft Growth and Alters Tumor Heterogeneity. Front. Oncol. 2020, 10, 1012. [Google Scholar] [CrossRef]
- Kuroshima, K.; Yoshino, H.; Okamura, S.; Tsuruda, M.; Osako, Y.; Sakaguchi, T.; Sugita, S.; Tatarano, S.; Nakagawa, M.; Enokida, H. Potential New Therapy of Rapalink-1, a New Generation Mammalian Target of Rapamycin Inhibitor, against Sunitinib-resistant Renal Cell Carcinoma. Cancer Sci. 2020, 111, 1607–1618. [Google Scholar] [CrossRef]
- Gazi, M.; Moharram, S.A.; Marhäll, A.; Kazi, J.U. The Dual Specificity PI3K/MTOR Inhibitor PKI-587 Displays Efficacy against T-Cell Acute Lymphoblastic Leukemia (T-ALL). Cancer Lett. 2017, 392, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.P.; Reynolds, C.P.; Kang, M.H. Modulation of Glucocorticoid Resistance in Pediatric T-Cell Acute Lymphoblastic Leukemia by Increasing BIM Expression with the PI3K/MTOR Inhibitor BEZ235. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Ewald, F.; Grabinski, N.; Grottke, A.; Windhorst, S.; Nörz, D.; Carstensen, L.; Staufer, K.; Hofmann, B.T.; Diehl, F.; David, K.; et al. Combined Targeting of AKT and MTOR Using MK-2206 and RAD001 Is Synergistic in the Treatment of Cholangiocarcinoma. Int. J. Cancer 2013, 133, 2065–2076. [Google Scholar] [CrossRef]
- Grabinski, N.; Ewald, F.; Hofmann, B.T.; Staufer, K.; Schumacher, U.; Nashan, B.; Jücker, M. Combined Targeting of AKT and MTOR Synergistically Inhibits Proliferation of Hepatocellular Carcinoma Cells. Mol. Cancer 2012, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Formisano, L.; Napolitano, F.; Rosa, R.; D’Amato, V.; Servetto, A.; Marciano, R.; De Placido, P.; Bianco, C.; Bianco, R. Mechanisms of Resistance to MTOR Inhibitors. Crit. Rev. Oncol. Hematol. 2020, 147, 102886. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Molecular Basis for Class Side Effects Associated with PI3K/AKT/MTOR Pathway Inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. [Google Scholar] [CrossRef]
Figure 1.
Domain structure of mTOR and components of mTORC1 and mTORC2. DEPTOR, DEP domain-containing mTOR-interacting protein; FAT, FRAP/ATM/TRRAP; FATC, FRAP/ATM/TRRAP/Carboxy terminal; FKBP-12, FK506-binding protein-12; FRB, FKBP12-rapamycin-binding; HEAT, Huntingtin/Elongation factor 3/A subunit of protein phosphatase-2A/TOR1; mLST8, mammalian lethal with SEC13 protein 8; mSIN1, mammalian stress-activated protein kinase interacting protein 1; mTOR, mechanistic target of rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; PRAS40, proline-rich AKT substrate 40 kDa; Protor, protein observed with RICTOR; RAPTOR, regulatory-associated protein of mTOR; RICTOR, rapamycin-insensitive companion of mTOR. The picture was modified from [53].
Figure 1.
Domain structure of mTOR and components of mTORC1 and mTORC2. DEPTOR, DEP domain-containing mTOR-interacting protein; FAT, FRAP/ATM/TRRAP; FATC, FRAP/ATM/TRRAP/Carboxy terminal; FKBP-12, FK506-binding protein-12; FRB, FKBP12-rapamycin-binding; HEAT, Huntingtin/Elongation factor 3/A subunit of protein phosphatase-2A/TOR1; mLST8, mammalian lethal with SEC13 protein 8; mSIN1, mammalian stress-activated protein kinase interacting protein 1; mTOR, mechanistic target of rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; PRAS40, proline-rich AKT substrate 40 kDa; Protor, protein observed with RICTOR; RAPTOR, regulatory-associated protein of mTOR; RICTOR, rapamycin-insensitive companion of mTOR. The picture was modified from [53].

Figure 2.
The overview of phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR signaling pathway and inhibitors.
Figure 2.
The overview of phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR signaling pathway and inhibitors.


{kind=link}
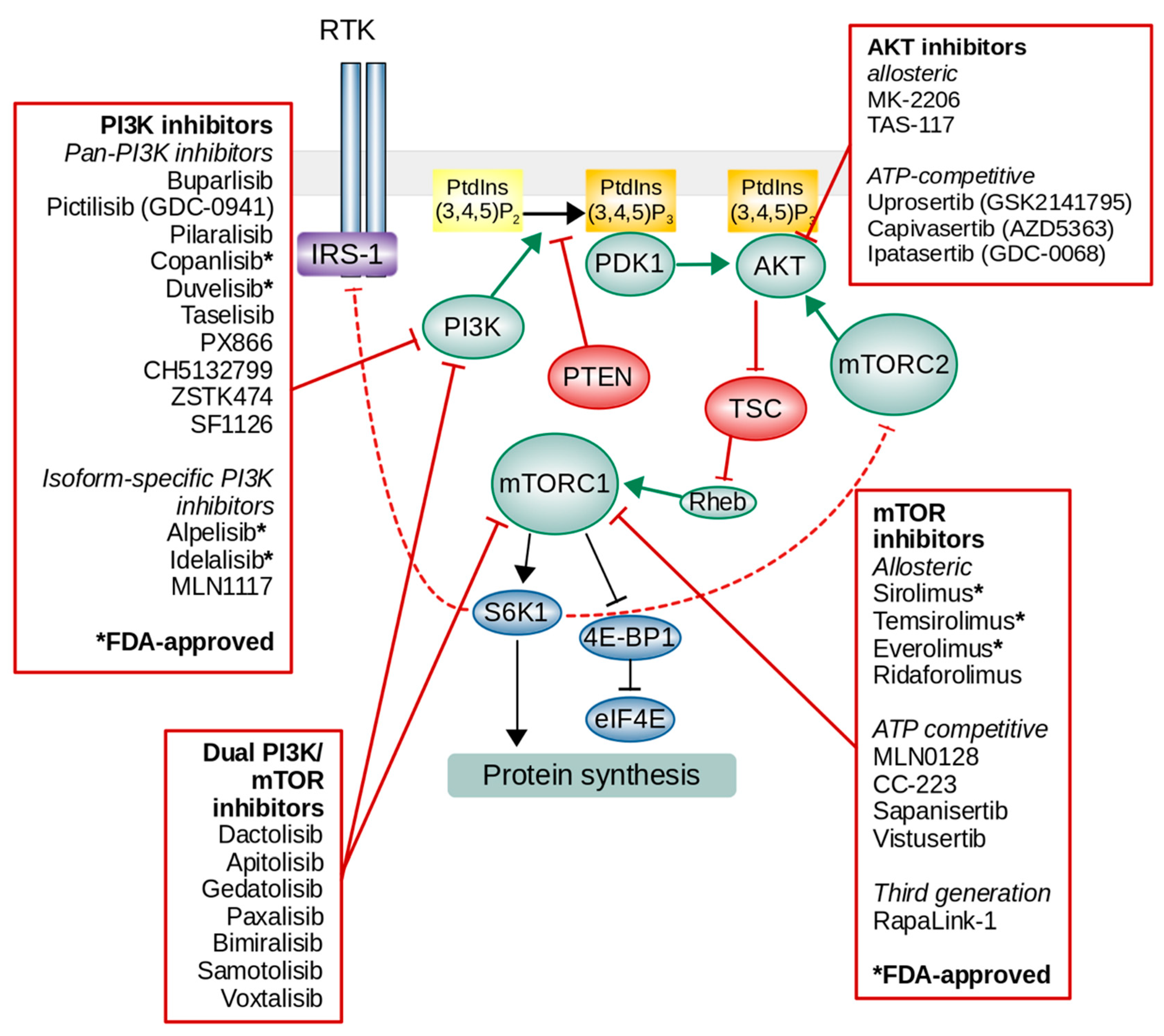{kind=link}
Table 1.
Genetic alterations of the PIK3CA gene in human cancers. HER: human epidermal growth factor receptor;
Table 1.
Genetic alterations of the PIK3CA gene in human cancers. HER: human epidermal growth factor receptor;
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 1/883 (0.11%) | 1/267 (0.37%) | 1/267 (0.37%) | 1/79 (1.27%) | - |
| Autonomic ganglia | 7/1605 (0.44%) | - | - | - | - |
| Biliary tract | 180/2501 (7.2%) | - | - | - | - |
| Bone | 44/1277 (3.45%) | - | - | - | - |
| Breast | 6559/22748 (28.83%) | 27/1492 (1.81%) | - | 97/1104 (8.79%) | 8/1104 (0.72%) |
| – ductal carcinoma | 1335/5021 (26.59%) | 0/315 (0%) | 0/315 (0%) | - | - |
| – ER-positive carcinoma | 619/1592 (38.88%) | - | - | - | - |
| – HER-positive carcinoma | 325/1455 (22.34%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 136/932 (14.59%) | - | - | - | - |
| – ER-PR-positive carcinoma | 211/656 (32.16%) | - | - | - | - |
| Central nervous system | 422/5741 (7.35%) | 13/1035 (1.26%) | - | 38/679 (5.45%) | - |
| Cervix | 347/2530 (13.72%) | 61/299 (20.4%) | - | 105/307 (34.2%) | 7/307 (2.28%) |
| Endometrium | 1277/4662 (27.39%) | 24/586 (4.1%) | - | 85/602 (14.12%) | - |
| – endometrioid carcinoma | 698/1964 (35.54%) | 19/530 (3.58%) | - | 71/545 (13.03%) | - |
| Eye | 1/372 (0.27%) | - | 1/80 (1.25%) | - | - |
| Gastrointestinal tract | 42/958 (4.38%) | - | - | - | - |
| Hematopoietic and Lymphoid | 110/8604 (1.28%) | 1/661 (0.15%) | - | 12/221 (5.43%) | 3/221 (1.36%) |
| Kidney | 68/3166 (2.15%) | 3/995 (0.3%) | 1/995 (0.1%) | 14/600 (2.33%) | 4/600 (0.67%) |
| Large intestine | 3234/22,967 (14.08%) | 1/718 (0.14%) | - | 25/610 (4.1%) | 4/610 (0.66%) |
| Liver | 177/3513 (5.04%) | 2/663 (0.3%) | - | 21/373 (5.63%) | 2/373 (0.54%) |
| Lung | 805/16,563 (4.86%) | 187/1006 (18.59%) | - | 329/1019 (32.29%) | 8/1019 (0.79%) |
| – adenocarcinoma | 295/7542 (3.91%) | 6/375 (1.6%) | - | 34/378 (8.99%) | 5/378 (1.32%) |
| – non-small cell carcinoma | 125/3728 (3.35%) | - | - | - | - |
| – squamous cell carcinoma | 230/2832 (8.12%) | 181/500 (36.2%) | - | 279/502 (55.58%) | - |
| Meninges | 100/478 (20.92%) | - | - | - | - |
| Esophagus | 314/3866 (8.12%) | 21/510 (4.12%) | - | 45/125 (36%) | 1/125 (0.8%) |
| Ovary | 531/4615 (11.51%) | 58/684 (8.48%) | - | 131/266 (29.25%) | - |
| – clear cell carcinoma | 254/763 (33.29%) | - | - | - | - |
| – endometrioid carcinoma | 79/327 (24.16%) | - | - | - | - |
| – serous carcinoma | 52/1634 (3.18%) | 58/568 (10.21%) | - | 131/266 (49.25%) | - |
| Pancreas | 122/3893 (3.13%) | 1/898 (0.11%) | - | 8/179 (4.47%) | 2/179 (1.12%) |
| Penis | 38/233 (16.31%) | - | - | - | - |
| Peritoneum | 7/188 (3.72%) | - | - | - | - |
| Pituitary | 12/467 (2.57%) | - | - | - | - |
| Pleura | 7/639 (1.1%) | 1/87 (1.15%) | 1/87 (1.15%) | - | - |
| Prostate | 296/4124 (7.18%) | 5/949 (0.53%) | - | 15/498 (3.01%) | - |
| Salivary gland | 89/707 (12.59%) | - | - | - | - |
| Skin | 378/4887 (7.73%) | 1/587 (0.17%) | 1/587 (0.17%) | 24/473 (5.07%) | |
| Small intestine | 24/434 (5.53%) | - | - | - | - |
| Soft tissue | 288/3718 (7.75%) | - | - | 12/263 (4.56%) | - |
| Stomach | 484/4584 (10.56%) | 13/472 2.75%) | - | 23/285 (8.07%) | - |
| Testis | 10/628 (1.59%) | 2/149 (1.34%) | - | - | - |
| Thymus | 8/442 (1.81%) | - | - | - | - |
| Thyroid | 210/5263 (3.99%) | 1/490 (0.2%) | - | 17/513 (3.31%) | 3/513 (0.58%) |
| Upper aerodigestive tract | 507/4898 (10.35%) | 75/520 (14.42%) | - | 142/522 (27.2%) | 3/522 (0.57%) |
| Urinary tract | 552/2732 (20.2%) | 11/399 (2.76%) | - | 39/408 (9.56%) | 3/408 (0.74%) |
| Vulva | 27/142 (19.01%) | - | - | - | - |
1 Mutated samples/total number of analyzed samples (%); -, no data. The data were derived from Catalogue Of Somatic Mutations in Cancer (COSMIC) v92 (released 27-AUG-20). ER, estrogen receptor; HER, human epidermal growth factor receptor; PR, progesterone receptor.
Table 2.
Genetic alterations of the AKT1 gene in human cancers.
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 0/790 (0%) | - | - | 9/79 (11.39%) | - |
| Autonomic ganglia | 0/1442 (0%) | - | - | - | - |
| Biliary tract | 20/1898 (1.05%) | - | - | - | - |
| Bone | 16/890 (1.8%) | - | - | - | - |
| Breast | 490/12,455 (3.93%) | 5/1492 (0.34%) | 2/1492 (0.13%) | 69/1104 (6.25%) | 3/1104 (0.27%) |
| – ductal carcinoma | 186/3743 (4.97%) | - | - | - | - |
| – ER-positive carcinoma | 71/1475 (4.81%) | - | - | - | - |
| – HER-positive carcinoma | 1/301 (0.33%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 9/553 (1.63%) | - | - | - | - |
| – ER-PR-positive carcinoma | 22/364 (6.04%) | - | - | - | - |
| Central nervous system | 15/4711 (0.32%) | 4/1035 (0.39%) | 1/1035 (0.1%) | 22/697 (3.16%) | 43/697 (6.17%) |
| Cervix | 11/901 (1.22%) | 1/299 (0.33%) | - | 15/307 (4.89%) | 7/307 (2.28%) |
| Endometrium | 58/1575 (3.68%) | 2/586 (0.34%) | - | 49/602 (8.14%) | 6/602 (1%) |
| – endometrioid carcinoma | 44/954 (4.61%) | 2/530 (0.38%) | - | 39/545 (7.16%) | 6/545 (1.1%) |
| Eye | 1/370 (0.27%) | - | - | - | - |
| Gastrointestinal tract | 0/1 (0%) | - | - | - | - |
| Hematopoietic and lymphoid | 33/8040 (0.41%) | - | 1/661 (0.15%) | 7/221 (3.17%) | 2/221 (0.9%) |
| Kidney | 10/3008 (0.33%) | - | 4/995 (0.4%) | 26/600 (4.33%) | 27/600 (4.5%) |
| Large intestine | 147/8066 (1.82%) | - | - | 18/610 (2.95%) | 44/610 (7.21%) |
| Liver | 31/2822 (1.1%) | 1/663 (0.15%) | 1/663 (0.15%) | 24/373 (6.43%) | 24/373 (6.43%) |
| Lung | 96/11467 (0.84%) | 10/1006 (0.99%) | 2/1006 (0.2%) | 89/1019 (8.73%) | 46/1019 (4.51%) |
| – adenocarcinoma | 41/6799 (0.6%) | 4/375 (1.07%) | - | 44/378 (11.64%) | 12/378 (3.17%) |
| – non-small cell carcinoma | 4/644 (0.62%) | - | - | - | - |
| – squamous cell carcinoma | 18/2138 (0.84%) | 6/500 (1.2%) | - | 37/502 (7.37%) | 26/502 (5.18%) |
| Meninges | 269/1766 (15.23%) | - | - | - | - |
| Esophagus | 14/2601 (0.54%) | 1/510 (0.2%) | - | 9/125 (7.2%) | - |
| Ovary | 32/2012 (1.59%) | 8/684 (1.17%) | - | 33/266 (12.41%) | 6/266 (2.26%) |
| Pancreas | 29/3155 (0.92%) | - | - | 10/179 (5.59%) | 8/179 (4.47%) |
| Penis | 1/101 (0.99%) | - | - | - | - |
| Peritoneum | 3/201 (1.49%) | - | - | - | - |
| Pituitary | 0/88 (0%) | - | - | - | - |
| Pleura | 1/511 (0.2%) | - | - | - | - |
| Prostate | 161/4202 (3.83%) | - | - | 22/498 (4.42%) | 5/498 (1%) |
| Salivary gland | 7/535 (1.31%) | - | - | - | - |
| Skin | 128/3730 (3.43%) | - | - | 21/473 (4.44%) | - |
| Small intestine | 2/315 (0.63%) | - | - | - | - |
| Soft tissue | 35/2336 (1.5%) | 1/264 (0.38%) | - | 28/263 (10.65%) | 2/263 (0.76%) |
| Stomach | 42/3628 (1.16%) | - | - | 15/285 (5.26%) | 10/285 (3.51%) |
| Testis | 2/602 (0.33%) | 1/149 (0.67%) | - | - | - |
| Thymus | 1/379 (0.26%) | - | - | - | - |
| Thyroid | 39/3657 (1.07%) | - | - | 24/513 (4.68%) | 1/513 (0.19%) |
| Upper aerodigestive tract | 27/2621 (1.03%) | 2/520 (0.38%) | - | 63/522 (12.07%) | 23/522 (4.41%) |
| Urinary tract | 47/1749 (2.69%) | - | 2/399 (0.5%) | 21/408 (5.15%) | 12/408 (2.94%) |
| Vulva | 0/64 (0%) | - | - | - | - |
1 Mutated samples/total number of sample analyzed (%); -, no data. The data were derived from COSMIC v92 (released 27-AUG-20). ER, estrogen receptor; HER, human epidermal growth factor receptor; PR, progesterone receptor.
Table 3.
Genetic alterations of EGFR in human cancers.
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 14/1112 (1.26%) | - | 1/267 (0.37%) | 4/79 (5.06%) | - |
| Breast | 304/10,708 (2.84%) | 14/1492 (0.94%) | 2/1492 (0.13%) | 65/1104 (5.89%) | - |
| – ductal carcinoma | 157/3346 (4.69%) | - | - | - | - |
| – ER-positive carcinoma | 8/466 (1.72%) | - | - | - | - |
| – HER-positive carcinoma | 33/609 (5.42%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 19/1182 (1.61%) | - | - | - | - |
| – ER-PR-positive carcinoma | 2/312 (0.64%) | - | - | - | - |
| Central nervous system | 581/6021 (9.65%) | 280/1035 (27.05%) | - | 142/697 (20.37%) | - |
| Cervix | 15/1038 (1.45%) | 6/299 (2.01%) | - | 27/307 (8.79%) | - |
| Endometrium | 91/1432 (6.35%) | 3/586 (0.51%) | - | 41/602 (6.81%) | - |
| – endometrioid carcinoma | 51/819 (6.23%) | 3/530 (0.57%) | - | 41/545 (7.52%) | - |
| Hematopoietic and lymphoid | 193/8438 (2.29%) | 1/661 (0.15%) | 1/661 (0.15%) | 18/221 (8.14%) | - |
| Kidney | 56/3554 (1.58%) | 2/995 (0.2%) | 3/995 (0.3%) | 20/600 (3.33%) | - |
| Large intestine | 345/9946 (3.47%) | 13/718 (1.81%) | 1/718 (0.14%) | 61/610 (10%) | 5/610 (0.82%) |
| Liver | 225/3015 (7.46%) | 2/663 (0.3%) | - | 20/373 (5.36%) | - |
| Lung | 26,499/99,694 (26.58%) | 53/1006 (5.27%) | 1/1006 (0.1%) | 148/1019 (14.52%) | - |
| – adenocarcinoma | 14,832/48,449 (30.61%) | 14/375 (3.73%) | - | 58/378 (15.34%) | - |
| – non-small cell carcinoma | 9016/35,920 (25.1%) | - | - | - | - |
| – squamous cell carcinoma | 413/5824 (7.09%) | 32/500 (6.4%) | 1/500 (0.2%) | 65/502 (12.95%) | - |
| Meninges | 150/394 (38.07%) | - | - | - | - |
| Esophagus | 134/3500 (3.83%) | 15/510 (2.94%) | 1/510 (0.2%) | 26/125 (20.08%) | - |
| Ovary | 89/2481 (3.59%) | 2/684 (0.29%) | 1/684 (0.15%) | 26/266 (9.77%) | - |
| Pancreas | 136/3681 (3.69%) | - | 1/898 (0.11%) | 4/179 (2.23%) | - |
| Prostate | 282/3949 (7.14%) | 1/949 (0.11%) | 1/949 (0.11%) | 23/498 (4.62%) | - |
| Skin | 242/3858 (6.27%) | 4/587 (0.68%) | - | 10/473 (2.11%) | - |
| Soft tissue | 66/2966 (2.23%) | 3/264 (1.14%) | 2/264 (0.76%) | 30/263 (11.41%) | - |
| Stomach | 146/3335 (4.38%) | 19/472 (4.03%) | 2/472 (0.42%) | 33/285 (11.58%) | 1/285 (0.35%) |
| Thyroid | 33/3623 (0.91%) | - | - | 27/513 (5.26%) | - |
| Upper aerodigestive tract | 153/4981 (3.07%) | 44/520 (8.46%) | 1/520 (0.19%) | 78/522 (14.94%) | - |
| Urinary tract | 51/1649 (3.09%) | 19/399 (4.76%) | 1/399 (0.25%) | 42/408 (10.29%) | - |
1 Affected samples/total number of samples analyzed (%); -, no data. The data were derived from COSMIC v92 (released 27-AUG-20). ER, estrogen receptor; HER, human epidermal growth factor receptor; PR, progesterone receptor.
Table 4.
Genetic alterations of PTEN in human cancers.
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 1/816 (0.12%) | - | - | 6/79 (7.59%) | 2/79 (2.53%) |
| Autonomic ganglia | 3/1404 (0.21%) | - | - | - | - |
| Biliary tract | 92/1922 (4.81%) | - | - | - | - |
| Bone | 11/889 (1.24%) | - | - | - | - |
| Breast | 567/10,325 (5.49%) | 1/1492 (0.07%) | 31/1492 (2.08%) | 21/1104 (1.9%) | 47/1104 (4.26%) |
| – ductal carcinoma | 214/2832 (7.56%) | - | - | - | - |
| – ER-positive carcinoma | 94/1477 (6.36%) | - | - | - | - |
| – HER-positive carcinoma | 11/275 (4%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 21/469 (4.48%) | - | - | - | - |
| – ER-PR-positive carcinoma | 12/403 (2.98%) | - | - | - | - |
| Central nervous system | 1040/7783 (13.36%) | - | 46/1035 (4.44%) | 19/697 (2.73%) | 146/679 (20.95%) |
| Cervix | 74/1598 (4.63%) | - | 12/299 (4.01%) | 13/307 (4.23%) | 21/307 (6.84%) |
| Endometrium | 1565/3944 (39.68%) | 1/586 (0.17%) | 19/586 (3.24%) | 50/602 (8.31) | 37/602 (6.15%) |
| – endometrioid carcinoma | 1170/2100 (55.71%) | 1/530 (0.19%) | 17/530 (3.21%) | 34/545 (6.24%) | 27/545 (4.95%) |
| Eye | 15/401 (3.74%) | - | - | - | - |
| Hematopoietic and lymphoid | 388/14,559 (2.67%) | - | - | 9/221 (4.07%) | 1/221 (0.45%) |
| Kidney | 132/3451 (3.82%) | - | 4/995 (0.4%) | 19/600 (3.17%) | 33/600 (5.5%) |
| Large intestine | 476/8913 (5.34%) | - | 11/718 (1.53%) | 14/610 (2.3%) | 46/610 (7.54%) |
| Liver | 184/2979 (6.18%) | 1/663 (0.15%) | 10/663 (1.51%) | 10/373 (2.68%) | 13/373 (3.49%) |
| Lung | 319/9054 (3.52%) | 1/1006 (0.1%) | 21/1006 (2.09%) | 28/1019 (2.75%) | 83/1019 (8.15%) |
| – adenocarcinoma | 94/4362 (2.15%) | - | 2/375 (0.53%) | 13/378 (3.44%) | 23/378 (6.08%) |
| – non-small cell carcinoma | 22/1521 (1.45%) | - | - | - | - |
| – squamous cell carcinoma | 113/1554 (7.27%) | 1/500 (0.2%) | 18/500 (3.6%) | 12/502 (2.39%) | 57/502 (11.35%) |
| Meninges | 30/513 (5.85%) | - | - | - | - |
| Esophagus | 63/2117 (2.98%) | - | 4/510 (0.78%) | 9/125 (7.2%) | 10/125 (8%) |
| Ovary | 141/2831 (4.98%) | 2/684 (0.29%) | 13/684 (1.9%) | 7/266 (2.63%) | 47/266 (17.67%) |
| Pancreas | 103/3192 (3.23%) | 1/898 (0.11%) | 1/898 (0.11%) | 11/179 (6.15%) | 4/179 (2.23%) |
| Penis | 2/74 (2.7%) | - | - | - | - |
| Peritoneum | 1/225 (0.44%) | - | - | - | - |
| Pituitary | 0/89 (0%) | - | - | - | - |
| Pleura | 2/530 (0.38%) | - | 1/87 (1.15%) | - | - |
| Prostate | 393/4517 (8.7%) | 1/949 (0.11%) | 71/949 (7.48%) | 16/498 (3.21%) | 91/498 (18.27%) |
| Salivary gland | 22/623 (3.53%) | - | - | - | - |
| Skin | 309/3679 (8.4%) | - | 28/587 (4.77%) | 17/473 (3.59%) | 30/473 (6.34%) |
| Small intestine | 2/326 (0.61%) | - | - | - | - |
| Soft tissue | 59/2514 (2.35%) | - | 14/264 (5.3%) | 5/263 (1.9%) | 15/263 (5.7%) |
| Stomach | 130/2980 (4.36%) | - | 15/472 (3.18%) | 7/285 (2.46%) | 26/285 (9.12%) |
| Testis | 7/639 (1.1%) | - | 1/149 (0.67%) | - | - |
| Thymus | 0/372 (0%) | - | - | - | - |
| Thyroid | 110/3936 (2.79%) | - | 1/490 (0.2%) | 25/513 (4.87%) | 9/513 (1.75%) |
| Upper aerodigestive tract | 126/3441 (3.66%) | - | 12/520 (2.31%) | 17/522 (3.26%) | 16/522 (3.07%) |
| Urinary tract | 45/1614 (2.79%) | 1/399 (0.25%) | 8/399 (2.01%) | 7/408 (1.72%) | 20/408 (4.9%) |
| Vulva | 12/157 (7.64%) | - | - | - | - |
1 Mutated samples/total number of sample analyzed (%); -, no data. The data were derived from COSMIC v92 (released 27-AUG-20).
Table 5.
Genetic alterations of MTOR in human cancers.
| Primary Tumor Tissues | Mutations (%) 1 | Copy Number Variations (%) 1 | Gene Expression (%) 1 | ||
|---|---|---|---|---|---|
| Gain | Loss | Over | Under | ||
| Adrenal gland | 0/658 (0%) | - | 4/267 (1.5%) | 1/79 (1.27%) | - |
| Autonomic ganglia | 0/1225 (0%) | - | - | - | - |
| Biliary tract | 51/1388 (3.67%) | - | - | - | - |
| Bone | 3/725 (0.41%) | - | - | - | - |
| Breast | 258/7666 (3.37%) | 1/1492 (0.07%) | 2/1492 (0.13%) | 39/1104 (3.53%) | 32/1104 (2.9%) |
| – ductal carcinoma | 120/2483 (4.83%) | - | - | - | - |
| – ER-positive carcinoma | 29/1363 (2.13%) | - | - | - | - |
| – HER-positive carcinoma | 8/205 (3.9%) | - | - | - | - |
| – basal (triple-negative) carcinoma | 5/372 (1.34%) | - | - | - | - |
| ER-PR-positive carcinoma | 6/308 (1.95%) | - | - | - | - |
| Central nervous system | 56/3499 (1.6%) | 1/1035 (0.1%) | - | 21/697 (3.01%) | 91/697 (13.06%) |
| Cervix | 21/388 (5.41%) | - | - | 18/307 (5.86%) | 5/307 (1.63%) |
| Endometrium | 103/988 (10.43%) | - | - | 27/602 (4.49%) | 9/602 (1.5%) |
| – endometrioid carcinoma | 82/649 (12.63%) | - | - | 23/545 (4.22%) | - |
| Eye | 3/179 (1.68%) | - | - | - | - |
| Hematopoietic and lymphoid | 118/6030 (1.96%) | - | - | 2/221 (0.9%) | 5/221 (2.26%) |
| Kidney | 165/2998 (5.5%) | - | - | 20/600 (3.33%) | 12/600 (2%) |
| Large intestine | 344/4526 (7.6%) | - | 1/718 (0.14%) | 16/610 (2.62%) | 35/610 (5.74%) |
| Liver | 175/2381 (7.35%) | - | - | 5/373 (1.34%) | - |
| Lung | 228/4930 (4.62%) | 3/1006 (0.3%) | 2/1006 (0.2%) | 44/1019 (4.32%) | 17/1019 (1.67%) |
| – adenocarcinoma | 126/2724 (4.63%) | 1/375 (0.27%) | 1/375 (0.27%) | 23/378 (6.08%) | 12/378 (3.17%) |
| – non-small cell carcinoma | 4/124 (3.23%) | - | - | - | - |
| – squamous cell carcinoma | 44/1004 (4.38%) | 1/500 (0.2%) | 1/500 (0.2%) | 11/502 (2.19%) | 5/502 (1%) |
| Meninges | 57/306 (18.63%) | - | - | - | - |
| Esophagus | 79/1673 (4.72%) | 1/510 (0.2%) | - | 5/125 (4%) | - |
| Ovary | 63/1396 (4.51%) | 1/684 (0.15%) | - | 26/266 (9.77%) | 11/266 (4.14%) |
| Pancreas | 83/2505 (3.31%) | - | - | 6/179 (3.35%) | 10/179 (5.59%) |
| Penis | 2/17 (11.76%) | - | - | - | - |
| Peritoneum | 1/43 (2.33%) | - | - | - | - |
| Pituitary | 0/89 (0%) | - | - | - | - |
| Pleura | 0/470 (0%) | - | - | - | - |
| Prostate | 217/3317 (6.54%) | - | 1/949 (0.11%) | 14/498 (2.81%) | - |
| Salivary gland | 7/410 (1.71%) | - | - | - | - |
| Skin | 236/2248 (10.5%) | 2/587 (0.34%) | - | 39/473 (8.25%) | 24/473 (5.07%) |
| Small intestine | 5/252 (1.98%) | - | - | - | - |
| Soft tissue | 18/1489 (1.21%) | - | - | - | - |
| Stomach | 124/1912 (6.49%) | 2/472 (0.42%) | - | 18/285 (6.32%) | 4/285 (1.4%) |
| Testis | 9/662 (1.36%) | - | - | - | - |
| Thymus | 0/173 (0%) | - | - | - | - |
| Thyroid | 42/2131 (1.97%) | - | - | 20/513 (3.9%) | 7/513 (1.36%) |
| Upper aerodigestive tract | 44/1836 (2.4%) | - | - | 26/522 (4.98%) | 5/522 (0.96%) |
| Urinary tract | 72/1209 (5.96%) | - | - | 34/408 (8.33%) | 2/408 (0.49%) |
| Vulva | 1/30 (3.33%) | - | - | - | - |
1 Mutated samples/total number of sample analyzed (%); -, no data. The data were derived from COSMIC v92 (released 27-AUG-20).
Table 6.
AKT inhibitors in phase III clinical trials 1.
| Inhibitor | Target | Condition or Disease | ClinicalTrials.gov Identifier |
|---|---|---|---|
| Ipatasertib and Paclitaxel | AKT and microtubules | Locally advanced or metastatic TNBC; locally advanced or metastatic HR+/HER2– breast adenocarcinoma | NCT03337724 Study start date: January 2018 Estimated study completion date: December 2021 |
| Ipatasertib and Abiraterone | AKT and CYP17 | Metastatic castrate-resistant prostate cancer | NCT03072238 Study start date: June 2017 Estimated study completion date: November 2023 |
| Ipatasertib, Palbociclib, and Fulvestrant | AKT, CDK4/6, and ER | HR+ and HER2– locally advanced unresectable or metastatic breast cancer | NCT04060862 Study start date: November 2019 Estimated study completion date: January 2026 |
| Ipatasertib, Paclitaxel, and Atezolizumab | AKT, microtubules, and PD-L1 | Locally advanced or metastatic TNBC | NCT04177108 Study start date: November 2019 Estimated study completion date: October 2025 |
| Ipatasertib and Fulvestrant | AKT and ER | Advanced HER2– and ER+ breast cancer | NCT04650581 Estimated study start date: December 2020 Estimated study completion date: December 2026 |
| Capivasertib and Paclitaxel | AKT and microtubules | Locally advanced or metastatic TNBC | NCT03997123 Study start date: June 2019 Estimated study completion date: January 2023 |
| Capivasertib and Abiraterone | AKT and CYP17 | Hormone-sensitive prostate cancer | NCT04493853 Study start date: July 2020 Estimated study completion date: November 2025 |
| Capivasertib and Fulvestrant | AKT and ER | Locally advanced (inoperable) or metastatic HR+/HER2– breast cancer | NCT04305496 Study start date: April 2020 Estimated study completion date: July 2024 |
1 Clinical trial data obtained from https://clinicaltrials.gov at 25 December 2020. CDK, cyclin-dependent kinase; HER2–, human epidermal growth factor receptor 2 negative; HR+, hormone receptor positive; ER+, estrogen receptor positive; TNBC, triple-negative breast cancer.
Table 7.
Allosteric mTOR inhibitors approved by the Food and Drug Administration (FDA) for human cancers.
Table 7.
Allosteric mTOR inhibitors approved by the Food and Drug Administration (FDA) for human cancers.
| Inhibitor (Trade Name) | Target | Indications | Approval Date |
|---|---|---|---|
| Sirolimus (Rapamune) | mTORC1 | Treatment of patients with lymphangioleiomyomatosis | August 2000 |
| Temsirolimus (Torisel) | mTORC1 | Treatment of advanced renal cell carcinoma | May 2007 |
| Everolimus (Afinitor) | mTORC1 |
| March 2009 August 2012 February 2016 |
Table 8.
Other mTOR inhibitors in Phase 2/3 clinical trials 1.
| Inhibitor | Target | Phase | Condition or Disease | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| Allosteric | ||||
| Ridaforolimus | mTORC1 | 3 | Metastatic soft-tissue sarcomas and metastatic bone sarcomas | NCT00538239 Study start date: October 2007 Study completion date: December 2012 |
| mTORC1 | 2 | Advanced sarcoma | NCT00093080 Study start date: October 2004 Study completion date: November 2008 | |
| mTORC1 | 2 | Relapsed or refractory hematologic malignancies | NCT00086125 Study start date: June 2004 Study completion date: June 2006 | |
| mTORC1 | 2 | Endometrial cancer | NCT00122343 Study start date: August 2005 Study completion date: January 2008 | |
| mTORC1 | 2 | Breast cancer | NCT00736970 Study start date: July 2008 Study completion date: May 2011 | |
| ATP-Competitive | ||||
| MLN0128 | mTORC1/2 | 2 | Metastatic castration-resistant prostate cancer | NCT02091531 Study start date: March 2014 Study completion date: October 2018 |
| mTORC1/2 | 2 | Metastatic anaplastic thyroid cancer | NCT02244463 Study start date: July 2015 Estimated study completion date: January 2022 | |
| CC-223 | mTORC1/2 | 1/2 | Multiple Myeloma Diffuse large B-cell lymphoma Glioblastoma multiforme Hepatocellular carcinoma non-small cell lung cancer Neuroendocrine tumors of non-pancreatic origin Hormone receptor-positive breast cancer | NCT01177397 Study start date: July 2010 Study completion date: December 2016 |
| Sapanisertib (TAK-228 or MLN0128) | mTORC1/2 | 2 | Estrogen receptor-positive breast cancer | NCT02988986 Study start date: April 2017 Study completion date: March 2019 |
| mTORC1/2 | 2 | Soft-tissue sarcoma | NCT02987959 Study start date: February 2017 Estimated study completion date: September 2020 | |
| mTORC1/2 | 2 | Locally advanced or metastatic bladder cancer | NCT03047213 Study start date: December 2016 Estimated study completion date: June 2021 | |
| Vistusertib (AZD2014) | mTORC1/2 | 2 | Progressive or symptomatic meningioma | NCT02831257 Study start date: August 2016 Study completion date: October 2020 |
| mTORC1/2 | 2 | Estrogen receptor positive breast cancer | NCT02216786 Study start date: January 2014 Estimated study completion date: July 2020 | |
| mTORC1/2 | 2 | Meningioma | NCT03071874 Study start date: October 2017 Estimated study completion date: July 2024 | |
| Dual PI3K/mTOR | ||||
| Dactolisib (BEZ235) | PI3K/mTOR | 2 | Pancreatic neuroendocrine tumors (pNET) | NCT01658436 Study start date: November 2012 Study completion date: July 2015 |
| Samotolisib (LY3023414) | PI3K/mTOR | 2 | Metastatic castration resistant prostate cancer | NCT02407054 Study start date: April 2015 Study completion date: April 2020 |
| Bimiralisib (PQR309) | PI3K/mTOR | 2 | Primary central nervous system lymphoma | NCT02669511 Study start date: November 2015 Study completion date: January 2018 |
| PI3K/mTOR | 2 | Relapsed or refractory lymphoma | NCT02249429 Study start date: May 2015 Study completion date: September 2018 | |
| Gedatolisib(PKI587) and Talazoparib | PI3K/mTORandPARP | 2 | Triple-Negative Breast Cancer | NCT03911973 Study start date: April 2019 Estimated study completion date: May 2022 |
| Apitolisib (GDC-0980) | PI3K/mTOR | 2 | Prostate Cancer | NCT01485861 Study start date: January 2019 Estimated study completion date: April 2021 |
| Voxtalisib (SAR245409, XL765) | PI3K/mTOR | 2 | Ovarian Cancer | NCT01936363 Study start date: September 2013 Study completion date: November 2017 |
| PI3K/mTOR | 2 | Lymphoma | NCT01403636 Study start date: October 2011 Study completion date: September 2014 | |
| Paxalisib (GDC-0084) | PI3K/mTOR | 2 | Glioblastoma | NCT03522298 Study start date: May 2018 Estimated study completion date: December 2020 |
1 Clinical trial data obtained from https://clinicaltrials.gov at 25 December 2020.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041743
AMA Style
Popova NV, Jücker M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. International Journal of Molecular Sciences. 2021; 22(4):1743. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041743
Chicago/Turabian StylePopova, Nadezhda V., and Manfred Jücker. 2021. "The Role of mTOR Signaling as a Therapeutic Target in Cancer" International Journal of Molecular Sciences 22, no. 4: 1743. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041743
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.