
Analysis of Stop Codons within Prokaryotic Protein-Coding Genes Suggests Frequent Readthrough Events

 ,
,  and
and
Abstract
:1. Introduction
2. Results
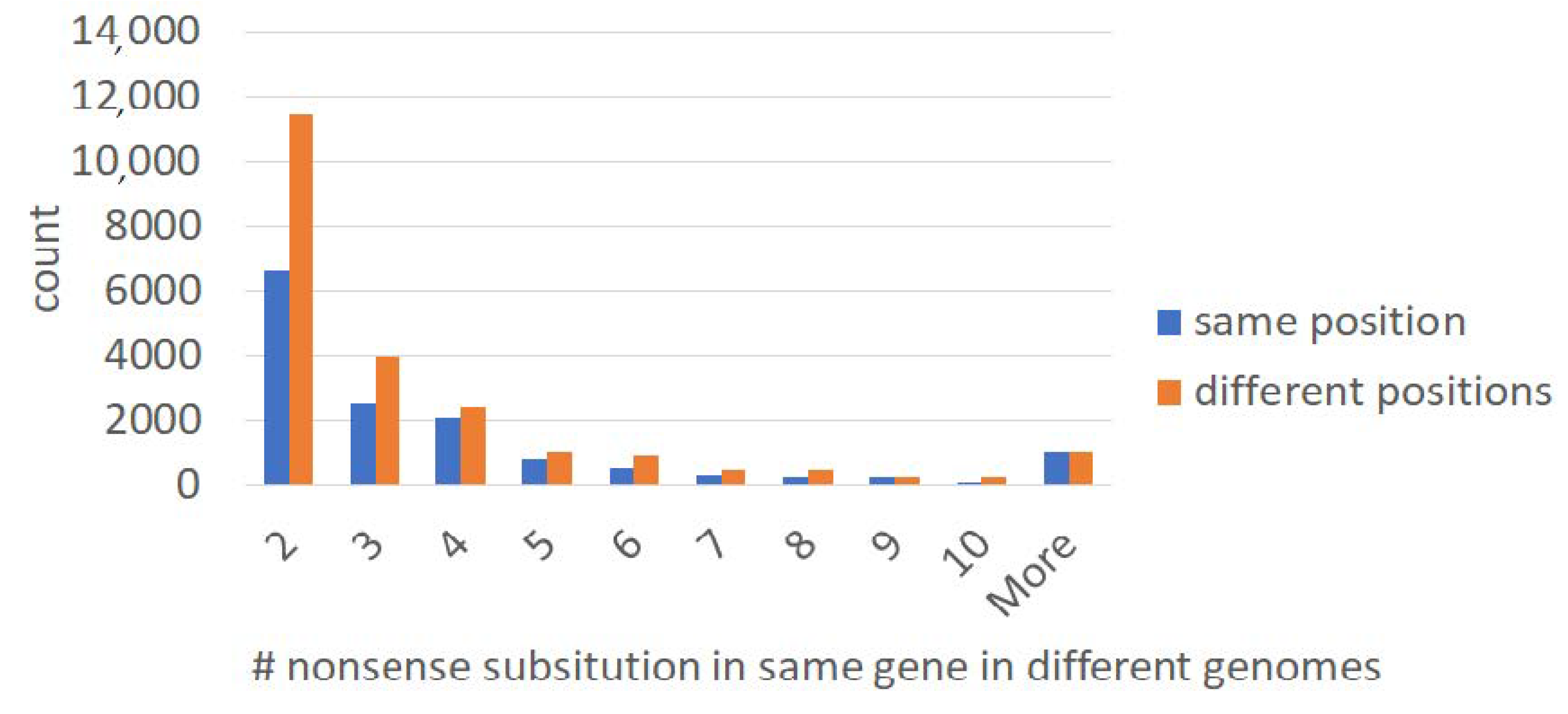
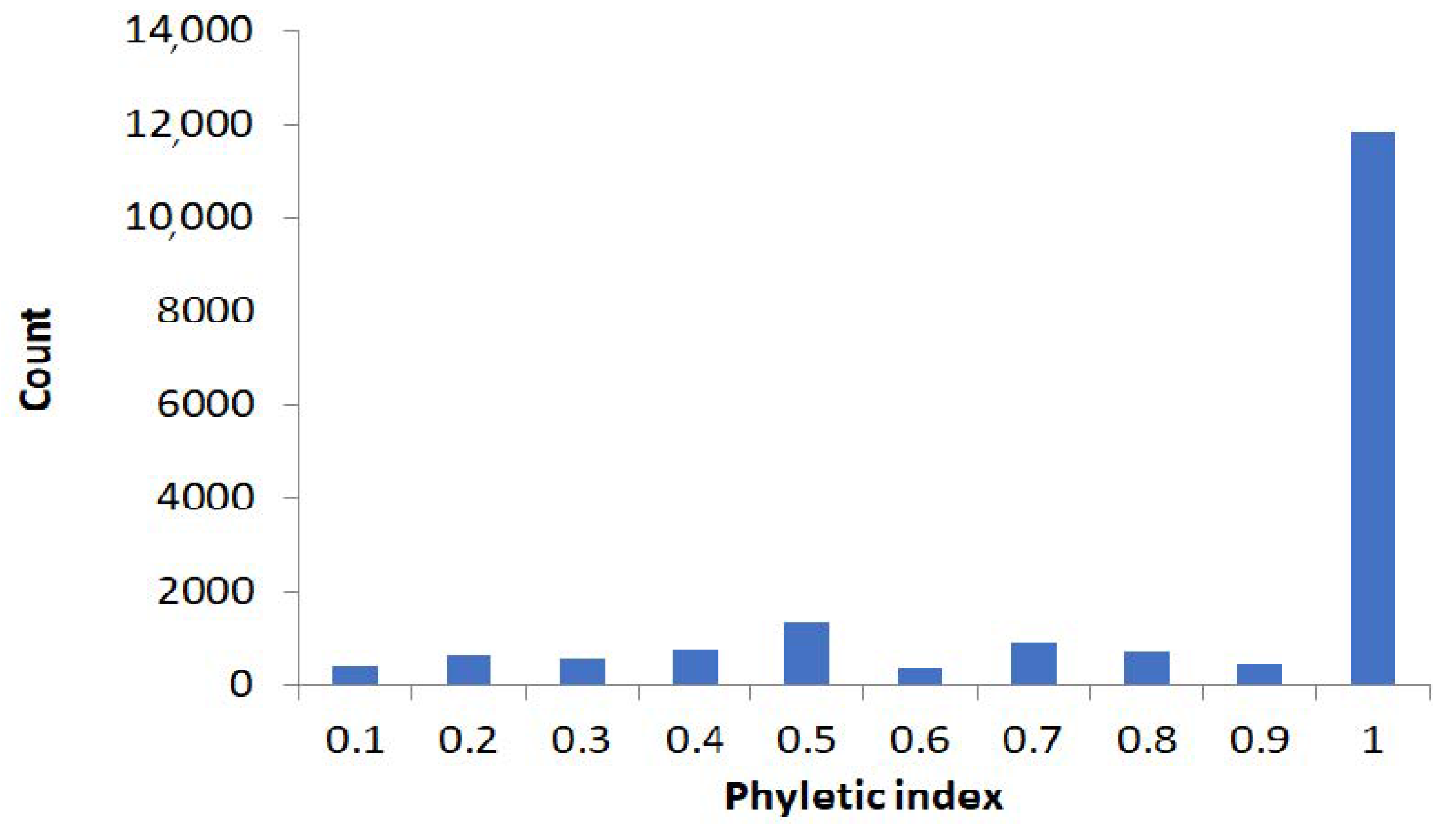
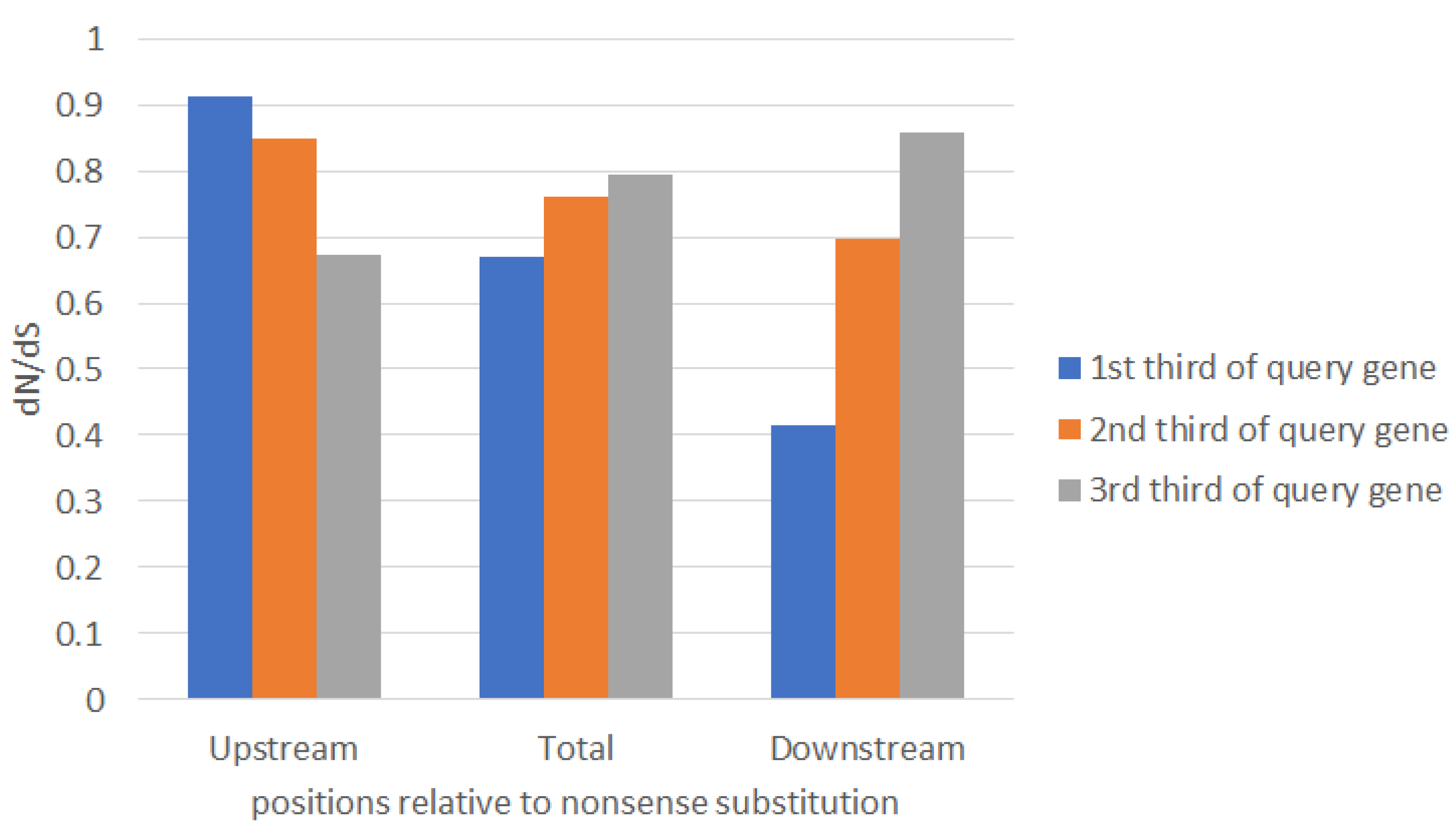
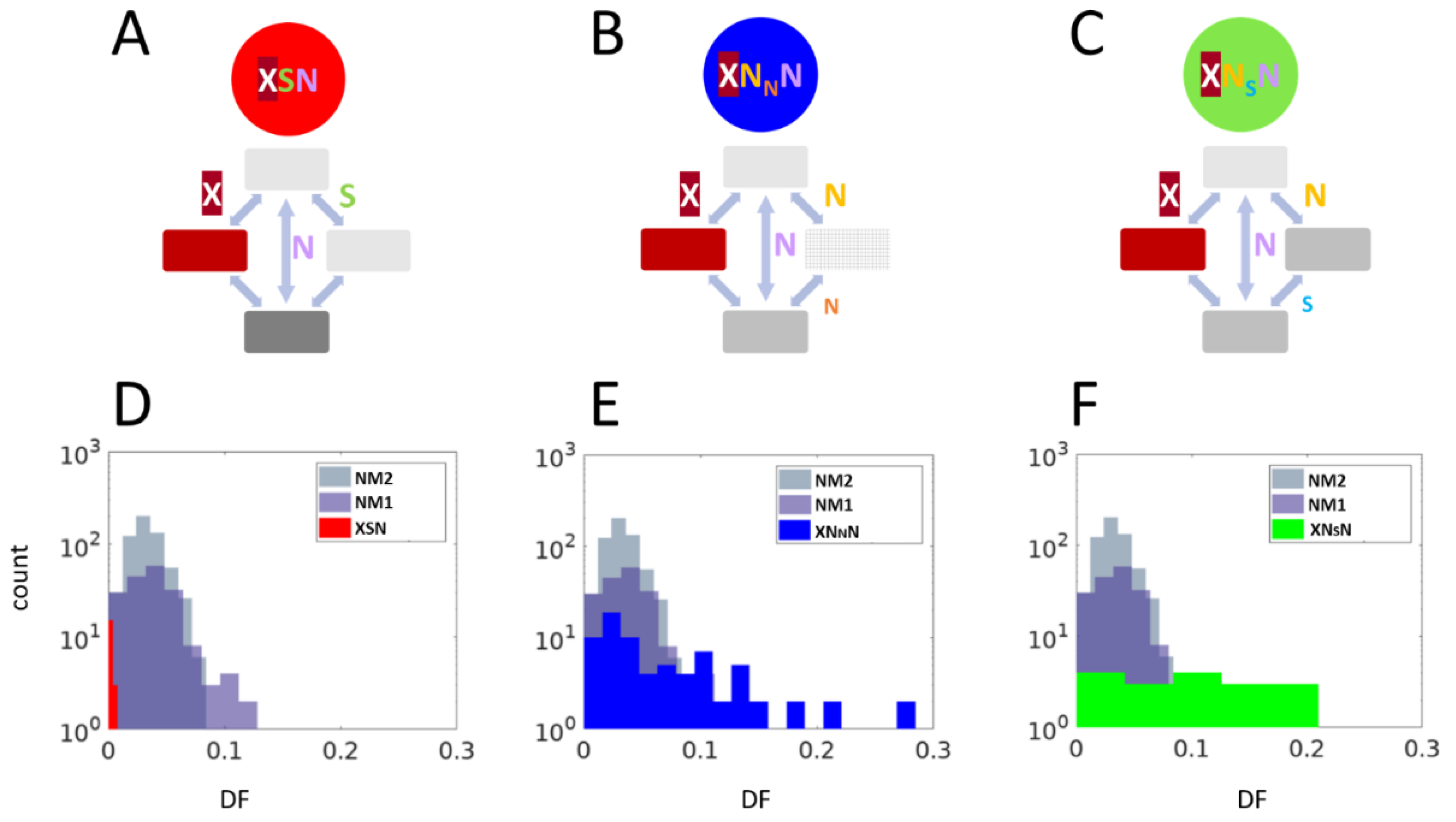
2.1. Large-Scale Study: Analysis of Complete Bacterial Genomes
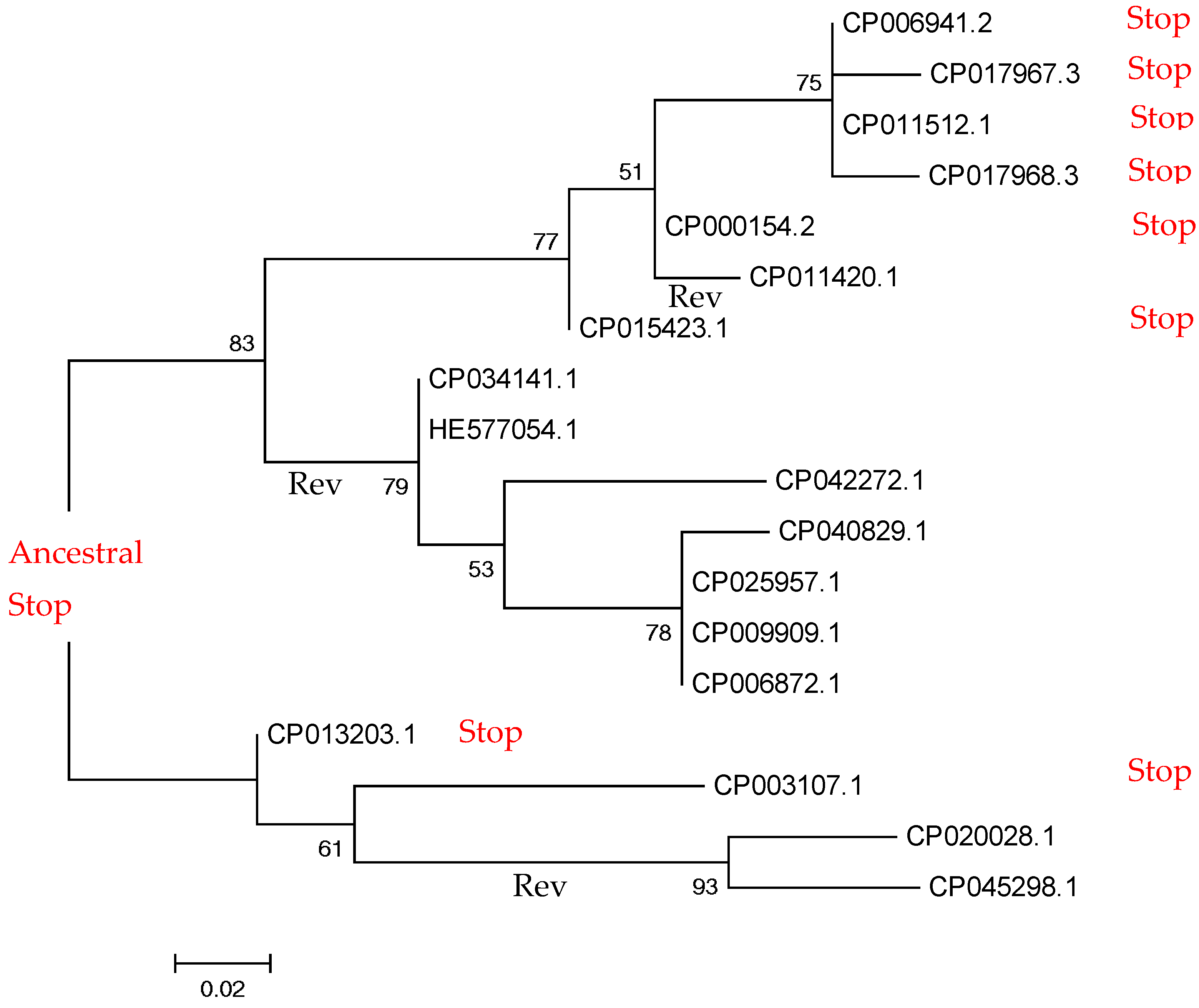
2.2. Small-Scale Study: Analysis of Stop Codons in Triplets of Species
3. Discussion
4. Materials and Methods
4.1. Identification of Nonsense Substitutions in Protein-Coding Genes
4.2. Phylogenetic Analysis and dN/dS Calculations
4.3. Double Substitution with Stop Intermediate Classification
4.4. Estimation of Selection on Double Substitutions with Stop Intermediates
4.5. Analysis of Stop Codons within Protein-Coding Genes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Koonin, E.V.; Rogozin, I.B. Getting positive about selection. Genome Biol. 2003, 4, 331. [Google Scholar] [CrossRef] [Green Version]
- Andersson, J.O.; Andersson, S.G. Insights into the evolutionary process of genome degradation. Curr. Opin. Genet. Dev. 1999, 9, 664–671. [Google Scholar] [CrossRef]
- Goodhead, I.; Darby, A.C. Taking the pseudo out of pseudogenes. Curr. Opin. Microbiol. 2015, 23, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Holt, K.E.; Thomson, N.R.; Wain, J.; Langridge, G.C.; Hasan, R.; Bhutta, Z.A.; Quail, M.A.; Norbertczak, H.; Walker, D.; Simmonds, M.; et al. Pseudogene accumulation in the evolutionary histories of Salmonella enterica serovars Paratyphi A and Typhi. BMC Genom. 2009, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Lerat, E.; Ochman, H. Recognizing the pseudogenes in bacterial genomes. Nucleic Acids Res. 2005, 33, 3125–3132. [Google Scholar] [CrossRef]
- Balakirev, E.S.; Ayala, F.J. Pseudogenes: Are they “junk” or functional DNA? Annu. Rev. Genet. 2003, 37, 123–151. [Google Scholar] [CrossRef] [Green Version]
- Schrimpe-Rutledge, A.C.; Jones, M.B.; Chauhan, S.; Purvine, S.O.; Sanford, J.A.; Monroe, M.E.; Brewer, H.M.; Payne, S.H.; Ansong, C.; Frank, B.C.; et al. Comparative omics-driven genome annotation refinement: Application across Yersiniae. PLoS ONE 2012, 7, e33903. [Google Scholar] [CrossRef]
- Mikkola, R.; Kurland, C.G. Selection of laboratory wild-type phenotype from natural isolates of Escherichia coli in chemostats. Mol. Biol. Evol. 1992, 9, 394–402. [Google Scholar]
- Bezerra, A.R.; Simoes, J.; Lee, W.; Rung, J.; Weil, T.; Gut, I.G.; Gut, M.; Bayes, M.; Rizzetto, L.; Cavalieri, D.; et al. Reversion of a fungal genetic code alteration links proteome instability with genomic and phenotypic diversification. Proc. Natl. Acad. Sci. USA 2013, 110, 11079–11084. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.; O’Donoghue, P.; Söll, D. Genetic code flexibility in microorganisms: Novel mechanisms and impact on physiology. Nat. Rev. Microbiol. 2015, 13, 707–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T. Adaptive translation as a mechanism of stress response and adaptation. Annu. Rev. Genet. 2013, 47, 121–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas de Pouplana, L.; Santos, M.A.; Zhu, J.H.; Farabaugh, P.J.; Javid, B. Protein mistranslation: Friend or foe? Trends Biochem. Sci. 2014, 39, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Javid, B.; Sorrentino, F.; Toosky, M.; Zheng, W.; Pinkham, J.T.; Jain, N.; Pan, M.; Deighan, P.; Rubin, E.J. Mycobacterial mistranslation is necessary and sufficient for rifampicin phenotypic resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 1132–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.W.; Zhu, J.H.; Li, H.; Cai, R.J.; Ealand, C.; Wang, X.; Chen, Y.X.; Kayani, M.U.; Zhu, T.F.; Moradigaravand, D.; et al. The essential mycobacterial amidotransferase GatCAB is a modulator of specific translational fidelity. Nat. Microbiol. 2016, 1, 16147. [Google Scholar] [CrossRef]
- Fan, Y.; Wu, J.; Ung, M.H.; De Lay, N.; Cheng, C.; Ling, J. Protein mistranslation protects bacteria against oxidative stress. Nucleic Acids Res. 2015, 43, 1740–1748. [Google Scholar] [CrossRef]
- Fredriksson, A.; Ballesteros, M.; Peterson, C.N.; Persson, O.; Silhavy, T.J.; Nystrom, T. Decline in ribosomal fidelity contributes to the accumulation and stabilization of the master stress response regulator sigmaS upon carbon starvation. Genes Dev. 2007, 21, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Evans, C.R.; Barber, K.W.; Banerjee, K.; Weiss, K.J.; Margolin, W.; Igoshin, O.A.; Rinehart, J.; Ling, J. Heterogeneity of stop codon readthrough in single bacterial cells and implications for population fitness. Mol. Cell 2017, 67, 826–836. [Google Scholar] [CrossRef] [Green Version]
- Osawa, S.; Jukes, T.H. Codon reassignment (codon capture) in evolution. J. Mol. Evol. 1989, 28, 271–278. [Google Scholar] [CrossRef]
- Ivanova, N.N.; Schwientek, P.; Tripp, H.J.; Rinke, C.; Pati, A.; Huntemann, M.; Visel, A.; Woyke, T.; Kyrpides, N.C.; Rubin, E.M. Stop codon reassignments in the wild. Science 2014, 344, 909–913. [Google Scholar] [CrossRef]
- Záhonová, K.; Kostygov, A.; Ševčíková, T.; Yurchenko, V.; Eliáš, M. An unprecedented non-canonical nuclear genetic code with all three termination codons reassigned as sense codons. Curr. Biol. 2016, 26, 2364–2369. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Wang, C.; Xu, J.; Schultz, M.D.; Schmitz, R.J.; Ecker, J.R.; Wang, L. Release factor one is nonessential in Escherichia coli. ACS Chem. Biol. 2012, 7, 1337–1344. [Google Scholar] [CrossRef]
- Li, L.; Linning, R.M.; Kondo, K.; Honda, B.M. Differential expression of individual suppressor tRNA(Trp) gene gene family members in vitro and in vivo in the nematode Caenorhabditis elegans. Mol. Cell Biol. 1998, 18, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Bienz, M.; Kubli, E. Wild-type tRNATyrG reads the TMV RNA stop codon, but Q base-modified tRNATyrQ does not. Nature 1981, 294, 188–190. [Google Scholar] [CrossRef]
- Hoesl, M.G.; Budisa, N. Recent advances in genetic code engineering in Escherichia coli. Curr. Opin. Biotechnol. 2012, 23, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Rother, M.; Krzycki, J.A. Selenocysteine, pyrrolysine, and the unique energy metabolism of methanogenic archaea. Archaea 2010, 2010, 453642. [Google Scholar] [CrossRef] [Green Version]
- Pasari, N.; Gupta, M.; Eqbal, D.; Yazdani, S.S. Genome analysis of Paenibacillus polymyxa A18 gives insights into the features associated with its adaptation to the termite gut environment. Sci. Rep. 2019, 9, 6091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olendzenski, L.; Gogarten, J.P. Evolution of genes and organisms: The tree/web of life in light of horizontal gene transfer. Ann. N. Y. Acad. Sci. 2009, 1178, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Brocchieri, L.; Karlin, S. Protein length in eukaryotic and prokaryotic proteomes. Nucleic Acids Res. 2005, 33, 3390–3400. [Google Scholar] [CrossRef] [Green Version]
- Kryazhimskiy, S.; Plotkin, J.B. The population genetics of dN/dS. PLoS Genet. 2008, 4, e1000304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogozin, I.B.; Spiridonov, A.N.; Sorokin, A.V.; Wolf, Y.I.; Jordan, I.K.; Tatusov, R.L.; Koonin, E.V. Purifying and directional selection in overlapping prokaryotic genes. Trends Genet. 2002, 18, 228–232. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Belinky, F.; Pavlenko, V.; Shabalina, S.A.; Kristensen, D.M.; Koonin, E.V. Evolutionary switches between two serine codon sets are driven by selection. Proc. Natl. Acad. Sci. USA 2016, 113, 13109–13113. [Google Scholar] [CrossRef] [Green Version]
- Belinky, F.; Rogozin, I.B.; Koonin, E.V. Selection on start codons in prokaryotes and potential compensatory nucleotide substitutions. Sci. Rep. 2017, 7, 12422. [Google Scholar] [CrossRef] [Green Version]
- Belinky, F.; Sela, I.; Rogozin, I.B.; Koonin, E.V. Crossing fitness valleys via double substitutions within codons. BMC Biol. 2019, 17, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belinky, F.; Babenko, V.N.; Rogozin, I.B.; Koonin, E.V. Purifying and positive selection in the evolution of stop codons. Sci. Rep. 2018, 8, 9260. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Pavlov, Y.I.; Bebenek, K.; Matsuda, T.; Kunkel, T.A. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat. Immunol. 2001, 2, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Gordenin, D.A. Clusters of multiple mutations: Incidence and molecular mechanisms. Annu. Rev. Genet. 2015, 49, 243–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.M.; Ferec, C.; Cooper, D.N. Complex multiple-nucleotide substitution mutations causing human inherited disease reveal novel insights into the action of translesion synthesis DNA polymerases. Hum. Mutat. 2015, 36, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.G.; Kurland, C.G. Codon preferences in free-living microorganisms. Microbiol. Rev. 1990, 54, 198–210. [Google Scholar] [CrossRef]
- Eggertsson, G.; Soll, D. Transfer ribonucleic acid-mediated suppression of termination codons in Escherichia coli. Microbiol. Rev. 1988, 52, 354–374. [Google Scholar] [CrossRef]
- Parker, J. Errors and alternatives in reading the universal genetic code. Microbiol. Rev. 1989, 53, 273–298. [Google Scholar] [CrossRef]
- Roth, J.R. UGA nonsense mutations in Salmonella typhimurium. J. Bacteriol. 1970, 102, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Karow, M.L.; Rogers, E.J.; Lovett, P.S.; Piggot, P.J. Suppression of TGA mutations in the Bacillus subtilis spoIIR gene by prfB mutations. J. Bacteriol. 1998, 180, 4166–4170. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; Tharp, J.M.; Liu, W.R. Pyrrolysyl-tRNA synthetase: An ordinary enzyme but an outstanding genetic code expansion tool. Biochim. Biophys. Acta 2014, 1844, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotini, S.B.; Peske, F.; Rodnina, M.V. Partitioning between recoding and termination at a stop codon-selenocysteine insertion sequence. Nucleic Acids Res. 2015, 43, 6426–6438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Flores, J.N.; Shetty, S.P.; Dubey, A.; Copeland, P.R. The molecular biology of selenocysteine. Biomol. Concepts 2013, 4, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Serio, T.R.; Lindquist, S.L. [PSI+]: An epigenetic modulator of translation termination efficiency. Annu. Rev. Cell Dev. Biol. 1999, 15, 661–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeling, K.M.; Lanier, J.; Du, M.; Salas-Marco, J.; Gao, L.; Kaenjak-Angeletti, A.; Bedwell, D.M. Leaky termination at premature stop codons antagonizes nonsense-mediated mRNA decay in S. cerevisiae. RNA 2004, 10, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Kramarski, L.; Arbely, E. Translational read-through promotes aggregation and shapes stop codon identity. Nucleic Acids Res. 2020, 48, 3747–3760. [Google Scholar] [CrossRef]
- Kondrashov, F.A.; Rogozin, I.B.; Wolf, Y.I.; Koonin, E.V. Selection in the evolution of gene duplications. Genome Biol. 2002, 3, RESEARCH0008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogozin, I.B. Complexity of gene expression evolution after duplication: Protein dosage rebalancing. Genet. Res. Int. 2014, 2014, 516508. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhou, J.; Omelchenko, M.V.; Beliaev, A.S.; Venkateswaran, A.; Stair, J.; Wu, L.; Thompson, D.K.; Xu, D.; Rogozin, I.B.; et al. Transcriptome dynamics of Deinococcus radiodurans recovering from ionizing radiation. Proc. Natl. Acad. Sci. USA 2003, 100, 4191–4196. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S. Positive and negative regulators of the metallothionein gene (review). Mol. Med. Rep. 2015, 12, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojo, D.; Rodriguez, D.; Wei, F.; Bane, A.; Tang, D. Downregulation of CYB5D2 is associated with breast cancer progression. Sci. Rep. 2019, 9, 6624. [Google Scholar] [CrossRef]
- Havis, E.; Duprez, D. EGR1 transcription factor is a multifaceted regulator of matrix production in tendons and other connective tissues. Int. J. Mol. Sci. 2020, 21, 1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peredo, E.L.; Cardon, Z.G. Shared up-regulation and contrasting down-regulation of gene expression distinguish desiccation-tolerant from intolerant green algae. Proc. Natl. Acad. Sci. USA 2020, 117, 17438–17445. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Gertz, E.M.; Baranov, P.V.; Poliakov, E.; Schaffer, A.A. Genome-wide changes in protein translation efficiency are associated with autism. Genome Biol. Evol. 2018, 10, 1902–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolowski, M.B. Functional testing of ASD-associated genes. Proc. Natl. Acad. Sci. USA 2020, 117, 26–28. [Google Scholar] [CrossRef]
- Ji, X.; Kember, R.L.; Brown, C.D.; Bucan, M. Increased burden of deleterious variants in essential genes in autism spectrum disorder. Proc. Natl. Acad. Sci. USA 2016, 113, 15054–15059. [Google Scholar] [CrossRef] [Green Version]
- Bobay, L.M.; Touchon, M.; Rocha, E.P. Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 12127–12132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czajkowski, R. May the phage be with you? Prophage-like elements in the genomes of soft rot Pectobacteriaceae: Pectobacterium spp. and Dickeya spp. Front. Microbiol. 2019, 10, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Gordon, E.; Shean, R.C.; Idle, A.; Deng, X.; Greninger, A.L.; Delwart, E. CrAssphage and its bacterial host in cat feces. Sci. Rep. 2021, 11, 815. [Google Scholar] [CrossRef]
- Baranov, P.V.; Gesteland, R.F.; Atkins, J.F. P-site tRNA is a crucial initiator of ribosomal frameshifting. RNA 2004, 10, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lainé, S.; Thouard, A.; Komar, A.A.; Rossignol, J.M. Ribosome can resume the translation in both +1 or −1 frames after encountering an AGA cluster in Escherichia coli. Gene 2008, 412, 95–101. [Google Scholar] [CrossRef]
- Kondrashov, A.S.; Rogozin, I.B. Context of deletions and insertions in human coding sequences. Hum. Mutat. 2004, 23, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Zhang, J. A simple method for estimating the strength of natural selection on overlapping genes. Genome Biol. Evol. 2014, 7, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, D.M.; Wolf, Y.I.; Koonin, E.V. ATGC database and ATGC-COGs: An updated resource for micro- and macro-evolutionary studies of prokaryotic genomes and protein family annotation. Nucleic Acids Res. 2017, 45, D210–D218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, J.O.; Andersson, S.G. Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol. Biol. Evol. 2001, 18, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Ejigu, G.F.; Jung, J. Review on the computational genome annotation of sequences obtained by next-generation sequencing. Biology 2020, 9, 295. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dN/dS | dN | dS | p Value | |
|---|---|---|---|---|
| TAA | 0.7444 | 0.0023 | 0.0031 | <0.001 |
| TAG | 0.8153 | 0.0023 | 0.0029 | <0.001 |
| TGA | 0.6602 | 0.0028 | 0.0043 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belinky, F.; Ganguly, I.; Poliakov, E.; Yurchenko, V.; Rogozin, I.B. Analysis of Stop Codons within Prokaryotic Protein-Coding Genes Suggests Frequent Readthrough Events. Int. J. Mol. Sci. 2021, 22, 1876. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041876
Belinky F, Ganguly I, Poliakov E, Yurchenko V, Rogozin IB. Analysis of Stop Codons within Prokaryotic Protein-Coding Genes Suggests Frequent Readthrough Events. International Journal of Molecular Sciences. 2021; 22(4):1876. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041876
Chicago/Turabian StyleBelinky, Frida, Ishan Ganguly, Eugenia Poliakov, Vyacheslav Yurchenko, and Igor B. Rogozin. 2021. "Analysis of Stop Codons within Prokaryotic Protein-Coding Genes Suggests Frequent Readthrough Events" International Journal of Molecular Sciences 22, no. 4: 1876. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041876