
Loss of FADD and Caspases Affects the Response of T-Cell Leukemia Jurkat Cells to Anti-Cancer Drugs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
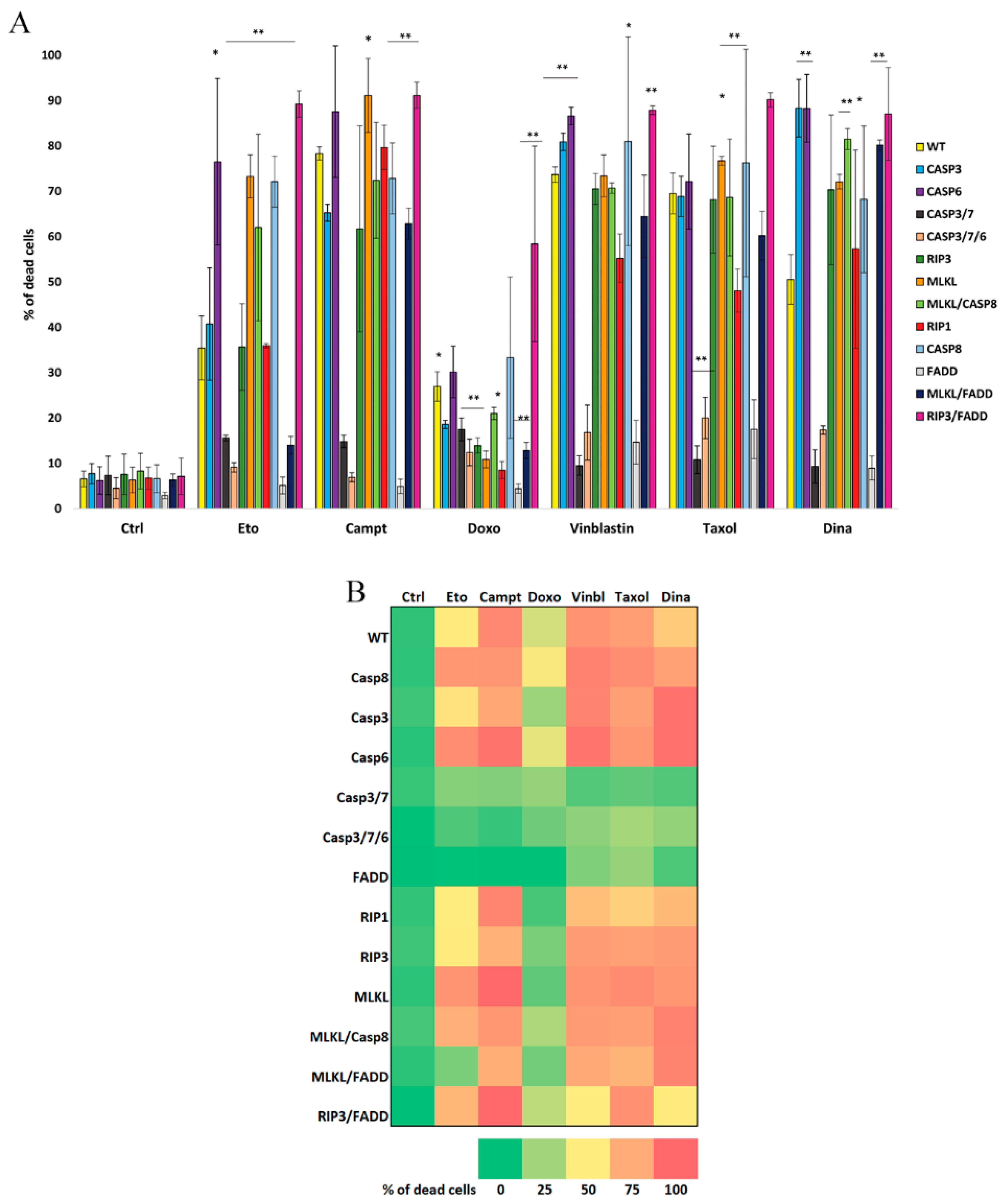
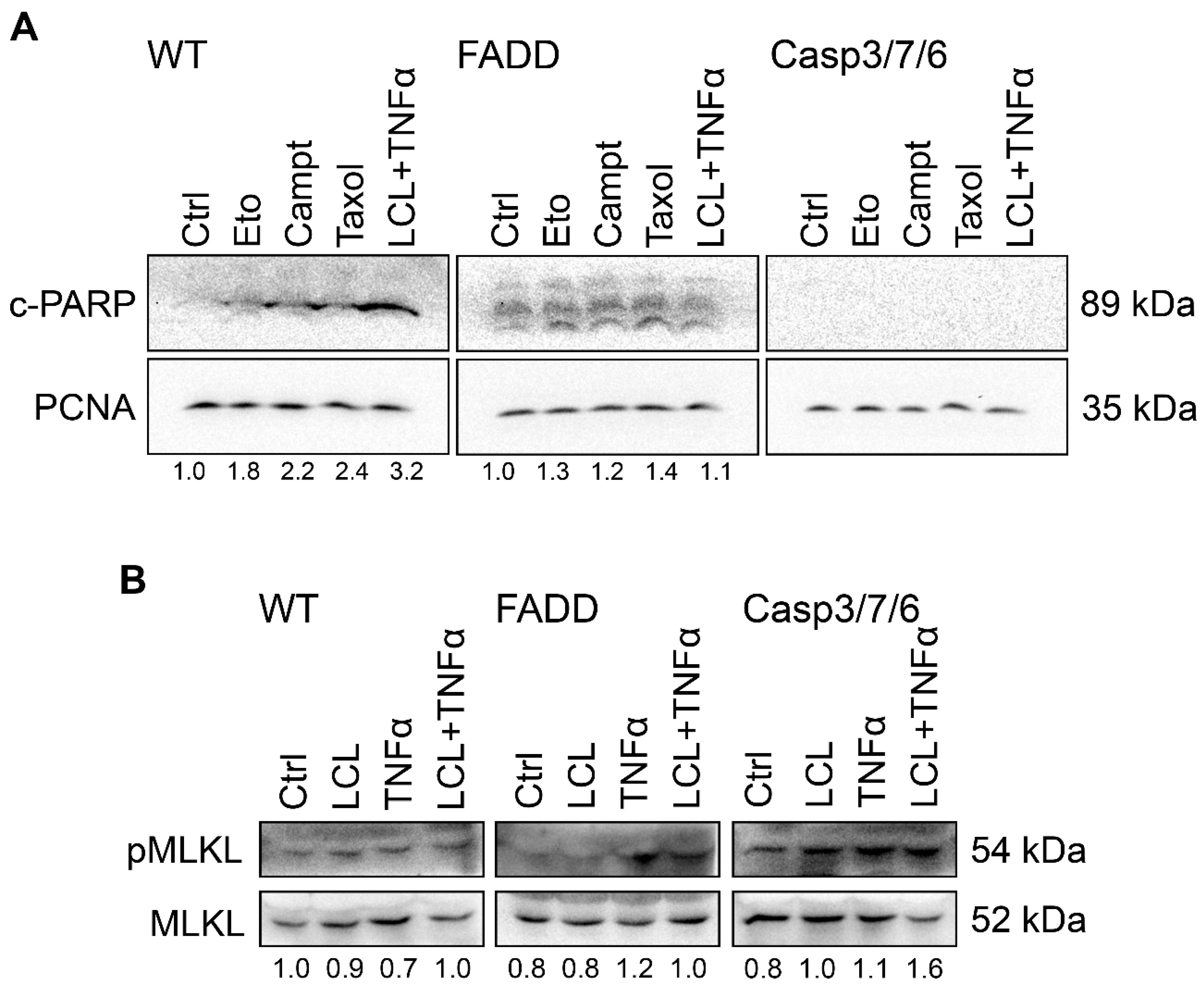
2.1. FADD and Executioner Caspases Are Indispensable for the Cell Death upon Anti-Cancer Drugs Treatment
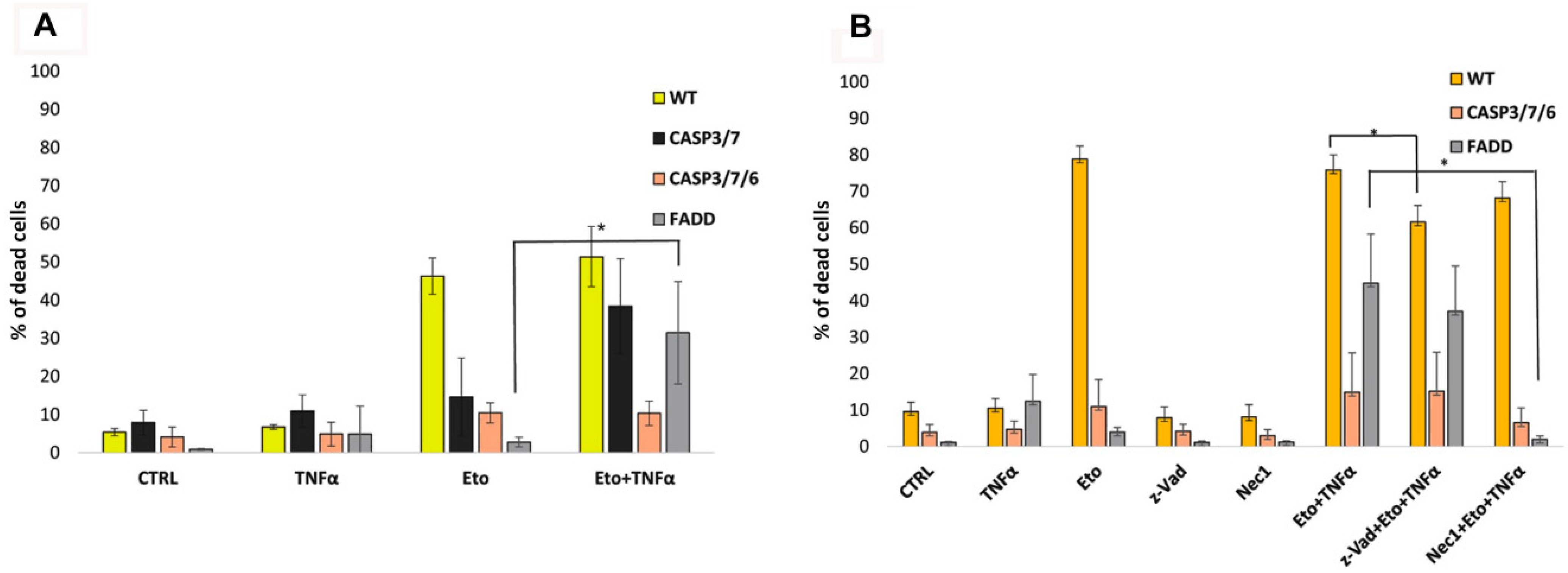
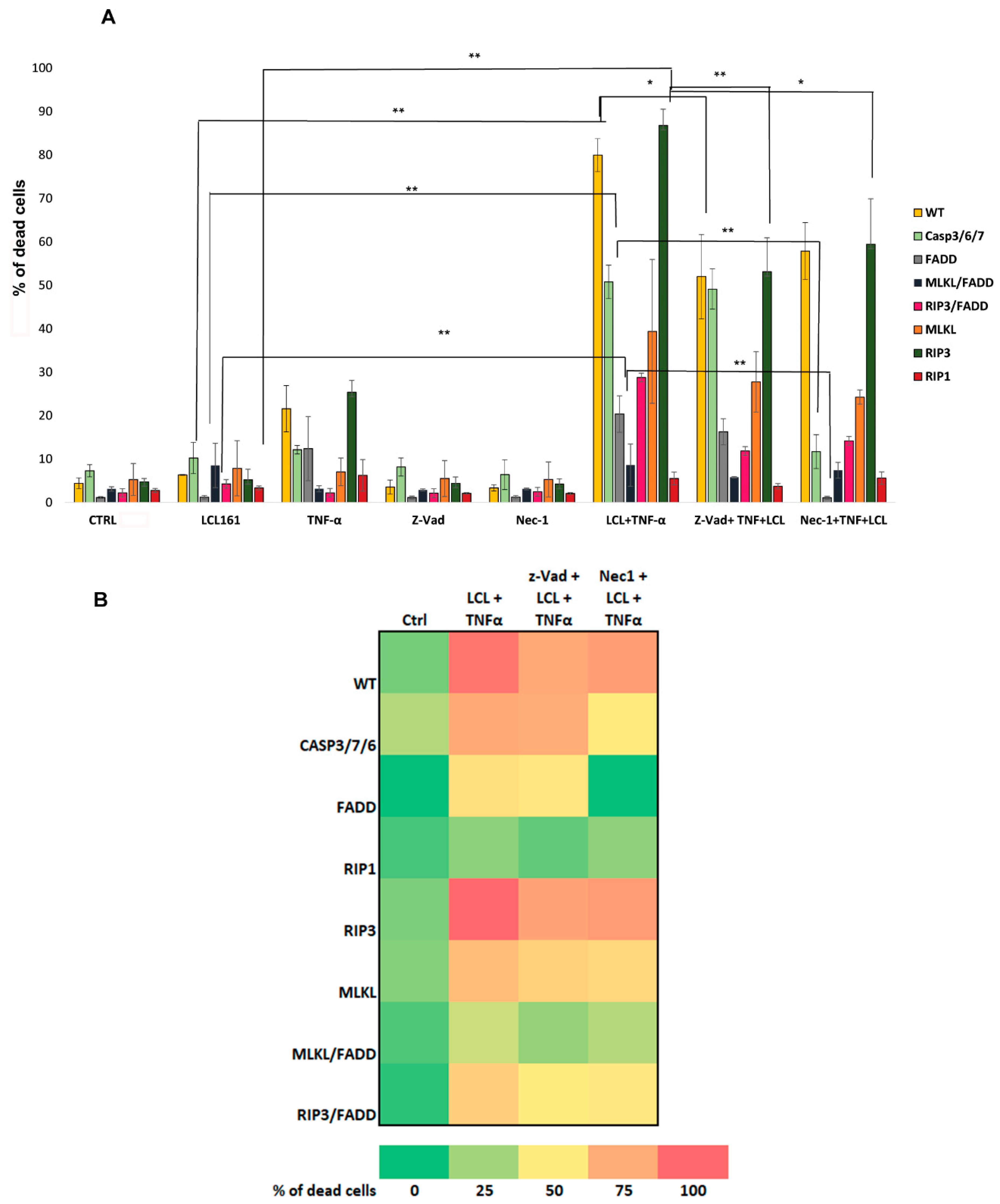
2.2. TNF-α and Smac Mimetic Overcome Drug Resistance of CASP 3/7/6-, CASP3/7-, and FADD-Deficient Cells
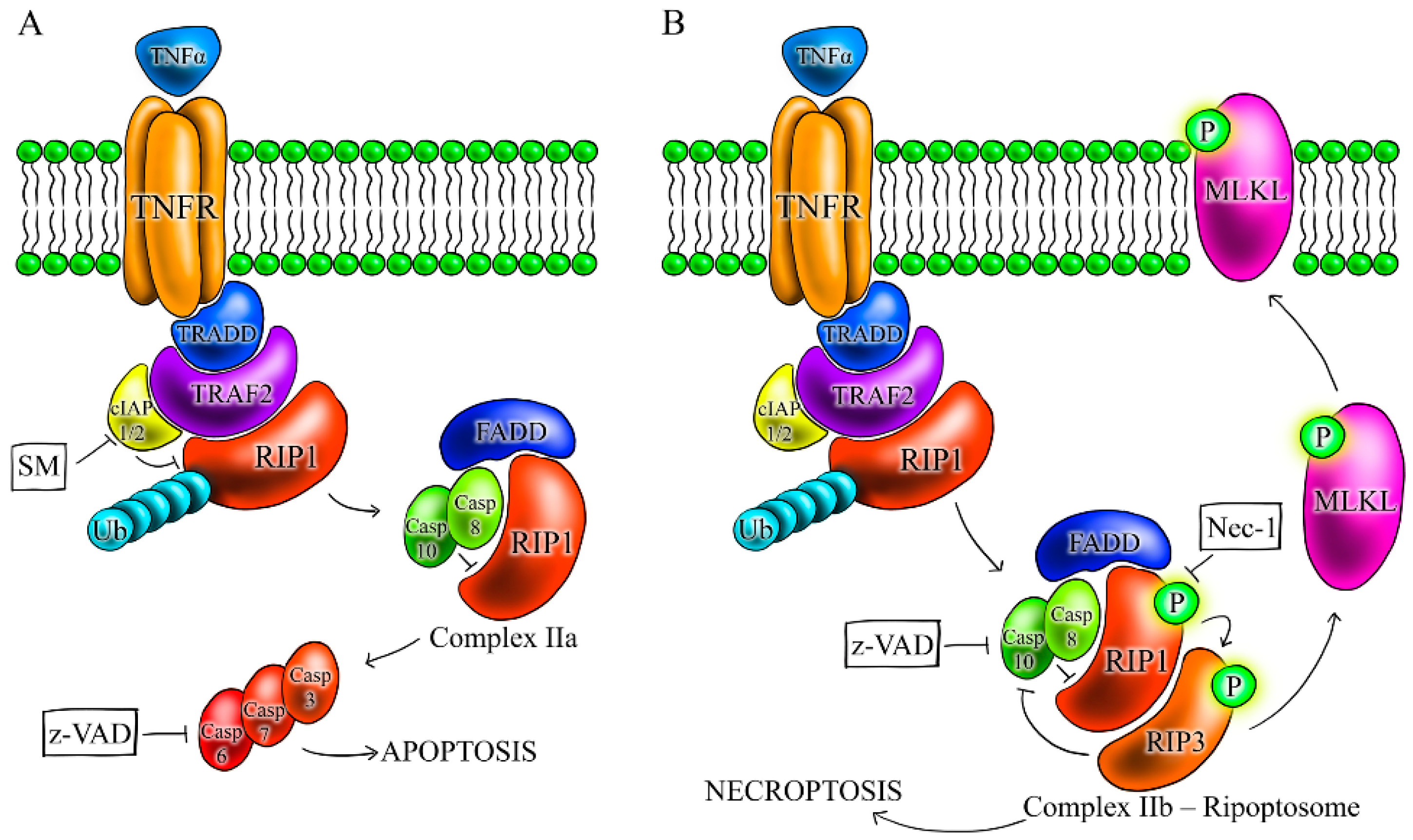
2.3. Apoptosis Is the Primary Cell Death in WT Cells, While FADD- and CASP3/7/6-Deficient Cells Die by RIP1-Dependent Necroptosis
2.4. Surviving CASP3/7/6 and FADD-Deficient Cells Stop Dividing and Change Morphology
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Cultivation Conditions
4.3. Propidium Iodide Exclusion Assay
4.4. SDS-PAGE and Western Blot Analysis
4.5. Long Time Live Cell Imaging
4.6. Transmission Electron Microscopy
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847–851. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Chan, F.K.-M.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Christofferson, D.E.; Li, Y.; Yuan, J. Control of Life-or-Death Decisions by RIP1 Kinase. Annu. Rev. Physiol. 2014, 76, 129–150. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-J.; Cao, Q.; Liu, X.; Wang, K.-T.; Mi, W.; Zhang, Y.; Li, L.-F.; LeBlanc, A.C.; Su, X.-D. Crystal structures of human caspase 6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep. 2010, 11, 841–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-α. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafont, E.; Milhas, D.; Teissié, J.; Therville, N.; Andrieu-Abadie, N.; Levade, T.; Benoist, H.; Ségui, B. Caspase-10-dependent cell death in Fas/CD95 signalling is not abrogated by caspase inhibitor zVAD-fmk. PLoS ONE 2010, 5, e13638. [Google Scholar] [CrossRef] [Green Version]
- Filomenko, R.; Prévotat, L.; Rébé, C.; Cortier, M.; Jeannin, J.-F.; Solary, E.; Bettaieb, A. Caspase-10 involvement in cytotoxic drug-induced apoptosis of tumor cells. Oncogene 2006, 25, 7635–7645. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J.; Wu, C.-H.; Gordon, J.D.; Zhong, X.; Emami, A.; Safa, A.R. Taxol Induces Caspase-10-dependent Apoptosis. J. Biol. Chem. 2004, 279, 51057–51067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanzer, M.C.; Khan, N.; Rickard, J.A.; Etemadi, N.; Lalaoui, N.; Spall, S.K.; Hildebrand, J.M.; Segal, D.; Miasari, M.; Chau, D.; et al. Combination of IAP antagonist and IFNγ activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 2017, 24, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Wang, L.; Wang, Z.; Wu, S.; Zhu, X.; Hu, S.; Wang, Y.; Yu, J.; Chen, G. TRADD mediates the tumor necrosis factor-induced apoptosis of L929 cells in the absence of RIP3. Sci. Rep. 2017, 7, 16111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.-W.; Kim, J.-H.; Ahn, Y.-H.; Seo, J.; Ko, A.; Jeong, M.; Kim, S.-J.; Ro, J.Y.; Park, K.-M.; Lee, H.-W.; et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat. Commun. 2012, 3, 978. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.-W.; Seo, J.H.; Jeong, M.; Lee, S.S.; Song, J.W. The roles of FADD in extrinsic apoptosis and necroptosis. BMB Rep. 2012, 45, 496–508. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, H.; Shimizu, Y.; Ohsawa, Y.; Kawahara, A.; Uchiyama, Y.; Nagata, S. Necrotic death pathway in Fas receptor signaling. J. Cell Biol. 2000, 151, 1247–1256. [Google Scholar] [CrossRef]
- Kawahara, A.; Ohsawa, Y.; Matsumura, H.; Uchiyama, Y.; Nagata, S. Caspase-independent Cell Killing by Fas-associated Protein with Death Domain. J. Cell Biol. 1998, 143, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irrinki, K.M.; Mallilankaraman, K.; Thapa, R.J.; Chandramoorthy, H.C.; Smith, F.J.; Jog, N.R.; Gandhirajan, R.K.; Kelsen, S.G.; Houser, S.R.; May, M.J.; et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol. Cell. Biol. 2011, 31, 3745–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.-L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Bertrand, M.J.M.; Bogaert, P.; Vandenabeele, P.; Vanden Berghe, T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011, 2, e230. [Google Scholar] [CrossRef]
- Feldmann, F.; Schenk, B.; Martens, S.; Vandenabeele, P.; Fulda, S. Sorafenib inhibits therapeutic induction of necroptosis in acute leukemia cells. Oncotarget 2017, 8, 68208–68220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannes, S.; Abhari, B.A.; Fulda, S. Smac mimetic triggers necroptosis in pancreatic carcinoma cells when caspase activation is blocked. Cancer Lett. 2016, 380, 31–38. [Google Scholar] [CrossRef]
- McComb, S.; Aguadé-Gorgorió, J.; Harder, L.; Marovca, B.; Cario, G.; Eckert, C.; Schrappe, M.; Stanulla, M.; von Stackelberg, A.; Bourquin, J.-P.; et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci. Transl. Med. 2016, 8, 339ra70. [Google Scholar] [CrossRef]
- Abhari, B.A.; Cristofanon, S.; Kappler, R.; von Schweinitz, D.; Humphreys, R.; Fulda, S. RIP1 is required for IAP inhibitor-mediated sensitization for TRAIL-induced apoptosis via a RIP1/FADD/caspase-8 cell death complex. Oncogene 2013, 32, 3263–3273. [Google Scholar] [CrossRef] [PubMed]
- Löder, S.; Fakler, M.; Schoeneberger, H.; Cristofanon, S.; Leibacher, J.; Vanlangenakker, N.; Bertrand, M.J.M.; Vandenabeele, P.; Jeremias, I.; Debatin, K.-M.; et al. RIP1 is required for IAP inhibitor-mediated sensitization of childhood acute leukemia cells to chemotherapy-induced apoptosis. Leukemia 2012, 26, 1020–1029. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Laukens, B.; Jennewein, C.; Schenk, B.; Vanlangenakker, N.; Schier, A.; Cristofanon, S.; Zobel, K.; Deshayes, K.; Vucic, D.; Jeremias, I.; et al. Smac mimetic bypasses apoptosis resistance in FADD- or caspase-8-deficient cells by priming for tumor necrosis factor α-induced necroptosis. Neoplasia 2011, 13, 971–979. [Google Scholar] [CrossRef]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Karpinich, N.O.; Tafani, M.; Schneider, T.; Russo, M.A.; Farber, J.L. The course of etoposide-induced apoptosis in Jurkat cells lacking p53 and Bax. J. Cell. Physiol. 2006, 208, 55–63. [Google Scholar] [CrossRef]
- Amin, P.; Florez, M.; Najafov, A.; Pan, H.; Geng, J.; Ofengeim, D.; Dziedzic, S.A.; Wang, H.; Barrett, V.J.; Ito, Y.; et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5944–E5953. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mrkvová, Z.; Portešová, M.; Slaninová, I. Loss of FADD and Caspases Affects the Response of T-Cell Leukemia Jurkat Cells to Anti-Cancer Drugs. Int. J. Mol. Sci. 2021, 22, 2702. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052702
Mrkvová Z, Portešová M, Slaninová I. Loss of FADD and Caspases Affects the Response of T-Cell Leukemia Jurkat Cells to Anti-Cancer Drugs. International Journal of Molecular Sciences. 2021; 22(5):2702. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052702
Chicago/Turabian StyleMrkvová, Zuzana, Michaela Portešová, and Iva Slaninová. 2021. "Loss of FADD and Caspases Affects the Response of T-Cell Leukemia Jurkat Cells to Anti-Cancer Drugs" International Journal of Molecular Sciences 22, no. 5: 2702. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052702