
The Role of Hypoxia-Induced Mitogenic Factor in Organ-Specific Inflammation in the Lung and Liver: Key Concepts and Gaps in Knowledge Regarding Molecular Mechanisms of Acute or Immune-Mediated Liver Injury


Abstract
:1. Introduction
2. Methods
2.1. PRISMA Criteria
2.2. Protocol and Registration
2.3. Eligibility Criteria
2.4. Information Sources
2.5. Search
2.6. Study Selection
2.7. Data Collection Process
2.8. Data Items
2.9. Risk of Bias in Individual Studies
2.10. Summary Measures
2.11. Synthesis of Results
2.12. Risk of Bias across Studies
2.13. Additional Analyses
3. Results
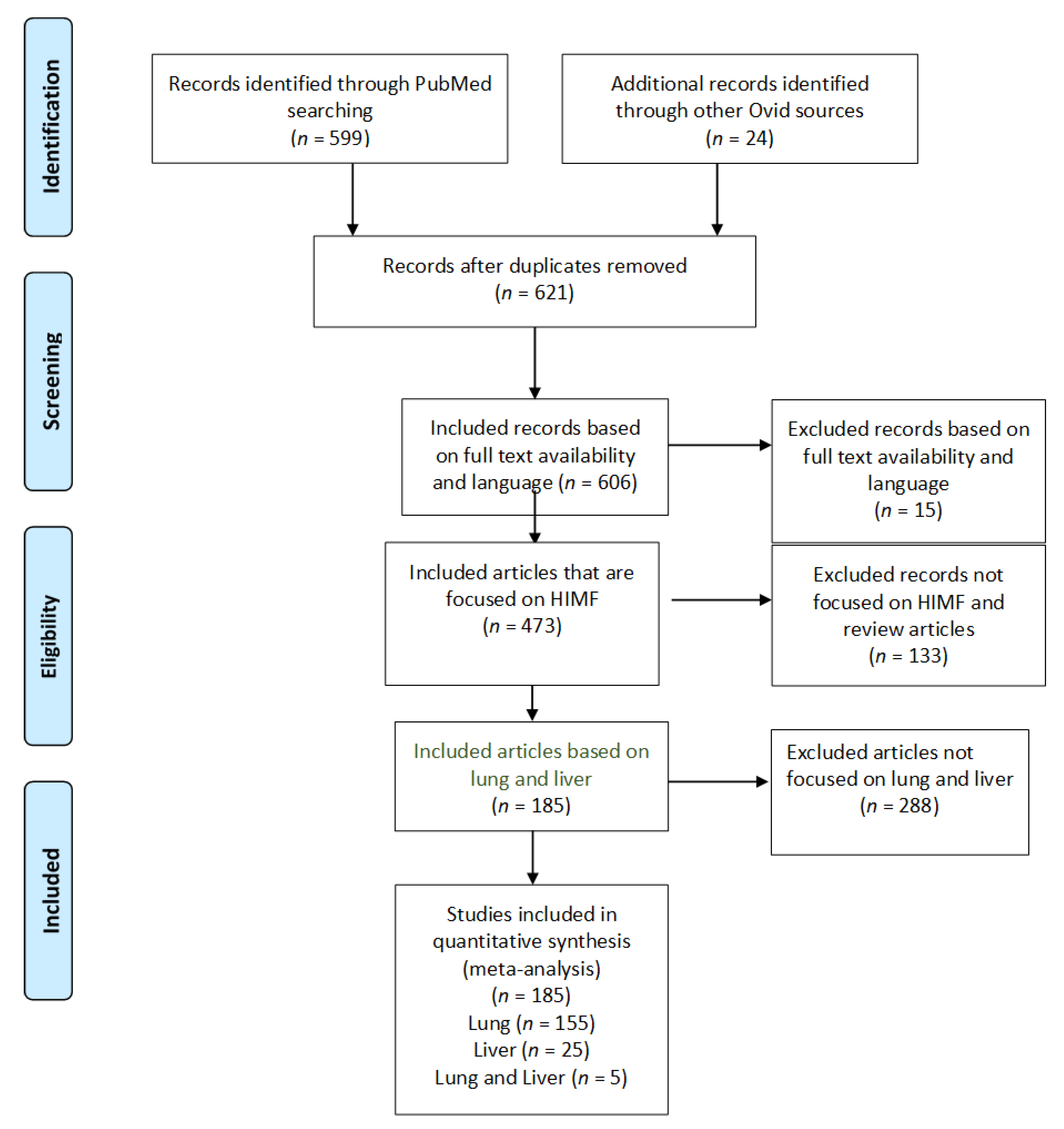
3.1. Study Selection
3.2. Effects of HIMF on the Pulmonary System
3.2.1. Human Subjects—Pulmonary Hypertension and Pulmonary Fibrosis
3.2.2. Human Subjects—Asthmatic/Allergic Inflammation
3.2.3. Murine Model—Pulmonary Hypertension
3.2.4. Murine Model—Pulmonary Fibrosis
3.2.5. Murine Model—Asthmatic/Allergic Inflammation
3.2.6. Murine Model—Parasitic Infection
3.2.7. Murine Model—Other Roles/Information Not Previously Described
3.3. Effects of HIMF on the Liver
3.3.1. Murine Model—Parasitic Infection
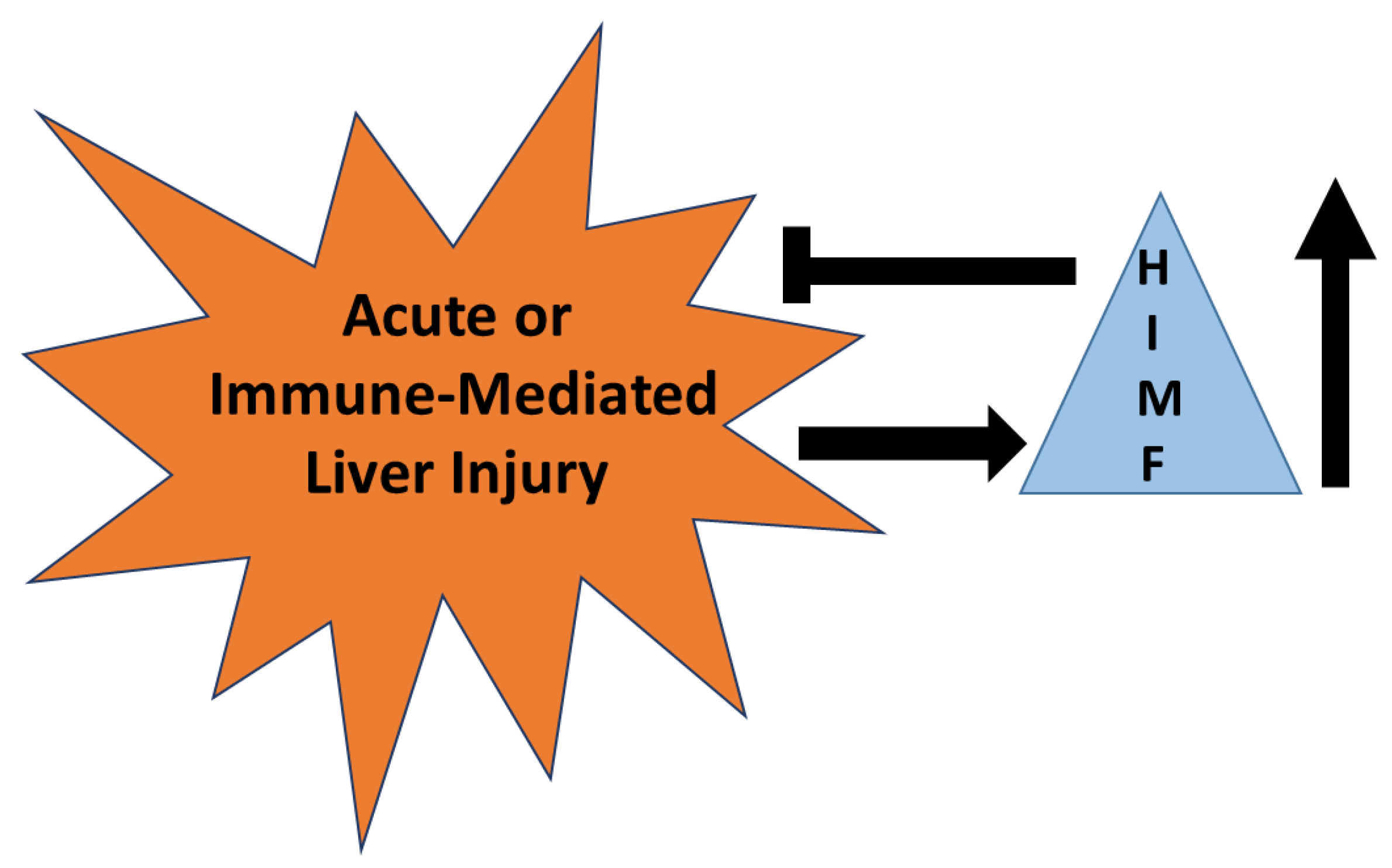
3.3.2. Murine Models—Liver Injury
3.4. Risk of Bias Assessments
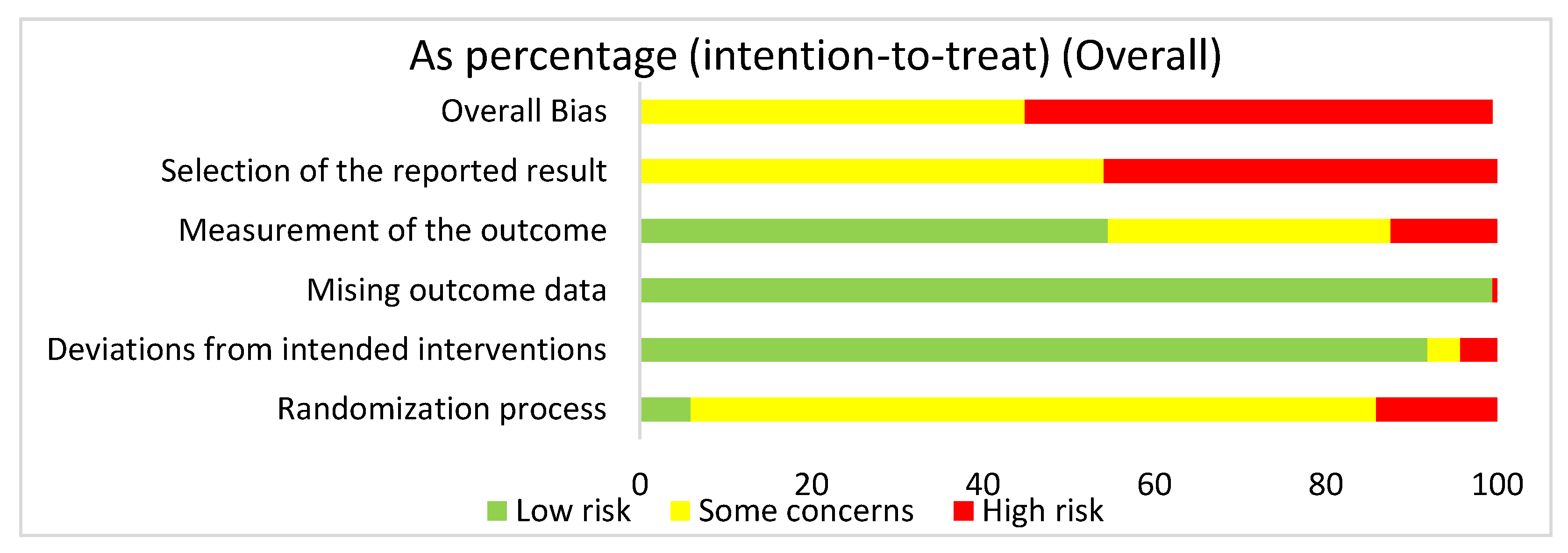
3.4.1. Overall Risk of Bias Assessment for ALL Articles
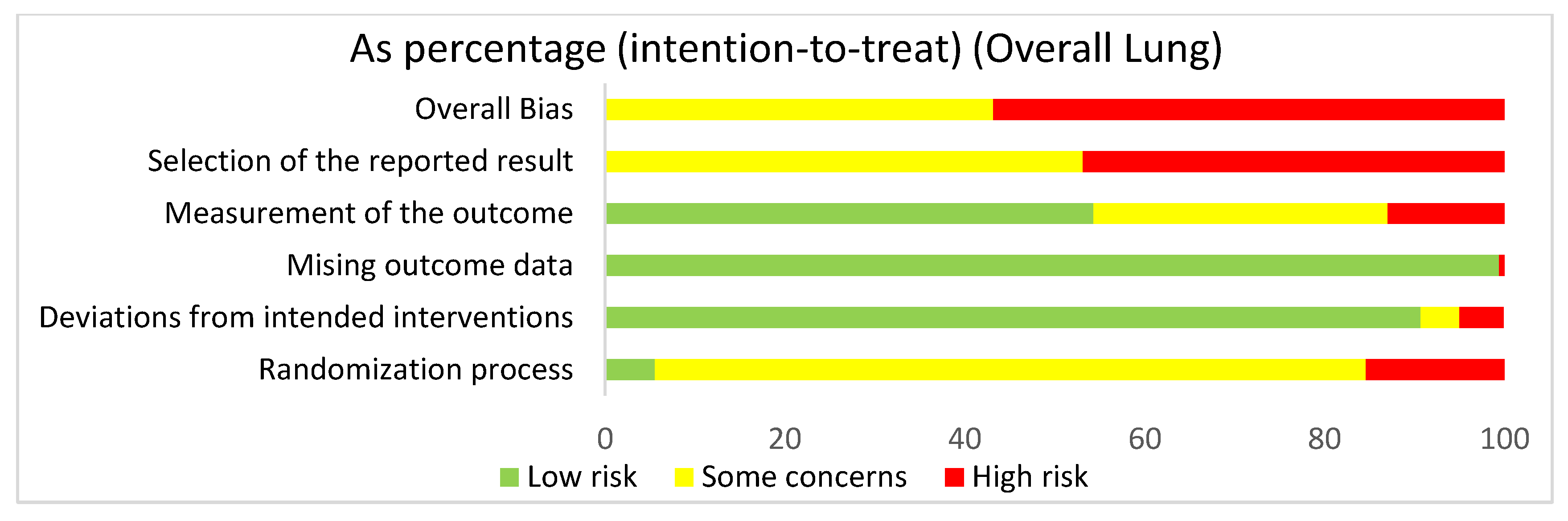
3.4.2. Lung: Overall Risk of Bias Assessment
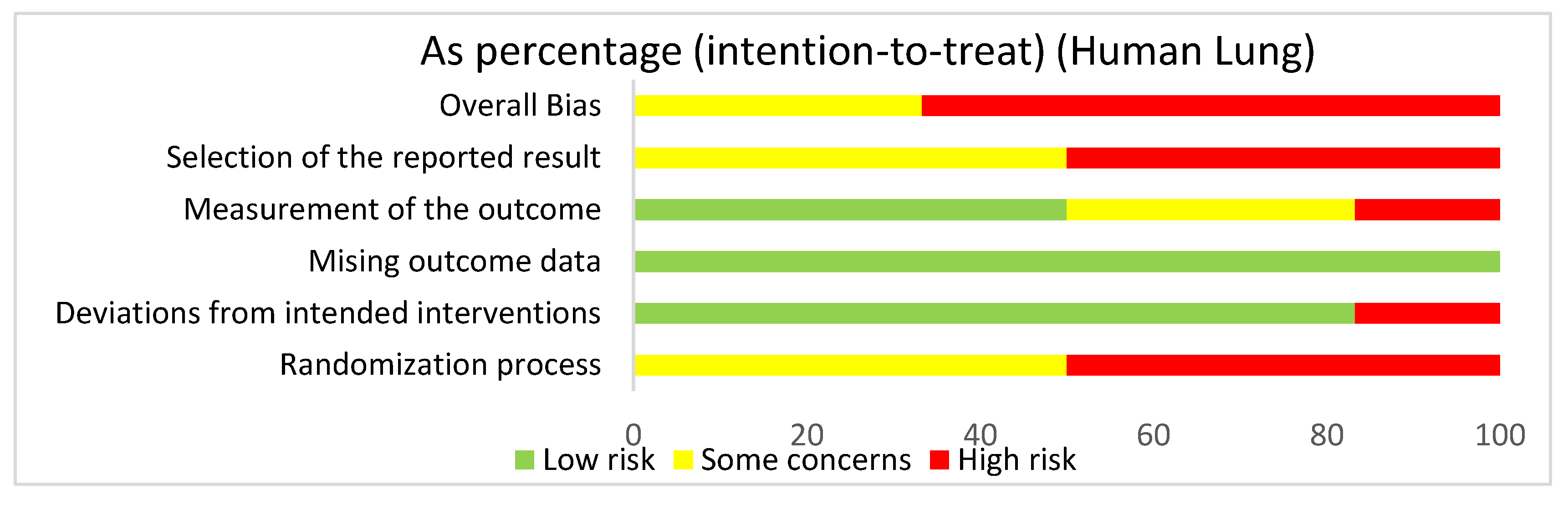
3.4.3. Lung: Human Studies Risk of Bias Assessment
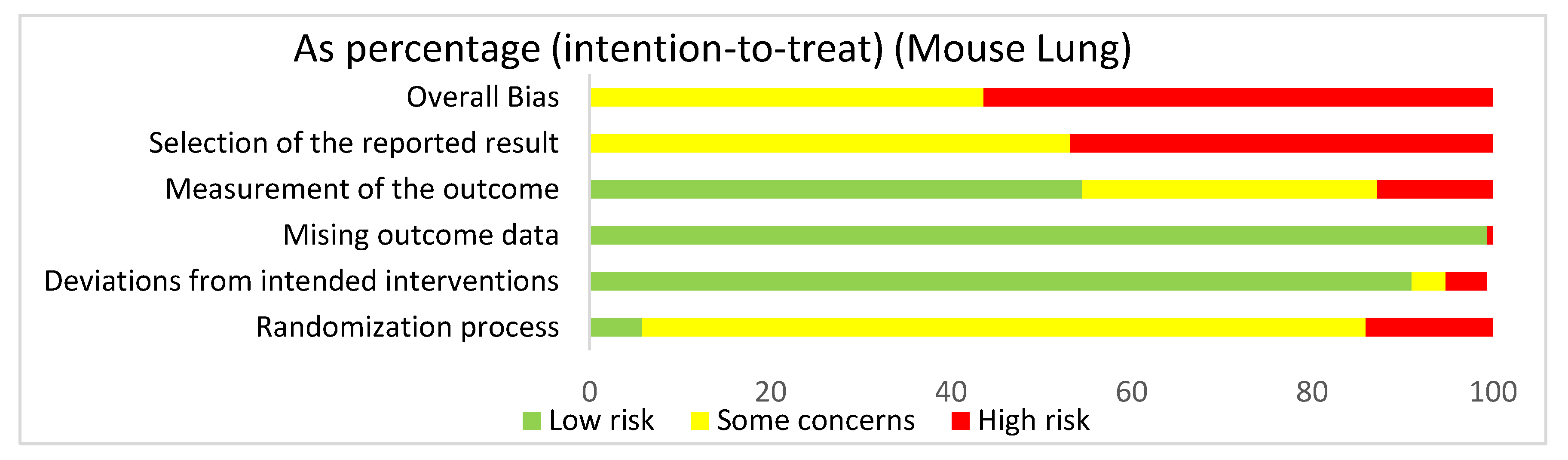
3.4.4. Lung: Mouse Studies Risk of Bias Assessment
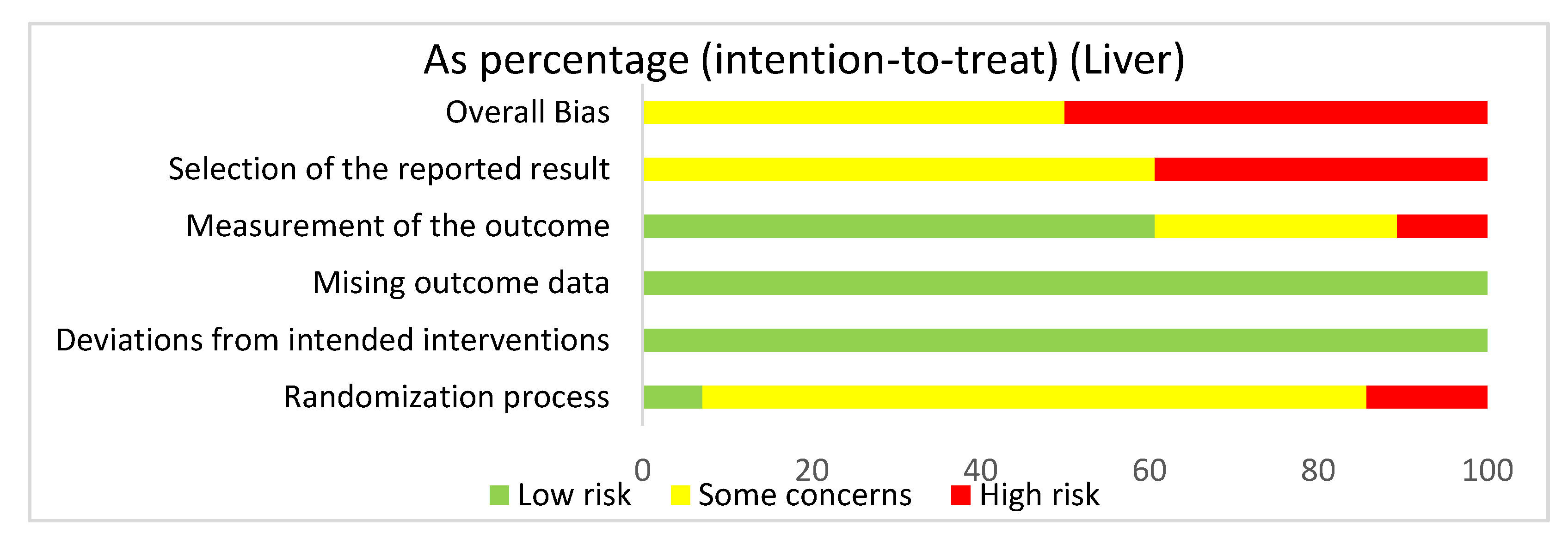
3.4.5. Liver: Overall Risk of Bias Assessment
3.4.6. Summary of Risk of Bias Assessments
4. Discussion
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Holcomb, I.N.; Kabakoff, R.C.; Chan, B.; Baker, T.W.; Gurney, A.; Henzel, W.; Nelson, C.; Lowman, H.B.; Wright, B.D.; Skelton, N.J.; et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000, 19, 4046–4055. [Google Scholar] [CrossRef]
- Teng, X.; Li, D.; Champion, H.C.; Johns, R.A. FIZZ1/RELMα, a Novel Hypoxia-Induced Mitogenic Factor in Lung With Vasoconstrictive and Angiogenic Properties. Circ. Res. 2003, 92, 1065–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Q.; Zhou, Y.; Johns, R.A. Bruton’s tyrosine kinase (BTK) is a binding partner for hypoxia induced mitogenic factor (HIMF/FIZZ1) and mediates myeloid cell chemotaxis. FASEB J. 2007, 21, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Ormrod, D.J.; Miller, T.E. The anti-inflammatory activity of a low molecular weight component derived from the milk of hyperimmunized cows. Inflamm. Res. 1991, 32, 160–166. [Google Scholar] [CrossRef]
- Stelwagen, K.; Ormrod, D.J. An anti-inflammatory component derived from milk of hyperimmunised cows reduces tight junction permeability in vitro. Inflamm. Res. 1998, 47, 384–388. [Google Scholar] [CrossRef]
- Steppan, C.M.; Brown, E.J.; Wright, C.M.; Bhat, S.; Banerjee, R.R.; Dai, C.Y.; Enders, G.H.; Silberg, D.G.; Wen, X.; Wu, G.D.; et al. A family of tissue-specific resistin-like molecules. Proc. Natl. Acad. Sci. USA 2001, 98, 502–506. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.-Z.; Huang, Q.; Xu, A.; McLenithan, J.C.; A Eison, J.; Shuldiner, A.R.; Alkan, S.; Gong, D.-W. Comparative studies of resistin expression and phylogenomics in human and mouse. Biochem. Biophys. Res. Commun. 2003, 310, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Su, Q.; Yamaji-Kegan, K.; Fan, C.; Skinner, J.T.; Champion, H.C.; Crow, M.T.; Johns, R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMα) induces the vascular and hemodynamic changes of pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L582–L593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Yu, H.; Ullenbruch, M.; Jin, H.; Ito, T.; Wu, Z.; Liu, J.; Phan, S.H. The In Vivo Fibrotic Role of FIZZ1 in Pulmonary Fibrosis. PLoS ONE 2014, 9, e88362. [Google Scholar] [CrossRef]
- Yamaji-Kegan, K.; Su, Q.; Angelini, D.J.; Champion, H.C.; Johns, R.A. Hypoxia-induced mitogenic factor has proangiogenic and proinflammatory effects in the lung via VEGF and VEGF receptor-2. Am. J. Physiol. Cell. Mol. Physiol. 2006, 291, L1159–L1168. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.L.; Yin, L.J.; Sharma, S.; Kierstein, S.; Wu, H.F.; Eid, G.; Haczku, A.; Corrigan, C.J.; Ying, S. Resistin-like molecule-β (RELM-β) targets airways fibroblasts to effect remodelling in asthma: From mouse to man. Clin. Exp. Allergy 2014, 45, 940–952. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Meng, Q.; Wu, H.; Eid, G.; Zhang, G.; Zhang, X.; Yang, S.; Huang, K.; Lee, T.H.; Corrigan, S.; et al. Resistin-like molecule-β is a human airway remodelling mediator. Eur. Respir. J. 2012, 39, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Su, Q.; Li, Y.; Liang, L.; Angelini, D.J.; Guggino, W.B.; Johns, R.A. Hypoxia-induced mitogenic factor/FIZZ1 induces intracellular calcium release through the PLC-IP3 pathway. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L263–L270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Fu, Z.; Su, Q.; Angelini, D.J.; Van Eyk, J.; Johns, R.A. S100A11 Mediates Hypoxia-induced Mitogenic Factor (HIMF)-induced Smooth Muscle Cell Migration, Vesicular Exocytosis, and Nuclear Activation. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-M.; Wang, I.-C.; Liu, S.-C.; Sun, Y.; Jin, L.; Wang, S.-W.; Lee, H.-P.; Tseng, W.-P.; Tang, C.-H. Hypoxia induced mitogenic factor (HIMF) triggers angiogenesis by increasing interleukin-18 production in myoblasts. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Wang, G.; Zheng, N.; Cheng, W.; Ouyang, K.; Lin, H.; Liao, Y.; Liu, J. HIMF (Hypoxia-Induced Mitogenic Factor)-IL (Interleukin)-6 Signaling Mediates Cardiomyocyte-Fibroblast Crosstalk to Promote Cardiac Hypertrophy and Fibrosis. Hypertension 2019, 73, 1058–1070. [Google Scholar] [CrossRef]
- Zheng, L.-D.; Tong, Q.-S.; Weng, M.-X.; He, J.; Lv, Q.; Pu, J.-R.; Jiang, G.-S.; Cai, J.-B.; Liu, Y.; Hou, X.-H. Enhanced Expression of Resistin-like Molecule Beta in Human Colon Cancer and Its Clinical Significance. Dig. Dis. Sci. 2008, 54, 274–281. [Google Scholar] [CrossRef]
- Propheter, D.C.; Chara, A.L.; Harris, T.A.; Ruhn, K.A.; Hooper, L.V. Resistin-like molecule β is a bactericidal protein that promotes spatial segregation of the microbiota and the colonic epithelium. Proc. Natl. Acad. Sci. USA 2017, 114, 11027–11033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Wang, M.-L.; Jiang, H.-Q.; Steppan, C.M.; E Shin, M.; Thurnheer, M.; Cebra, J.J.; A Lazar, M.; Wu, G.D. Bacterial colonization leads to the colonic secretion of RELMβ/FIZZ2, a novel goblet cell-specific protein. Gastroenterology 2003, 125, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, A.-C.D.; Sun, R.; Choi, H.; Laskin, J.D.; Laskin, D.L. Role of Galectin-3 in Classical and Alternative Macrophage Activation in the Liver following Acetaminophen Intoxication. J. Immunol. 2012, 189, 5934–5941. [Google Scholar] [CrossRef] [Green Version]
- Okubo, H.; Kushiyama, A.; Sakoda, H.; Nakatsu, Y.; Iizuka, M.; Taki, N.; Fujishiro, M.; Fukushima, T.; Kamata, H.; Nagamachi, A.; et al. Involvement of resistin-like molecule β in the development of methionine-choline deficient diet-induced non-alcoholic steatohepatitis in mice. Sci. Rep. 2016, 6, 20157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesce, J.T.; Ramalingam, T.R.; Wilson, M.S.; Mentink-Kane, M.M.; Thompson, R.W.; Cheever, A.W.; Wynn, T.A. Retnla (Relmα/Fizz1) Suppresses Helminth-Induced Th2-Type Immunity. PLOS Pathog. 2009, 5, e1000393. [Google Scholar] [CrossRef]
- Pesce, J.; Kaviratne, M.; Ramalingam, T.R.; Thompson, R.W.; Urban, J.F.; Cheever, A.W.; Young, D.A.; Collins, M.; Grusby, M.J.; Wynn, T.A. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J. Clin. Investig. 2006, 116, 2044–2055. [Google Scholar] [CrossRef]
- Tong, Q.; Zheng, L.; Lin, L.; Li, B.; Wang, D.; Huang, C.; Matuschak, G.M.; Li, D. Participation of the PI-3K/Akt-NF-κB signaling pathways in hypoxia-induced mitogenic factor-stimulated Flk-1 expression in endothelial cells. Respir. Res. 2006, 7, 101. [Google Scholar] [CrossRef] [Green Version]
- Sterne, J.A.C.; Savović, J.; Page, M.J.; Elbers, R.G.; Blencowe, N.S.; Boutron, I.; Cates, C.J.; Cheng, H.-Y.; Corbett, M.S.; Eldridge, S.M.; et al. RoB 2: A revised tool for assessing risk of bias in randomised trials. BMJ 2019, 366, l4898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renigunta, A.; Hild, C.; Rose, F.; Klepetko, W.; Grimminger, F.; Seeger, W.; Hänze, J. Human RELMβ is a mitogenic factor in lung cells and induced in hypoxia. FEBS Lett. 2006, 580, 900–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, D.J.; Su, Q.; Yamaji-Kegan, K.; Fan, C.; Teng, X.; Hassoun, P.M.; Yang, S.C.; Champion, H.C.; Tuder, R.M.; Johns, R.A. Resistin-Like Molecule-β in Scleroderma-Associated Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2009, 41, 553–561. [Google Scholar] [CrossRef]
- Liu, T.; Baek, H.A.; Yu, H.; Lee, H.J.; Park, B.-H.; Ullenbruch, M.; Liu, J.; Nakashima, T.; Choi, Y.Y.; Wu, G.D.; et al. FIZZ2/RELM-β Induction and Role in Pulmonary Fibrosis. J. Immunol. 2011, 187, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Fan, C.; Gomez-Arroyo, J.; Van Raemdonck, K.; Meuchel, L.W.; Skinner, J.T.; Everett, A.D.; Fang, X.; Macdonald, A.A.; Yamaji-Kegan, K.; et al. HIMF (Hypoxia-Induced Mitogenic Factor) Signaling Mediates the HMGB1 (High Mobility Group Box 1)-Dependent Endothelial and Smooth Muscle Cell Crosstalk in Pulmonary Hypertension. Arter. Thromb. Vasc. Biol. 2019, 39, 2505–2519. [Google Scholar] [CrossRef]
- Lin, Q.; Fan, C.; Skinner, J.T.; Hunter, E.N.; Macdonald, A.A.; Illei, P.B.; Yamaji-Kegan, K.; Johns, R.A. RELMα Licenses Macrophages for Damage-Associated Molecular Pattern Activation to Instigate Pulmonary Vascular Remodeling. J. Immunol. 2019, 203, 2862–2871. [Google Scholar] [CrossRef]
- St-Laurent, J.; Turmel, V.; Boulet, L.-P.; Bissonnette, E. Alveolar Macrophage Subpopulations in Bronchoalveolar Lavage and Induced Sputum of Asthmatic and Control Subjects. J. Asthma 2009, 46, 1–8. [Google Scholar] [CrossRef]
- Grainge, C.; Dulay, V.; Ward, J.; Sammut, D.; Davies, E.; Green, B.; Lau, L.; Cottey, L.; Haitchi, H.-M.; Davies, D.E.; et al. Resistin-like molecule-β is induced following bronchoconstriction of asthmatic airways. Respirology 2012, 17, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.D.; Han, S.; Harmsen, A.; Shampeny, K.; Harmsen, A.G. Pulmonary Hypertension Can Be a Sequela of Prior Pneumocystis Pneumonia. Am. J. Pathol. 2007, 171, 790–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Wang, M.; Schlotman, J.; Nikolaidis, N.M.; Debrosse, C.W.; Karow, M.L.; Rothenberg, M.E. Resistin-like molecule-β is an allergen-induced cytokine with inflammatory and remodeling activity in the murine lung. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L305–L313. [Google Scholar] [CrossRef] [Green Version]
- Daley, E.; Emson, C.; Guignabert, C.; Malefyt, R.D.W.; Louten, J.; Kurup, V.P.; Hogaboam, C.; Taraseviciene-Stewart, L.; Voelkel, N.F.; Rabinovitch, M.; et al. Pulmonary arterial remodeling induced by a Th2 immune response. J. Exp. Med. 2008, 205, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Su, Q.; Kolosova, I.A.; Fan, C.; Skinner, J.T.; Yamaji-Kegan, K.; Collector, M.; Sharkis, S.J.; Johns, R.A. Hypoxia-Induced Mitogenic Factor (HIMF/FIZZ1/RELMα) Recruits Bone Marrow-Derived Cells to the Murine Pulmonary Vasculature. PLoS ONE 2010, 5, e11251. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Su, Q.; Yamaji-Kegan, K.; Fan, C.; Skinner, J.T.; Poloczek, A.; El-Haddad, H.; Cheadle, C.; A Johns, R. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMα) in chronic hypoxia- and antigen-mediated pulmonary vascular remodeling. Respir. Res. 2013, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Zhu, L.; Xiao, R.; Liu, B.; Sun, M.; Liu, F.; Hao, Q.; Lu, Y.; Zhang, J.; Li, J.; et al. Hypoxia-Induced Mitogenic Factor Acts as a Nonclassical Ligand of Calcium-Sensing Receptor, Therapeutically Exploitable for Intermittent Hypoxia-Induced Pulmonary Hypertension. Hypertension 2017, 69, 844–854. [Google Scholar] [CrossRef]
- Yamaji-Kegan, K.; Su, Q.; Angelini, D.J.; Myers, A.C.; Cheadle, C.; Johns, R.A. Hypoxia-Induced Mitogenic Factor (HIMF/FIZZ1/RELMα) Increases Lung Inflammation and Activates Pulmonary Microvascular Endothelial Cells via an IL-4–Dependent Mechanism. J. Immunol. 2010, 185, 5539–5548. [Google Scholar] [CrossRef] [Green Version]
- Johns, R.A.; Takimoto, E.; Meuchel, L.W.; Elsaigh, E.; Zhang, A.; Heller, N.M.; Semenza, G.L.; Yamaji-Kegan, K. Hypoxia-Inducible Factor 1α Is a Critical Downstream Mediator for Hypoxia-Induced Mitogenic Factor (FIZZ1/RELMα)–Induced Pulmonary Hypertension. Arter. Thromb. Vasc. Biol. 2016, 36, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Njoku, D.B.; Li, Z.; Washington, N.D.; Mellerson, J.L.; Talor, M.V.; Sharma, R.; Rose, N.R. Suppressive and pro-inflammatory roles for IL-4 in the pathogenesis of experimental drug-induced liver injury. Eur. J. Immunol. 2009. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Dhanasekaran, S.M.; Jin, H.; Hu, B.; Tomlins, S.A.; Chinnaiyan, A.M.; Phan, S.H. FIZZ1 Stimulation of Myofibroblast Differentiation. Am. J. Pathol. 2004, 164, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Hu, B.; Choi, Y.Y.; Chung, M.; Ullenbruch, M.; Yu, H.; Lowe, J.B.; Phan, S.H. Notch1 Signaling in FIZZ1 Induction of Myofibroblast Differentiation. Am. J. Pathol. 2009, 174, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Madala, S.K.; Edukulla, R.; Davis, K.R.; Schmidt, S.; Davidson, C.; A Kitzmiller, J.; Hardie, W.D.; Korfhagen, T.R. Resistin-like molecule alpha1 (Fizz1) recruits lung dendritic cells without causing pulmonary fibrosis. Respir. Res. 2012, 13, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karo-Atar, D.; Moshkovits, I.; Eickelberg, O.; Königshoff, M.; Munitz, A. Paired Immunoglobulin-Like Receptor–B Inhibits Pulmonary Fibrosis by Suppressing Profibrogenic Properties of Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2013, 48, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Wang, S.-J.; Camoretti-Mercado, B.; Li, H.-J.; Chen, M.; Bi, W.-X. FIZZ1 Plays a Crucial Role in Early Stage Airway Remodeling of OVA-Induced Asthma. J. Asthma 2008, 45, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.Q.; Fillet, M.; De Seny, M.; Meuwis, M.-A.; Marée, R.; Crahay, C.; Paulissen, G.; Rocks, N.; Guéders, M.; Wehenkel, L.; et al. Biomarker discovery in asthma-related inflammation and remodeling. Proteomics 2009, 9, 2163–2170. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, M.; Kang, X.; Boontheung, P.; Li, N.; Nel, A.E.; Loo, J.A. Oxidative Stress and Asthma: Proteome Analysis of Chitinase-like Proteins and FIZZ1 in Lung Tissue and Bronchoalveolar Lavage Fluid. J. Proteome Res. 2009, 8, 1631–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuberi, R.I.; Na Ge, X.; Jiang, S.; Bahaie, N.S.; Na Kang, B.; Hosseinkhani, R.M.; Frenzel, E.M.; Fuster, M.M.; Esko, J.D.; Rao, S.P.; et al. Deficiency of Endothelial Heparan Sulfates Attenuates Allergic Airway Inflammation. J. Immunol. 2009, 183, 3971–3979. [Google Scholar] [CrossRef] [Green Version]
- Doherty, T.A.; Khorram, N.; Sugimoto, K.; Sheppard, D.; Rosenthal, P.; Cho, J.Y.; Pham, A.; Miller, M.; Croft, M.; Broide, D.H. Alternaria Induces STAT6-Dependent Acute Airway Eosinophilia and Epithelial FIZZ1 Expression That Promotes Airway Fibrosis and Epithelial Thickness. J. Immunol. 2012, 188, 2622–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-R.; Shim, D.; Yoon, J.; Jang, H.S.; Oh, S.-W.; Suh, S.H.; Choi, J.-H.; Oh, G.T. Retnla Overexpression Attenuates Allergic Inflammation of the Airway. PLoS ONE 2014, 9, e112666. [Google Scholar] [CrossRef]
- LeMessurier, K.S.; Palipane, M.; Tiwary, M.; Gavin, B.; Samarasinghe, A.E. Chronic features of allergic asthma are enhanced in the absence of resistin-like molecule-beta. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Tong, X.; An, Y. BAFF gene silencing attenuates allergic airway inflammation by promoting the generation of Tregs via activating pro-Treg cytokines. Life Sci. 2020, 241, 117172. [Google Scholar] [CrossRef]
- Stütz, A.M.; Pickart, L.A.; Trifilieff, A.; Baumruker, T.; Prieschl-Strassmayr, E.; Woisetschläger, M. The Th2 cell cytokines IL-4 and IL-13 regulate found in inflammatory zone 1/resistin-like molecule α gene expression by a STAT6 and CCAAT/enhancer-binding protein-dependent mechanism. J. Immunol. 2003, 170, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Chapoval, S.P.; Smith, E.P.; Keegan, A.D. Transfer of in vivo primed transgenic T cells supports allergic lung inflammation and FIZZ1 and Ym1 production in an IL-4Rα and STAT6 dependent manner. BMC Immunol. 2011, 12, 60. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, J.; Li, H.; Han, X. Found in Inflammatory Zone 1 Induces Angiogenesis in Murine Models of Asthma. Lung 2008, 186, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Chen, W.-C.; Hoffman, C.; Marsh, L.M.; West, J.; Grunig, G. Modification of Hemodynamic and Immune Responses to Exposure with a Weak Antigen by the Expression of a Hypomorphic BMPR2 Gene. PLoS ONE 2013, 8, e55180. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Jiao, X.; Wu, J.; Wang, J.; Gong, W.; Liu, F.; Liu, W.; Bi, W.; Dong, L. FIZZ1 Promotes Airway Remodeling in Asthma Through the PTEN Signaling Pathway. Inflammation 2015, 38, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-L.; Cai, P.-C.; Xiong, X.-Z.; Ye, H. Exercise training attenuated chronic cigarette smoking-induced up-regulation of FIZZ1/RELMα in lung of rats. Acta Acad. Med. Wuhan 2013, 33, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Gordon, W.; Galitovskiy, V.; Edwards, R.; Andersen, B.; Grando, S.A. The tobacco carcinogen nitrosamine induces a differential gene expression response in tumour susceptible A/J and resistant C3H mouse lungs. Eur. J. Cancer 2013, 49, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Loke, P.; Gallagher, I.; Nair, M.G.; Zang, X.; Brombacher, F.; Mohrs, M.; Allison, J.P.; Allen, J.E. Alternative Activation Is an Innate Response to Injury That Requires CD4+T Cells to be Sustained during Chronic Infection. J. Immunol. 2007, 179, 3926–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, M.G.; Du, Y.; Perrigoue, J.G.; Zaph, C.; Taylor, J.J.; Goldschmidt, M.; Swain, G.P.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.; et al. Alternatively activated macrophage-derived RELM-α is a negative regulator of type 2 inflammation in the lung. J. Exp. Med. 2009, 206, 937–952. [Google Scholar] [CrossRef]
- Burke, M.L.; McGarvey, L.; McSorley, H.J.; Bielefeldt-Ohmann, H.; McManus, D.P.; Gobert, G.N. Migrating Schistosoma japonicum schistosomula induce an innate immune response and wound healing in the murine lung. Mol. Immunol. 2011, 49, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.-Y.; Lewkowich, I.P.; Dawson, L.A.; Downey, J.; Yang, Y.; Smith, D.E.; Herbert, D.R. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc. Natl. Acad. Sci. USA 2012, 110, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Bouffi, C.; Rochman, M.; Zust, C.B.; Stucke, E.M.; Kartashov, A.; Fulkerson, P.C.; Barski, A.; Rothenberg, M.E. IL-33 markedly activates murine eosinophils by an NF-κB–Dependent mechanism differentially dependent upon an IL-4–Driven autoinflammatory loop. J. Immunol. 2013, 191, 4317–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meester, I.; Rosas-Taraco, A.G.; Salinas-Carmona, M.C. Retnla down-regulation and IL-13-rich environment correlate with inflammation severity in experimental actinomycetoma byNocardia brasiliensis. Pathog. Dis. 2013, 67, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Batugedara, H.M.; Li, J.; Chen, G.; Lu, D.; Patel, J.J.; Jang, J.C.; Radecki, K.C.; Burr, A.C.; Lo, D.D.; Dillman, A.R.; et al. Hematopoietic cell-derived RELMα regulates hookworm immunity through effects on macrophages. J. Leukoc. Biol. 2018, 104, 855–869. [Google Scholar] [CrossRef]
- Sutherland, T.E.; Rückerl, D.; Logan, N.; Duncan, S.; Wynn, T.A.; Allen, J.E. Ym1 induces RELMα and rescues IL-4Rα deficiency in lung repair during nematode infection. PLoS Pathog. 2018, 14, e1007423. [Google Scholar] [CrossRef] [Green Version]
- Krljanac, B.; Schubart, C.; Naumann, R.; Wirtz, S.; Culemann, S.; Krönke, G.; Voehringer, D. RELMα-expressing macrophages protect against fatal lung damage and reduce parasite burden during helminth infection. Sci. Immunol. 2019, 4, eaau3814. [Google Scholar] [CrossRef]
- Wagner, K.F.; Hellberg, A.K.; Balenger, S.; Depping, R.; Dodd-O, J.; Johns, R.A.; Li, D. Hypoxia-induced mitogenic factor has antiapoptotic action and is upregulated in the developing lung: Coexpression with hypoxia-inducible factor-2α. Am. J. Respir. Cell Mol. Biol. 2004, 31, 276–282. [Google Scholar] [CrossRef]
- Li, D.; Fernandez, L.G.; Dodd-O, J.; Langer, J.; Wang, D.; Laubach, V.E. Upregulation of Hypoxia-Induced Mitogenic Factor in Compensatory Lung Growth after Pneumonectomy. Am. J. Respir. Cell Mol. Biol. 2005, 32, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Zheng, L.; Dodd-O, J.; Langer, J.; Wang, D.; Li, D. Hypoxia-Induced Mitogenic Factor Modulates Surfactant Protein B and C Expression in Mouse Lung. Am. J. Respir. Cell Mol. Biol. 2006, 34, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, M.J.; Liu, T.; Ullenbruch, M.R.; Phan, S.H. Antiapoptotic effect of found in inflammatory zone (FIZZ)1 on mouse lung fibroblasts. J. Pathol. 2007, 212, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Lager, J.; Wang, D.; Li, D. Ets-1 participates in and facilitates developmental expression of hypoxia-induced mitogenic factor in mouse lung. Front. Biosci. 2007, 12, 2269–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolosova, I.A.; Angelini, D.; Fan, C.; Skinner, J.; Cheadle, C.; Johns, R.A. Resistin-Like Molecule α Stimulates Proliferation of Mesenchymal Stem Cells While Maintaining Their Multipotency. Stem Cells Dev. 2013, 22, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Tong, Q.; Zheng, L.; Kang, Q.; Dodd-O, J.; Langer, J.; Li, B.; Wang, D.; Li, D. Upregulation of hypoxia-induced mitogenic factor in bacterial lipopolysaccharide-induced acute lung injury. FEBS Lett. 2006, 580, 2207–2215. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Xu, J.; Qu, J.; Sai, Y.; Chen, C.; Yu, L.; Li, D.; Guo, X. A therapeutic role for mesenchymal stem cells in acute lung injury independent of hypoxia-induced mitogenic factor. J. Cell. Mol. Med. 2011, 16, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, S.; O’Neill, S.M.; Sekiya, M.; Mulcahy, G.; Dalton, J.P. Thioredoxin Peroxidase Secreted by Fasciola hepatica Induces the Alternative Activation of Macrophages. Infect. Immun. 2005, 73, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Reiman, R.M.; Thompson, R.W.; Feng, C.G.; Hari, D.; Knight, R.; Cheever, A.W.; Rosenberg, H.F.; Wynn, T.A. Interleukin-5 (IL-5) Augments the Progression of Liver Fibrosis by Regulating IL-13 Activity. Infect. Immun. 2006, 74, 1471–1479. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.D.; Raymond, T.; Coelho, A.L.; Kunkel, S.L.; Hogaboam, C.M. A systemic granulomatous response to Schistosoma mansoni eggs alters responsiveness of bone marrow-derived macrophages to Toll-like receptor agonists. J. Leukoc. Biol. 2008, 83, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Rutitzky, L.I.; Smith, P.M.; Stadecker, M.J. T-bet protects against exacerbation of schistosome egg-induced immunopathology by regulating Th17-mediated inflammation. Eur. J. Immunol. 2009, 39, 2470–2481. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Huang, W.; Wang, D.; Wang, H.; Nie, K. The absence of MyD88 has no effect on the induction of alternatively activated macrophage during Fasciola hepatica infection. BMC Immunol. 2011, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truscott, M.; Evans, D.A.; Gunn, M.; Hoffmann, K.F. Schistosoma mansoni Hemozoin Modulates Alternative Activation of Macrophages via Specific Suppression of Retnla Expression and Secretion. Infect. Immun. 2012, 81, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.N.; Aldridge, A.; Vukman, K.V.; Donnelly, S.; O’Neill, S.M. Fasciola hepatica tegumental antigens indirectly induce an M2 macrophage-like phenotype in vivo. Parasite Immunol. 2014, 36, 531–539. [Google Scholar] [CrossRef]
- Carasi, P.; Rodríguez, E.; Da Costa, V.; Frigerio, S.; Brossard, N.; Noya, V.; Robello, C.; Anegón, I.; Freire, T. Heme-Oxygenase-1 Expression Contributes to the Immunoregulation Induced by Fasciola hepatica and Promotes Infection. Front. Immunol. 2017, 8, 883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.M.; Holt, V.V.; Malpass, L.R.; Hines, I.N.; Wheeler, M.D. Fatty acid-binding protein 5 limits the anti-inflammatory response in murine macrophages. Mol. Immunol. 2015, 67, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.I.; Zhai, D.S.; Ruan, B.J.; Xu, C.M.; Ye, Z.C.; Lu, H.Y.; Jiang, Y.-H.; Wang, Z.Y.; Xiang, A.; Yang, Y.; et al. Quaking Deficiency Amplifies Inflammation in Experimental Endotoxemia via the Aryl Hydrocarbon Receptor/Signal Transducer and Activator of Transcription 1–NF-κB Pathway. Front. Immunol. 2017, 8, 1754. [Google Scholar] [CrossRef] [Green Version]
- McMahan, R.H.; Wang, X.X.; Cheng, L.L.; Krisko, T.; Smith, M.; El Kasmi, K.; Pruzanski, M.; Adorini, L.; Golden-Mason, L.; Levi, M.; et al. Bile Acid Receptor Activation Modulates Hepatic Monocyte Activity and Improves Nonalcoholic Fatty Liver Disease. J. Biol. Chem. 2013, 288, 11761–11770. [Google Scholar] [CrossRef] [Green Version]
- Raes, G.; De Baetselier, P.; Noël, W.; Beschin, A.; Brombacher, F.; Gh, G.H. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J. Leukoc. Biol. 2002, 71, 597–602. [Google Scholar] [PubMed]
- Sun, Y.-Y.; Li, X.-F.; Meng, X.-M.; Huang, C.; Zhang, L.; Li, J. Macrophage Phenotype in Liver Injury and Repair. Scand. J. Immunol. 2016, 85, 166–174. [Google Scholar] [CrossRef]
- Danan, G.; Teschke, R. RUCAM in Drug and Herb Induced Liver Injury: The Update. Int. J. Mol. Sci. 2015, 17, 14. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Intervention | Comparison Intervention | Outcome Measure |
|---|---|---|---|
| Mouse Mice Rat Squirrel Hamster Guinea Pig Rodent Human Patient BALB/C C57BL/6 Knockout | Resistin-like molecules HIMF RELM-a FIZZ1 Retnla RELM-beta | Liver Lungs | Proinflammatory Inflammatory Protection Pathogenesis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pai, S.; Njoku, D.B. The Role of Hypoxia-Induced Mitogenic Factor in Organ-Specific Inflammation in the Lung and Liver: Key Concepts and Gaps in Knowledge Regarding Molecular Mechanisms of Acute or Immune-Mediated Liver Injury. Int. J. Mol. Sci. 2021, 22, 2717. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052717
Pai S, Njoku DB. The Role of Hypoxia-Induced Mitogenic Factor in Organ-Specific Inflammation in the Lung and Liver: Key Concepts and Gaps in Knowledge Regarding Molecular Mechanisms of Acute or Immune-Mediated Liver Injury. International Journal of Molecular Sciences. 2021; 22(5):2717. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052717
Chicago/Turabian StylePai, Sananda, and Dolores B. Njoku. 2021. "The Role of Hypoxia-Induced Mitogenic Factor in Organ-Specific Inflammation in the Lung and Liver: Key Concepts and Gaps in Knowledge Regarding Molecular Mechanisms of Acute or Immune-Mediated Liver Injury" International Journal of Molecular Sciences 22, no. 5: 2717. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052717