
A Detailed Catalogue of Multi-Omics Methodologies for Identification of Putative Biomarkers and Causal Molecular Networks in Translational Cancer Research




Abstract
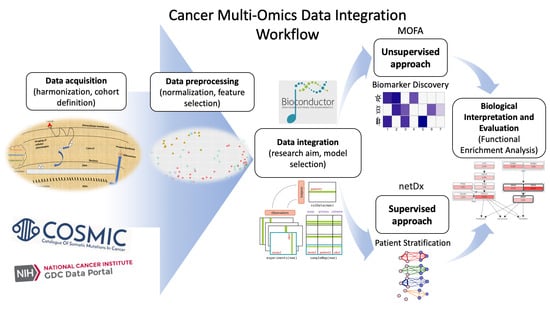
:
1. Introduction
1.1. Limitations of Single-Omics Approaches in Complex Phenotypes
1.2. Multi-Omics Concept Introduction and Background
1.3. Public Cancer Multi-Omics Data Repositories
- GDC: The Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/) from the National Cancer Institute (NCI) is the largest scale consortium that enables the retrieval, download, comprehensive analysis and exploitation of multimodal cancer genomics studies. Within the GDC, scientists can access over 3 petabytes of data from programs like the NCI’s Clinical Proteomic Tumor Analysis Consortium (CPTAC), the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative and The Cancer Genome Atlas (TCGA). Both harmonized datasets and legacy data on older genome versions are available. Furthermore, it includes various data visualization tools to enhance the exploration of specific projects and cancer types, and available bioinformatics pipelines. Based on the latest data summary (October 27, 2020), this data-driven platform contains 67 projects, 68 different cancer types with more than 84 thousand cases, along with clinical data [32].
- ICGC: The International Cancer Genome Consortium was established in 2008 as an international effort to harmonize the large number of ongoing and future projects on cancer genomics. Members include the NIH, the Wellcome Trust Sanger Institute, Cancer Research UK, RIKEN, and many more. Its data portal (https://dcc.icgc.org/) currently holds 86 projects with more than 80 million somatic mutations (data release 28). Its flagship projects are the Pan-Cancer Analysis of Whole Genomes (PCAWG) and ARGO (Accelerating Research in Genomic Oncology). Launched in 2019, ARGO is the next phase of ICGC, which aims to uniformly analyze specimens from 100,000 cancer patients with high quality clinical data to address outstanding questions in genomic cancer research (https://www.icgc-argo.org/).
- PCAWG: The Pan-Cancer Analysis of Whole Genomes is the latest ICGC initiative with data released in 2020, and one of the biggest international collaborative studies, including 13 research institutes and more than 700 scientists from individual TCGA and ICGC working groups. The major aim of the PCAWG consortium is to identify common mutational patterns and to investigate the nature and consequences of somatic and germline mutations of 38 cancer types collected from 48 TCGA & ICGC projects. The project data is available from five major resources [33]:
- The ICGC Data Portal (https://dcc.icgc.org/repositories) as the main dissemination platform for ICGC
- UCSC Xena (https://xenabrowser.net/) is the main exploration tool for the included multi-omics resource data, in order to identify any putative correlations among all primary results. Additionally, it includes the possibility of performing survival analyses
- EBI Expression Atlas (https://www.ebi.ac.uk/gxa/) is an open science resource hub hosted by EMBL/EBI. It provides information about gene and protein expression across species and biological conditions such as different tissues, cell types, developmental stages and diseases.
- BSC PCAWG Scout (https://pcawgscout.bsc.es) is another analysis platform to visualize and explore PCAWG data. It consists of a portal that presents the original omics data and sample annotation along with the results from different analysis working-groups, whereas its main focus lies on providing information on driver mutations and resulting proteins.
- Chromothripsis Explorer (http://compbio.med.harvard.edu/chromothripsis/) is a tool that provides highly interactive Circos plots for all tumors in the PCAWG cohort. Each Circos plot reports the point mutations, small insertions and deletions, structural variations and copy number profiles detected in each tumor. On this premise, the user can exploit large-scale alterations such as chromosome arm deletions, and complex mutational patterns such as chromothripsis.
- CCLE: The Cancer Cell Line Encyclopedia (https://portals.broadinstitute.org/ccle) is an ongoing collaborative project between the Broad Institute and the Novartis Institutes for BioMedical Research. Established in 2008, its main goal is to conduct a thorough genetic and pharmacologic characterization of a large panel of 1457 cancer cell lines. Its aims are to capture the genomic heterogeneity of the preclinical models and link them with the molecular heterogeneity in cancer patients. Additionally, it can unravel clinically actionable molecular targets that might be associated with drug response and ultimately link them to cancer survival, enhancing personalized medicine. Collectively, the multi-omics cell lines dataset includes gene expression from microarray and RNA-Sequencing experiments, reverse-phase protein arrays, copy number, gene methylation and mutation data. In parallel, the database also stores legacy data, which include pharmacological profiles of 24 anticancer drugs across 504 cell lines. Besides data the web page also includes tools for data visualization, including box plots, scatter plots and bubble maps for methylation data [34].
- cBioPortal: The cBioPortal for Cancer Genomics (https://www.cbioportal.org/) is an open-source resource platform developed at Memorial Sloan Kettering Cancer Center, whereas the software is developed and maintained by various research institutes. Its main goal lies in the interrogation, interactive visualization and integrated analysis of clinical and complex multimodal cancer genomics datasets. While the major focus of the platform lies on genomic alterations (non-synonymous somatic mutations, DNA copy-number variations), it also hosts mRNA and microRNA expression, protein and phosphoprotein level data (RPPA or mass spectrometry based), DNA methylation and microbiome data, especially for TCGA data. Additionally to TCGA projects, cBioPortal includes other large-scale cancer genomics projects to advance translational cancer research, such as immunogenomic and pan-cancer studies. Overall, whereas cBioPortal is considered mainly as an exploratory analysis tool, GDC would be a more appropriate choice if the user requires full access to raw data from various cancer projects (TCGA, TARGET). Additionally, cBioPortal is currently using only data aligned to the hg19/GRCh37 reference genome, and it doesn’t provide normal tissue samples for any study [35,36].
- COSMIC: Besides cBioPortal, the Catalogue Of Somatic Mutations In Cancer (COSMIC)–developed at the Wellcome Sanger Institute—is the largest and most comprehensive resource for mining publicly available cancer sequence data, aiming to investigate the impact of somatic mutations on cancer progression and pathophysiology [37]. The latest version (COSMIC v. 92, August 2020) contains more than 37 million coding mutations and other clinical information from more than 1500 cancer types both on GRCh38 (hg38) and GRCh37 genomes. The impact of somatic variants can be summarized on various levels and across projects, such as clinical actionability (drug resistance), mutational processes associated with cancer progression (mutational signatures) and more. Furthermore, COSMIC includes the Cell Lines Project (CLP), a multidimensional dataset containing a detailed molecular characterization of more than 1000 cancer cell lines with copy number variation and gene expression data, including also other previously published moderate scale sequencing projects, such as the NCI-60 Human Tumor Cell Lines Screen dataset [38].
1.4. Platforms and Packages for Leveraging Multi-Omics Data Retrieval
1.5. Challenges Integrating Multi-Omics Experiments
1.6. Research Outlook: Single-Cell Multimodal Analysis
2. Results
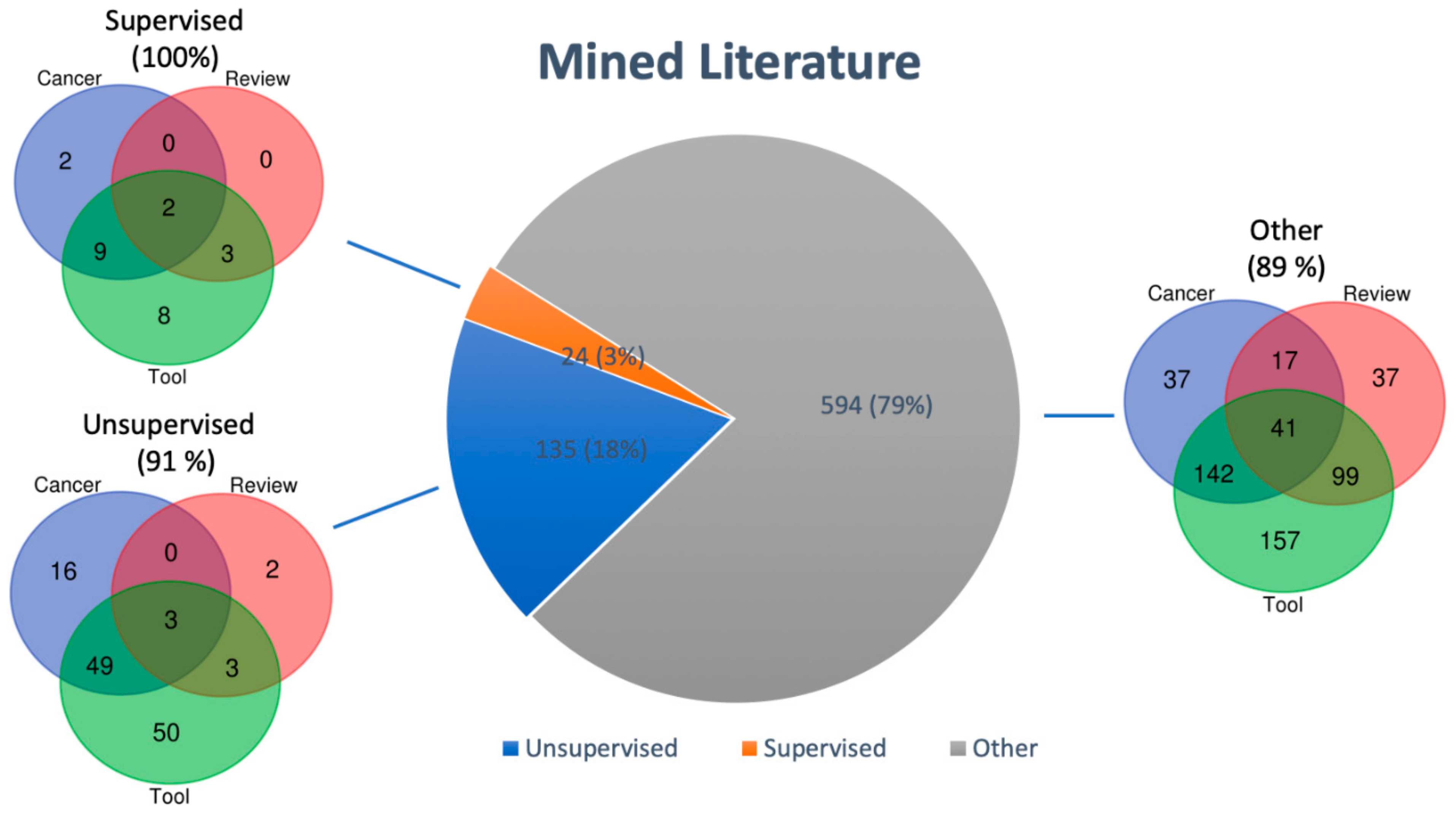
2.1. Literature Mining
2.2. Methodologies and Motivation
2.2.1. Patient Stratification
2.2.2. Biomarker Discovery
2.2.3. Pathway Analysis
2.2.4. Drug Analysis
2.2.5. Cancer Subtype Classification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Nature Orientation | Tool Name | Programming Language | Integration Method | Used Omics | Reference |
|---|---|---|---|---|---|
| Unsupervised | mixKernel | R | Multiple Kernel learning | mRNA, miRNA, MET | [136] |
| Unsupervised | iClusterBayes | R | Bayesian clustering | mRNA, CNV, MUT, MET | [137] |
| Unsupervised | SNF | R, Matlab | Network fusion | mRNA, miRNA, MET | [138] |
| Unsupervised | iCluster | R | Matrix factorization | mRNA, CNV | [139] |
| Unsupervised | iCluster Plus | Matlab | Matrix factorization | mRNA, CNV, MUT | [140] |
| Unsupervised | JIVE | Matlab | Matrixfactorization | mRNA, miRNA | [141] |
| Unsupervised | PSDF | Matlab | Bayesian | mRNAs, CNV | [142] |
| Unsupervised | BCC | R | Bayesian | mRNA, miRNA, MET, PROT | [143] |
| Unsupervised | SCFA | R | Multi-step analysis | mRNA, miRNA, MET | [144] |
| Supervised | MAUI | Python | Autoencoder | mRNA, CNV, MUT | [145] |
2.2.6. Multi-Omics Data Discovery
- Research aim: Firstly, the most important aspect is the research question: what is the purpose of the specific study, or which biological insights are aimed for regarding a specific cancer type or cohort? This can significantly inform the selection of the most suitable tools that match the specific research goal, as no single tool or pipeline covered above can address a complex disease like cancer in its entirety. In addition, except for the initial selection (i.e., unsupervised or supervised methodologies), benchmark studies can be further utilized and considered as guidelines for narrowing the candidate tools [156].
- Another crucial criterion is the experimental design and the interrogated datasets: which is the relative sample size? For example, usually ML-based approaches require a higher number of samples for model training and validation in comparison to unsupervised methodologies. Also, can the relative tool cope with the percentage of missing values and/or the nature of omics layers (continuous vs. sparse genetic data)? When a large percentage of missing values is present in both omic layers and clinical data, researchers should consider various published studies covering different methodologies for missing value imputation [157].
- Furthermore, the third selection criterion that is often underestimated is the presence of extensive documentation that accompanies an available tool. Despite the fact that an approach might be well suited for a specific research scenario, the absence of a rich vignette and detailed reproducible examples poses a significant constraint on the utilization of the respective methodology [158].
2.3. Rationale for Selection of Tools and Datasets
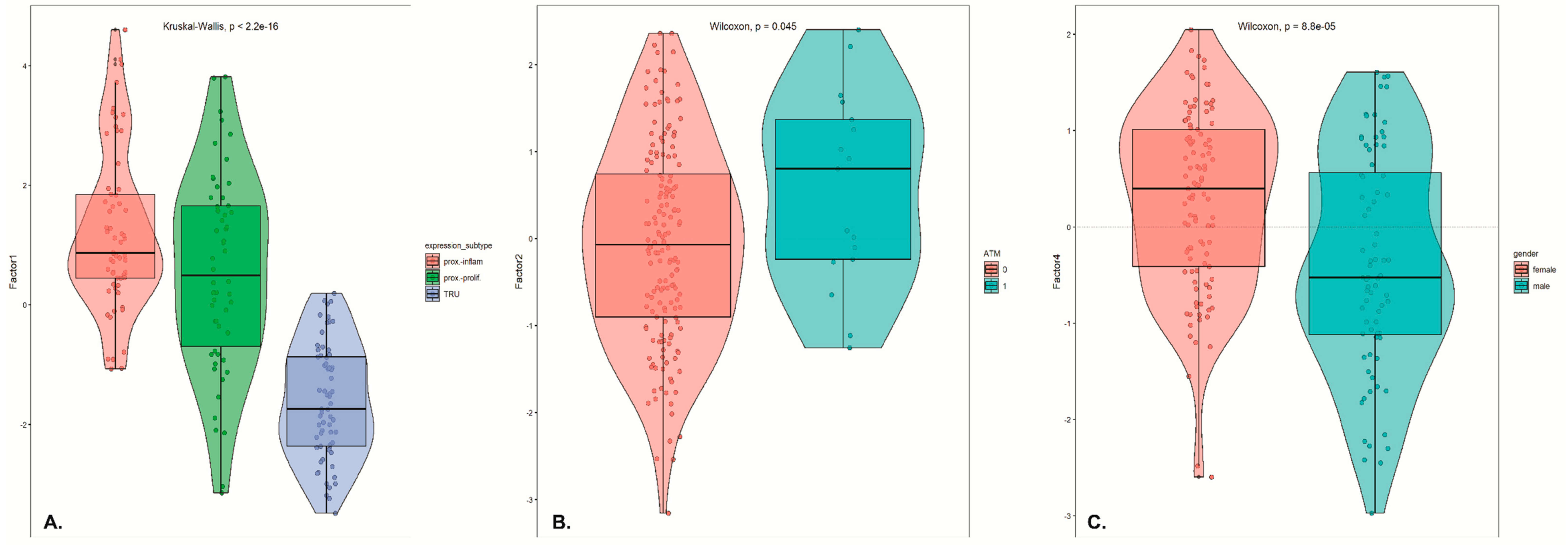
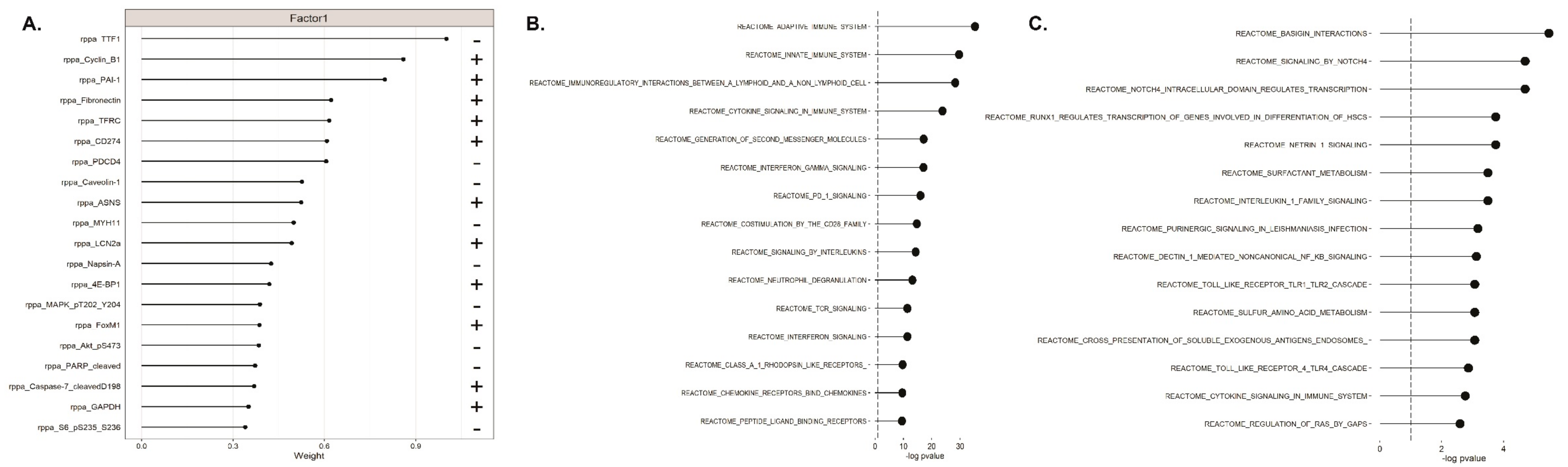
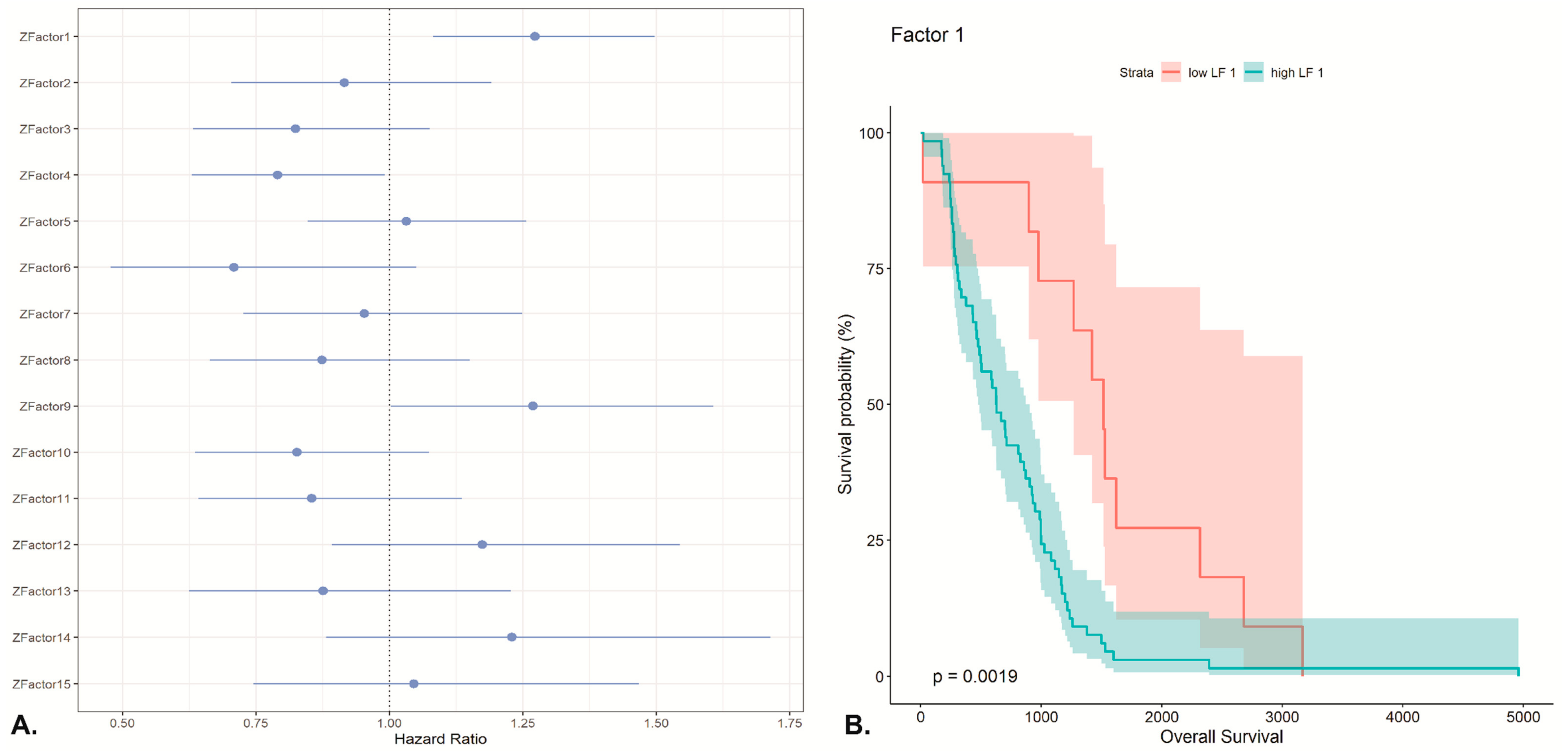
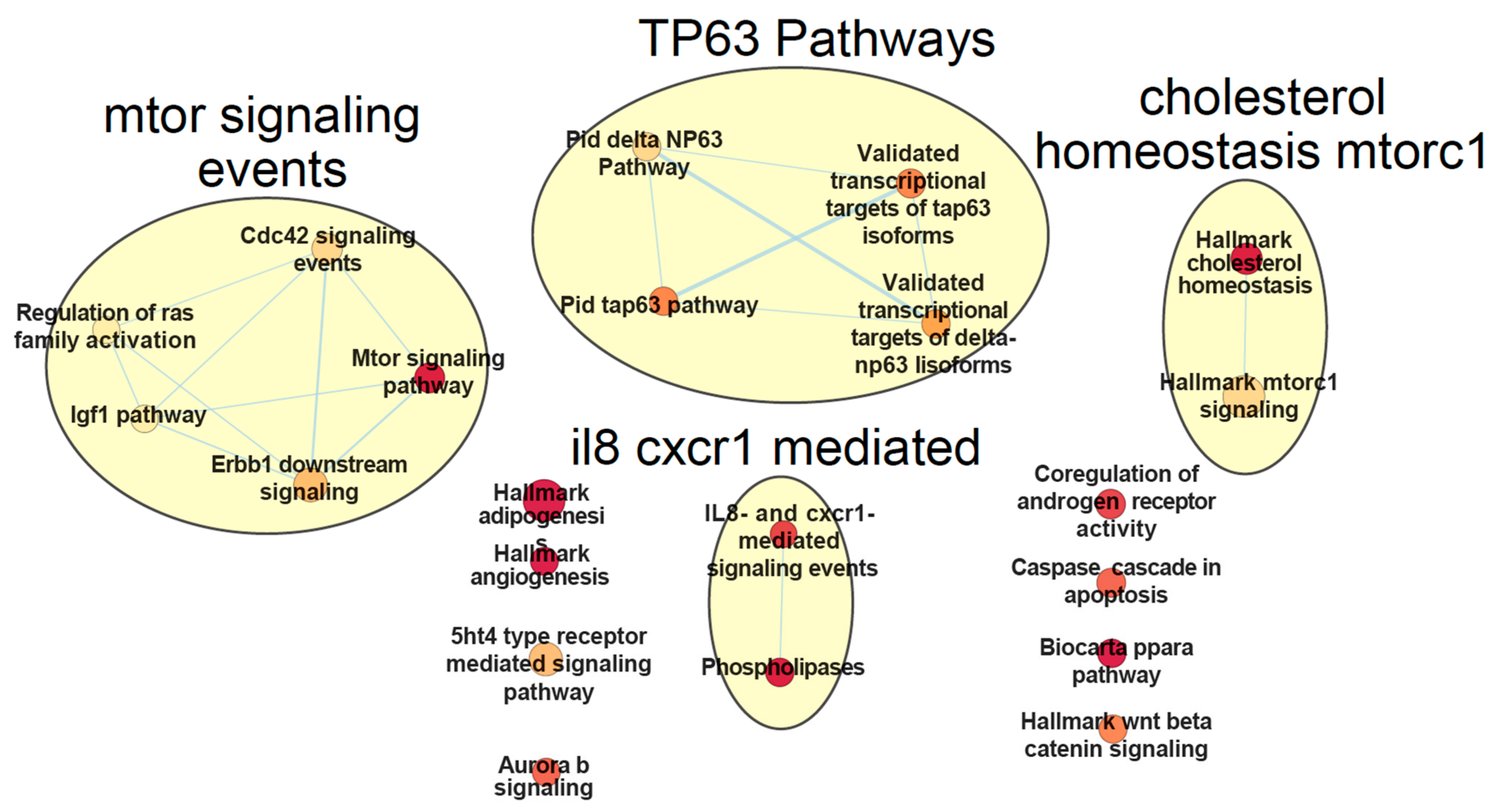
2.4. Unsupervised Multimodal Data Integration Case Study with MOFA
2.5. Supervised Multimodal Classification Case Study with netDx
3. Discussion
- Initially, one important aspect that influences the integration part includes the pre-processing steps prior to integrating any multimodal data: initially, appropriate normalization or transformation is essential to remove any technical confounders related to each omic data layer. For example, for count based data like RNA-Seq, size factor normalization and variance stabilization are generally recommended. Also, significant differences in size in at least one of the interrogated omics layers could inflate the data integration model to capture non-biological variation associated with this specific data layer, while downweighting more subtle sources of variation. In addition, it is well known that most genomic studies suffer from the “curse of dimensionality”, that is the number of features being substantially higher than the number of samples. Hence, a feature selection step like selecting the top most variable features per omic modality is essential, both in supervised and unsupervised approaches. However, filtering is not trivial especially when dealing with somatic mutations or copy number alterations, where a more “sophisticated” filtering is needed. Somatic mutations can be very sparse with the vast majority of cancer genes being of low prevalence, cancer-specific and not shared among all patients of the same cancer. Intratumoral diversification adds further complexity to the application of a simple reduction based on the frequency of no events [191]. Instead, clinical data portals with prior biological knowledge should be used along with computational frameworks to identify putative driver genes, aiming to reduce the CNV/somatic mutations feature space. A quality control step is critical to investigate the percentage and distribution of missing values relative to the number of total samples. In the near future, improvements of the human reference genome (GRCh38) could increase completeness of multi-omics studies. For example, applied telomere to telomere long-read sequencing has started to fill unresolved gaps in the human reference genome for the X Chromosome [192]. Further ongoing efforts will reveal new functional landscapes by creating a human pan-genome, which would include diverse sets of individuals in order to catch the genomic variation across different populations [193]. This will provide the opportunity to study genetic similarities and differences among human populations within genomic or multi-omics studies of complex diseases.
- Additionally, another crucial part lies in the biological interpretation of the integrative analysis: it is vital to associate any findings to molecular mechanisms and perturbed pathways, in order to identify any causal regulatory relationships between the profiled entities. For this purpose there are various recent tools and databases that perform pathway analysis and provide prior knowledge on molecular biology to construct intracellular communication networks well-suited for multi-omics functional annotation. These include but are not limited to SignaLink 2.0 [123], OmniPath [124,194], ReactomeGSA [195] and ActivePathways [196]. Moreover, incorporation of prior knowledge from clinical data portals could further facilitate the prioritization of features or signatures from multimodal studies, which could serve as putative biomarkers. A representative example is the Variant Interpretation for Cancer Consortium (VICC) meta-knowledgebase [197], a harmonized effort for cancer variant interpretation by encapsulating multiple different cancer variant annotation databases. VICC can be utilized for the validation of putative biomarkers from multi-omics cancer studies. Consequently, the development of multi-omics data integration methodologies that incorporate such prior biological knowledge should be enhanced as well, in order to delineate more readable causal networks between the perturbed omics in cancer manifestation. COSMOS (Causal Oriented Search of Multi-Omics Space) for example integrates phosphoproteomics, transcriptomics, and metabolomics data sets with prior knowledge such as protein-protein interactions to create hypotheses about causal links between signaling kinase cascades, transcriptional factors and metabolites [198].
- Finally, a researcher should strongly consider to follow specific protocols such as the FAIR guiding principles (findability, accessibility, interoperability, and reusability) [199] when publishing multi-omics cancer data, and to take into account important bioethics considerations when sharing cancer patient data [200].
4. Materials and Methods
4.1. Literature Review Workflow
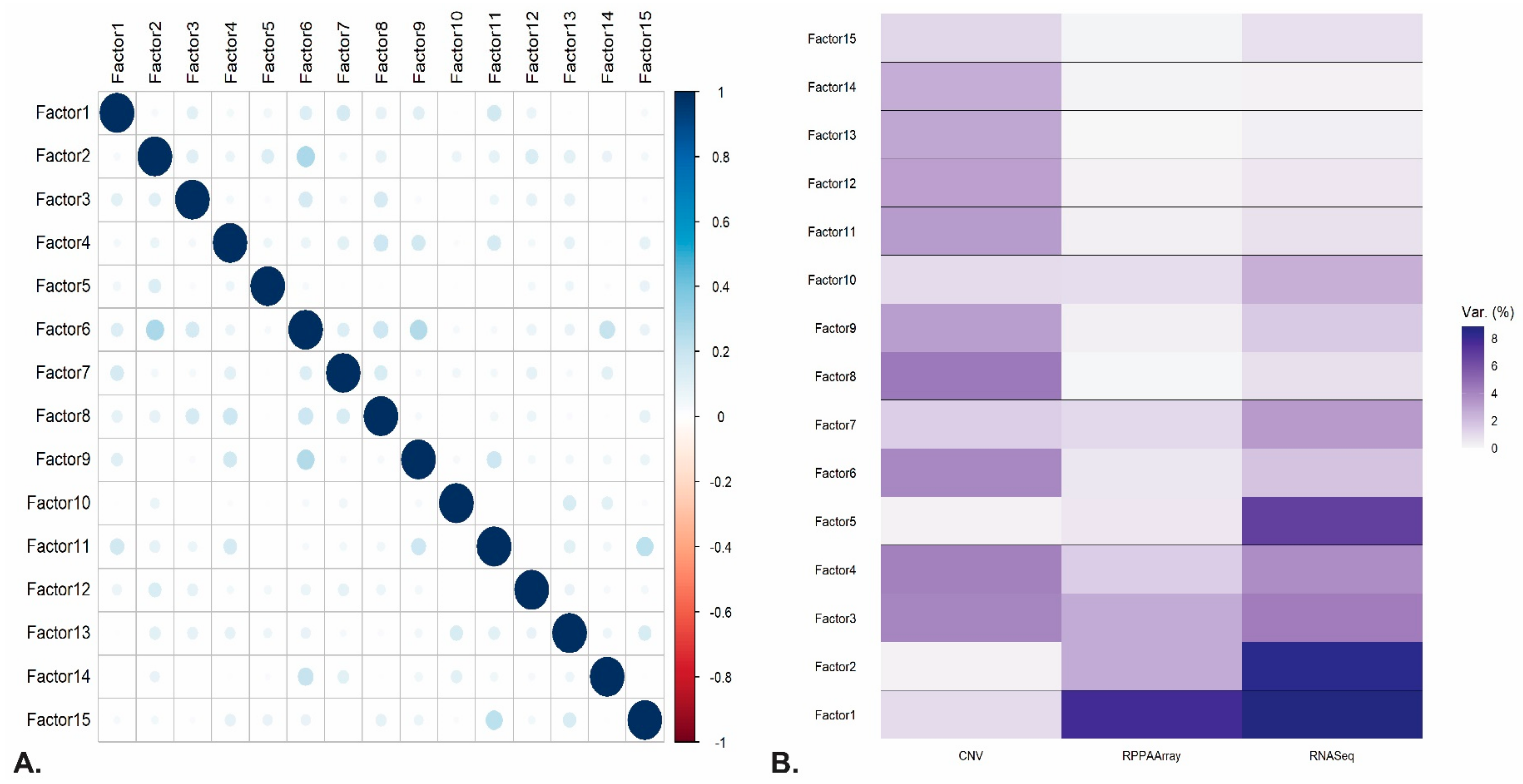
4.2. LUAD Dataset and MOFA Analysis
4.3. CLL Dataset and netDx Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oliver, S. Guilt-by-association goes global. Nature 2000, 403, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Bunnik, E.M.; Le Roch, K.G. An introduction to functional genomics and systems biology. Adv. Wound Care 2013, 2, 490–498. [Google Scholar] [CrossRef] [Green Version]
- Perakakis, N.; Yazdani, A.; Karniadakis, G.E.; Mantzoros, C. Omics, big data and machine learning as tools to propel understanding of biological mechanisms and to discover novel diagnostics and therapeutics. Metabolism 2018, 87, A1–A9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, A.D.; Landweber, L.F. What is a genome? PLoS Genet. 2016, 12, e1006181. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. Rna-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Timp, W.; Timp, G. Beyond mass spectrometry, the next step in proteomics. J. Sci. Adv. 2020, 6, eaax8978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.-Y.; Fauman, E.B.; Petersen, A.-K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.-P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stricker, S.H.; Köferle, A.; Beck, S. From profiles to function in epigenomics. Nat. Rev. Genet. 2017, 18, 51–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Org, E.; Parks, B.W.; Joo, J.W.; Emert, B.; Schwartzman, W.; Kang, E.Y.; Mehrabian, M.; Pan, C.; Knight, R.; Gunsalus, R.; et al. Genetic and environmental control of host-gut microbiota interactions. Genome Res. 2015, 25, 1558–1569. [Google Scholar] [CrossRef] [Green Version]
- Raghu, P. Functional diversity in a lipidome. Proc. Natl. Acad. Sci. USA 2020, 117, 11191–11193. [Google Scholar] [CrossRef]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association between brca1 and brca2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012, 307, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385.e318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, S.; Lin, S.; Li, Y.; Zhao, W.; Mills, G.B.; Sahni, N. Functional variomics and network perturbation: Connecting genotype to phenotype in cancer. Nat. Rev. Genet. 2017, 18, 395–410. [Google Scholar] [CrossRef]
- Francies, H.E.; McDermott, U.; Garnett, M.J. Genomics-guided pre-clinical development of cancer therapies. Nat. Cancer 2020, 1, 482–492. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef]
- Rheinbay, E. The genomic landscape of advanced cancer. Nat. Cancer 2020, 1, 372–373. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Ebrahim, A.; Brunk, E.; Tan, J.; O’Brien, E.J.; Kim, D.; Szubin, R.; Lerman, J.A.; Lechner, A.; Sastry, A.; Bordbar, A.; et al. Multi-omic data integration enables discovery of hidden biological regularities. Nat. Commun. 2016, 7, 13091. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, E.; Han, J.; Luo, W.; Zhou, J.; An, L.; Liu, J.; Zhang, H.H.; Li, H. On fusion methods for knowledge discovery from multi-omics datasets. Comput. Struct. Biotechnol. J. 2020, 18, 509–517. [Google Scholar] [CrossRef]
- Shannon, C.P.; Blimkie, T.M.; Ben-Othman, R.; Gladish, N.; Amenyogbe, N.; Drissler, S.; Edgar, R.D.; Chan, Q.; Krajden, M.; Foster, L.J.; et al. Multi-omic data integration allows baseline immune signatures to predict hepatitis b vaccine response in a small cohort. Front. Immunol. 2020, 11, 578801. [Google Scholar] [CrossRef]
- Wang, B.; Lunetta, K.L.; Dupuis, J.; Lubitz, S.A.; Trinquart, L.; Yao, L.; Ellinor, P.T.; Benjamin, E.J.; Lin, H. Integrative omics approach to identifying genes associated with atrial fibrillation. Circ. Res. 2020, 126, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Jiang, H.; Liang, Z.H.; Ju, H. Integrating multi-omics data to identify novel disease genes and single-neucleotide polymorphisms. Front. Genet. 2019, 10, 1336. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.G.; Choi, J.-H.; Yoon, S.; Jee, B.A.; Cho, E.J.; Lee, J.-H.; Yu, S.J.; Yoon, J.-H.; Yi, N.-J.; Lee, K.-W.; et al. Integrative analysis of genomic and epigenomic regulation of the transcriptome in liver cancer. Nat. Commun. 2017, 8, 839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Song, N.; Shen, R.; Arora, A.; Machiela, M.J.; Song, L.; Landi, M.T.; Ghosh, D.; Chatterjee, N.; Baladandayuthapani, V.; et al. Integrating clinical and multiple omics data for prognostic assessment across human cancers. Sci. Rep. 2017, 7, 16954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlandi, E.; Iacovelli, N.A.; Tombolini, V.; Rancati, T.; Polimeni, A.; De Cecco, L.; Valdagni, R.; De Felice, F. Potential role of microbiome in oncogenesis, outcome prediction and therapeutic targeting for head and neck cancer. Oral Oncol. 2019, 99, 104453. [Google Scholar] [CrossRef] [Green Version]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Lee, R.J.; Kalinich, M.; LiCausi, J.A.; Zheng, Y.; Chen, T.; Milner, J.D.; Emmons, E.; Ho, U.; Broderick, K.; et al. An rna-based digital circulating tumor cell signature is predictive of drug response and early dissemination in prostate cancer. J. Cancer Discov. 2018, 8, 288–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, T.; Andrieux, G.; Ahmed, M.; Chakraborty, S. Integration of online omics-data resources for cancer research. Front. Genet. 2020, 11, 578345. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. J. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. J. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. Cosmic: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abaan, O.D.; Polley, E.C.; Davis, S.R.; Zhu, Y.J.; Bilke, S.; Walker, R.L.; Pineda, M.; Gindin, Y.; Jiang, Y.; Reinhold, W.C.; et al. The exomes of the nci-60 panel: A genomic resource for cancer biology and systems pharmacology. J. Cancer Res. 2013, 73, 4372–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the field in multi-omics research: From computational needs to data mining and sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Ghemawat, S.; et al. Tensorflow: Large-scale machine learning on heterogeneous systems. arXiv 2016, arXiv:1603.04467. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Kileen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. Pytorch: An imperative style, high-performance deep learning library. In Advances in Neural Information Processing Systems; Curran Associates Inc.: New York, NY, USA, 2019; Volume 32, pp. 8024–8035. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M. Genomic Data Commons: NIH/NCI Genomic Data Commons Access; Bioconductor: Seattle, WA, USA, 2016. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. Tcgabiolinks: An r/bioconductor package for integrative analysis of tcga data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.; Waldron, L.; Schiffer, L.; Geistlinger, L.; Obenchain, V.; Morgan, M. Curatedtcgadata: Curated Data from the Cancer Genome Atlas (TCGA) as Multiassay Experiment Objects, Bioconductor: Seattle, WA, USA, 2020. [CrossRef]
- Ramos, M.; Geistlinger, L.; Oh, S.; Schiffer, L.; Azhar, R.; Kodali, H.; de Bruijn, I.; Gao, J.; Carey, V.J.; Morgan, M.; et al. Multiomic integration of public oncology databases in bioconductor. JCO Clin. Cancer Inform. 2020, 4, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.; Schiffer, L.; Re, A.; Azhar, R.; Basunia, A.; Rodriguez, C.; Chan, T.; Chapman, P.; Davis, S.R.; Gomez-Cabrero, D.; et al. Software for the integration of multiomics experiments in bioconductor. J. Cancer Res. 2017, 77, e39–e42. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Amrollahi, F.; Amgad, M.; Dong, C.; Lewis, J.E.; Song, C.; Gutman, D.A.; Halani, S.H.; Velazquez Vega, J.E.; Brat, D.J.; et al. Predicting clinical outcomes from large scale cancer genomic profiles with deep survival models. Sci. Rep. 2017, 7, 11707. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. The ucsc xena platform for public and private cancer genomics data visualization and interpretation. bioRvix 2019, 326470. [Google Scholar]
- Nhat, T.; pyup.io bot; DeepSource Bot; Moss, S. Biomecis-Lab/Openomics: Bug Fixes from Pyopensci Reviewer 2, Zenodo: Meyrin, Switzerland, 2021.
- Nalisnik, M.; Amgad, M.; Lee, S.; Halani, S.H.; Velazquez Vega, J.E.; Brat, D.J.; Gutman, D.A.; Cooper, L.A.D. Interactive phenotyping of large-scale histology imaging data with histomicsml. Sci. Rep. 2017, 7, 14588. [Google Scholar] [CrossRef] [Green Version]
- Levings, D.C.; Wang, X.; Kohlhase, D.; Bell, D.A.; Slattery, M. A distinct class of antioxidant response elements is consistently activated in tumors with nrf2 mutations. Redox Biol. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Giwa, A.; Fatai, A.; Gamieldien, J.; Christoffels, A.; Bendou, H. Identification of novel prognostic markers of survival time in high-risk neuroblastoma using gene expression profiles. Oncotarget 2020, 11, 4293–4305. [Google Scholar] [CrossRef] [PubMed]
- Ostrovsky, A.; Hillman-Jackson, J.; Bouvier, D.; Clements, D.; Afgan, E.; Blankenberg, D.; Schatz, M.C.; Nekrutenko, A.; Taylor, J.; Team, t.G.; et al. Using galaxy to perform large-scale interactive data analyses—an update. Curr. Protoc. 2021, 1, e31. [Google Scholar]
- McGowan, T.; Johnson, J.E.; Kumar, P.; Sajulga, R.; Mehta, S.; Jagtap, P.D.; Griffin, T.J. Multi-omics visualization platform: An extensible galaxy plug-in for multi-omics data visualization and exploration. GigaScience 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef]
- Haas, R.; Zelezniak, A.; Iacovacci, J.; Kamrad, S.; Townsend, S.; Ralser, M. Designing and interpreting ‘multi-omic’ experiments that may change our understanding of biology. Curr. Opin. Syst. Biol. 2017, 6, 37–45. [Google Scholar] [CrossRef]
- Iacovacci, J.; Peluso, A.; Ebbels, T.; Ralser, M.; Glen, R.C. Extraction and integration of genetic networks from short-profile omic data sets. Metabolites 2020, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Vitrinel, B.; Koh, H.W.L.; Mujgan Kar, F.; Maity, S.; Rendleman, J.; Choi, H.; Vogel, C. Exploiting interdata relationships in next-generation proteomics analysis *. Mol. Cell. Proteom. 2019, 18, S5–S14. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.S.; Kester, L.; Spanjaard, B.; Bienko, M.; van Oudenaarden, A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015, 33, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M.; et al. G&t-seq: Parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar]
- Reyes, M.; Billman, K.; Hacohen, N.; Blainey, P.C. Simultaneous profiling of gene expression and chromatin accessibility in single cells. Adv. Biosyst. 2019, 3, 1900065. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, Y.; Zhang, Y.; Kubo, N.; Yu, M.; Fang, R.; Kellis, M.; Ren, B. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods 2019, 16, 991–993. [Google Scholar] [CrossRef]
- Lee, D.-S.; Luo, C.; Zhou, J.; Chandran, S.; Rivkin, A.; Bartlett, A.; Nery, J.R.; Fitzpatrick, C.; O’Connor, C.; Dixon, J.R.; et al. Simultaneous profiling of 3d genome structure and DNA methylation in single human cells. Nat. Methods 2019, 16, 999–1006. [Google Scholar] [CrossRef]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R.; et al. Perturb-seq: Dissecting molecular circuits with scalable single-cell rna profiling of pooled genetic screens. Cell 2016, 167, 1853–1866.e1817. [Google Scholar] [CrossRef] [Green Version]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nuñez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y.; et al. A multiplexed single-cell crispr screening platform enables systematic dissection of the unfolded protein response. Cell 2016, 167, 1867–1882.e1821. [Google Scholar] [CrossRef] [Green Version]
- Jaitin, D.A.; Weiner, A.; Yofe, I.; Lara-Astiaso, D.; Keren-Shaul, H.; David, E.; Salame, T.M.; Tanay, A.; van Oudenaarden, A.; Amit, I. Dissecting immune circuits by linking crispr-pooled screens with single-cell rna-seq. Cell 2016, 167, 1883–1896.e1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Preissl, S.; Ren, B. Single-cell multimodal omics: The power of many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef]
- Ma, A.; McDermaid, A.; Xu, J.; Chang, Y.; Ma, Q. Integrative methods and practical challenges for single-cell multi-omics. Trends Biotechnol. 2020, 38, 1007–1022. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-cell multiomics: Technologies and data analysis methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in idh-mutant gliomas by single-cell rna-seq. J. Sci. 2017, 355, eaai8478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Dong, X.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in b lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. USA 2019, 116, 9014–9019. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dang, M.; Harada, K.; Han, G.; Wang, F.; Pool Pizzi, M.; Zhao, M.; Tatlonghari, G.; Zhang, S.; Hao, D.; et al. Single-cell dissection of intratumoral heterogeneity and lineage diversity in metastatic gastric adenocarcinoma. Nat. Med. 2021, 27, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 2020, 182, 497–514.e422. [Google Scholar] [CrossRef]
- Efremova, M.; Teichmann, S.A. Computational methods for single-cell omics across modalities. Nat. Methods 2020, 17, 14–17. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902.e1821. [Google Scholar] [CrossRef]
- Samir, J.; Rizzetto, S.; Gupta, M.; Luciani, F. Exploring and analysing single cell multi-omics data with vdjview. BMC Med. Genom. 2020, 13, 29. [Google Scholar] [CrossRef] [Green Version]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, D.J.; Campbell, K.R.; Lun, A.T.; Wills, Q.F. Scater: Pre-processing, quality control, normalization and visualization of single-cell rna-seq data in r. Bioinformatics 2017, 33, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, V.Y.; Kirschner, K.; Schaub, M.T.; Andrews, T.; Yiu, A.; Chandra, T.; Natarajan, K.N.; Reik, W.; Barahona, M.; Green, A.R.; et al. Sc3: Consensus clustering of single-cell rna-seq data. Nat. Methods 2017, 14, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, J.D.; Hartemink, A.J.; Prins, J.F. Matcher: Manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol. 2017, 18, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Huang, Y.; Singh, R.; Vert, J.-P.; Noble, W.S. Jointly embedding multiple single-cell omics measurements. BioRxiv 2019, 644310. [Google Scholar] [CrossRef]
- Simidjievski, N.; Bodnar, C.; Tariq, I.; Scherer, P.; Andres Terre, H.; Shams, Z.; Jamnik, M.; Liò, P. Variational autoencoders for cancer data integration: Design principles and computational practice. Front. Genet. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K.R.; Steif, A.; Laks, E.; Zahn, H.; Lai, D.; McPherson, A.; Farahani, H.; Kabeer, F.; O’Flanagan, C.; Biele, J.; et al. Clonealign: Statistical integration of independent single-cell rna and DNA sequencing data from human cancers. Genome Biol. 2019, 20, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Sun, Z.; Zhang, Y.; Xu, Z.; Xin, H.; Huang, H.; Duerr, R.H.; Chen, K.; Ding, Y.; Chen, W. Brem-sc: A bayesian random effects mixture model for joint clustering single cell multi-omics data. Nucleic Acids Res. 2020, 48, 5814–5824. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. J. Sci. 2018, 361, 1380–1385. [Google Scholar] [CrossRef] [Green Version]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018, 36, 70–80. [Google Scholar] [CrossRef]
- Edsgärd, D.; Johnsson, P.; Sandberg, R. Identification of spatial expression trends in single-cell gene expression data. Nat. Methods 2018, 15, 339–342. [Google Scholar] [CrossRef]
- Svensson, V.; Teichmann, S.A.; Stegle, O. Spatialde: Identification of spatially variable genes. Nat. Methods 2018, 15, 343–346. [Google Scholar] [CrossRef]
- Zuo, C.; Chen, L. Deep-joint-learning analysis model of single cell transcriptome and open chromatin accessibility data. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Yang, K.D.; Belyaeva, A.; Venkatachalapathy, S.; Damodaran, K.; Katcoff, A.; Radhakrishnan, A.; Shivashankar, G.V.; Uhler, C. Multi-domain translation between single-cell imaging and sequencing data using autoencoders. Nat. Commun. 2021, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Setty, M.; Tadmor, M.D.; Reich-Zeliger, S.; Angel, O.; Salame, T.M.; Kathail, P.; Choi, K.; Bendall, S.; Friedman, N.; Pe’er, D. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol. 2016, 34, 637–645. [Google Scholar] [CrossRef]
- Welch, J.D.; Kozareva, V.; Ferreira, A.; Vanderburg, C.; Martin, C.; Macosko, E.Z. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 2019, 117, 1873–1887.e1817. [Google Scholar] [CrossRef] [PubMed]
- Duren, Z.; Chen, X.; Zamanighomi, M.; Zeng, W.; Satpathy, A.T.; Chang, H.Y.; Wang, Y.; Wong, W.H. Integrative analysis of single-cell genomics data by coupled nonnegative matrix factorizations. Proc. Natl. Acad. Sci. USA 2018, 115, 7723–7728. [Google Scholar] [CrossRef] [Green Version]
- Argelaguet, R.; Arnol, D.; Bredikhin, D.; Deloro, Y.; Velten, B.; Marioni, J.C.; Stegle, O. Mofa+: A statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol 2020, 21, 111. [Google Scholar] [CrossRef]
- Duan, B.; Zhou, C.; Zhu, C.; Yu, Y.; Li, G.; Zhang, S.; Zhang, C.; Ye, X.; Ma, H.; Qu, S.; et al. Model-based understanding of single-cell crispr screening. Nat. Commun. 2019, 10, 2233. [Google Scholar] [CrossRef]
- KwameForbes. Integratewithsinglecell. Available online: https://rdrr.io/bioc/DESeq2/man/integrateWithSingleCell.html (accessed on 4 March 2021).
- Huang, S.; Chaudhary, K.; Garmire, L.X. More is better: Recent progress in multi-omics data integration methods. Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [Green Version]
- Nicora, G.; Vitali, F.; Dagliati, A.; Geifman, N.; Bellazzi, R. Integrated multi-omics analyses in oncology: A review of machine learning methods and tools. Front. Oncol 2020, 10, 1030. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.J.; Lock, E.F.R. Jive for exploration of multi-source molecular data. Bioinformatics 2016, 32, 2877–2879. [Google Scholar] [CrossRef] [PubMed]
- Beal, J.; Montagud, A.; Traynard, P.; Barillot, E.; Calzone, L. Personalization of logical models with multi-omics data allows clinical stratification of patients. Front. Physiol 2018, 9, 1965. [Google Scholar] [CrossRef]
- Huang, Z.; Zhan, X.; Xiang, S.; Johnson, T.S.; Helm, B.; Yu, C.Y.; Zhang, J.; Salama, P.; Rizkalla, M.; Han, Z.; et al. Salmon: Survival analysis learning with multi-omics neural networks on breast cancer. Front. Genet. 2019, 10, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, S.; Hui, S.; Isserlin, R.; Shah, M.A.; Kaka, H.; Bader, G.D. Netdx: Interpretable patient classification using integrated patient similarity networks. Mol. Syst. Biol. 2019, 15, e8497. [Google Scholar] [CrossRef] [PubMed]
- Velten, B.; Huber, W. Adaptive penalization in high-dimensional regression and classification with external covariates using variational bayes. Biostatistics 2019. [Google Scholar] [CrossRef] [Green Version]
- Vlachavas, E.-I.; Pilalis, E.; Papadodima, O.; Koczan, D.; Willis, S.; Klippel, S.; Cheng, C.; Pan, L.; Sachpekidis, C.; Pintzas, A.; et al. Radiogenomic analysis of f-18-fluorodeoxyglucose positron emission tomography and gene expression data elucidates the epidemiological complexity of colorectal cancer landscape. Comput. Struct. Biotechnol. J. 2019, 17, 177–185. [Google Scholar] [CrossRef] [PubMed]
- González-Reymúndez, A.; Vázquez, A.I. Multi-omic signatures identify pan-cancer classes of tumors beyond tissue of origin. Sci. Rep. 2020, 10, 8341. [Google Scholar] [CrossRef]
- Ray, P.; Zheng, L.; Lucas, J.; Carin, L. Bayesian joint analysis of heterogeneous genomics data. Bioinformatics 2014, 30, 1370–1376. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Ji, J.; Gleason, K.J.; Yang, F.; Martignetti, J.A.; Chen, L.S.; Wang, P. Insights into impact of DNA copy number alteration and methylation on the proteogenomic landscape of human ovarian cancer via a multi-omics integrative analysis. Mol. Cell Proteom. 2019, 18, S52–S65. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Zhang, J.; Li, J.; Calhoun, V.D.; Deng, H.W.; Wang, Y.P. Group sparse canonical correlation analysis for genomic data integration. Bmc Bioinform. 2013, 14, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, S.; Liu, C.C.; Zhou, X.J. Identifying multi-layer gene regulatory modules from multi-dimensional genomic data. Bioinformatics 2012, 28, 2458–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louhimo, R.; Hautaniemi, S. Cnamet: An r package for integrating copy number, methylation and expression data. Bioinformatics 2011, 27, 887–888. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Baladandayuthapani, V.; Morris, J.S.; Broom, B.M.; Manyam, G.; Do, K.A. Ibag: Integrative bayesian analysis of high-dimensional multiplatform genomics data. Bioinformatics 2013, 29, 149–159. [Google Scholar] [CrossRef]
- Ovaska, K.; Laakso, M.; Haapa-Paananen, S.; Louhimo, R.; Chen, P.; Aittomaki, V.; Valo, E.; Nunez-Fontarnau, J.; Rantanen, V.; Karinen, S.; et al. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2010, 2, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Zheng, Y.; Huang, D.S. Capsule network based modeling of multi-omics data for discovery of breast cancer-related genes. IEEE Acm Trans. Comput. Biol. Bioinform 2020, 17, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2019, 48, D498–D503. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodchenkov, I.; Babur, O.; Luna, A.; Aksoy, B.A.; Wong, J.V.; Fong, D.; Franz, M.; Siper, M.C.; Cheung, M.; Wrana, M.; et al. Pathway commons 2019 update: Integration, analysis and exploration of pathway data. Nucleic Acids Res. 2019, 48, D489–D497. [Google Scholar] [CrossRef] [Green Version]
- Fazekas, D.; Koltai, M.; Türei, D.; Módos, D.; Pálfy, M.; Dúl, Z.; Zsákai, L.; Szalay-Bekő, M.; Lenti, K.; Farkas, I.J.; et al. Signalink 2—a signaling pathway resource with multi-layered regulatory networks. BMC Syst. Biol. 2013, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Türei, D.; Valdeolivas, A.; Gul, L.; Palacio-Escat, N.; Ivanova, O.; Gábor, A.; Módos, D.; Korcsmáros, T.; Saez-Rodriguez, J. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. bioRvix 2020. [Google Scholar] [CrossRef]
- Amar, D.; Shamir, R. Constructing module maps for integrated analysis of heterogeneous biological networks. Nucleic. Acids Res. 2014, 42, 4208–4219. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakopoulos, C.; Hindupur, S.K.; Hafliger, L.; Behr, J.; Montazeri, H.; Hall, M.N.; Beerenwinkel, N. Network-based integration of multi-omics data for prioritizing cancer genes. Bioinformatics 2018, 34, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, Q.; Liu, J.; Zhou, X.J. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microrna-gene regulatory modules. Bioinformatics 2011, 27, i401–i409. [Google Scholar] [CrossRef]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using paradigm. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef]
- Seoane, J.A.; Day, I.N.M.; Gaunt, T.R.; Campbell, C. A pathway-based data integration framework for prediction of disease progression. Bioinformatics 2013, 30, 838–845. [Google Scholar] [CrossRef] [Green Version]
- Savage, S.R.; Shi, Z.; Liao, Y.; Zhang, B. Graph algorithms for condensing and consolidating gene set analysis results. Mol. Cell Proteom. 2019, 18, S141–S152. [Google Scholar] [CrossRef] [Green Version]
- Martini, P.; Chiogna, M.; Calura, E.; Romualdi, C. Mosclip: Multi-omic and survival pathway analysis for the identification of survival associated gene and modules. Nucleic Acids Res. 2019, 47, e80. [Google Scholar] [CrossRef] [Green Version]
- Lawson, J.T.; Smith, J.P.; Bekiranov, S.; Garrett-Bakelman, F.E.; Sheffield, N.C. Cocoa: Coordinate covariation analysis of epigenetic heterogeneity. Genome Biol 2020, 21, 240. [Google Scholar] [CrossRef]
- Turanli, B.; Karagoz, K.; Bidkhori, G.; Sinha, R.; Gatza, M.L.; Uhlen, M.; Mardinoglu, A.; Arga, K.Y. Multi-omic data interpretation to repurpose subtype specific drug candidates for breast cancer. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Noghabi, H.; Zolotareva, O.; Collins, C.C.; Ester, M. Moli: Multi-omics late integration with deep neural networks for drug response prediction. Bioinformatics 2019, 35, i501–i509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhang, S. Integrative analysis for identifying joint modular patterns of gene-expression and drug-response data. Bioinformatics 2016, 32, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Mariette, J.; Villa-Vialaneix, N. Unsupervised multiple kernel learning for heterogeneous data integration. Bioinformatics 2018, 34, 1009–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, Q.; Shen, R.; Guo, C.; Vannucci, M.; Chan, K.S.; Hilsenbeck, S.G. A fully bayesian latent variable model for integrative clustering analysis of multi-type omics data. Biostatistics 2018, 19, 71–86. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Mo, Q.; Wang, S.; Seshan, V.E.; Olshen, A.B.; Schultz, N.; Sander, C.; Powers, R.S.; Ladanyi, M.; Shen, R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. USA 2013, 110, 4245–4250. [Google Scholar] [CrossRef] [Green Version]
- Lock, E.F.; Hoadley, K.A.; Marron, J.S.; Nobel, A.B. Joint and individual variation explained (jive) for integrated analysis of multiple data types. Ann. Appl Stat. 2013, 7, 523–542. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Savage, R.S.; Markowetz, F. Patient-specific data fusion defines prognostic cancer subtypes. PloS Comput. Biol. 2011, 7, e1002227. [Google Scholar] [CrossRef] [Green Version]
- Lock, E.F.; Dunson, D.B. Bayesian consensus clustering. Bioinformatics 2013, 29, 2610–2616. [Google Scholar] [CrossRef]
- Tran, D.; Nguyen, H.; Le, U.; Bebis, G.; Luu, H.N.; Nguyen, T. A novel method for cancer subtyping and risk prediction using consensus factor analysis. Front. Oncol 2020, 10, 1052. [Google Scholar] [CrossRef]
- Ronen, J.; Hayat, S.; Akalin, A. Evaluation of colorectal cancer subtypes and cell lines using deep learning. Life Sci. Alliance 2019, 2, e201900517. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.; Kuster, B.; Culhane, A.C.; Gholami, A.M. A multivariate approach to the integration of multi-omics datasets. BMC Bioinform. 2014, 15, 162. [Google Scholar] [CrossRef] [Green Version]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. Mixomics: An r package for ‘omics feature selection and multiple data integration. PloS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, E.; Calzone, L.; Michoel, T. Integrative multi-omics module network inference with lemon-tree. PloS Comput. Biol. 2015, 11, e1003983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabasova, E.; Reid, J.; Wernisch, L. Clusternomics: Integrative context-dependent clustering for heterogeneous datasets. PloS Comput. Biol. 2017, 13, e1005781. [Google Scholar] [CrossRef] [Green Version]
- Champion, M.; Brennan, K.; Croonenborghs, T.; Gentles, A.J.; Pochet, N.; Gevaert, O. Module analysis captures pancancer genetically and epigenetically deregulated cancer driver genes for smoking and antiviral response. EBioMedicine 2018, 27, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.W.L.; Fermin, D.; Vogel, C.; Choi, K.P.; Ewing, R.M.; Choi, H. Iomicspass: Network-based integration of multiomics data for predictive subnetwork discovery. Npj. Syst. Biol. Appl. 2019, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.; Basunia, A.; Peters, B.; Gholami, A.M.; Kuster, B.; Culhane, A.C. Mogsa: Integrative single sample gene-set analysis of multiple omics data. Mol. Cell Proteom. 2019, 18, S153–S168. [Google Scholar] [CrossRef] [Green Version]
- Lemsara, A.; Ouadfel, S.; Frohlich, H. Pathme: Pathway based multi-modal sparse autoencoders for clustering of patient-level multi-omics data. BMC Bioinform. 2020, 21, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Brunell, D.; Stephan, C.; Mancuso, J.; Yu, X.; He, B.; Thompson, T.C.; Zinner, R.; Kim, J.; Davies, P.; et al. Driver network as a biomarker: Systematic integration and network modeling of multi-omics data to derive driver signaling pathways for drug combination prediction. Bioinformatics 2019, 35, 3709–3717. [Google Scholar] [CrossRef] [Green Version]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-omics factor analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Velten, B.; Braunger, J.M.; Arnol, D.; Argelaguet, R.; Stegle, O. Identifying temporal and spatial patterns of variation from multi-modal data using mefisto. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cantini, L.; Zakeri, P.; Hernandez, C.; Naldi, A.; Thieffry, D.; Remy, E.; Baudot, A. Benchmarking joint multi-omics dimensionality reduction approaches for the study of cancer. Nat. Commun. 2021, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Greenbaum, J.; Luttrell, J.; Zhou, W.; Wu, C.; Shen, H.; Gong, P.; Zhang, C.; Deng, H.-W. A review of integrative imputation for multi-omics datasets. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Kumuthini, J.; Chimenti, M.; Nahnsen, S.; Peltzer, A.; Meraba, R.; McFadyen, R.; Wells, G.; Taylor, D.; Maienschein-Cline, M.; Li, J.-L.; et al. Ten simple rules for providing effective bioinformatics research support. PloS Comput. Biol. 2020, 16, e1007531. [Google Scholar] [CrossRef]
- Alcala, N.; Leblay, N.; Gabriel, A.A.G.; Mangiante, L.; Hervas, D.; Giffon, T.; Sertier, A.S.; Ferrari, A.; Derks, J.; Ghantous, A.; et al. Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nat. Commun. 2019, 10, 3407. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.C.H.; Sterne-Weiler, T.; Morris, Q.; Weatheritt, R.J.; Blencowe, B.J. Differential contribution of transcriptomic regulatory layers in the definition of neuronal identity. Nat. Commun. 2021, 12, 335. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, P.; Verstockt, B.; Cremer, J.; Verstockt, S.; Sabino, J.; Ferrante, M.; Vermeire, S. Understanding the molecular drivers of disease heterogeneity in crohn’s disease using multi-omic data integration and network analysis. Inflamm. Bowel Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Forcato, M.; Romano, O.; Bicciato, S. Computational methods for the integrative analysis of single-cell data. Brief. Bioinform. 2020, 22, 20–29. [Google Scholar] [CrossRef]
- Pai, S.; Weber, P.; Isserlin, R.; Kaka, H.; Hui, S.; Shah, M.; Giudice, L.; Giugno, R.; Nøhr, A.; Baumbach, J.; et al. Netdx: Software for building interpretable patient classifiers by multi-’omic data integration using patient similarity networks [version 2; peer review: 2 approved with reservations]. F1000Research 2021, 9. [Google Scholar] [CrossRef]
- Weber, A.M.; Drobnitzky, N.; Devery, A.M.; Bokobza, S.M.; Adams, R.A.; Maughan, T.S.; Ryan, A.J. Phenotypic consequences of somatic mutations in the ataxia-telangiectasia mutated gene in non-small cell lung cancer. Oncotarget 2016, 7, 60807–60822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, G.; Li, J.; Huang, Y.-Y.; Li, Y.; Lin, J.; Chen, L.-Z.; Lu, J.-P.; Wang, Y.-Q.; Wang, C.-X.; et al. Association of tumor protein p53 and ataxia-telangiectasia mutated comutation with response to immune checkpoint inhibitors and mortality in patients with non–small cell lung cancer. Jama Netw. Open 2019, 2, e1911895. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting mapk signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Escors, D.; Gato-Cañas, M.; Zuazo, M.; Arasanz, H.; García-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of pd-l1 in cancer cells. Signal. Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Liu, D.; Li, L. Pd-1/pd-l1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Hopewell, E.L.; Zhao, W.; Fulp, W.J.; Bronk, C.C.; Lopez, A.S.; Massengill, M.; Antonia, S.; Celis, E.; Haura, E.B.; Enkemann, S.A.; et al. Lung tumor nf-κb signaling promotes t cell–mediated immune surveillance. J. Clin. Invest. 2013, 123, 2509–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westhoff, B.; Colaluca, I.N.; D’Ario, G.; Donzelli, M.; Tosoni, D.; Volorio, S.; Pelosi, G.; Spaggiari, L.; Mazzarol, G.; Viale, G.; et al. Alterations of the notch pathway in lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 22293–22298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Wu, H.; Han, N.; Xu, H.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Notch signaling and emt in non-small cell lung cancer: Biological significance and therapeutic application. J. Hematol. Oncol. 2014, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Hothorn, T.; Lausen, B. On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal. 2003, 43, 121–137. [Google Scholar] [CrossRef]
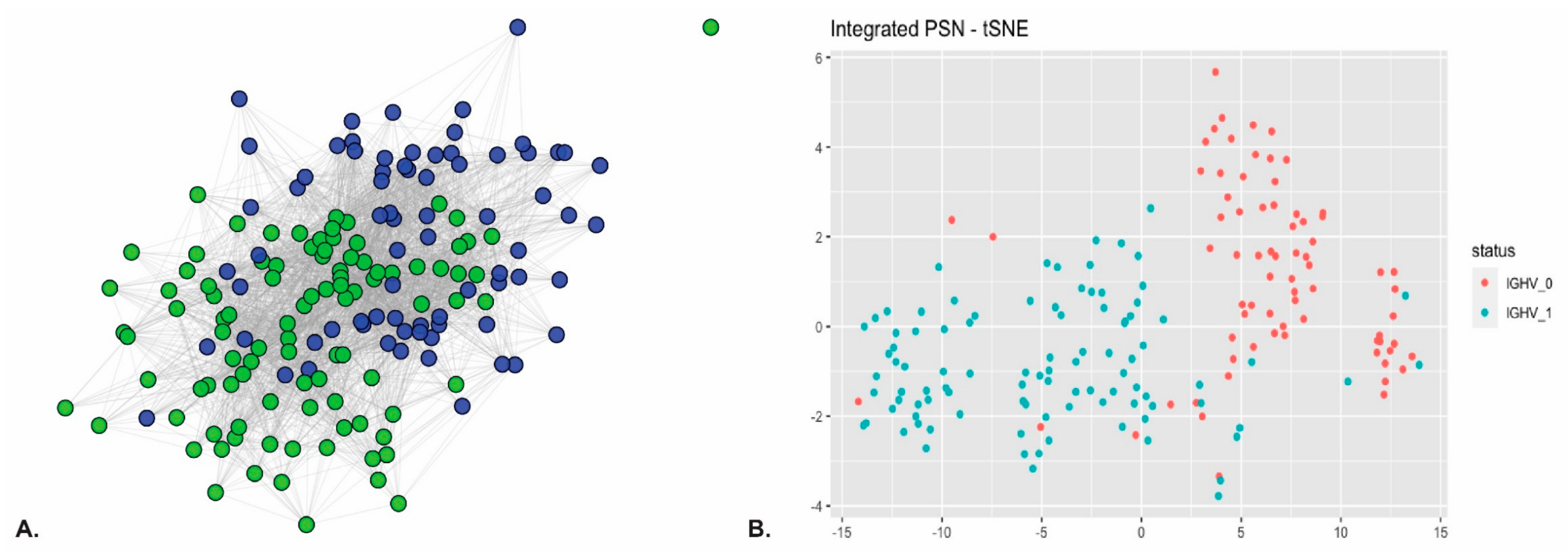
- Rozovski, U.; Keating, M.J.; Estrov, Z. Why is the immunoglobulin heavy chain gene mutation status a prognostic indicator in chronic lymphocytic leukemia? Acta Haematol. 2018, 140, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Kucera, M.; Isserlin, R.; Arkhangorodsky, A.; Bader, G. Autoannotate: A cytoscape app for summarizing networks with semantic annotations [version 1; peer review: 2 approved]. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Sheng, X.; Mittelman, S.D. The role of adipose tissue and obesity in causing treatment resistance of acute lymphoblastic leukemia. Front. Pediatr 2014, 2, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankanagoudar, S.; Singh, G.; Mahapatra, M.; Kumar, L.; Chandra, N.C. Cholesterol homeostasis in isolated lymphocytes: A differential correlation between male control and chronic lymphocytic leukemia subjects. Asian Pac. J. Cancer Prev. 2017, 18, 23–30. [Google Scholar]
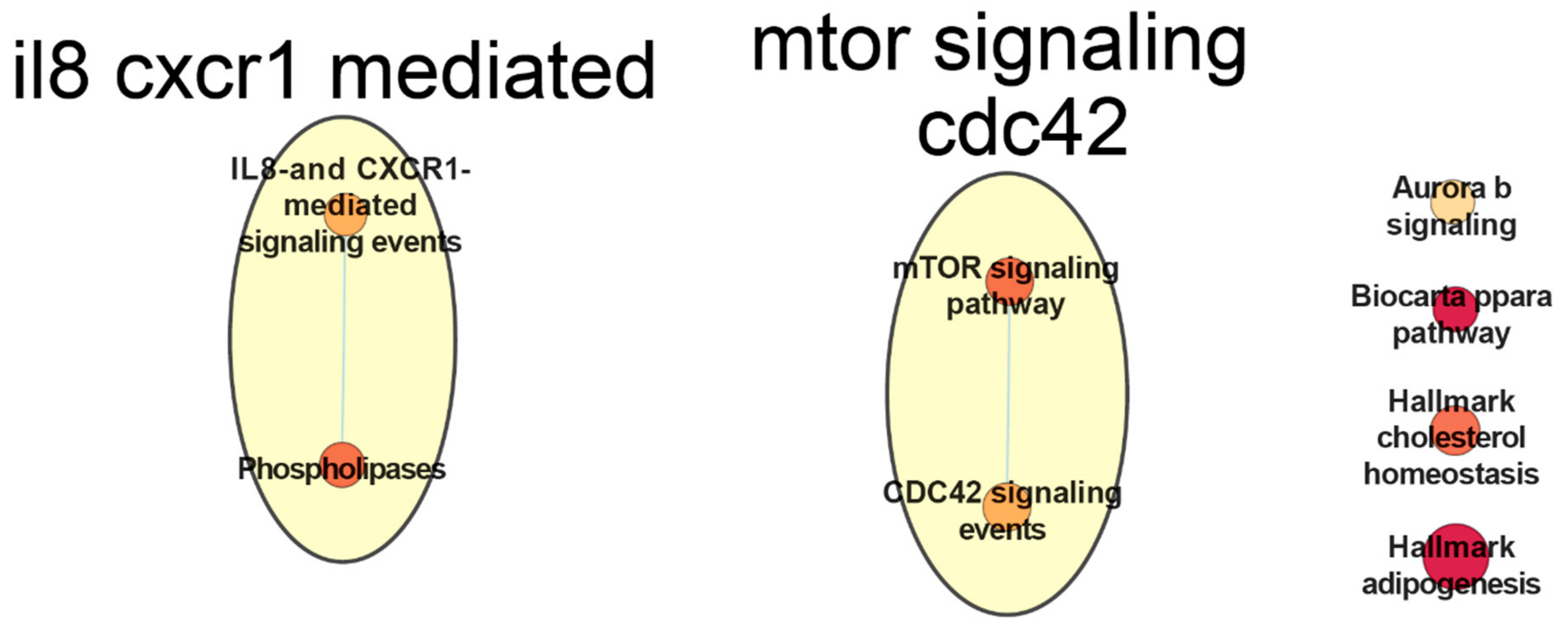
- Ha, H.; Debnath, B.; Neamati, N. Role of the cxcl8-cxcr1/2 axis in cancer and inflammatory diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Risnik, D.; Podaza, E.; Almejún, M.B.; Colado, A.; Elías, E.E.; Bezares, R.F.; Fernández-Grecco, H.; Cranco, S.; Sánchez-Ávalos, J.C.; Borge, M.; et al. Revisiting the role of interleukin-8 in chronic lymphocytic leukemia. Sci. Rep. 2017, 7, 15714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabilii, S.; Ricciardi, M.R.; Piedimonte, M.; Gianfelici, V.; Bianchi, M.P.; Tafuri, A. Biological aspects of mtor in leukemia. Int. J. Mol. Sci. 2018, 19, 2396. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, A.G.; Evans, R.; Kiaii, S.; Svensson, L.; Hogg, N.; Gribben, J.G. Chronic lymphocytic leukemia cells induce defective lfa-1-directed t-cell motility by altering rho gtpase signaling that is reversible with lenalidomide. Blood 2013, 121, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Humphries, L.A.; Godbersen, J.C.; Danilova, O.V.; Kaur, P.; Christensen, B.C.; Danilov, A.V. Pro-apoptotic tp53 homolog tap63 is repressed via epigenetic silencing and b-cell receptor signalling in chronic lymphocytic leukaemia. Br. J. Haematol. 2013, 163, 590–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.-W.; Reva, B.; et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 2020, 182, 200–225.e235. [Google Scholar] [CrossRef]
- AbdulJabbar, K.; Raza, S.E.A.; Rosenthal, R.; Jamal-Hanjani, M.; Veeriah, S.; Akarca, A.; Lund, T.; Moore, D.A.; Salgado, R.; Al Bakir, M.; et al. Geospatial immune variability illuminates differential evolution of lung adenocarcinoma. Nat. Med. 2020, 26, 1054–1062. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, B.; Hu, M.; Lou, Y.; Lu, J.; Zhang, X.; Wang, H.; Qian, J.; Chu, T.; Han, B. Cxcl9 as a prognostic inflammatory marker in early-stage lung adenocarcinoma patients. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Pocha, K.; Mock, A.; Rapp, C.; Dettling, S.; Warta, R.; Geisenberger, C.; Jungk, C.; Martins, L.R.; Grabe, N.; Reuss, D.; et al. Surfactant expression defines an inflamed subtype of lung adenocarcinoma brain metastases that correlates with prolonged survival. J. Clin. Cancer Res. 2020, 26, 2231–2243. [Google Scholar] [CrossRef]
- Tini, G.; Marchetti, L.; Priami, C.; Scott-Boyer, M.-P. Multi-omics integration—a comparison of unsupervised clustering methodologies. Brief. Bioinform. 2017, 20, 1269–1279. [Google Scholar] [CrossRef]
- Haider, S.; Yao, C.Q.; Sabine, V.S.; Grzadkowski, M.; Stimper, V.; Starmans, M.H.W.; Wang, J.; Nguyen, F.; Moon, N.C.; Lin, X.; et al. Pathway-based subnetworks enable cross-disease biomarker discovery. Nat. Commun. 2018, 9, 4746. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human x chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef]
- Sherman, R.M.; Salzberg, S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Türei, D.; Korcsmáros, T.; Saez-Rodriguez, J. Omnipath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 2016, 13, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Griss, J.; Viteri, G.; Sidiropoulos, K.; Nguyen, V.; Fabregat, A.; Hermjakob, H. Reactomegsa-efficient multi-omics comparative pathway analysis. Mol. Cell. Proteom. 2020, 19, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Paczkowska, M.; Barenboim, J.; Sintupisut, N.; Fox, N.S.; Zhu, H.; Abd-Rabbo, D.; Mee, M.W.; Boutros, P.C.; Abascal, F.; Amin, S.B.; et al. Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun. 2020, 11, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.H.; Walsh, B.; Mayfield, G.; Tamborero, D.; Sonkin, D.; Krysiak, K.; Deu-Pons, J.; Duren, R.P.; Gao, J.; McMurry, J.; et al. A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer. Nat. Genet. 2020, 52, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Dugourd, A.; Kuppe, C.; Sciacovelli, M.; Gjerga, E.; Emdal, K.B.; Bekker-Jensen, D.B.; Kranz, J.; Bindels, E.M.J.; Costa, A.S.H.; Olsen, J.V.; et al. Causal integration of multi-omics data with prior knowledge to generate mechanistic hypotheses. J. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Boeckhout, M.; Zielhuis, G.A.; Bredenoord, A.L. The fair guiding principles for data stewardship: Fair enough? Eur J. Hum. Genet. 2018, 26, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupras, C.; Bunnik, E.M. Toward a framework for assessing privacy risks in multi-omic research and databases. Am. J. Bioeth. 2021, 1–32. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information, Entrez Programming Utilities Help; National Center for Biotechnology Information: Bethesda, MD, USA, 2010.
- Li, Y.; Wu, F.X.; Ngom, A. A review on machine learning principles for multi-view biological data integration. Brief. Bioinform 2018, 19, 325–340. [Google Scholar] [CrossRef]
- Sun, S. A survey of multi-view machine learning. Neural Comput. Appl. 2013, 23, 2031–2038. [Google Scholar] [CrossRef]
- Rappoport, N.; Shamir, R. Multi-omic and multi-view clustering algorithms: Review and cancer benchmark. Nucleic Acids Res. 2019, 47, 1044. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, Á.; Chan, J.M.; Yu, H.A.; Pe’er, D.; Sawyers, C.L.; Sen, T.; Rudin, C.M. Lineage plasticity in cancer: A shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 2020, 17, 360–371. [Google Scholar] [CrossRef]
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global epidemiology of lung cancer. Ann. Glob. Health 2019, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- John, C.R.; Watson, D.; Russ, D.; Goldmann, K.; Ehrenstein, M.; Pitzalis, C.; Lewis, M.; Barnes, M. M3c: Monte carlo reference-based consensus clustering. Sci. Rep. 2020, 10, 1816. [Google Scholar] [CrossRef] [Green Version]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The cosmic cancer gene census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Frost, H.R.; Li, Z.; Moore, J.H. Principal component gene set enrichment (pcgse). Biodata Min. 2015, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2017, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (msigdb) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Gaidano, G.; Rossi, D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematol. Am. Soc. Hematol. Educ. Program. 2017, 2017, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Holroyd, A.K.; Michie, A.M. The role of mtor-mediated signaling in the regulation of cellular migration. Immunol. Lett. 2018, 196, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondrisova, L.; Mraz, M. Genetic and non-genetic mechanisms of resistance to bcr signaling inhibitors in b cell malignancies. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, S.; Oleś, M.; Lu, J.; Sellner, L.; Anders, S.; Velten, B.; Wu, B.; Hüllein, J.; da Silva Liberio, M.; Walther, T.; et al. Drug-perturbation-based stratification of blood cancer. J. Clin. Invest. 2018, 128, 427–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]








| Level of Analysis | Definition | Method of Analysis |
|---|---|---|
| Genome [4] | Complete set of genes of an organism or its organelles | WGS, WES, DNA microarray |
| Transcriptome [5] | Complete set of messenger RNA molecules present in a cell, tissue of organ | RNA-Sequencing Expression microarray Spatially resolved transcriptomics |
| Proteome [6] | Complete set of protein molecules present in a cell, tissue or organ | Peptide/protein microarrays (RPPA) Mass spectrometry Imaging mass cytometry |
| Metabolome [7] | Complete set of metabolites (low-molecular-weight intermediates) in a cell, tissue or organ | Nuclear magnetic resonance spectrometry Mass spectrometry Infa-red spectroscopy |
| Methylome [8] | Complete set of methylation sites within a genome | Bisulfite-Sequencing, ChIP-Seq |
| Microbiome [9] | Complete set of genes of all microbes (bacteria, fungi, protozoa and viruses) in a cell, tissue or organ | DNA-Sequencing 16S rRNA-Sequencing |
| Lipidome [10] | Complete set of all biomolecules defined as lipids | Mass Spectrometry |
| Model Nature Orientation | Tool Name | Programming Language | Integration Method | Used Omics | Reference |
|---|---|---|---|---|---|
| Unsupervised | R.JIVE | R | Multi-step analysis | mRNA, miRNA, MET | [104] |
| Unsupervised | PROFILE | R, Python | Multi-step analysis | mRNA, CNV, MUT | [105] |
| Supervised | SALMON | Python | Gene co-expression Analysis | mRNA, miRNA, CNV | [106] |
| Supervised | netDx | R | Feature network aggregation | mRNA, miRNA, CNV, MUT, MET, PROT | [107] |
| Supervised | Graper | R | Bayesian | mRNA, DRUGre, MET | [108] |
| Model Nature Orientation | Tool Name | Programming Language | Integration Method | Used Omics | Reference |
|---|---|---|---|---|---|
| Unsupervised | Joint Bayes Factor | Matlab | Matrix factorization | mRNA, MET, CNV | [111] |
| Unsupervised | iProFun | R | Multiple-step analysis | mRNA, CNV, MET | [112] |
| Unsupervised | CCA-sparse group | Matlab | Canonical correlation analysis | mRNA, SNP | [113] |
| Supervised | sMBPLS | Matlab | Partial Least Squares | mRNA, miRNA, CNV, MET | [114] |
| Unsupervised | CNAmet | R | Multi-step analysis | mRNA, CNV, MET | [115] |
| Supervised | iBAG | R | Multi-step analysis | mRNA, CNV, MET | [116] |
| Supervised | Anduril | R, Python, Shell | Multi-step analysis | aCGH, mRNA, miRNA, SNP, MET | [117] |
| Supervised | CapsNetMMD | Python | Capsule network model | mRNA, CNV, MET | [118] |
| Model Nature Orientation | Tool Name | Programming Language | Integration Method | Used Omics | Reference |
|---|---|---|---|---|---|
| Unsupervised | ModMap | Java | Multi-step analysis | mRNA, miRNA, PROT | [125] |
| Unsupervised | NetICS | Matlab | Multi-step analysis | mRNA, miRNA, CNV, MUT, MET, PROT | [126] |
| Unsupervised | SNMNMF | Matlab | Matrix factorization | mRNA, miRNA | [127] |
| Unsupervised | PARADIGM | Web-app, Python | Probabilistic graphical models | mRNA, CNV | [128] |
| Supervised | FSMKL | Matlab | Multiple kernel learning | mRNA, CNV | [129] |
| Unsupervised | Sumer | R | Multi-step analysis | mRNA, PROT | [130] |
| Unsupervised | MOSClip | R | PCA | mRNA, CNV, MUT, MET | [131] |
| Supervised and Unsupervised | COCOA | R | Multi-step analysis | mRNA, ATAC-Seq, DRUGre, MUT, MET | [132] |
| Model Nature Orientation | Tool Name | Programming Language | Integration Method | Used Omics | Reference |
|---|---|---|---|---|---|
| Supervised | MOLI | Python | Neural networks | mRNA, CNV, MUT | [134] |
| Unsupervised | SNPLS | Matlab | Partial least squares | mRNA, DRUGre | [135] |
| Research Purpose | Model Nature Orientation | Tool Name | Programming Language | Integration Method | Used Omics | Reference |
|---|---|---|---|---|---|---|
| Biomarker discovery, Cancer subtype classification, Pathway analysis | Unsupervised | MCIA | R | Multi-step analysis | mRNA, PROT | [146] |
| Patient stratification, Cancer subtype analysis | Supervised | mixOmics | R | Feature transformation | mRNA, miRNA, PROT | [147] |
| Biomarker prediction, Pathway analysis | Unsupervised | Lemon-Tree | Java | Module network learning | mRNA, CNV | [148] |
| Patient stratification, Cancer subtype classification | Unsupervised | Clusternomics | R | Multi-step analysis | mRNA, miRNA, MET, PROT | [149] |
| Biomarker discovery, Pathway analysis | Unsupervised | AMARETTO | R | Multi-step analysis | mRNA, CNV, MET | [150] |
| Pathway analysis, Cancer subtype classification | Supervised | iOmicsPASS | C++ | Multi-step analysis | mRNAs, CNV, PROT | [151] |
| Biomarker discovery, Cancer subtype classification | Unsupervised | MOGSA | R | Matrix factorization | mRNA, CNV, Phosp, PROT | [152] |
| Patient stratification, Pathway analysis | Unsupervised | PathME | R, Python | Matrix factorization | mRNA, miRNA, CNV, MET | [153] |
| Drug analysis, Pathway analysis | Supervised | DrugComboExplorer | Java, Python | Multi-step analysis | DNA, mRNA, CNV, MET | [154] |
| Biomarker discovery and Patient stratification | Unsupervised | MOFA | R | Matrix Factorization | mRNA, MUT, MET, DRUGre | [155] |
| Search Round | Search Terms |
|---|---|
| General Search | (multi AND omics) OR multi-omics OR multiomics OR (multivariate AND genomic) OR (Algorithms AND integrative AND Cluster AND Analysis)) AND data AND integration |
| Searching for supervised methods | General Search + supervised |
| Searching for unsupervised methods | General Search + unsupervised OR cluster OR (Factor AND Analysis) NOT supervised |
| Searching for reviews | General Search + review [PTYP] OR review |
| Searching for tools | General Search + Tool OR Application OR Algorithm OR method |
| Searching for cancer | General Search + humans [MESH] AND cancer [MESH]) OR cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlachavas, E.I.; Bohn, J.; Ückert, F.; Nürnberg, S. A Detailed Catalogue of Multi-Omics Methodologies for Identification of Putative Biomarkers and Causal Molecular Networks in Translational Cancer Research. Int. J. Mol. Sci. 2021, 22, 2822. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062822
Vlachavas EI, Bohn J, Ückert F, Nürnberg S. A Detailed Catalogue of Multi-Omics Methodologies for Identification of Putative Biomarkers and Causal Molecular Networks in Translational Cancer Research. International Journal of Molecular Sciences. 2021; 22(6):2822. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062822
Chicago/Turabian StyleVlachavas, Efstathios Iason, Jonas Bohn, Frank Ückert, and Sylvia Nürnberg. 2021. "A Detailed Catalogue of Multi-Omics Methodologies for Identification of Putative Biomarkers and Causal Molecular Networks in Translational Cancer Research" International Journal of Molecular Sciences 22, no. 6: 2822. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062822