
Could Small Heat Shock Protein HSP27 Be a First-Line Target for Preventing Protein Aggregation in Parkinson’s Disease?

 , , ,
, , ,
Abstract
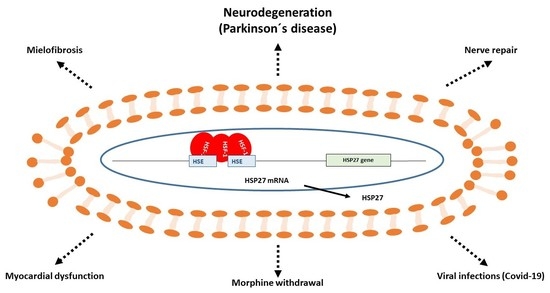
:
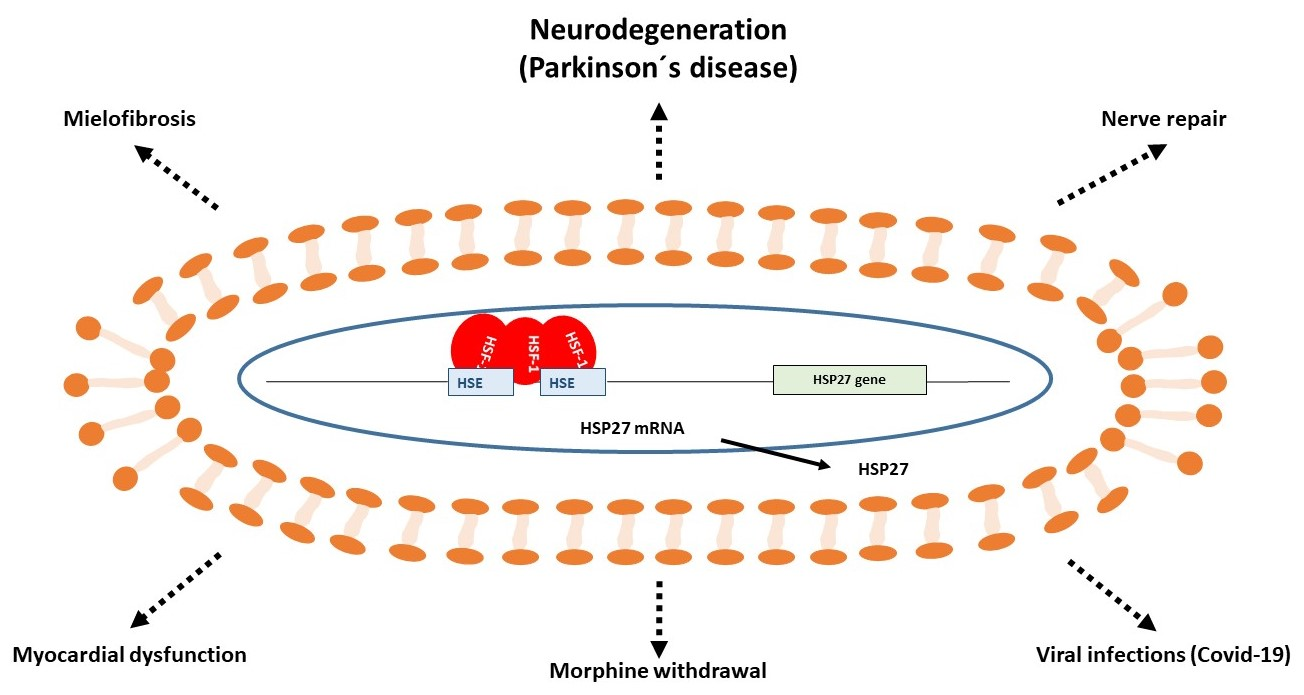{kind=link}
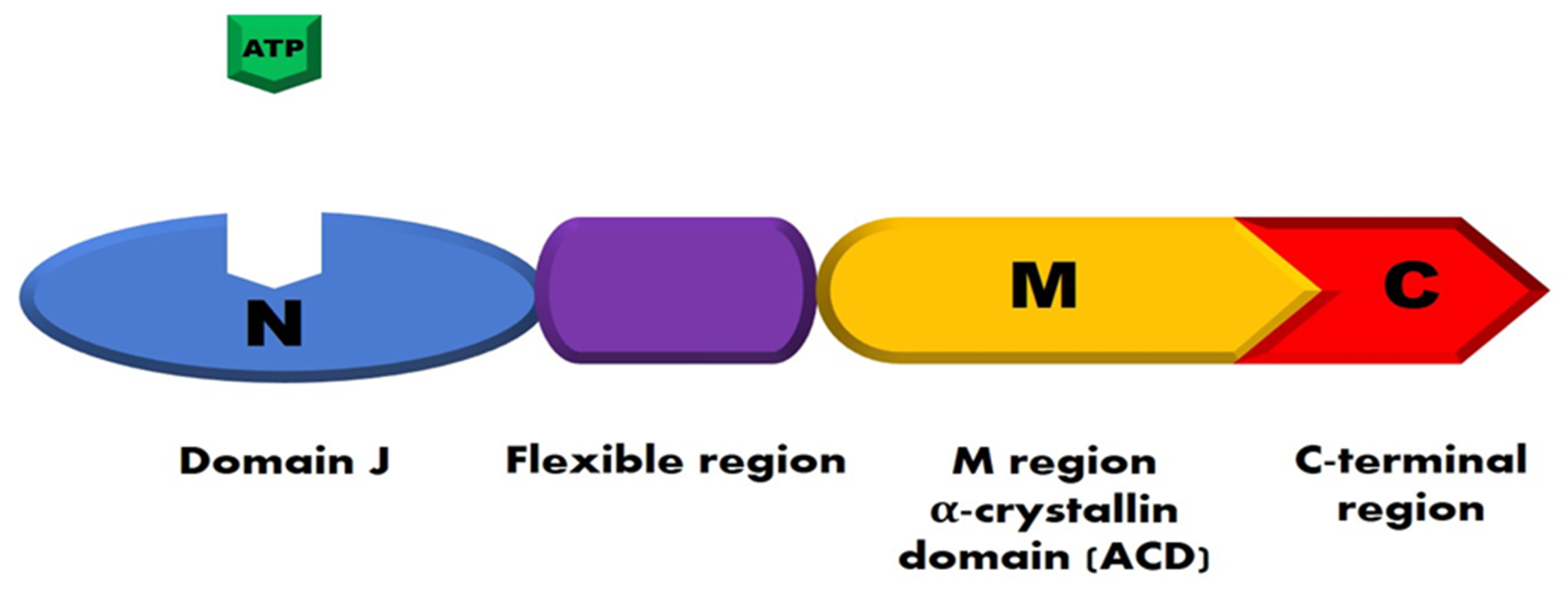{kind=link}
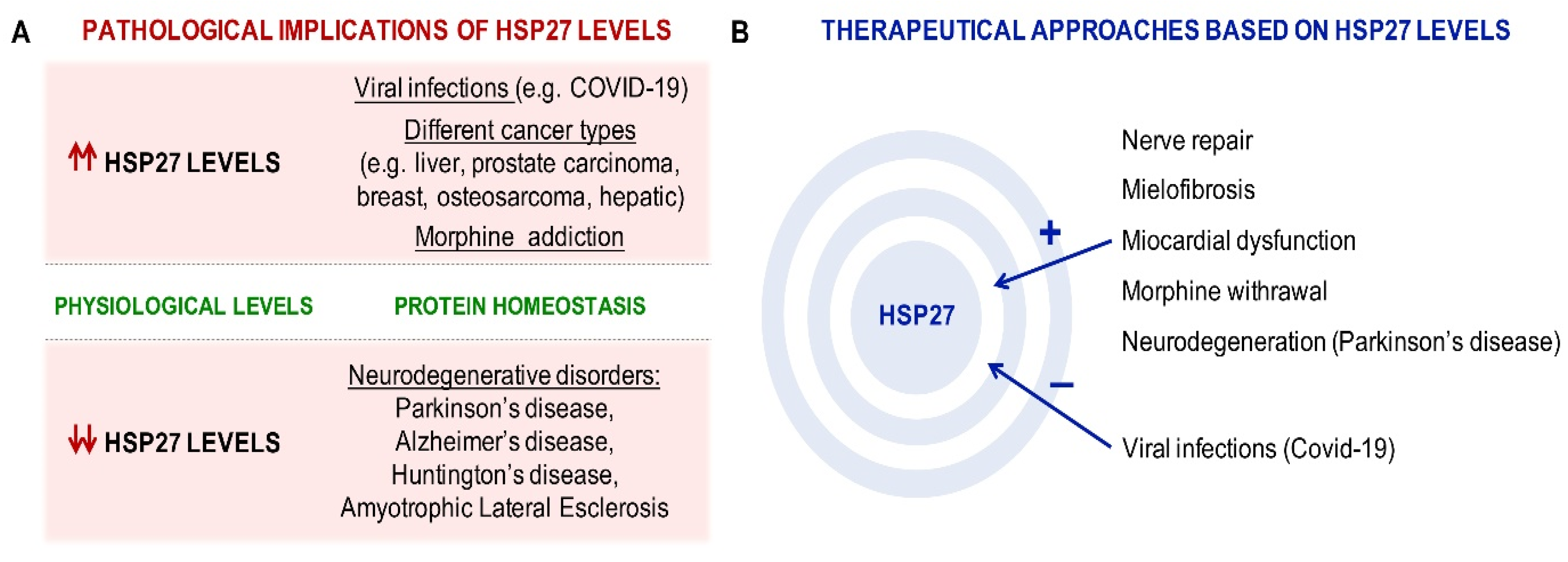{kind=link}
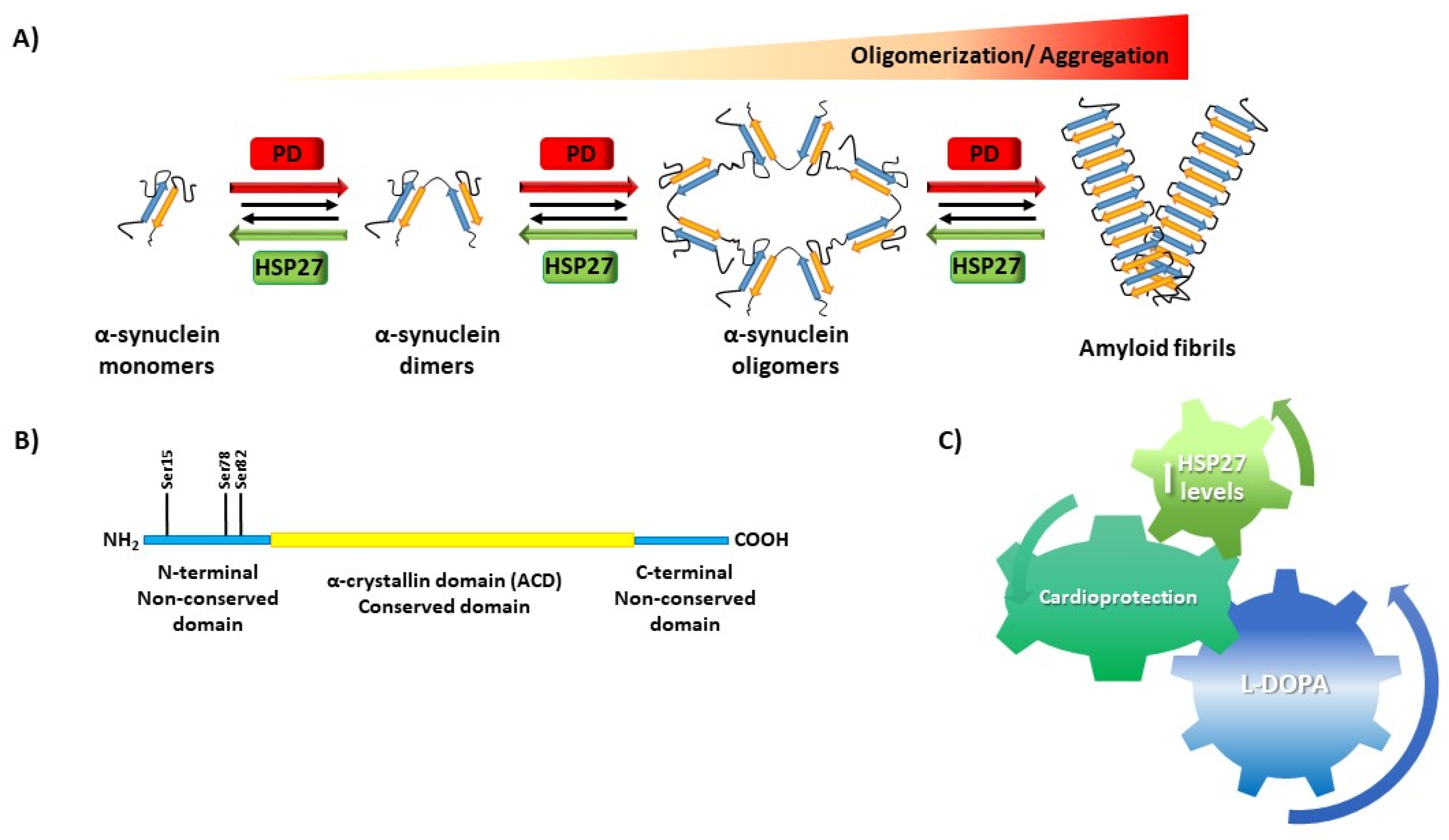{kind=link}
1. Introduction
2. Autonomic Dysfunction in Parkinson’s Disease
3. Structural Characteristics and Functions of Small Heat Shock Protein 27
4. Interaction between Small Heat Shock Protein 27 and α-Synuclein: Possible Treatment of Parkinson’s Disease
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACD | α-crystallin domain |
| CNS | Central nervous system |
| COMT | Catechol-o methyl transferase |
| DA | Dopamine |
| HSPs | Heat shock proteins |
| MAO | Monoaminoxidase |
| MGO | Methylglyoxal |
| NA | Noradrenaline |
| OH | Orthostatic hypotension |
| PD | Parkinson´s disease |
| SNpc | Substantia nigra pars compacta |
| TH | Tyrosine hydroxylase |
References
- Linquistm, S. The heat-shock response. Ann. Int. Rev. Biochem. 1986, 55, 1151–1191. [Google Scholar]
- Hartl, F.V.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Cetinbas, M.; Shakhnovich, E.I. Catalysis of protein folding by chaperones accelerates evolutionary dynamics in adapting cell populations. PLoS Comput. Biol. 2013, 9, e1003269. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ Proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 534–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muranova, L.K.; Ryzhavskaya, A.S.; Sudnitsyna, M.V.; Shatov, V.M.; Gusev, N.B. Small heat shock proteins and human neurodegenerative diseases. Biochemistry 2019, 84, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Ecroyd, H. The small heat shock proteins αB-crystallin (HSPB5) and Hsp27 (HSPB1) inhibit the intracellular aggregation of α-synuclein. Cell Stress Chaperones 2017, 22, 589–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebouvier, T.; Clairembault, T.; Devos, D.; Pallardy, A.; Coron, E.; Neunlist, M.; Derkinderen, P. Peripheral autonomic nervous system involvement in Gaucher-related parkinsonism. J. Parkinsons Dis. 2014, 4, 29–32. [Google Scholar] [CrossRef]
- Cuenca, L.; Gil-Martinez, A.L.; Cano-Fernández, L.; Sánchez-Rodrigo, C.; Estrada, C.; Fernández-Villalba, E.; Herrero, M.T. Parkinson’s disease: A short story of 200 years. Histol. Histopathol. 2019, 34, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 509–512. [Google Scholar] [CrossRef]
- Chen, Z.; Li, G.; Liu, J. Autonomic dysfunction in Parkinson’s disease: Implications for pathophysiology, diagnosis, and treatment. Neurobiol. Dis. 2020, 34, 104700. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.J.; Cheon, S.M.; Kim, J.W. Orthostatic hypotension in drug-naive patients with Parkinson’s disease. J. Mov. Disord. 2011, 4, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, A.; Atmis, V.; Cengiz, O.K.; Cinar, E.; Aras, S.; Varli, M.; Atli, T. Evaluation of cardiac autonomic functions in older Parkinson’s disease patients: A cross-sectional study. Aging Dis. 2016, 7, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S.; Aral-Becher, B.; Jost, W. Nondipping in Parkinson’s disease. Parkinson’s Dis. 2011, 2011, 897586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, D.S.; Pechnik, S.; Holmes, C.; Eldadah, B.; Sharabi, Y. Association between supine hypertension and orthostatic hypotension in autonomic failure. Hypertension 2003, 42, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Fanciulli, A.; Gobel, G.; Ndayisaba, J.P.; Granata, R.; Duerr, S.; Strano, S.; Colosimo, C.; Poewe, W.; Pontieri, F.E.; Wenning, G.K. Supine hypertension in Parkinson’s disease and multiple system atrophy. Clin. Auton. Res. 2016, 26, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Morita, Y.; Shimizu, T.; Takahashi, K.; Suzuki, N. Cardiac para-sympathetic dysfunction concurrent with cardiac sympathetic denervation in Parkinson’s disease. J. Neurol. Sci. 2009, 276, 79–83. [Google Scholar] [CrossRef]
- Goldstein, D.S. The sick-but-not-dead phenomenon applied to catecholamine deficiency in neurodegenerative diseases. Semin. Neurol. 2020, 40, 502–514. [Google Scholar] [CrossRef]
- Ma, L.Y.; Liu, G.L.; Wang, D.X.; Zhang, M.M.; Kou, W.Y.; Feng, T. Alpha-synuclein in peripheral tissues in Parkinson’s disease. ACS Chem. Neurosci. 2019, 10, 812–823. [Google Scholar] [CrossRef]
- Almela, P.; Cuenca-Bermejo, L.; Yuste, J.E.; Estrada, C.; De Pablos, V.; Bautista-Hernández, V.; Fernández-Villalba, E.; Laorden, M.L.; Herrero, M.T. Cardiac noradrenaline turnover and heat shock protein 27 phosphorylation in dyskinetic monkeys. Mov. Disord. 2020, 35, 698–703. [Google Scholar] [CrossRef]
- Carmona-Abellán, M.; Martínez-Valbuena, I.; Di Caudo, C.; Marcilla, I.; Luquin, M.R. Cardiac sympathetic innervation in the MPTP non-human primate model of Parkinson disease. Clin. Auton. Res. 2019, 29, 415–425. [Google Scholar] [CrossRef]
- Dickson, D.W.; Fujishiro, H.; DelleDonne, A.; Menke, J.; Ahmed, Z.; Klos, K.J.; Josephs, K.A.; Frigerio, R.; Burnett, M.; Parisi, J.E.; et al. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol. 2008, 115, 437–444. [Google Scholar] [CrossRef]
- Fujishiro, H.; Frigerio, R.; Burnett, M.; Klos, K.J.; Josephs, K.A.; Delledonne, A.; Parisi, J.E.; Ahlskog, J.E.; Dickson, D.W. Cardiac sympathetic denervation correlates with clinical and pathologic stages of Parkinson’s disease. Mov. Disord. 2008, 23, 1085–1092. [Google Scholar] [CrossRef]
- Cuenca-Bermejo, L.; Almela, P.; Gallo-Soljancic, P.; Yuste, J.E.; de Pablos, V.; Bautista-Hernández, V.; Fernández-Villalba, M.; Laorden, M.L.; Herrero, M.T. Cardiac tyrosine hydroxylase activation and MB-COMT in dyskinetic monkeys. Sci. Rep. 2021. [Google Scholar] [CrossRef]
- Christopher, L.; Duff-Canning, S.; Koshimori, Y.; Segura, B.; Boileau, I.; Chen, R.; Lang, A.E.; Houle, S.; Rusjan, P.; Strafella, A.P. Salience network and parahippocampal dopamine dysfunction in memory-impaired Parkinson disease. Ann. Neurol. 2015, 77, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pablo-Fernández, E.; Tur, C.; Revesz, T.; Lees, A.J.; Holton, J.L.; Warner, T.T. Association of autonomic dysfunction with disease progression and survival in parkinson disease. JAMA Neurol. 2017, 74, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. Spinal cord lesions in sporadic Parkinson’s disease. Acta Neuropathol. 2012, 124, 643–664. [Google Scholar] [CrossRef] [PubMed]
- Oinas, M.; Paetau, A.; Myllykangas, L.; Notkola, I.L.; Kalimo, H.; Polvikoski, T. Alpha-Synuclein pathology in the spinal cord autonomic nuclei associates with alpha-synuclein pathology in the brain: A population-based Vantaa 85+ study. Acta Neuropathol. 2010, 119, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Sastre, M.; Bohl, J.R.; de Vos, R.A.; Del Tredici, K. Parkinson’s disease: Lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. 2007, 113, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Gold, A.; Turkalp, Z.T.; Munoz, D.G. Enteric alpha-synuclein expression is increased in Parkinson’s disease but not Alzheimer’s disease. Mov. Disord. 2013, 28, 237–240. [Google Scholar] [CrossRef]
- Bourdenx, M.; Nioche, A.; Dovero, S.; Arotcarena, M.L.; Camus, S.; Porras, G.; Thiolat, M.L.; Rougier, N.P.; Prigent, A.; Aubert, P.; et al. Identification of distinct pathological signatures induced by patient-derived α-synuclein structures in nonhuman primates. Sci. Adv. 2020, 6, eaaz9165. [Google Scholar] [CrossRef] [PubMed]
- Arotcarena, M.L.; Dovero, S.; Prigent, A.; Bourdenx, M.; Camus, S.; Porras, G.; Thiolat, M.L.; Tasselli, M.; Aubert, P.; Kruse, N. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain 2020, 143, 1462–1475. [Google Scholar] [CrossRef]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M.; et al. Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain 2020, 143, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Ulusoy, A.; Phillips, R.; Helwig, M.; Klinkenberg, M.; Powley, T.L.; Di Monte, D.A. Brain-to-stomach transfer of alpha-synuclein via vagal preganglionic projections. Acta Neuropathol. 2017, 133, 381–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musgrove, R.E.; Helwig, M.; Bae, E.J.; Aboutalebi, H.; Lee, S.J.; Ulusoy, A.; Di Monte, D.A. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular alpha-synuclein transfer. J. Clin. Investig. 2019, 129, 3738–3753. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, H.; Gram, T.W.; Mikkelsen, A.K.O.; Alstrup, N.; Casadei, P.; Tsung-Pin, O.; Riess, J.R.; Nyengaard, G.; Tamguney, P.H.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.S.; Sharabi, Y. The heart of PD: Lewy body diseases as neurocardiologic disorders. Brain Res. 2019, 1702, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Holmes, C.; Kopin, I.J.; Sharabi, Y. Intraneuronal vesicular uptake of catecholamines is decresed in patients with Lewy body diseases. J. Clin. Investig. 2011, 121, 3320–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.J.M.M.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 1–11. [Google Scholar] [CrossRef]
- Guerrero-Rojas, R.; Guerrero-Fonsecaz, R. Molecular mechanisms of heat shock peoteins (HSPs) involved in neoplasm development. Rev. Cient. Salud Uninorte 2018, 34, 455–474. [Google Scholar] [CrossRef]
- Stamler, R.; Kappe, G.; Boelens, W.; Slingsby, C. Wrapping the apha-crystallin domain fold in a chaperone assembly. J. Mol. Biol. 2005, 353, 68–69. [Google Scholar] [CrossRef] [Green Version]
- Jäättelä, M. Heat shock proteins as cellular lifeguards. Ann. Med. 1999, 31, 261–271. [Google Scholar] [CrossRef]
- Vos, M.J.; Hageman, J.; Carra, S.; Kampinga, H.H. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry 2008, 47, 7001–7011. [Google Scholar] [CrossRef]
- Mainz, A.; Peschek, J.; Stavropoulou, M.; Back, K.C.; Bardiaux, B.; Asami, S.; Prade, K.; Peters, C.; Weinkauf, S.; Buchner, J.; et al. The chaperone alphaB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 2015, 22, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Carper, S.W.; Rocheleau, T.A.; Storm, F.K. cDNA sequence of a human heat shock protein HSP27. Nucleic Acids Res. 1990, 18, 6457–6460. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Shinohara, H.; Goto, S.; Inaguma, Y.; Morishita, R.; Asano, J. Copurification of small heat shock protein with alpha B crystallin from human skeletal muscle. Biol. Chem. 1992, 267, 7718–7725. [Google Scholar] [CrossRef]
- Zhu, Z.; Reiser, G. The small heat shock proteins, especially HspB4 and HspB5 are promising protectants in neurodegenerative diseases. Neurochem. Int. 2018, 115, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Hino, M.; Kurogi, K.; Okubo, M.A.; Murata-Hori, M.; Hosoya, H. Small heat shock protein 27 (HSP27) associates with tubulin/microtubules in HeLa cells. Biochem. Biophys. Res. Commun. 2000, 271, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Camandona, V.L.; Rios-Anjos, R.M.; Alegria, T.G.P.; Pereira, F.; Bicev, R.N.; da Cunha, F.M.; Digiampietri, L.A.; de Barros, M.H.; Netto, L.E.S.; Ferreira-Junior, J.R. Expression of human HSP27 in yeast extends replicative lifespan and uncovers a hormetic response. Biogerontology 2020, 21, 559–575. [Google Scholar] [CrossRef]
- Gobbo, J.; Gaucher-Di-Stasio, C.; Weidmann, S.; Guzzo, J.; Garrido, C. Quantification of HSP27 and HSP70 molecular chaperone activities. Methods Mol. Biol. 2011, 787, 137–143. [Google Scholar] [CrossRef]
- Bukach, O.V.; Glukhova, A.E.; Seit-Nebi, A.S.; Gusev, N.B. Heterooligomeric complexes formed by human small heat shock proteins HspB1 (Hsp27) and HspB6 (Hsp20). Biochem. Biophys. Acta 2009, 1794, 486–495. [Google Scholar] [CrossRef]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat shock protein 27 and 70: Anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006, 5, 2592–2601. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Paul, C.; Seigneuric, R.; Kampinga, H. The small heat shock proteins family: The long forgotten chaperones. Int. J. Mol. Cell Biol. 2012, 44, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110, 357–368. [Google Scholar]
- Bolhassani, A.; Agi, E. Heat shock proteins in infection. Clin. Chim. Acta 2019, 498, 90–100. [Google Scholar] [CrossRef]
- Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of heat shock proteins in apoptosis, oxidative Stress, human inflammatory diseases, and cancer. Pharmaceuticals 2017, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Song, D.; LI, H.; He, M.L. Stress proteins: The biological functions in virus infection, present and challenges for target-based antiviral drug. Sign. Trans. Targ. Ther. 2020, 5, 125–132. [Google Scholar] [CrossRef]
- Paul, C.; Simon, S.; Gibert, B.; Virot, S.; Manero, F.; Arrigo, A.P. Dynamic processes that reflect anti-apoptotic strategies set up by HspB1 (Hsp27). Exp. Cell Res. 2010, 316, 1535–1552. [Google Scholar] [CrossRef]
- Shevtsov, M.; Balogi, Z.; Khachatryan, W.; Gao, H.; Vígh, L.; Multhoff, G. Membrane-associated heat shock proteins in oncology: From basic research to new theragnostic targets. Cells 2020, 9, 1263. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Laorden, E.; Navarro-Zaragoza, J.; Milanés, M.V.; Laorden, M.L.; Almela, P. Cardiac protective role of heat shock protein 27 in the stress induced by drugs of abuse. Int. J. Mol. Sci. 2020, 21, 3623. [Google Scholar] [CrossRef] [PubMed]
- Vahabpour, R.; Soleymani, S.; Roohvand, F.; Zabihollahi, R.; Bolhassani, A. In vitro anti-viral effects of small heat shock proteins 20 and 27: A novel therapeutic approach. Curr. Pharm. Biotechnol. 2019, 20, 1011–1017. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef]
- Asthana, P.; Zhangb, G.; Sheikhb, K.A.; Ma, C.H.E. Heat shock protein is a key therapeutic target for nerve repair in autoimmune peripheral neuropathy and severe peripheral nerve injury. Brain Behav. Immun. 2020, 25, 31011–31014. [Google Scholar] [CrossRef]
- Lucijanic, M.; Livun, A.; Tupek, K.M.; Stoos-Veic, T.; Aralica, G.; Gecek, I.; Pejsa, V.; Kusec, R. Heat shock protein 27 (HSP27/HSPB1) expression is increased in patients with primary and secondary myelofibrosis and may be affecting their survival Leuk. Lymphoma 2017, 58, 2497–2500. [Google Scholar] [CrossRef]
- Lu, X.Y.; Chen, L.; Cai, X.L.; Yang, H.T. Overexpression of heat shock protein 27 protects against ischaemia/reperfusion-induced cardiac dysfunction via stabilization of troponin I and T. Cardiovasc. Res. 2008, 79, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, M.E.; Szegedi, V.; Varga, E.; Juhász, G.; Horváth, J.; Borbély, E.; Csibrány, E.; Alföldi, R.; Lénárt, N.; Penke, B.; et al. Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/P,S1 mice. Cell Stress Chaperones 2013, 18, 759–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, N.; Wu, Y.C.; Brunet, M.; Garrido, C.; Chung, F.L.; Dai, C.; Mi, L. Sulforaphane activates heat shock response and enhances proteasome activity through up-regulation of Hsp27. J. Biol. Chem. 2010, 285, 35528–35536. [Google Scholar] [CrossRef] [Green Version]
- Chondrogianni, N.; Voutetakis, K.; Kapetanou, M.; Delitsikou, V.; Papaevgeniou, N.; Sakellari, M.; Lefaki, M.; Filippopoulou, K.; Gonos, E.S. Proteasome activation: An innovative promising approach for delaying aging and retarding age-related diseases. Ageing Res. Rev. 2015, 23, 37–55. [Google Scholar] [CrossRef]
- Ryden, L.E.; Lewis, S.J.G. Parkinson’s disease in the era of personalised medicine: One size does not fit all. Drugs Aging 2018, 36, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Lenka, A.; Padmakumar, C.; Pal, P.K. Treatment of older Parkinson’s disease. Int. Rev. Neurobiol. 2017, 132, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Selig, E.E.; Zlatic, C.O.; Cox, D.; Mok, Y.F.; Gooley, P.R.; Ecroyd, H.; Griffin, M.D.W. N-and C-terminal regions of B-cristalyn and Hsp27 mediate inhibition of amyloid nucleation, fibril binding, and fibril disaggregation. J. Biol. Chem. 2020, 295, 9838–9854. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White, C.H.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.M.; Rey, J.; Martí, M.J.; Hernández, I.; Valldeoriola, F.; Reñé, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. 2014, 29, 1010–1018. [Google Scholar] [CrossRef]
- Burre, J. The synaptic function of a-synuclein. J. Parkinsons Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Renkawek, K.; Stege, G.J.; Bosman, G.J. Dementia, gliosis and expression of the small heat shock proteins hsp27 and alpha B-crystallin in Parkinson’s disease. Neuroreport 1999, 10, 2273–2276. [Google Scholar] [CrossRef]
- Cox, D.; Whiten, D.R.; Brown, J.W.P.; Horrocks, M.H.; Gil, R.S.; Dobson, C.M.; Klenerman, D.; Oijen, A.M.V.; Ecroyd, H. The small heat shock protein Hsp27 binds a-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef] [Green Version]
- Baughman, H.E.R.; Clouser, A.F.; Klevit, R.E.; Nath, A. HspB1 and Hsc70 chaperones engage distinct Tau species and have different inhibitory effects on amyloid formation. J. Biol. Chem. 2018, 293, 2687–2700. [Google Scholar] [CrossRef] [Green Version]
- Yerbury, J.J.; Gower, D.; Vanags, L.; Roberts, K.; Lee, J.A.; Ecroyd, H. The small heat shock proteins aB-crystallin and Hsp27 suppress SOD1 aggregation in vitro. Cell Stress Chaperones 2013, 18, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; Priya, S. Expanding role of molecular chaperones in regulating α-synuclein misfolding, implications in Parkinson’s disease. Cell. Mol. Life Sci. 2017, 74, 617–629. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Klucken, J.; Strathearn, K.E.; Liu, F.; Nguyen, P.; Rochet, J.C.; Hyman, B.T.; McLean, P.J. Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem. Biophys. Res. Commun. 2006, 351, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Aquilina, J.A.; Benesch, J.L.P.; Bateman, O.A.; Slingsby, C.; Robinson, C.V. Polydispersity of a mammalian chaperone: Mass spectrometry reveals the population of oligomers in Bcrystallin. Proc. Natl. Acad. Sci. USA 2003, 100, 10611–10616. [Google Scholar] [CrossRef] [Green Version]
- Jehle, S.; Vollmar, B.S.; Bardiaux, B.; Dove, K.K.; Rajagopal, P.; Gonen, T.; Oschkinat, H.; Klevit, R.E. N-terminal domain of alphaB-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proc. Natl. Acad. Sci. USA 2011, 108, 6409–6414. [Google Scholar] [CrossRef] [Green Version]
- Aquilina, J.A.; Benesch, J.L.P.; Ding, L.L.; Yaron, O.; Horwitz, J.; Robinson, C.V. Phosphorylation of αB-Crystallin Alters Chaperone Function through Loss of Dimeric Substructure. J. Biol. Chem. 2004, 279, 28675–28680. [Google Scholar] [CrossRef] [Green Version]
- Hochberg, H.; Liu, C.; Cox, D.; Cascio, D.; Sawaya, M.R.; Collier, M.P.; Stroud, J.; Carver, J.A.; Baldwin, A.J.; Robinson, C.V.; et al. The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, E1562–E1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannini, B.; Cascella, R.; Zampagni, M.; Waarde-Verhagen, M.; van Meehan, S.; Roodveldt, C.; Campioni, S.; Boninsegna, M.; Penco, A.; Relini, A.; et al. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA 2012, 109, 12479–12484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binger, K.J.; Ecroyd, H.; Yang, S.; Carver, J.A.; Howlett, G.J.; Griffin, M.D.W. Avoiding the oligomeric state: αB-crystallin inhibits fragmentation and induces dissociation of apolipoprotein CII amyloid fibrils. FASEB J. 2013, 27, 1214–1222. [Google Scholar] [CrossRef]
- Miranda, H.V.; Chegão, A.; Oliveira, M.S.; Gomes, B.F.; Enguita, F.J.; Outeiro, T.F. Hsp27 reduces glycation-induced toxicity and aggregation of alpha-synuclein. FASEB J. 2020, 34, 6718–6728. [Google Scholar] [CrossRef] [Green Version]
- Rai, S.N.; Singh, P.; Varshney, R.; Chaturvedi, V.K.; Vamanu, E.; Singh, M.P.; Singh, B.K. Promising drug targets and associated therapeutic interventions in Parkinson’s disease. Neural Regen. Res. 2021, 16, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Todorova, A.; Jenner, P.; Chaudhuri, K.R. Non-motor Parkinson’s: Integral to motor Parkinson’s, yet often neglected. Pract. Neurol. 2014, 14, 1–13. [Google Scholar] [CrossRef]
- Pires, A.O.; Teixeira, F.G.; Mendes-Pinheiro, B.; Serra, S.C.; Sousa, N.; Salgado, A.J. Old and New Challenges in Parkinson’s Disease Therapeutics. Prog. Neurobiol. 2017, 156, 69–89. [Google Scholar] [CrossRef]
- Paredes-Rodríguez, E.; Vargas-Suarez, S.; Morera-Herrera, T.; De Deurwaerdere, P.; Miguelez, C. The noradrenergic system in Parkinson’s disease. Front. Pharmacol. 2020, 11, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Palma, J.A.; Kaufmann, H. Treatment of autonomic dysfunction in Parkinson disease and other synucleinopathies. Mov. Disord. 2018, 33, 372–390. [Google Scholar] [CrossRef] [PubMed]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.M.; Höllerhage, M.; Höglinger, G.U.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 865–873. [Google Scholar] [CrossRef]
- Singh, S.K.; Dutta, A.; Modi, G. Alpha-Synuclein aggregation modulation: An emerging approach for the treatment of Parkinson’s disease. Future Med. Chem. 2017, 9, 1039–1053. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The synucleinopathies: Twenty years on. J. Parkinsons. Dis. 2017, 7, S53–S71. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Dave, K.D.; Kordower, J.H. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235. [Google Scholar] [CrossRef]
- Peart, J.N.; Gross, E.R.; Headrick, J.P.; Gross, G.J. Impaired p38 MAPK/HSP27 signaling underlies aging-related failure in opioid-mediated cardioprotection. J. Mol. Cell. Cardiol. 2007, 49, 972–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, M.E.; Oddo, S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penke, B.; Bogár, F.; Crul, T.; Sántha, M.; Tóth, M.E.; Vígh, L. Heat shock proteins and autophagy pathways in neuroprotection: From molecular bases to pharmacological interventions. Int. J. Mol. Sci. 2018, 19, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escriba, P.V.; Busquets, X.; Inokuchi, J.; Balogh, G.; Torok, Z.; Horvath, I.; Harwood, J.L.; Vigh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Critchley, B.J.; Isalan, M.; Mielcarek, M. Neuro-mechanisms in Huntington’s disease and other neurodegenerative disorders. Front. Physiol. 2018, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro-Zaragoza, J.; Cuenca-Bermejo, L.; Almela, P.; Laorden, M.-L.; Herrero, M.-T. Could Small Heat Shock Protein HSP27 Be a First-Line Target for Preventing Protein Aggregation in Parkinson’s Disease? Int. J. Mol. Sci. 2021, 22, 3038. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063038
Navarro-Zaragoza J, Cuenca-Bermejo L, Almela P, Laorden M-L, Herrero M-T. Could Small Heat Shock Protein HSP27 Be a First-Line Target for Preventing Protein Aggregation in Parkinson’s Disease? International Journal of Molecular Sciences. 2021; 22(6):3038. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063038
Chicago/Turabian StyleNavarro-Zaragoza, Javier, Lorena Cuenca-Bermejo, Pilar Almela, María-Luisa Laorden, and María-Trinidad Herrero. 2021. "Could Small Heat Shock Protein HSP27 Be a First-Line Target for Preventing Protein Aggregation in Parkinson’s Disease?" International Journal of Molecular Sciences 22, no. 6: 3038. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063038