
Cytokines Stimulated by IL-33 in Human Skin Mast Cells: Involvement of NF-κB and p38 at Distinct Levels and Potent Co-Operation with FcεRI and MRGPRX2


Abstract
:1. Introduction
2. Results
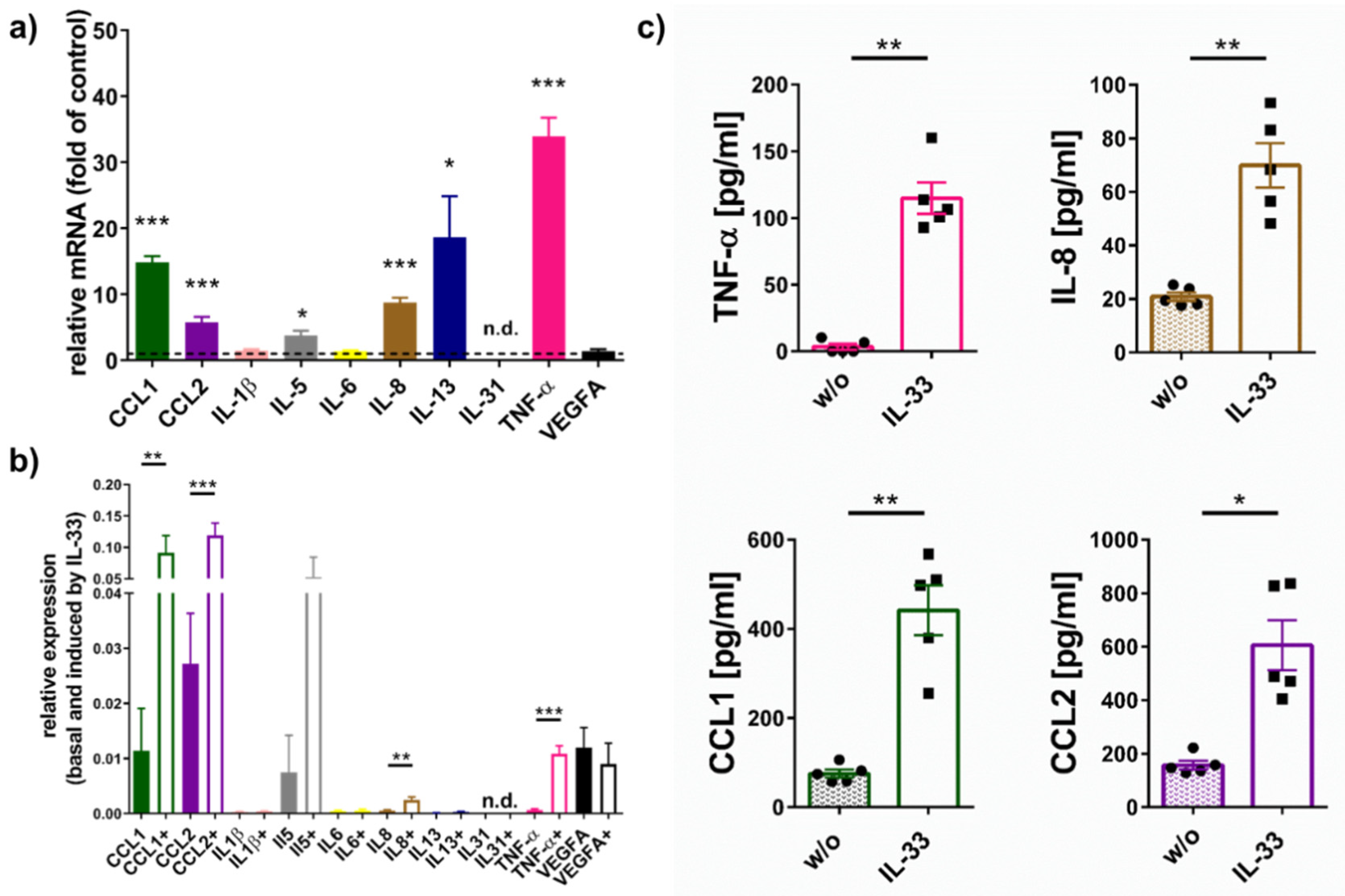
2.1. IL-33 Elicits Cytokine Production in Skin MCs
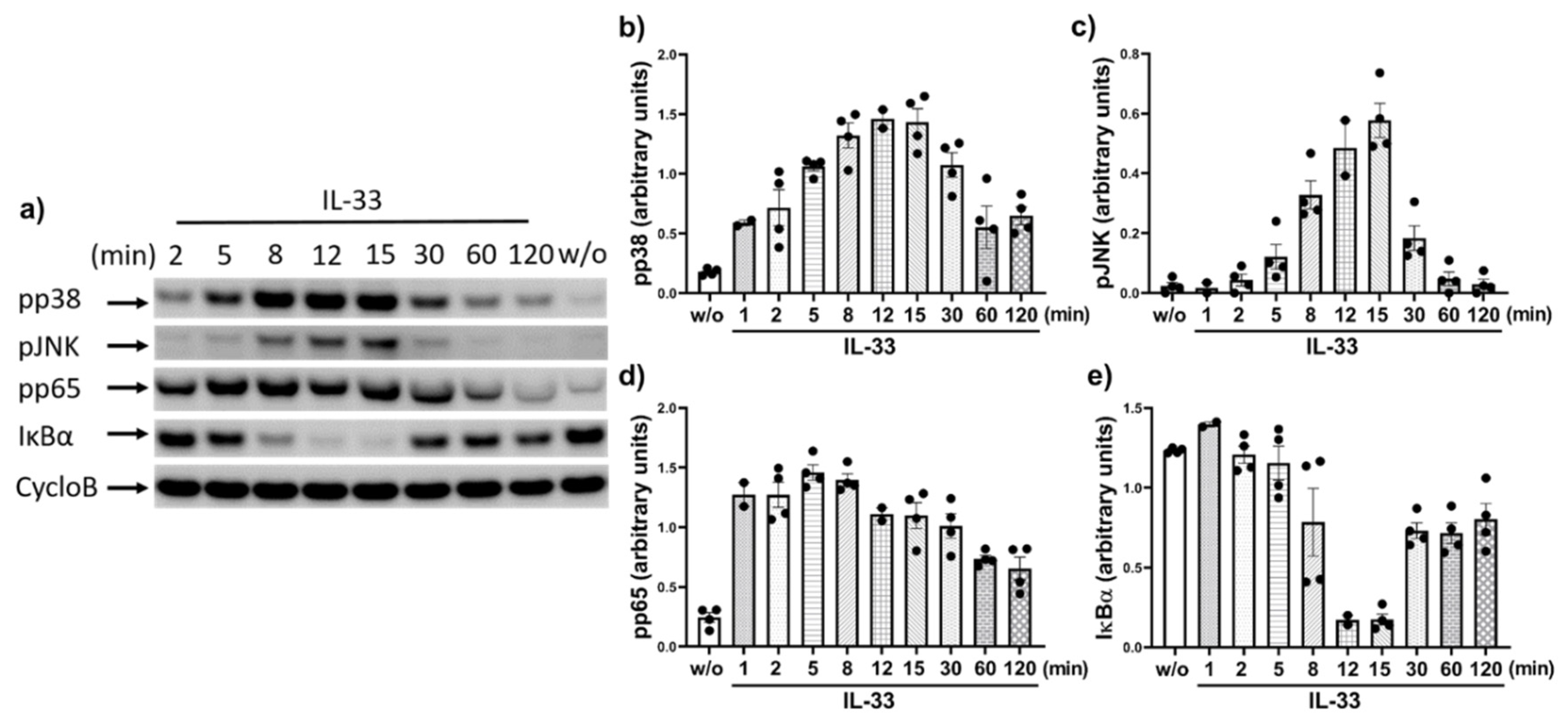
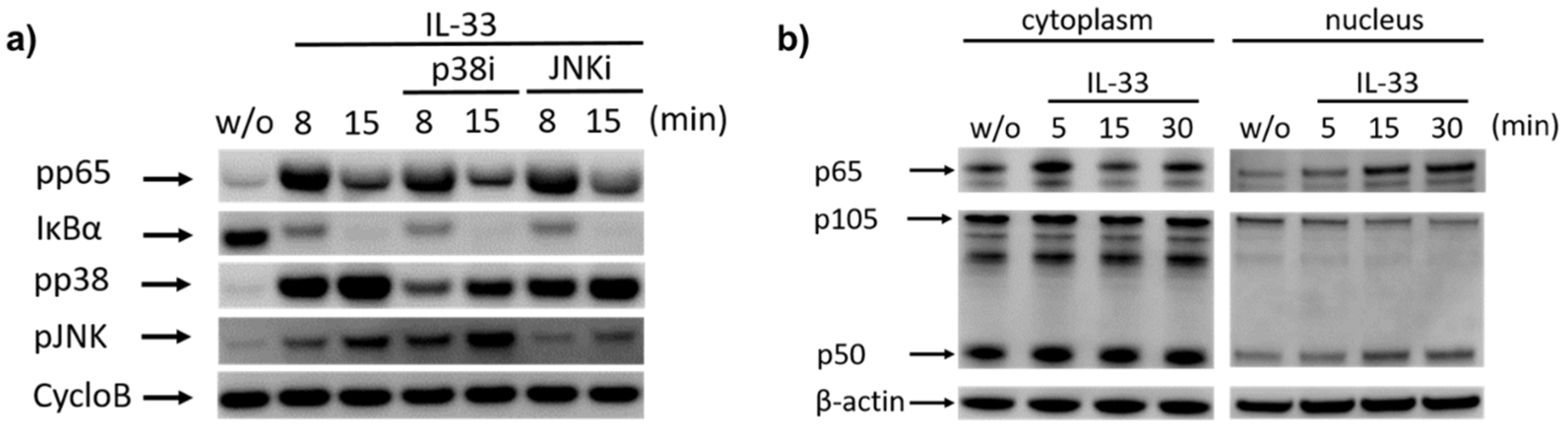
2.2. IL-33 Prompts the Phosphorylation of p38, JNK, and Potently Activates the NF-κB Pathway in Cutaneous MCs
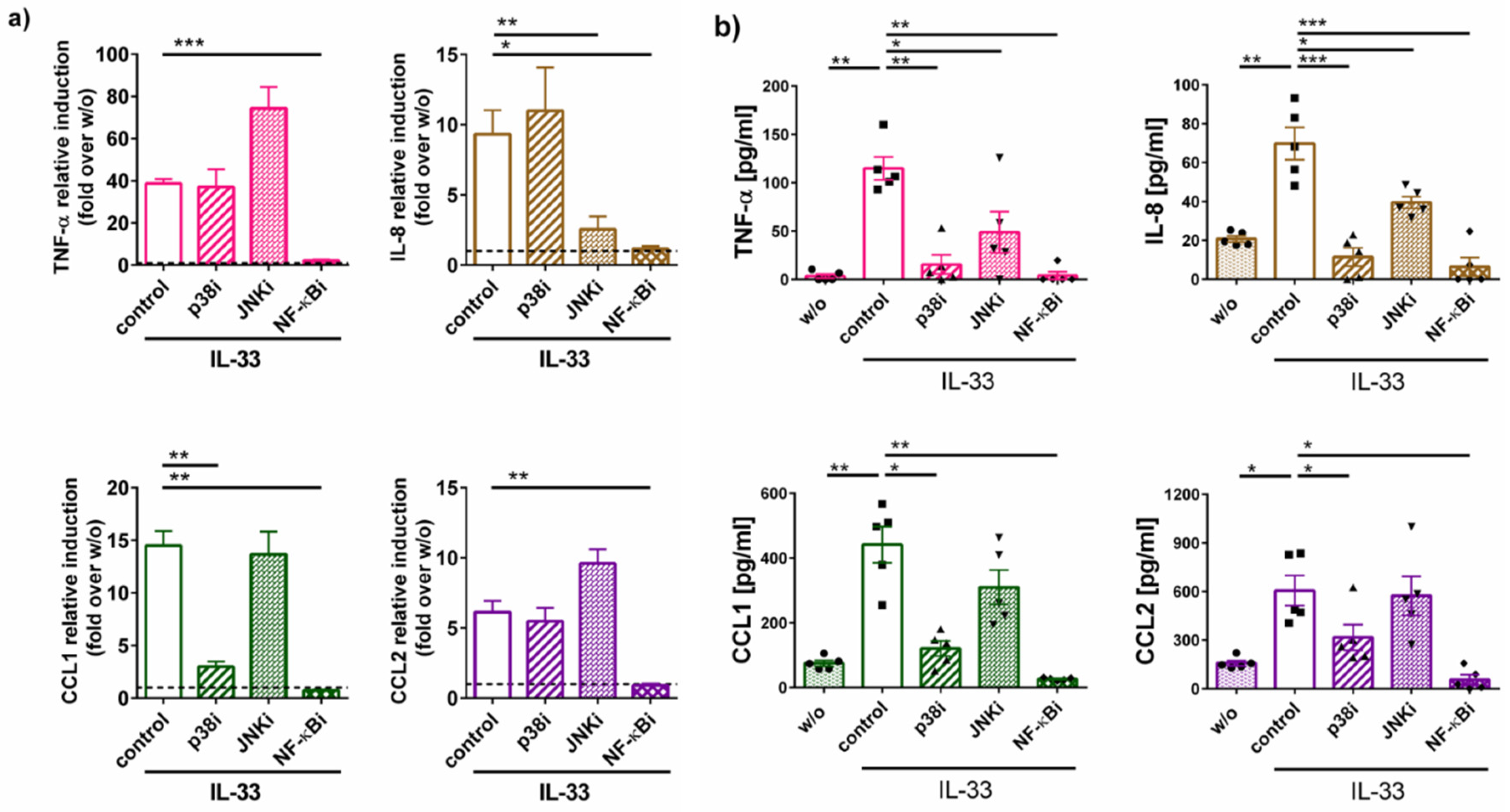
2.3. Cytokine Generation by IL-33 Chiefly Depends on NF-κB and p38: Differences between RNA and Protein Induction
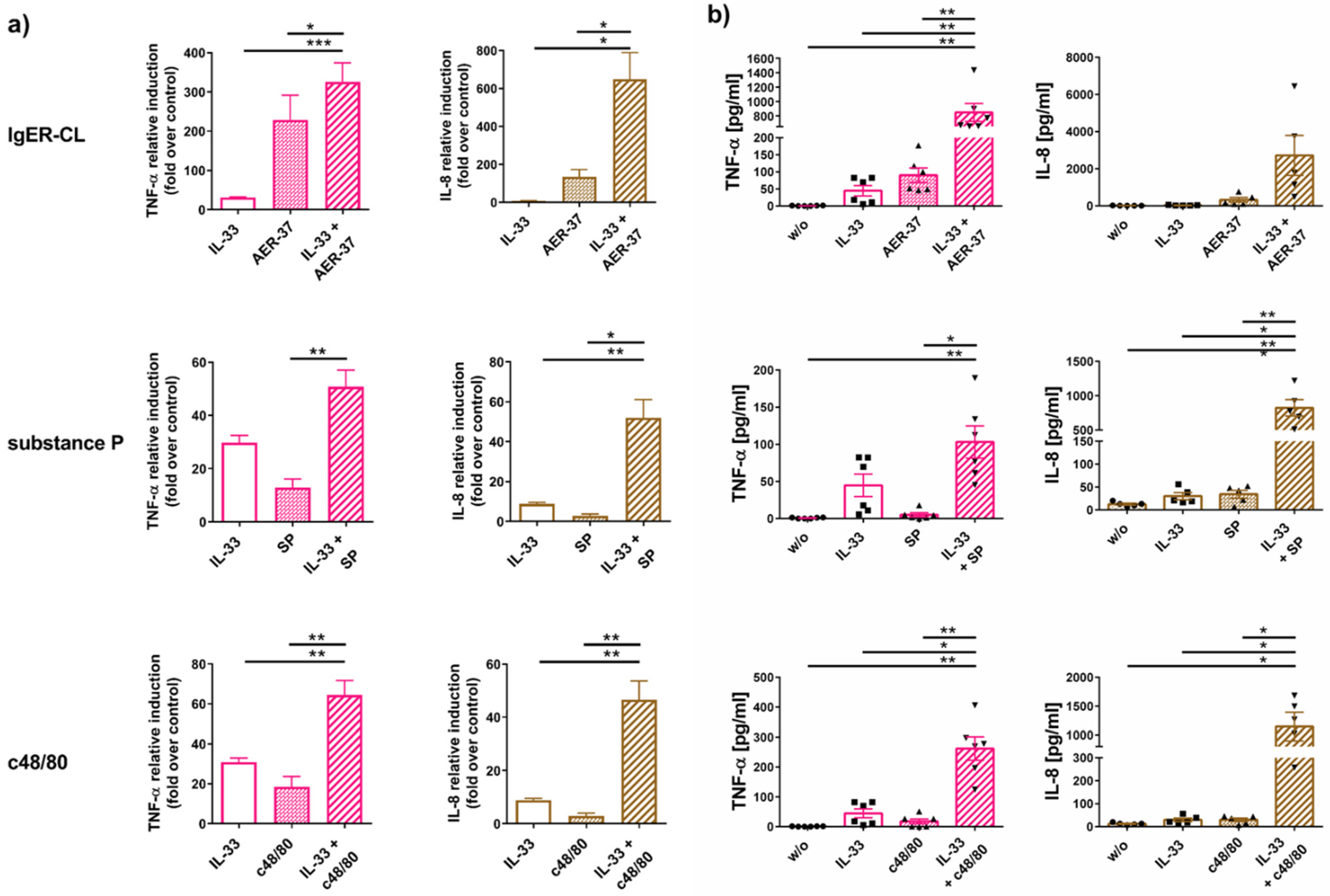
2.4. IL-33 Efficiently Synergizes with FcεRI and MRGPRX2 to Elicit Cytokine Production
3. Discussion
4. Materials and Methods
4.1. Cells and Treatments
4.2. Reverse Transcription-Quantitative PCR (RT-PCR)
4.3. ELISA
4.4. Immunoblot Analysis
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chackerian, A.A.; Oldham, E.R.; Murphy, E.E.; Schmitz, J.; Pflanz, S.; Kastelein, R.A. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J. Immunol. 2007, 179, 2551–2555. [Google Scholar] [CrossRef] [PubMed]
- Oboki, K.; Ohno, T.; Kajiwara, N.; Saito, H.; Nakae, S. IL-33 and IL-33 receptors in host defense and diseases. Allergol. Int. 2010, 59, 143–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, F.Y. IL-33: A Janus cytokine. Ann. Rheum. Dis. 2012, 71, i101–i104. [Google Scholar] [CrossRef]
- Saluja, R.; Ketelaar, M.E.; Hawro, T.; Church, M.K.; Maurer, M.; Nawijn, M.C. The role of the IL-33/IL-1RL1 axis in mast cell and basophil activation in allergic disorders. Mol. Immunol. 2015, 63, 80–85. [Google Scholar] [CrossRef]
- Valitutti, S.; Joulia, R.; Espinosa, E. The Mast Cell Antibody-Dependent Degranulatory Synapse. Methods Mol. Biol. 2017, 1584, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.O.; Pullen, N.A. Beyond IgE: Alternative Mast Cell Activation Across Different Disease States. Int. J. Mol. Sci. 2020, 21, 1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.X.; Kaieda, S.; Ameri, S.; Fishgal, N.; Dwyer, D.; Dellinger, A.; Kepley, C.L.; Gurish, M.F.; Nigrovic, P.A. IL-33/ST2 axis promotes mast cell survival via BCLXL. Proc. Natl. Acad. Sci. USA 2014, 111, 10281–10286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saluja, R.; Hawro, T.; Eberle, J.; Church, M.K.; Maurer, M. Interleukin-33 promotes the proliferation of mouse mast cells through ST2/MyD88 and p38 MAPK-dependent and Kit-independent pathways. J. Biol. Regul. Homeost. Agents 2014, 28, 575–585. [Google Scholar]
- Enoksson, M.; Moller-Westerberg, C.; Wicher, G.; Fallon, P.G.; Forsberg-Nilsson, K.; Lunderius-Andersson, C.; Nilsson, G. Intraperitoneal influx of neutrophils in response to IL-33 is mast cell-dependent. Blood 2013, 121, 530–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klonowska, J.; Glen, J.; Nowicki, R.J.; Trzeciak, M. New Cytokines in the Pathogenesis of Atopic Dermatitis-New Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 3086. [Google Scholar] [CrossRef] [Green Version]
- Moffatt, M.F.; Gut, I.G.; Demenais, F.; Strachan, D.P.; Bouzigon, E.; Heath, S.; von Mutius, E.; Farrall, M.; Lathrop, M.; Cookson, W.; et al. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 2010, 363, 1211–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, D.H.; Lee, E.; Lee, B.C.; Choi, M.J.; Kwon, T.R.; Hong, J.; Mun, S.K.; Lee, K.; Kim, S.; Na, J.; et al. Therapeutic potential of topically administered gamma-AlOOH on 2,4-dinitrochlorobenzene-induced atopic dermatitis-like lesions in Balb/c mice. Exp. Dermatol. 2018. [Google Scholar] [CrossRef]
- Qiao, Y.; Chen, J. Serum levels of IL-31, IL-33 and ST2 in allergic rhinitis of children in China. Cell Mol. Biol. 2018, 64, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Lee, J.B.; Liu, B.; Ohta, S.; Wang, P.Y.; Kartashov, A.V.; Mugge, L.; Abonia, J.P.; Barski, A.; Izuhara, K.; et al. Induction of Interleukin-9-Producing Mucosal Mast Cells Promotes Susceptibility to IgE-Mediated Experimental Food Allergy. Immunity 2015, 43, 788–802. [Google Scholar] [CrossRef] [Green Version]
- Galand, C.; Leyva-Castillo, J.M.; Yoon, J.; Han, A.; Lee, M.S.; McKenzie, A.N.J.; Stassen, M.; Oyoshi, M.K.; Finkelman, F.D.; Geha, R.S. IL-33 promotes food anaphylaxis in epicutaneously sensitized mice by targeting mast cells. J. Allergy Clin. Immunol. 2016, 138, 1356–1366. [Google Scholar] [CrossRef] [Green Version]
- Khodoun, M.V.; Tomar, S.; Tocker, J.E.; Wang, Y.H.; Finkelman, F.D. Prevention of food allergy development and suppression of established food allergy by neutralization of thymic stromal lymphopoietin, IL-25, and IL-33. J. Allergy Clin. Immunol. 2018, 141, 171–179.e1. [Google Scholar] [CrossRef] [Green Version]
- Theoharides, T.C.; Zhang, B.; Kempuraj, D.; Tagen, M.; Vasiadi, M.; Angelidou, A.; Alysandratos, K.D.; Kalogeromitros, D.; Asadi, S.; Stavrianeas, N.; et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. USA 2010, 107, 4448–4453. [Google Scholar] [CrossRef] [Green Version]
- Balato, A.; Lembo, S.; Mattii, M.; Schiattarella, M.; Marino, R.; De Paulis, A.; Balato, N.; Ayala, F. IL-33 is secreted by psoriatic keratinocytes and induces pro-inflammatory cytokines via keratinocyte and mast cell activation. Exp. Dermatol. 2012, 21, 892–894. [Google Scholar] [CrossRef] [PubMed]
- Savinko, T.; Matikainen, S.; Saarialho-Kere, U.; Lehto, M.; Wang, G.; Lehtimaki, S.; Karisola, P.; Reunala, T.; Wolff, H.; Lauerma, A.; et al. IL-33 and ST2 in atopic dermatitis: Expression profiles and modulation by triggering factors. J. Investig. Dermatol. 2012, 132, 1392–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Enoksson, M.; Lyberg, K.; Moller-Westerberg, C.; Fallon, P.G.; Nilsson, G.; Lunderius-Andersson, C. Mast cells as sensors of cell injury through IL-33 recognition. J. Immunol. 2011, 186, 2523–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drube, S.; Kraft, F.; Dudeck, J.; Muller, A.L.; Weber, F.; Gopfert, C.; Meininger, I.; Beyer, M.; Irmler, I.; Hafner, N.; et al. MK2/3 Are Pivotal for IL-33-Induced and Mast Cell-Dependent Leukocyte Recruitment and the Resulting Skin Inflammation. J. Immunol. 2016, 197, 3662–3668. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Metcalfe, D.D.; Peavy, R.D.; Gilfillan, A.M. Mechanisms of mast cell signaling in anaphylaxis. J. Allergy Clin. Immunol. 2009, 124, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potaczek, D.P.; Kabesch, M. Current concepts of IgE regulation and impact of genetic determinants. Clin. Exp. Allergy. 2012, 42, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Wang, Z.; Roy, S.; Guhl, S.; Franke, K.; Artuc, M.; Ali, H.; Zuberbier, T. MRGPRX2 Is the Codeine Receptor of Human Skin Mast Cells: Desensitization through beta-Arrestin and Lack of Correlation with the FcepsilonRI Pathway. J. Investig. Dermatol. 2020. [Google Scholar] [CrossRef]
- Kuhn, H.; Kolkhir, P.; Babina, M.; Dull, M.; Frischbutter, S.; Fok, J.S.; Jiao, Q.; Metz, M.; Scheffel, J.; Wolf, K.; et al. Mas-related G protein-coupled receptor X2 and its activators in dermatologic allergies. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Fujisawa, D.; Kashiwakura, J.; Kita, H.; Kikukawa, Y.; Fujitani, Y.; Sasaki-Sakamoto, T.; Kuroda, K.; Nunomura, S.; Hayama, K.; Terui, T.; et al. Expression of Mas-related gene X2 on mast cells is upregulated in the skin of patients with severe chronic urticaria. J. Allergy Clin. Immunol. 2014, 134, 622–633.e9. [Google Scholar] [CrossRef]
- Liu, R.; Che, D.; Zhao, T.; Pundir, P.; Cao, J.; Lv, Y.; Wang, J.; Ma, P.; Fu, J.; Wang, N.; et al. MRGPRX2 is essential for sinomenine hydrochloride induced anaphylactoid reactions. Biochem. Pharmacol. 2017, 146, 214–223. [Google Scholar] [CrossRef]
- Manorak, W.; Idahosa, C.; Gupta, K.; Roy, S.; Panettieri, R., Jr.; Ali, H. Upregulation of Mas-related G Protein coupled receptor X2 in asthmatic lung mast cells and its activation by the novel neuropeptide hemokinin-1. Respir. Res. 2018, 19, 1. [Google Scholar] [CrossRef] [Green Version]
- Babina, M.; Guhl, S.; Artuc, M.; Zuberbier, T. Allergic FcepsilonRI- and pseudo-allergic MRGPRX2-triggered mast cell activation routes are independent and inversely regulated by SCF. Allergy 2018, 73, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Theunis, J.; Black, D.; Degouy, A.; Schmitt, A.M.; Misery, L. Comparison of perceived itch induced by skin prick-tests with histamine and codeine. Acta Derm. Venereol. 2008, 88, 455–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, K.; Grize, L.; Schindler, C.; Surber, C.; Bircher, A.J. Reaction pattern to histamine and codeine in a human intradermal skin test model. Clin. Exp. Allergy. 2007, 37, 39–46. [Google Scholar] [CrossRef]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Gaudenzio, N.; Sibilano, R.; Marichal, T.; Starkl, P.; Reber, L.L.; Cenac, N.; McNeil, B.D.; Dong, X.; Hernandez, J.D.; Sagi-Eisenberg, R.; et al. Different activation signals induce distinct mast cell degranulation strategies. J. Clin. Investig. 2016, 126, 3981–3998. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Y.; Erlich, E.R.; Don, P.C. Skin prick test responses to codeine, histamine, and ragweed utilizing the Multitest device. Ann. Allergy 1990, 65, 222–226. [Google Scholar] [PubMed]
- Allakhverdi, Z.; Smith, D.E.; Comeau, M.R.; Delespesse, G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 2007, 179, 2051–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iikura, M.; Suto, H.; Kajiwara, N.; Oboki, K.; Ohno, T.; Okayama, Y.; Saito, H.; Galli, S.J.; Nakae, S. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab. Investig. 2007, 87, 971–978. [Google Scholar] [CrossRef] [Green Version]
- Moulin, D.; Donze, O.; Talabot-Ayer, D.; Mezin, F.; Palmer, G.; Gabay, C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 2007, 40, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Andrade, M.V.; Iwaki, S.; Ropert, C.; Gazzinelli, R.T.; Cunha-Melo, J.R.; Beaven, M.A. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur. J. Immunol. 2011, 41, 760–772. [Google Scholar] [CrossRef]
- Fux, M.; Pecaric-Petkovic, T.; Odermatt, A.; Hausmann, O.V.; Lorentz, A.; Bischoff, S.C.; Virchow, J.C.; Dahinden, C.A. IL-33 is a mediator rather than a trigger of the acute allergic response in humans. Allergy 2014, 69, 216–222. [Google Scholar] [CrossRef]
- Cop, N.; Ebo, D.G.; Bridts, C.H.; Elst, J.; Hagendorens, M.M.; Mertens, C.; Faber, M.A.; De Clerck, L.S.; Sabato, V. Influence of IL-6, IL-33, and TNF-alpha on human mast cell activation: Lessons from single cell analysis by flow cytometry. Cytom. B Clin. Cytom. 2018, 94, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Babina, M.; Wang, Z.; Franke, K.; Guhl, S.; Artuc, M.; Zuberbier, T. Yin-Yang of IL-33 in Human Skin Mast Cells: Reduced Degranulation, but Augmented Histamine Synthesis through p38 Activation. J. Investig. Dermatol. 2019, 139, 1516–1525.e3. [Google Scholar] [CrossRef]
- Cristinziano, L.; Poto, R.; Criscuolo, G.; Ferrara, A.L.; Galdiero, M.R.; Modestino, L.; Loffredo, S.; de Paulis, A.; Marone, G.; Spadaro, G.; et al. IL-33 and Superantigenic Activation of Human Lung Mast Cells Induce the Release of Angiogenic and Lymphangiogenic Factors. Cells 2021, 10, 145. [Google Scholar] [CrossRef]
- Joulia, R.; L’Faqihi, F.E.; Valitutti, S.; Espinosa, E. IL-33 fine tunes mast cell degranulation and chemokine production at the single-cell level. J. Allergy Clin. Immunol. 2017, 140, 497–509.e10. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Guhl, S.; Franke, K.; Artuc, M.; Zuberbier, T.; Babina, M. IL-33 and MRGPRX2-Triggered Activation of Human Skin Mast Cells-Elimination of Receptor Expression on Chronic Exposure, but Reinforced Degranulation on Acute Priming. Cells 2019, 8, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, M.R.; Margulis, A.; Wood, N.; Goldman, S.J.; Kasaian, M.; Chaudhary, D. IL-33 synergizes with IgE-dependent and IgE-independent agents to promote mast cell and basophil activation. Inflamm. Res. 2010, 59, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Cato, A.C.B.; Ainooson, G.K.; Freichel, M.; Tsvilovskyy, V.; Jessberger, R.; Riedlinger, E.; Sommerhoff, C.P.; Bischoff, S.C. Regulation of the pleiotropic effects of tissue-resident mast cells. J. Allergy Clin. Immunol. 2019, 144, S31–S45. [Google Scholar] [CrossRef] [Green Version]
- Motakis, E.; Guhl, S.; Ishizu, Y.; Itoh, M.; Kawaji, H.; de Hoon, M.; Lassmann, T.; Carninci, P.; Hayashizaki, Y.; Zuberbier, T.; et al. Redefinition of the human mast cell transcriptome by deep-CAGE sequencing. Blood 2014, 123, e58–e67. [Google Scholar] [CrossRef]
- Babina, M.; Guhl, S.; Starke, A.; Kirchhof, L.; Zuberbier, T.; Henz, B.M. Comparative cytokine profile of human skin mast cells from two compartments--strong resemblance with monocytes at baseline but induction of IL-5 by IL-4 priming. J. Leukoc. Biol. 2004, 75, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Guhl, S.; Lee, H.H.; Babina, M.; Henz, B.M.; Zuberbier, T. Evidence for a restricted rather than generalized stimulatory response of skin-derived human mast cells to substance P. J. Neuroimmunol. 2005, 163, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Guhl, S.; Motakis, E.; Artuc, M.; Hazzan, T.; Worm, M.; Forrest, A.R.; Zuberbier, T. Retinoic acid potentiates inflammatory cytokines in human mast cells: Identification of mast cells as prominent constituents of the skin retinoid network. Mol. Cell Endocrinol. 2015, 406, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Franke, K.; Zuberbier, T.; Babina, M. Cytokine production elicited via FceRI versus MRGPRX2 in human skin mast cells—Lower efficiency upon pseudo-allergic stimulation but equal dominance of ERK1/2 for both receptor systems. J. Investig. Derm. 2021. under review. [Google Scholar]
- FANTOM Consortium and the RIKEN PMI and CLST (DGT); Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Dimont, E.; Vitezic, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, S.; Arakawa, T.; Fukuda, S.; Furuno, M.; Hasegawa, A.; Hori, F.; Ishikawa-Kato, S.; Kaida, K.; Kaiho, A.; Kanamori-Katayama, M.; et al. FANTOM5 CAGE profiles of human and mouse samples. Sci. Data 2017, 4, 170112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHale, C.; Mohammed, Z.; Gomez, G. Human Skin-Derived Mast Cells Spontaneously Secrete Several Angiogenesis-Related Factors. Front. Immunol. 2019, 10, 1445. [Google Scholar] [CrossRef]
- Ho, L.H.; Ohno, T.; Oboki, K.; Kajiwara, N.; Suto, H.; Iikura, M.; Okayama, Y.; Akira, S.; Saito, H.; Galli, S.J.; et al. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J. Leukoc. Biol. 2007, 82, 1481–1490. [Google Scholar] [CrossRef] [Green Version]
- Drube, S.; Heink, S.; Walter, S.; Lohn, T.; Grusser, M.; Gerbaulet, A.; Berod, L.; Schons, J.; Dudeck, A.; Freitag, J.; et al. The receptor tyrosine kinase c-Kit controls IL-33 receptor signaling in mast cells. Blood 2010, 115, 3899–3906. [Google Scholar] [CrossRef] [Green Version]
- Ohto-Ozaki, H.; Hayakawa, M.; Kamoshita, N.; Maruyama, T.; Tominaga, S.I.; Ohmori, T. Induction of IkappaBzeta Augments Cytokine and Chemokine Production by IL-33 in Mast Cells. J. Immunol. 2020, 204, 2033–2042. [Google Scholar] [CrossRef]
- Ronnberg, E.; Ghaib, A.; Ceriol, C.; Enoksson, M.; Arock, M.; Safholm, J.; Ekoff, M.; Nilsson, G. Divergent Effects of Acute and Prolonged Interleukin 33 Exposure on Mast Cell IgE-Mediated Functions. Front. Immunol. 2019, 10, 1361. [Google Scholar] [CrossRef]
- Bawazeer, M.A.; Theoharides, T.C. IL-33 stimulates human mast cell release of CCL5 and CCL2 via MAPK and NF-kappaB, inhibited by methoxyluteolin. Eur. J. Pharmacol. 2019, 865, 172760. [Google Scholar] [CrossRef]
- Nakajima, S.; Ishimaru, K.; Kobayashi, A.; Yu, G.; Nakamura, Y.; Oh-Oka, K.; Suzuki-Inoue, K.; Kono, K.; Nakao, A. Resveratrol inhibits IL-33-mediated mast cell activation by targeting the MK2/3-PI3K/Akt axis. Sci. Rep. 2019, 9, 18423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yin, N.; Tao, W.; Wang, Q.; Fan, H.; Wang, Z. Berberine suppresses IL-33-induced inflammatory responses in mast cells by inactivating NF-kappaB and p38 signaling. Int. Immunopharmacol. 2019, 66, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Ndaw, V.S.; Abebayehu, D.; Spence, A.J.; Paez, P.A.; Kolawole, E.M.; Taruselli, M.T.; Caslin, H.L.; Chumanevich, A.P.; Paranjape, A.; Baker, B.; et al. TGF-beta1 Suppresses IL-33-Induced Mast Cell Function. J. Immunol. 2017, 199, 866–873. [Google Scholar] [CrossRef] [Green Version]
- Gopfert, C.; Andreas, N.; Weber, F.; Hafner, N.; Yakovleva, T.; Gaestel, M.; Kamradt, T.; Drube, S. The p38-MK2/3 Module Is Critical for IL-33-Induced Signaling and Cytokine Production in Dendritic Cells. J. Immunol. 2018, 200, 1198–1206. [Google Scholar] [CrossRef]
- Brint, E.K.; Fitzgerald, K.A.; Smith, P.; Coyle, A.J.; Gutierrez-Ramos, J.C.; Fallon, P.G.; O’Neill, L.A. Characterization of signaling pathways activated by the interleukin 1 (IL-1) receptor homologue T1/ST2. A role for Jun N-terminal kinase in IL-4 induction. J. Biol. Chem. 2002, 277, 49205–49211. [Google Scholar] [CrossRef] [Green Version]
- Saluja, R.; Khan, M.; Church, M.K.; Maurer, M. The role of IL-33 and mast cells in allergy and inflammation. Clin. Transl. Allergy 2015, 5, 33. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.M.; Xu, W.M.; Lin, J.C.; Mo, L.Q.; Hua, X.X.; Chen, P.X.; Wu, K.; Zheng, D.D.; Feng, J.Q. Activation of the p38 MAPK/NF-kappaB pathway contributes to doxorubicin-induced inflammation and cytotoxicity in H9c2 cardiac cells. Mol. Med. Rep. 2013, 8, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Baldassarre, F.; Mallardo, M.; Mezza, E.; Scala, G.; Quinto, I. Regulation of NF-kappa B through the nuclear processing of p105 (NF-kappa B1) in Epstein-Barr virus-immortalized B cell lines. J. Biol. Chem. 1995, 270, 31244–31248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, P.E.; Mitxitorena, I.; Carmody, R.J. The Ubiquitination of NF-kappaB Subunits in the Control of Transcription. Cells 2016, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermans, M.A.W.; Stigt, A.C.v.; Meerendonk, S.v.d.; Schrijver, B.; Daele, P.L.A.v.; Hagen, P.M.v.; Splunter, M.v.; Dik, W.A. Human mast cell line HMC1 Expresses functional Mas-related G-protein coupled receptor 2. Front. Immunol. 2021, 12, 5284. [Google Scholar] [CrossRef]
- Paranjape, A.; Chernushevich, O.; Qayum, A.A.; Spence, A.J.; Taruselli, M.T.; Abebayehu, D.; Barnstein, B.O.; McLeod, J.J.; Baker, B.; Bajaj, G.S.; et al. Dexamethasone rapidly suppresses IL-33-stimulated mast cell function by blocking transcription factor activity. J. Leukoc. Biol. 2016, 100, 1395–1404. [Google Scholar] [CrossRef]
- Espinosa-Riquer, Z.P.; Segura-Villalobos, D.; Ramirez-Moreno, I.G.; Perez Rodriguez, M.J.; Lamas, M.; Gonzalez-Espinosa, C. Signal Transduction Pathways Activated by Innate Immunity in Mast Cells: Translating Sensing of Changes into Specific Responses. Cells 2020, 9, 2411. [Google Scholar] [CrossRef] [PubMed]
- Katsoulis-Dimitriou, K.; Kotrba, J.; Voss, M.; Dudeck, J.; Dudeck, A. Mast Cell Functions Linking Innate Sensing to Adaptive Immunity. Cells 2020, 9, 2538. [Google Scholar] [CrossRef]
- Dwyer, D.F.; Barrett, N.A.; Austen, K.F.; Immunological Genome Project, C. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat. Immunol. 2016, 17, 878–887. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Beaven, M.A. Regulation of mast cell responses in health and disease. Crit. Rev. Immunol. 2011, 31, 475–529. [Google Scholar] [CrossRef]
- Bulfone-Paus, S.; Bahri, R. Mast Cells as Regulators of T Cell Responses. Front. Immunol. 2015, 6, 394. [Google Scholar] [CrossRef] [Green Version]
- Che, D.N.; Shin, J.Y.; Kang, H.J.; Cho, B.O.; Kim, Y.S.; Jang, S.I. Luteolin suppresses IL-31 production in IL-33-stimulated mast cells through MAPK and NF-kappaB signaling pathways. Int. Immunopharmacol. 2020, 83, 106403. [Google Scholar] [CrossRef] [PubMed]
- Taracanova, A.; Tsilioni, I.; Conti, P.; Norwitz, E.R.; Leeman, S.E.; Theoharides, T.C. Substance P and IL-33 administered together stimulate a marked secretion of IL-1beta from human mast cells, inhibited by methoxyluteolin. Proc. Natl. Acad. Sci. USA 2018, 115, E9381–E9390. [Google Scholar] [CrossRef] [Green Version]
- Altman, M.C.; Lai, Y.; Nolin, J.D.; Long, S.; Chen, C.C.; Piliponsky, A.M.; Altemeier, W.A.; Larmore, M.; Frevert, C.W.; Mulligan, M.S.; et al. Airway epithelium-shifted mast cell infiltration regulates asthmatic inflammation via IL-33 signaling. J. Clin. Investig. 2019, 129, 4979–4991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Kawakami, Y.; Kasakura, K.; Kawakami, T. Recent advances in mast cell activation and regulation. F1000Res 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, C.; Yao, S.Y.; Sriram, S. TLR3 Agonist Poly-IC Induces IL-33 and Promotes Myelin Repair. PLoS ONE 2016, 11, e0152163. [Google Scholar] [CrossRef] [PubMed]
- Ruisong, M.; Xiaorong, H.; Gangying, H.; Chunfeng, Y.; Changjiang, Z.; Xuefei, L.; Yuanhong, L.; Hong, J. The Protective Role of Interleukin-33 in Myocardial Ischemia and Reperfusion Is Associated with Decreased HMGB1 Expression and Up-Regulation of the P38 MAPK Signaling Pathway. PLoS ONE 2015, 10, e0143064. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Hirahara, K.; Iinuma, T.; Shinoda, K.; Tumes, D.J.; Asou, H.K.; Matsugae, N.; Obata-Ninomiya, K.; Yamamoto, H.; Motohashi, S.; et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity 2015, 42, 294–308. [Google Scholar] [CrossRef] [Green Version]
- Matta, B.M.; Turnquist, H.R. Expansion of Regulatory T Cells In Vitro and In Vivo by IL-33. Methods Mol. Biol. 2016, 1371, 29–41. [Google Scholar] [CrossRef]
- Sakai, N.; Van Sweringen, H.L.; Quillin, R.C.; Schuster, R.; Blanchard, J.; Burns, J.M.; Tevar, A.D.; Edwards, M.J.; Lentsch, A.B. Interleukin-33 is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology 2012, 56, 1468–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecaric-Petkovic, T.; Didichenko, S.A.; Kaempfer, S.; Spiegl, N.; Dahinden, C.A. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood 2009, 113, 1526–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, J.Y.; Wong, C.K.; Cheung, P.F.; Lam, C.W. Intracellular signaling mechanisms regulating the activation of human eosinophils by the novel Th2 cytokine IL-33: Implications for allergic inflammation. Cell Mol. Immunol. 2010, 7, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Tare, N.; Li, H.; Morschauser, A.; Cote-Sierra, J.; Ju, G.; Renzetti, L.; Lin, T.A. KU812 cells provide a novel in vitro model of the human IL-33/ST2L axis: Functional responses and identification of signaling pathways. Exp. Cell Res. 2010, 316, 2527–2537. [Google Scholar] [CrossRef]
- Eeles, D.G.; Hodge, J.M.; Singh, P.P.; Schuijers, J.A.; Grills, B.L.; Gillespie, M.T.; Myers, D.E.; Quinn, J.M. Osteoclast formation elicited by interleukin-33 stimulation is dependent upon the type of osteoclast progenitor. Mol. Cell Endocrinol. 2015, 399, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Fujita, J.; Kawaguchi, M.; Kokubu, F.; Ohara, G.; Ota, K.; Huang, S.K.; Morishima, Y.; Ishii, Y.; Satoh, H.; Sakamoto, T.; et al. Interleukin-33 induces interleukin-17F in bronchial epithelial cells. Allergy 2012, 67, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Heger, K.; Fierens, K.; Vahl, J.C.; Aszodi, A.; Peschke, K.; Schenten, D.; Hammad, H.; Beyaert, R.; Saur, D.; van Loo, G.; et al. A20-deficient mast cells exacerbate inflammatory responses in vivo. PLoS Biol. 2014, 12, e1001762. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.E.; Walker, S.R.; Savery, K.; Frank, D.A.; Gaudet, S. Fold change of nuclear NF-kappaB determines TNF-induced transcription in single cells. Mol. Cell 2014, 53, 867–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taracanova, A.; Alevizos, M.; Karagkouni, A.; Weng, Z.; Norwitz, E.; Conti, P.; Leeman, S.E.; Theoharides, T.C. SP and IL-33 together markedly enhance TNF synthesis and secretion from human mast cells mediated by the interaction of their receptors. Proc. Natl. Acad. Sci. USA 2017, 114, E4002–E4009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beenstock, J.; Ben-Yehuda, S.; Melamed, D.; Admon, A.; Livnah, O.; Ahn, N.G.; Engelberg, D. The p38beta mitogen-activated protein kinase possesses an intrinsic autophosphorylation activity, generated by a short region composed of the alpha-G helix and MAPK insert. J. Biol. Chem. 2014, 289, 23546–23556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef]
- Zhu, M.M.; Tong, J.L.; Xu, Q.; Nie, F.; Xu, X.T.; Xiao, S.D.; Ran, Z.H. Increased JNK1 signaling pathway is responsible for ABCG2-mediated multidrug resistance in human colon cancer. PLoS ONE 2012, 7, e41763. [Google Scholar] [CrossRef]
- McCarthy, P.C.; Phair, I.R.; Greger, C.; Pardali, K.; McGuire, V.A.; Clark, A.R.; Gaestel, M.; Arthur, J.S.C. IL-33 regulates cytokine production and neutrophil recruitment via the p38 MAPK-activated kinases MK2/3. Immunol. Cell Biol. 2019, 97, 54–71. [Google Scholar] [CrossRef]
- Available online: https://bioinfo.lifl.fr/NF-KB (accessed on 3 February 2021).
- Emi-Sugie, M.; Saito, H.; Matsumoto, K. Cultured human mast cells release various chemokines after stimulation with IL-33. Allergol. Int. 2021. [Google Scholar] [CrossRef]
- Hochdorfer, T.; Tiedje, C.; Stumpo, D.J.; Blackshear, P.J.; Gaestel, M.; Huber, M. LPS-induced production of TNF-alpha and IL-6 in mast cells is dependent on p38 but independent of TTP. Cell Signal. 2013, 25, 1339–1347. [Google Scholar] [CrossRef] [Green Version]
- Babina, M. The pseudo-allergic/neurogenic route of mast cell activation via MRGPRX2: Discovery, functional programs, regulation, relevance to disease, and relation with allergic stimulation. Itch 2020, 5, e32. [Google Scholar] [CrossRef]
- Tatemoto, K.; Nozaki, Y.; Tsuda, R.; Konno, S.; Tomura, K.; Furuno, M.; Ogasawara, H.; Edamura, K.; Takagi, H.; Iwamura, H.; et al. Immunoglobulin E-independent activation of mast cell is mediated by Mrg receptors. Biochem. Biophys. Res. Commun. 2006, 349, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, H.; Gupta, K.; Ali, H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J. Allergy Clin. Immunol. 2016, 138, 700–710. [Google Scholar] [CrossRef] [Green Version]
- Babina, M.; Wang, Z.; Franke, K.; Zuberbier, T. Thymic Stromal Lymphopoietin Promotes MRGPRX2-Triggered Degranulation of Skin Mast Cells in a STAT5-Dependent Manner with Further Support from JNK. Cells 2021, 10, 102. [Google Scholar] [CrossRef]
- Babina, M.; Wang, Z.; Artuc, M.; Guhl, S.; Zuberbier, T. MRGPRX2 is negatively targeted by SCF and IL-4 to diminish pseudo-allergic stimulation of skin mast cells in culture. Exp. Dermatol. 2018, 27, 1298–1303. [Google Scholar] [CrossRef]
- Hazzan, T.; Eberle, J.; Worm, M.; Babina, M. Apoptotic resistance of human skin mast cells is mediated by Mcl-1. Cell Death Discov. 2017, 3, 17048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastogi, S.; Willmes, D.M.; Nassiri, M.; Babina, M.; Worm, M. PGE2 deficiency predisposes to anaphylaxis by causing mast cell hyperresponsiveness. J. Allergy Clin. Immunol. 2020, 146, 1387–1396.e13. [Google Scholar] [CrossRef]
- Babina, M.; Artuc, M.; Guhl, S.; Zuberbier, T. Retinoic Acid Negatively Impacts Proliferation and MCTC Specific Attributes of Human Skin Derived Mast Cells, but Reinforces Allergic Stimulability. Int. J. Mol. Sci. 2017, 18, 525. [Google Scholar] [CrossRef] [Green Version]
- Guhl, S.; Neou, A.; Artuc, M.; Zuberbier, T.; Babina, M. Skin mast cells develop non-synchronized changes in typical lineage characteristics upon culture. Exp. Dermatol. 2014, 23, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Guhl, S.; Artuc, M.; Neou, A.; Babina, M.; Zuberbier, T. Long-term cultured human skin mast cells are suitable for pharmacological studies of anti-allergic drugs due to high responsiveness to FcepsilonRI cross-linking. Biosci. Biotechnol. Biochem. 2011, 75, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Tsang, C.M.; Ip, W.K.; Lam, C.W. Molecular mechanisms for the release of chemokines from human leukemic mast cell line (HMC)-1 cells activated by SCF and TNF-alpha: Roles of ERK, p38 MAPK, and NF-kappaB. Allergy 2006, 61, 289–297. [Google Scholar] [CrossRef]
- Tung, H.Y.; Plunkett, B.; Huang, S.K.; Zhou, Y. Murine mast cells secrete and respond to interleukin-33. J. Interferon. Cytokine. Res. 2014, 34, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.-W. Inhibitory Effects of PD98059, SB203580, and SP600125 on α- and δ-granule Release and Intracellular Ca2+ Levels in Human Platelets. Biomed. Sci. Lett. 2018, 24, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Mitomo, S.; Omatsu, T.; Tsuchiaka, S.; Nagai, M.; Furuya, T.; Mizutani, T. Activation of c-Jun N-terminal kinase by Akabane virus is required for apoptosis. Res. Vet. Sci. 2016, 107, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Guhl, S.; Artuc, M.; Trivedi, N.N.; Zuberbier, T. Phenotypic variability in human skin mast cells. Exp. Dermatol. 2016, 25, 434–439. [Google Scholar] [CrossRef]
- Hasel, K.W.; Glass, J.R.; Godbout, M.; Sutcliffe, J.G. An endoplasmic reticulum-specific cyclophilin. Mol. Cell Biol. 1991, 11, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Price, E.R.; Zydowsky, L.D.; Jin, M.J.; Baker, C.H.; McKeon, F.D.; Walsh, C.T. Human cyclophilin B: A second cyclophilin gene encodes a peptidyl-prolyl isomerase with a signal sequence. Proc. Natl. Acad. Sci. USA 1991, 88, 1903–1907. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Carrero, G.; Andrin, C.; de Vries, G.; Hendzel, M.J. Nucleoplasmic beta-actin exists in a dynamic equilibrium between low-mobility polymeric species and rapidly diffusing populations. J. Cell Biol. 2006, 172, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Shen, X. Nuclear actin and actin-related proteins in chromatin dynamics. Curr. Opin. Cell Biol. 2007, 19, 326–330. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Thuraisingam, T.; Morais, D.A.; Rola-Pleszczynski, M.; Radzioch, D. Nuclear translocation of beta-actin is involved in transcriptional regulation during macrophage differentiation of HL-60 cells. Mol. Biol. Cell 2010, 21, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serebryannyy, L.; de Lanerolle, P. Nuclear actin: The new normal. Mutat. Res. 2020, 821, 111714. [Google Scholar] [CrossRef] [PubMed]
- Serebryannyy, L.A.; Cruz, C.M.; de Lanerolle, P. A Role for Nuclear Actin in HDAC 1 and 2 Regulation. Sci. Rep. 2016, 6, 28460. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine (RNA) | IgER-CL | c48/80 | SP |
| TNF-α | 1.10 | 1.38 | 1.43 |
| IL-8 | 5.95 | 5.20 | 4.56 |
| CCL1 | 1.90 | 4.31 | 3.24 |
| CCL2 | 1.47 | 2.35 | 2.11 |
| Cytokine (Protein) | IgER-CL | c48/80 | SP |
| TNF-α | 8.12 | 5.67 | 2.72 |
| IL-8 | 7.59 | 15.71 | 13.50 |
| Gene | Forward 5′-3′ | Reverse 5′-3′ |
|---|---|---|
| TNF-α | TCTCGAACCCCGAGTGACAA | TCAGCCACTGGAGCTGCC |
| IL-8 | ATGACTTCCAAGCTGGCCGTGGCT | TCTCAGCCCTCTTCAAAAACTTCTC |
| CCL1 | TTGCGGAGCAAGAGATTCCC | GGCAGTGCCTCAGCATTTTT |
| CCL2 | CCCCAAGCAGAAGTGGGTTC | TTGGGTTGTGGAGTGAGTGTT |
| IL-1β | CGAGGGAGAAACTGGCAGAT | AAGCCATCATTTCACTGGCG |
| IL-5 | GAGTCAAACTGTGCAAGGGG | TGGCTGCAACAAACCAGTTT |
| IL-6 | ATGTAGCCGCCCCACACAGA | CATCCATCTTTTTCAGCCAT |
| IL-13 | CATCCGCTCCTCAATCCTCT | GATGCTCCATACCATGCTGC |
| IL-31 | CCCGTCCGTTTACTACGACC | TTGAGATATGCCCGGATGGC |
| VEGFA | GAAGAAGCAGCCCATGACAG | CTCACACACACACAACAAGG |
| β-actin | CTGGAACGGTGAAGGTGACA | AAGGGACTTCCTGTAACAATGCA |
| Cyclophilin B | AAGATGTCCCTGTGCCCTAC | ATGGCAAGCATGTGGTGTTT |
| GAPDH | ATCTCGCTCCTGGAAGATGG | AGGTCGGAGTCAACGGATTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franke, K.; Wang, Z.; Zuberbier, T.; Babina, M. Cytokines Stimulated by IL-33 in Human Skin Mast Cells: Involvement of NF-κB and p38 at Distinct Levels and Potent Co-Operation with FcεRI and MRGPRX2. Int. J. Mol. Sci. 2021, 22, 3580. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073580
Franke K, Wang Z, Zuberbier T, Babina M. Cytokines Stimulated by IL-33 in Human Skin Mast Cells: Involvement of NF-κB and p38 at Distinct Levels and Potent Co-Operation with FcεRI and MRGPRX2. International Journal of Molecular Sciences. 2021; 22(7):3580. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073580
Chicago/Turabian StyleFranke, Kristin, Zhao Wang, Torsten Zuberbier, and Magda Babina. 2021. "Cytokines Stimulated by IL-33 in Human Skin Mast Cells: Involvement of NF-κB and p38 at Distinct Levels and Potent Co-Operation with FcεRI and MRGPRX2" International Journal of Molecular Sciences 22, no. 7: 3580. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073580