
Comparative Transcriptomes of the Body Wall of Wild and Farmed Sea Cucumber Isostichopus badionotus

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
2.1. Samples
2.2. Transcriptome Sequencing
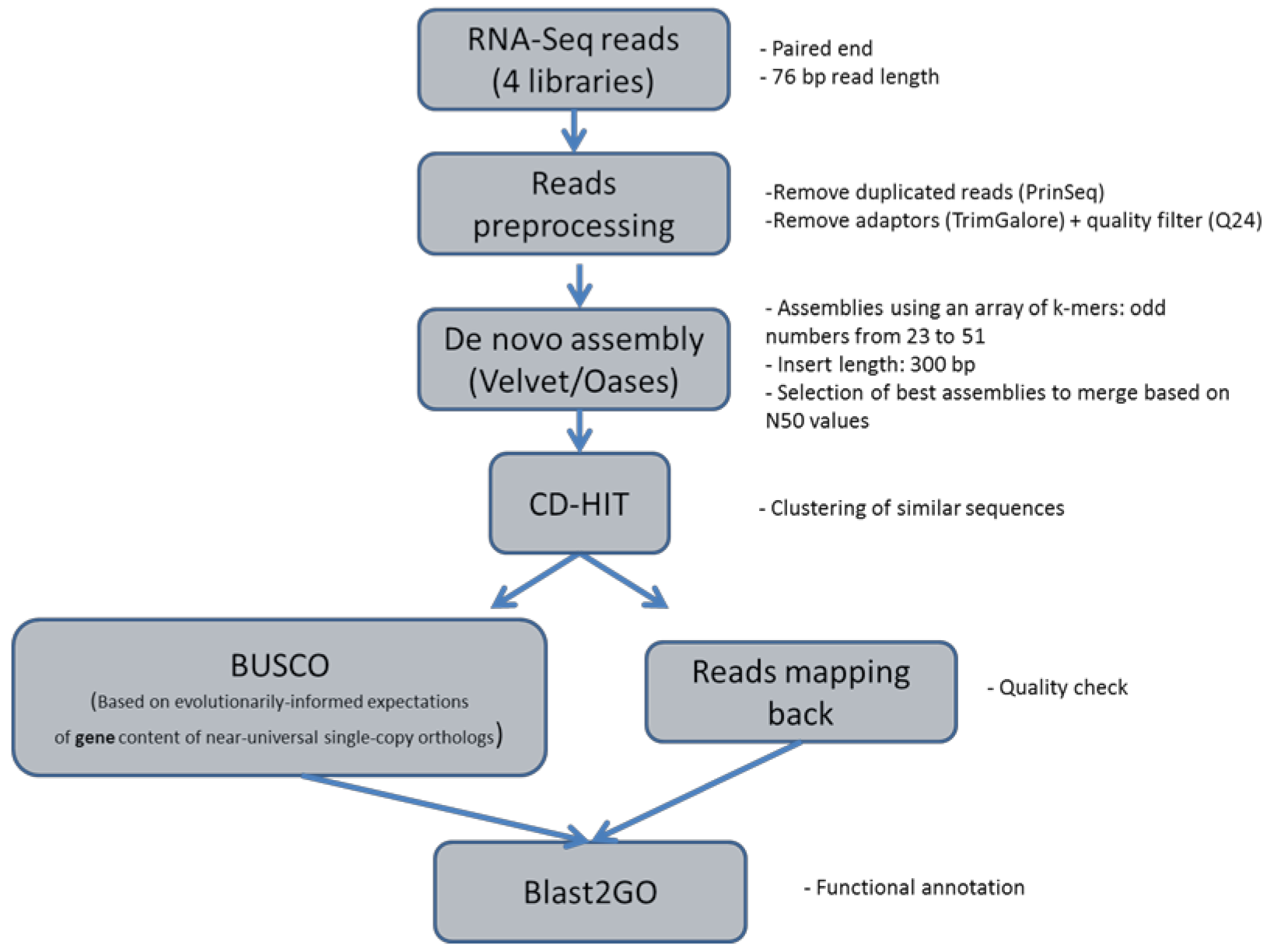
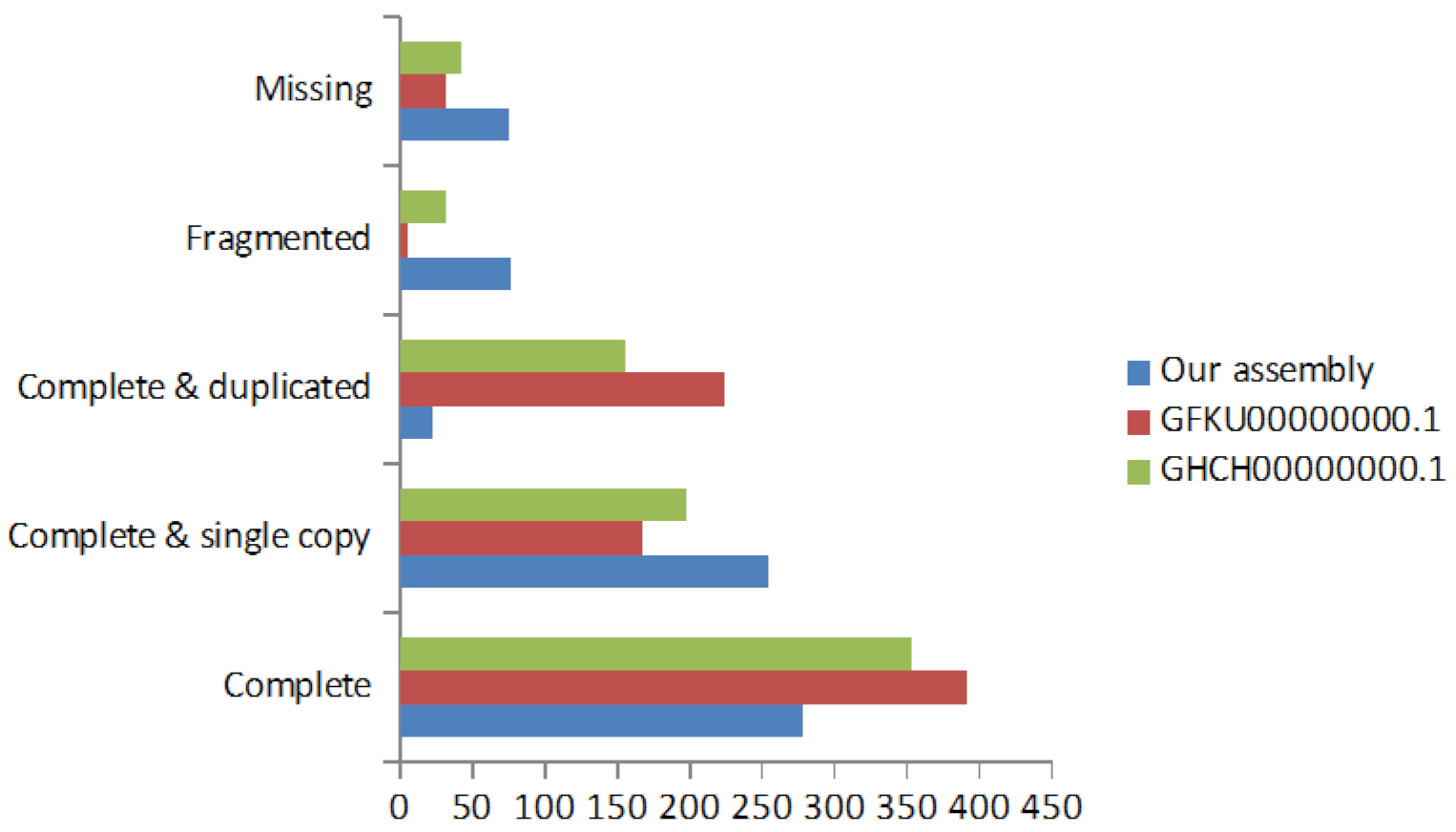
2.3. De Novo Transcriptome Assembly
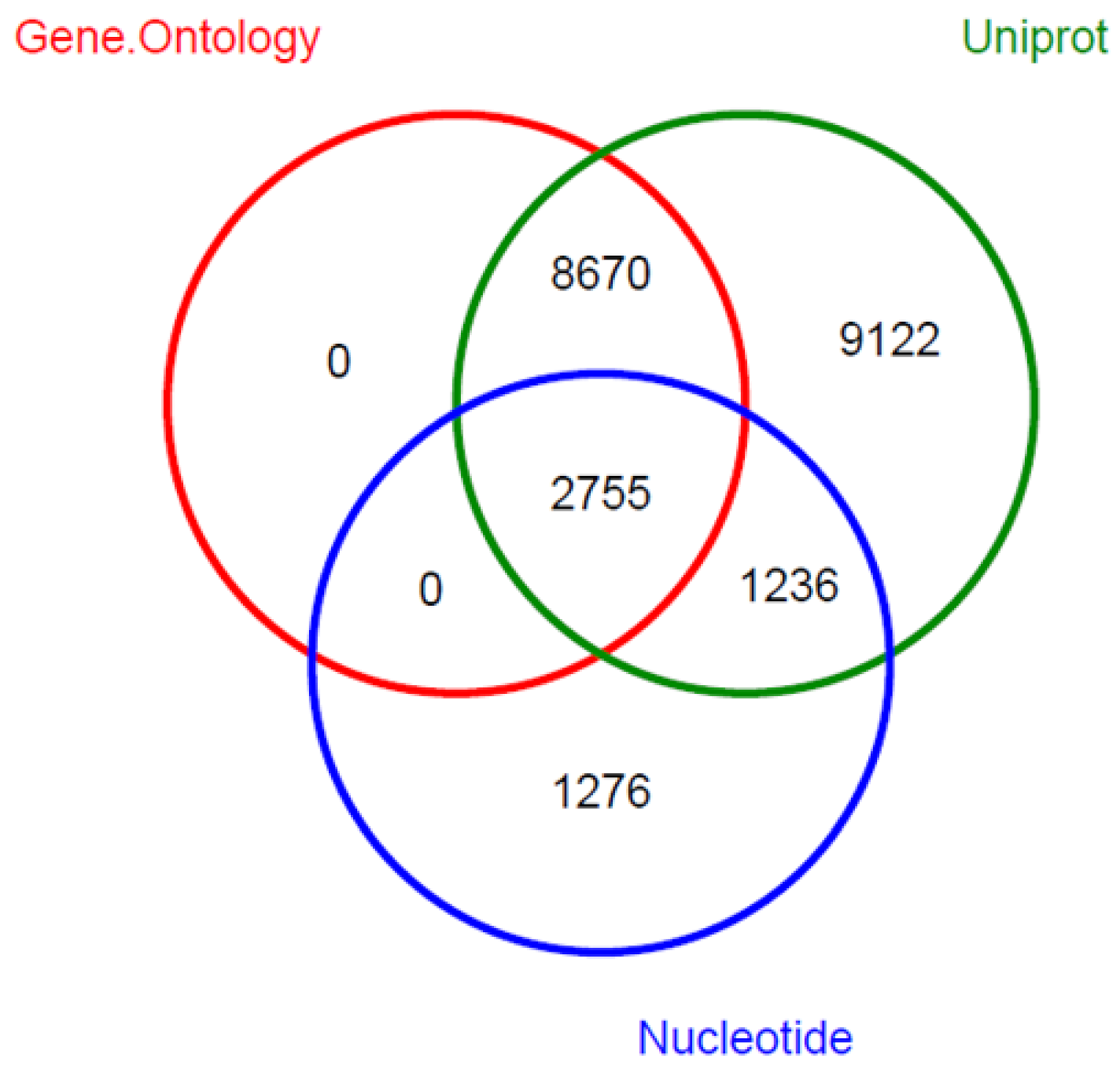
2.4. Transcriptome Annotation
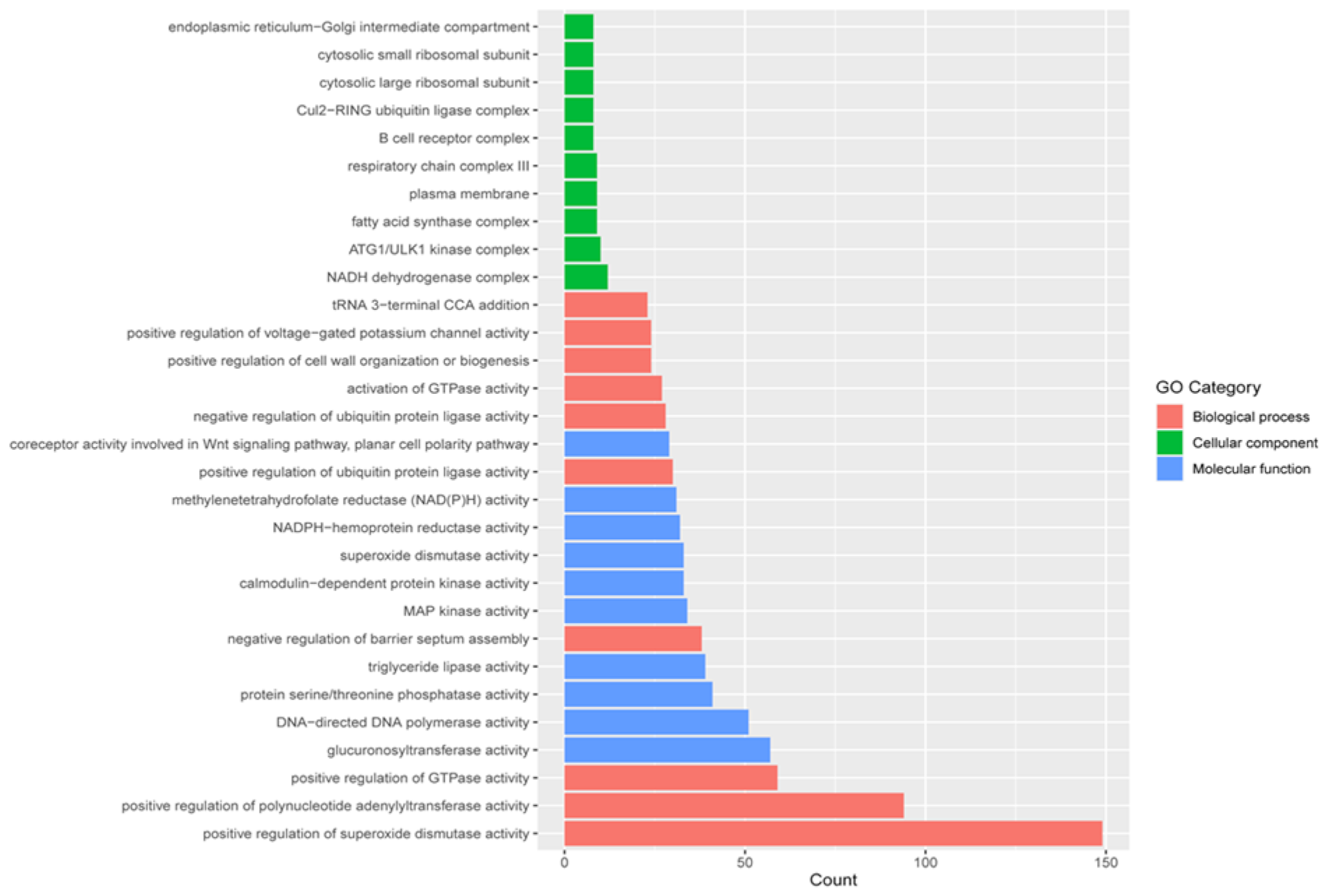
2.5. Gene Ontology (GO) Classification
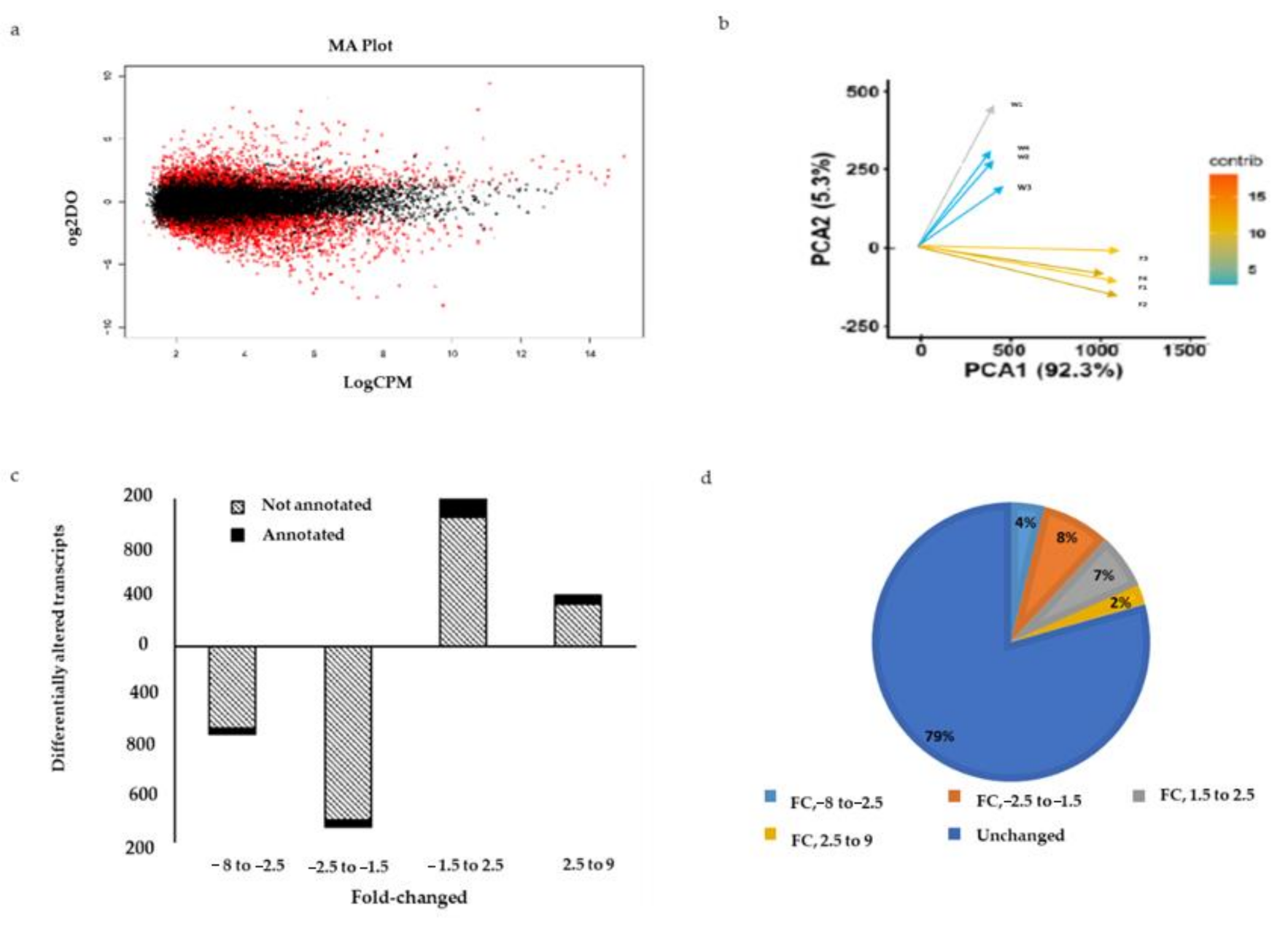
2.6. Differentially Expressed Genes between Wild and Farmed Sea Cucumbers
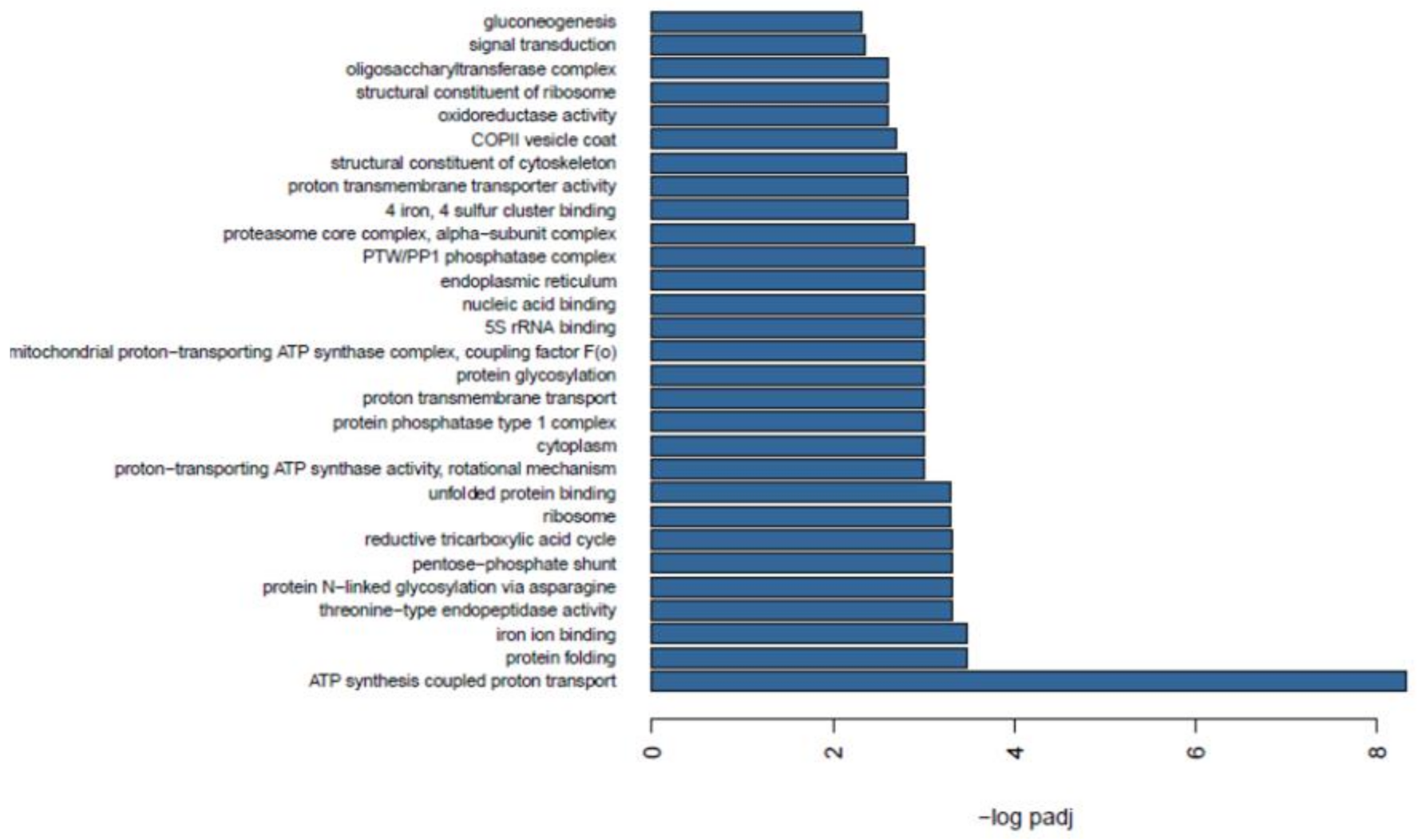
2.7. Functional Enrichment between Wild and Farmed Sea Cucumbers
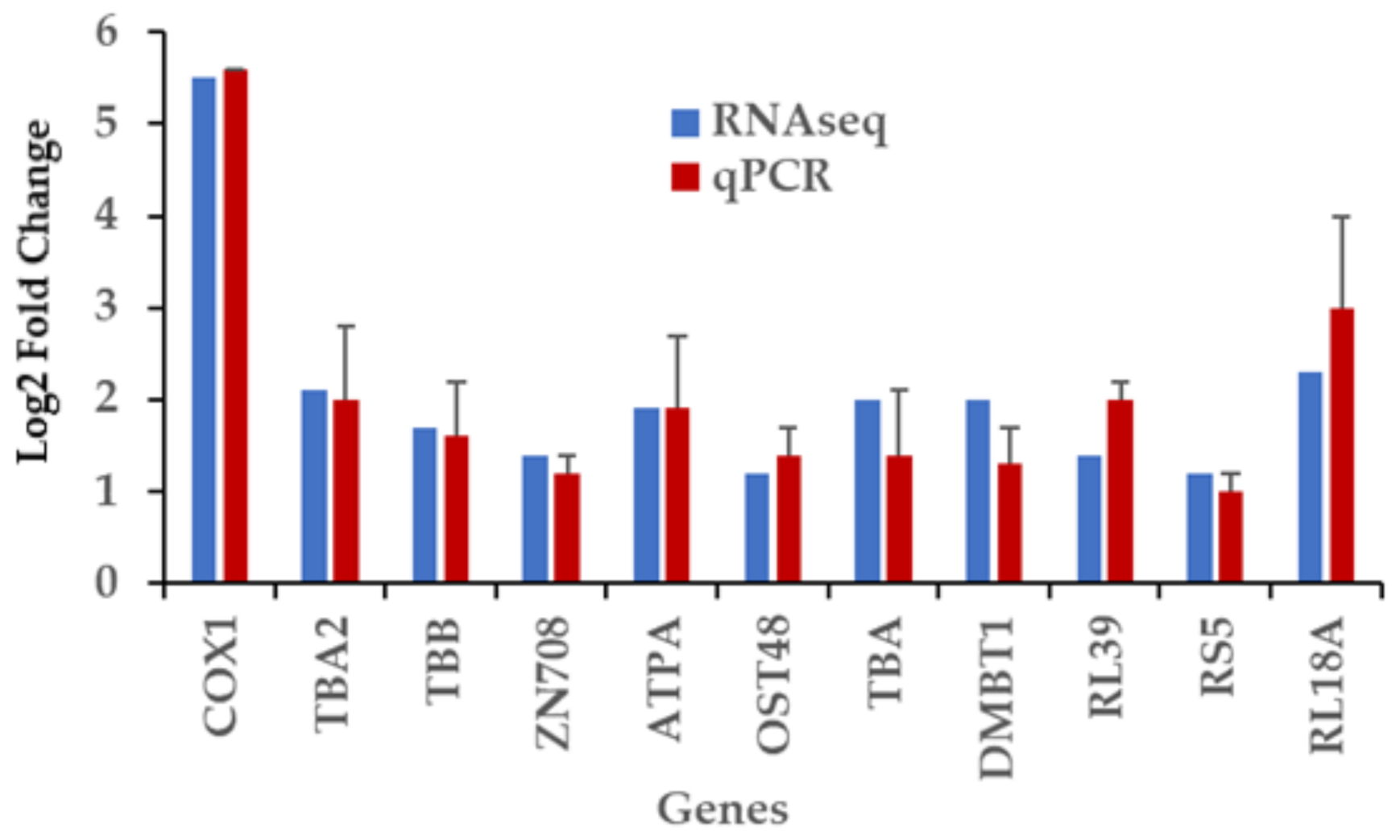
2.8. RT-qPCR Validation of Differentially Expressed Genes Induced by Captivity
3. Discussion
4. Materials and Methods
4.1. Collection of Wild Sea Cucumbers
4.2. Cultivation of Sea Cucumbers
4.3. RNA Isolation, cDNA Library, and Illumina Sequencing
4.4. Short Reads De Novo Assembly
4.5. Functional Annotation
4.6. Functional Enrichment
4.7. Differential Expression between Sea Cucumber Body Wall Transcriptomes, and Pathway Analysis
4.8. Gene Validation
4.9. Ethics Statement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATPA | ATP synthase subunit alpha |
| BLAST | Basic Local Alignment Search Tool |
| COX1 | cytochrome c oxidase subunit 1 |
| CREB1 | CAMP responsive element binding protein 1 |
| DEG | Differentially expressed gene |
| EIF2AK3 | Eukaryotic Translation Initiation Factor 2 Alpha Kinase 3 |
| FDR | False discovery rate |
| GO | Gene Ontology |
| KO | KEGG Orthology |
| RT-qPCR | Reverse transcription qualitative real-time polymerase chain rection |
| SRA | Sequence Read Archive |
References
- Pawson, D.L. Phylum Echinodermata. In Linnaeus Tercentenary: Progress in Invertebrate Taxonomy, Zootaxa 1668; Zhang, Z.-Q., Shear, W.A., Eds.; Magnolia Press: Auckland, New Zealand, 2007; pp. 649–764. [Google Scholar] [CrossRef]
- Khotimchenko, Y. Pharmacological potential of sea cucumbers. Int. J. Mol. Sci. 2018, 19, 1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pangestuti, R.; Arifin, Z. Medicinal and health benefit effects of functional sea cucumbers. J. Tradit. Complement. Med. 2018, 8, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Baker-Médard, M.; Ohl, K.N. Sea cucumber management strategies: Challenges and opportunities in a developing country context. Environ. Conserv. 2019, 46, 267–277. [Google Scholar] [CrossRef]
- Gamboa-Álvarez, M.Á.; López-Rocha, J.A.; Poot-López, G.R.; Aguilar-Perera, A.; Villegas-Hernández, H. Rise and decline of the sea cucumber fishery in Campeche Bank, Mexico. Ocean Coast. Manag. 2020, 184, 105011. [Google Scholar] [CrossRef]
- López-Rocha, J.A.; Velázquez-Abunader, I. Fast decline of the sea cucumber Isostichopus badionotus as a consequence of high exploitation in Yucatan, Mexico. Reg. Stud. Mar. Sci. 2019, 27, 100547. [Google Scholar] [CrossRef]
- Anderson, S.C.; Flemming, J.M.; Watson, R.; Lotze, H.K. Serial exploitation of global sea cucumber fisheries. Fish Fish. 2010, 12, 317–339. [Google Scholar] [CrossRef]
- Zacarías-Soto, M.; Tec-Tec, P.; Olvera-Novoa, M. Effect of diet on growth and body biochemical composition of juvenile four-sided sea cucumber Isostichopus badionotus (Selenka, 1867). Aquac. Res. 2018, 49, 939–946. [Google Scholar] [CrossRef]
- Zacarías-Soto, M.; Olvera-Novoa, M.A.; Pensamiento-Villarauz, S.; Sánchez-Tapia, I. Spawning and larval development of the four-sided sea cucumber, Isostichopus badionotus (Selenka 1867), under controlled conditions. J. World Aquac. Soc. 2013, 44, 694–705. [Google Scholar] [CrossRef]
- Sánchez-Tapia, I.A.; Slater, M.; Olvera-Novoa, M.A. Evaluation of the growth and survival rate of the Caribbean Sea cucumber, Isostichopus badionotus (Selenka, 1867), early juveniles produced in captivity. J. World Aquac. Soc. 2019, 50, 763–773. [Google Scholar] [CrossRef]
- Boyko, A.V.; Girich, A.S.; Tkacheva, E.S.; Dolmatov, I.Y. The Eupentacta fraudatrix transcriptome provides insights into regulation of cell transdifferentiation. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Chen, J.; Li, Y.; Zhang, H. Insights into high-pressure acclimation: Comparative transcriptome analysis of sea cucumber Apostichopus japonicus at different hydrostatic pressure exposures. BMC Genom. 2020, 21, 68. [Google Scholar] [CrossRef]
- Gao, K.; Wang, Z.; Qiu, X.; Song, J.; Wang, H.; Zhao, C.; Wang, X.; Chang, Y. Transcriptome analysis of body wall reveals growth difference between the largest and smallest individuals in the pure and hybrid populations of Apostichopus japonicus. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 31, 100591. [Google Scholar] [CrossRef]
- Xing, L.; Sun, L.; Liu, S.; Li, X.; Zhang, L.; Yang, H. De Novo assembly and comparative transcriptome analyses of purple and green morphs of Apostichopus japonicus during body wall pigmentation process. Comp. Biochem. Physiol. Part D Genom. Proteom. 2018, 28, 151–161. [Google Scholar] [CrossRef]
- Poot-Salazar, A.; Hernández-Flores, Á.; Ardisson, P.-L. Use of the SLW index to calculate growth function in the sea cucumber Isostichopus badionotus. Sci. Rep. 2014, 4, 5151. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction. Methods in Molecular Biology; Kollmar, M., Ed.; Humana: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Liu, X.; Yang, J.; Wang, S.; Sun, G.; Yang, J. Critical roles of sea cucumber C-type lectin in non-self recognition and bacterial clearance. Fish Shellfish. Immunol. 2015, 45, 791–799. [Google Scholar] [CrossRef]
- Lv, Z.; Guo, M.; Li, C.; Shao, Y.; Zhao, X.; Zhang, W. Divergent proteomics response of Apostichopus japonicus suffering from skin ulceration syndrome and pathogen infection. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 30, 196–205. [Google Scholar] [CrossRef]
- Zhong, M.; Chen, T.; Hu, C.; Ren, C. Isolation and Characterization of Collagen from the Body Wall of Sea Cucumber Stichopus monotuberculatus. J. Food Sci. 2015, 80, C671–C679. [Google Scholar] [CrossRef]
- Du, H.; Bao, Z.; Hou, R.; Wang, S.; Su, H.; Yan, J.; Tian, M.; Li, Y.; Wei, W.; Lu, W.; et al. Transcriptome sequencing and characterization for the sea cucumber Apostichopus japonicus (Selenka, 1867). PLoS ONE 2012, 7, e33311. [Google Scholar] [CrossRef]
- Ding, K.; Zhang, L.; Sun, L.; Lin, C.; Feng, Q.; Zhang, S.; Yang, H.; Brinkman, R.; Lin, G.; Huang, Z. Transcriptome analysis provides insights into the molecular mechanisms responsible for evisceration behavior in the sea cucumber Apostichopus japonicus. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 30, 143–157. [Google Scholar] [CrossRef]
- Li, C.; Fang, H.; Xu, D. Effect of seasonal high temperature on the immune response in Apostichopus japonicus by transcriptome analysis. Fish Shellfish. Immunol. 2019, 92, 765–771. [Google Scholar] [CrossRef]
- Gao, L.; He, C.; Bao, X.; Tian, M.; Ma, Z. Transcriptome analysis of the sea cucumber (Apostichopus japonicus) with variation in individual growth. PLoS ONE 2017, 12, e0181471. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Kong, X.; Chen, J.; Zhang, H. De novo sequencing and transcriptome analysis of Stichopus horrens to reveal genes related to biosynthesis of triterpenoids. Aquaculture 2018, 491, 358–367. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, L.; Xia, X.; Hu, W.; Zhou, P. Effect of geographic variation on the proteome of sea cucumber (Stichopus japonicus. Food Res. Int. 2020, 136, 109498. [Google Scholar] [CrossRef]
- Liu, R.; Liu, J.; Zhang, H. Positive selection analysis reveals the deep-sea adaptation of a hadal sea cucumber (Paelopatides sp.) to the Mariana Trench. J. Oceanol. Limnol. 2021, 39, 266–281. [Google Scholar] [CrossRef]
- Xue, Z.; Li, H.; Wang, X.; Li, X.; Liu, Y.; Sun, J.; Liu, C. A review of the immune molecules in the sea cucumber. Fish Shellfish. Immunol. 2015, 44, 1–11. [Google Scholar] [CrossRef]
- Xu, D.; Zhou, S.; Sun, L. RNA-seq based transcriptional analysis reveals dynamic genes expression profiles and immune-associated regulation under heat stress in Apostichopus japonicus. Fish Shellfish. Immunol. 2018, 78, 169–176. [Google Scholar] [CrossRef]
- Olivera-Castillo, L.; Grant, G.; Kantún-Moreno, N.; Barrera-Pérez, H.A.; Montero, J.; Olvera-Novoa, M.A.; Carrillo-Cocom, L.M.; Acevedo, J.J.; Puerto-Castillo, C.; Solís, V.M.; et al. A glycosaminoglycan-rich fraction from sea cucumber Isostichopus badionotus has potent anti-inflammatory properties in vitro and in vivo. Nutrients 2020, 12, 1698. [Google Scholar] [CrossRef]
- Olivera-Castillo, L.; Barrientos, R.G.; Legarreta, I.G.; Sámano, A.H.; Chi, Y.C. Sea Cucumber as a Source of Bioactive Compounds: Current Research on Isostichopus badionotus and Isostichopus fuscus from Mexico. Bioact. Compd. Mar. Foods Plant Anim. Sources 2013, 329–342. [Google Scholar] [CrossRef]
- Laguerre, H.; Raymond, G.; Plan, P.; Améziane, N.; Bailly, X.; Le Chevalier, P. First description of embryonic and larval development, juvenile growth of the black sea-cucumber Holothuria forskali (Echinodermata: Holothuroidea), a new species for aquaculture in the north-eastern Atlantic. Aquaculture 2020, 521, 734961. [Google Scholar] [CrossRef]
- Purcell, S.W.; Hair, C.A.; Mills, D.J. Sea cucumber culture, farming and sea ranching in the tropics: Progress, problems and opportunities. Aquaculture 2012, 368-369, 68–81. [Google Scholar] [CrossRef]
- Gamboa-Álvarez, M.Á.; Poot-López, G.R.; Olvera-Novoa, M.A.; Aguilar-Perera, A.; Ponce-Márquez, M.A.; López-Rocha, J.A. Survival and growth of wild-translocated individuals and released-cultured juveniles of sea cucumber Isostichopus badionotus off the northern Yucatan Peninsula, Mexico. Estuar. Coast. Shelf Sci. 2021, 252, 107273. [Google Scholar] [CrossRef]
- Dong, Y.; Li, Q.; Zhong, X.; Kong, L. Development of Gene-derived SNP Markers and Their Application for the Assessment of Genetic Diversity in Wild and Cultured Populations in Sea Cucumber, Apostichopus japonicus. J. World Aquac. Soc. 2016, 47, 873–888. [Google Scholar] [CrossRef]
- Dong, G.; Dong, S.; Wang, F.; Tian, X. Effects of light intensity on daily activity rhythm of juvenile sea cucumber, Apostichopus japonicus (Selenka). Aquac. Res. 2010, 41, 1640–1647. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Cao, X.; Song, S.; Li, J.; Wang, F.; Wang, L.; Liu, S. Effect of temperature on growth, digestion and immunity of green, white and purple sea cucumbers, Apostichopus japonicus (Selenka). Aquac. Res. 2019, 50, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Palomar-Abesamis, N.; Juinio-Meñez, M.A.; Slater, M.J. Effects of light and microhabitat on activity pattern and behaviour of wild and hatchery-reared juveniles of Stichopus cf. horrens. J. Mar. Biol. Assoc. UK 2018, 98, 1703–1713. [Google Scholar] [CrossRef]
- Gao, L.; Yuan, Z.; Yu, S.; Yang, Y.; Li, Y.; He, C. Genome-wide identification of HSP70/110 genes in sea cucumber Apostichopus japonicus and comparative analysis of their involvement in aestivation. Comp. Biochem. Physiol. Part D Genom. Proteom. 2018, 28, 162–171. [Google Scholar] [CrossRef]
- Zhang, L.; Feng, Q.; Sun, L.; Fang, Y.; Xu, D.; Zhang, T.; Yang, H. Differential gene expression in the body wall of the sea cucumber (Apostichopus japonicus) under strong lighting and dark conditions. Acta Oceanol. Sin. 2018, 37, 54–66. [Google Scholar] [CrossRef]
- Wang, Q.-L.; Yu, S.-S.; Qin, C.-X.; Dong, S.-L.; Dong, Y.-W. Combined effects of acute thermal and hypo-osmotic stresses on osmolality and hsp70, hsp90 and sod expression in the sea cucumber Apostichopus japonicus Selenka. Aquac. Int. 2014, 22, 1149–1161. [Google Scholar] [CrossRef]
- Dong, Y.-W.; Yu, S.-S.; Wang, Q.-L.; Dong, S.-L. Physiological responses in a variable environment: Relationships between metabolism, hsp and thermotolerance in an intertidal-subtidal species. PLoS ONE 2011, 6, e26446. [Google Scholar] [CrossRef] [Green Version]
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.-R.F.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Somasekharan, S.P.; et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 2013, 153, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, X.; Chen, M.; Li, W.; Zhang, P. Comparison of intestinal microbiota and activities of digestive and immune-related enzymes of sea cucumber Apostichopus japonicus in two habitats. J. Oceanol. Limnol. 2017, 36, 990–1001. [Google Scholar] [CrossRef]
- Xia, S.; Wang, X. Nutritional and medicinal value. Dev.Aquac. Fish. Sci. 2015, 39, 353–365. [Google Scholar] [CrossRef]
- Senadheera, T.R.; Dave, D.; Shahidi, F. Sea cucumber derived type i collagen: A comprehensive review. Mar. Drugs 2020, 18, 471. [Google Scholar] [CrossRef]
- Zhao, Z.; Jiang, J.; Pan, Y.; Dong, Y.; Chen, Z.; Zhang, G.; Gao, S.; Sun, H.; Guan, X.; Wang, B.; et al. Temporal dynamics of bacterial communities in the water and sediments of sea cucumber (Apostichopus japonicus) culture ponds. Aquaculture 2020, 528, 735498. [Google Scholar] [CrossRef]
- Günay, D.; Emiroğlu, D.; Suzer, C. Seasonal variations of digestive enzymes in sea cucumbers (Holothuria tubulosa, G. 1788) under culture conditions. J. Exp. Zool. A Ecol. Integr. Physiol. 2020, 333, 144–150. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.; Dong, S.; Wang, F.; Gao, Q. Effects of light intensity on larval development and juvenile growth of sea cucumber Apostichopus japonicus. Aquac. Res. 2019, 50, 2333–2340. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, L.; Pan, Y.; Lin, C.; Wang, F.; Kan, R.; Yang, H. Feeding behavior and digestive physiology in sea cucumber Apostichopus japonicus. Physiol. Behav. 2015, 139, 336–343. [Google Scholar] [CrossRef]
- Yingst, J.Y. The utilization of organic matter in shallow marine sediments by an epibenthic deposit-feeding holothurian. J. Exp. Mar. Biol. Ecol. 1976, 23, 55–69. [Google Scholar] [CrossRef]
- Fujiwara, R.; Yokoi, T.; Nakajima, M. Structure and protein–protein interactions of human UDP-glucuronosyltransferases. Front. Pharmacol. 2016, 7, 388. [Google Scholar] [CrossRef] [Green Version]
- Gullian Klanian, M.; Terrats Preciat, M. Effect of pH on temperature controlled degradation of reactive oxygen species, heat shock protein expression, and mucosal immunity in the sea cucumber Isostichopus badionotus. PLoS ONE 2017, 12, e0175812. [Google Scholar] [CrossRef] [Green Version]
- Puch-Hau, C.; Sánchez-Tapia, I.A.; Patiño-Suárez, V.; Olvera-Novoa, M.A.; Klanian, M.G.; Quintanilla-Mena, M.; Zapata-Pérez, O. Evaluation of two independent protocols for the extraction of DNA and RNA from different tissues of sea cucumber Isostichopus badionotus. MethodsX 2019, 6, 1627–1634. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 9 April 2019).
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinform 2012, 28, 1086–1092. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinform 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Agarwala, R.; Bolton, E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 47, D23–D28. [Google Scholar] [CrossRef] [Green Version]
- Petrov, A.I.; Kay, S.; Kalvari, I.; Howe, K.L.; Gray, K.A.; Bruford, E.A.; Kersey, P.J.; Cochrane, G.; Finn, R.D.; Bateman, A.; et al. RNAcentral: A comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 2017, 45, D128–D134. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinform 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Zamora-Briseño, J.A.; Ruiz-May, E.; Elizalde-Contreras, J.M.; Hernández-Velázquez, I.M.; Hernández-Pérez, A.; Fuentes-García, A.G.; Herrera-Salvatierra, N.; Briones-Fourzán, P.; Pascual-Jiménez, C.; Lozano-Álvarez, E.; et al. iTRAQ-Based Proteomic Profile Analysis of the Hepatopancreas of Caribbean Spiny Lobsters Infected With Panulirus argus Virus 1: Metabolic and Physiological Implications. Front. Microbiol. 2020, 11, 1084. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample * | Number of Paired Reads | Aligned Concordantly 0 Times | Aligned Concordantly Exactly 1 Time | Aligned Concordantly >1 Time | Overall Alignment Rate |
|---|---|---|---|---|---|
| W1 | 43,091,441 | 14,423,783 (33.5%) | 21,501,037 (49.9%) | 7,166,621 (16.6%) | 83.90% |
| W2 | 38,520,780 | 15,694,808 (40.7%) | 16,972,681 (44.1%) | 5,853,290 (15.2%) | 75.50% |
| W3 | 41,558,460 | 16,415,528 (39.5%) | 18,538,978 (44.6%) | 6,603,954 (15.9%) | 76.60% |
| W4 | 37,099,576 | 15,847,351 (42.7%) | 16,619,310 (44.8%) | 4,632,915 (12.5%) | 84.30% |
| F1 | 22,251,490 | 11,211,107 (50.4%) | 8,994,180 (40.4%) | 2,046,203 (9.2%) | 63.60% |
| F2 | 22,603,025 | 10,767,121 (47.6%) | 9,890,391 (43.7%) | 1,945,513 (8.6%) | 67.40% |
| F3 | 29,004,587 | 12,871,831 (44.4%) | 12,775,028 (44.0%) | 3,357,728 (11.6%) | 67.40% |
| F4 | 19,755,626 | 9,614,938 (48.7%) | 8,230,706 (41.7%) | 1,909,982 (9/7%) | 65.90% |
| Gene | Fold Change |
|---|---|
| Cytochrome c oxidase subunit I (COXI) | +6-fold |
| Eukaryotic translation initiation factor 2-alpha kinase 3 (EIF2AK3) | +2-fold |
| ATP synthase subunit alpha (APTA) | +2.2-fold |
| Mitogen-activated protein kinase 14 (MAPK14-1) | +2.9-fold |
| Heat Shock Protein-40 (HSP40) | +2.3-fold |
| Heat Shock protein cognate70 | +1.7-fold |
| * TRPM2 | +2.9-fold |
| Cyclic AMP-responsive element-binding protein 1 (CREB1) | −1.9-fold |
| Techylectin 5A | −2.9-fold |
| Suppressors of cytokine signaling 2 (SOCS2) | −2.6-fold |
| I. badionotus isolate FAO44 16S ribosomal RNA (mitochondrial) | −3.9-fold |
| Collagen mRNAs | ~−5-fold |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Hernández, R.; Rodríguez-Canul, R.; Kantún-Moreno, N.; Olvera-Novoa, M.A.; Medina-Contreras, O.; Garikoitz-Legarda, C.; Triviño, J.C.; Zamora-Briseño, J.A.; May-Solis, V.; Poot-Salazar, A.; et al. Comparative Transcriptomes of the Body Wall of Wild and Farmed Sea Cucumber Isostichopus badionotus. Int. J. Mol. Sci. 2021, 22, 3882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083882
Martín-Hernández R, Rodríguez-Canul R, Kantún-Moreno N, Olvera-Novoa MA, Medina-Contreras O, Garikoitz-Legarda C, Triviño JC, Zamora-Briseño JA, May-Solis V, Poot-Salazar A, et al. Comparative Transcriptomes of the Body Wall of Wild and Farmed Sea Cucumber Isostichopus badionotus. International Journal of Molecular Sciences. 2021; 22(8):3882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083882
Chicago/Turabian StyleMartín-Hernández, Roberto, Rossanna Rodríguez-Canul, Nuvia Kantún-Moreno, Miguel A. Olvera-Novoa, Oscar Medina-Contreras, Cristobal Garikoitz-Legarda, Juan Carlos Triviño, Jesús Alejandro Zamora-Briseño, Víctor May-Solis, Alicia Poot-Salazar, and et al. 2021. "Comparative Transcriptomes of the Body Wall of Wild and Farmed Sea Cucumber Isostichopus badionotus" International Journal of Molecular Sciences 22, no. 8: 3882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083882