
Enriched-GWAS and Transcriptome Analysis to Refine and Characterize a Major QTL for Anaerobic Germination Tolerance in Rice

 , , , and
, , , and
Abstract
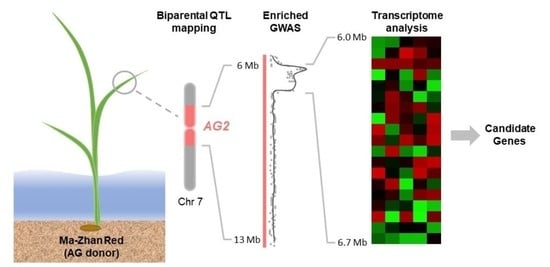
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
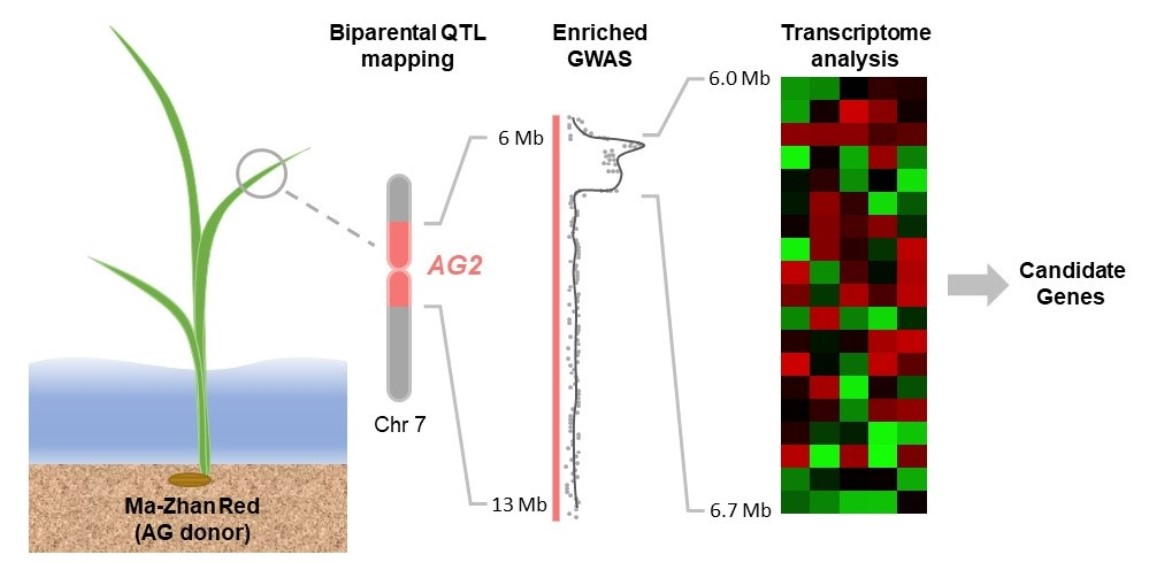
2.1. AG2-Enriched Panel Developed Based on SNP Variation between Ma-Zhan Red and IR42
2.2. Significant Phenotypic Variation Displayed within the Enriched-AG2 Rice Panel
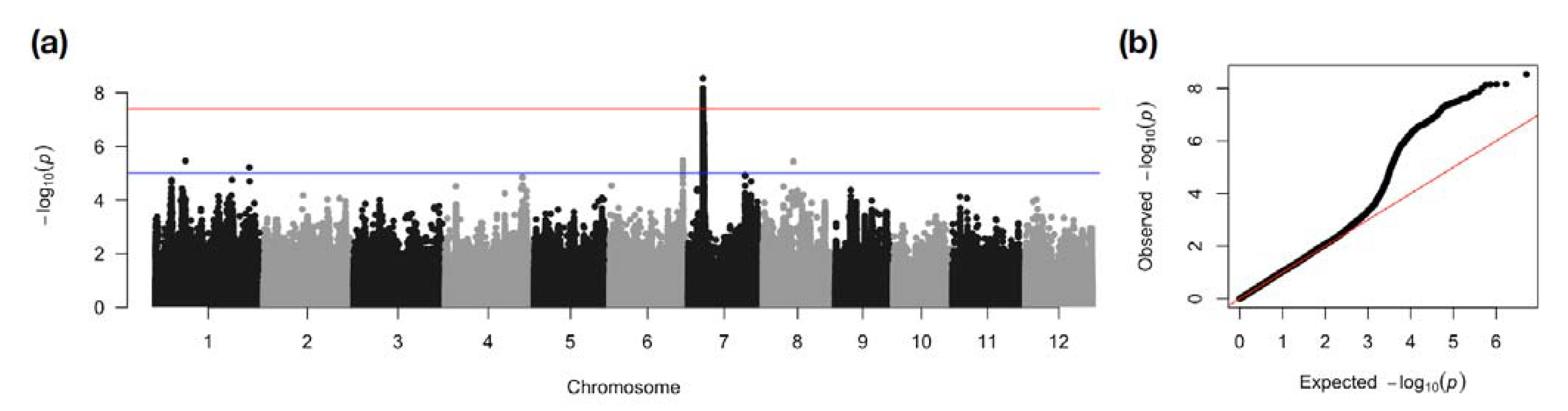
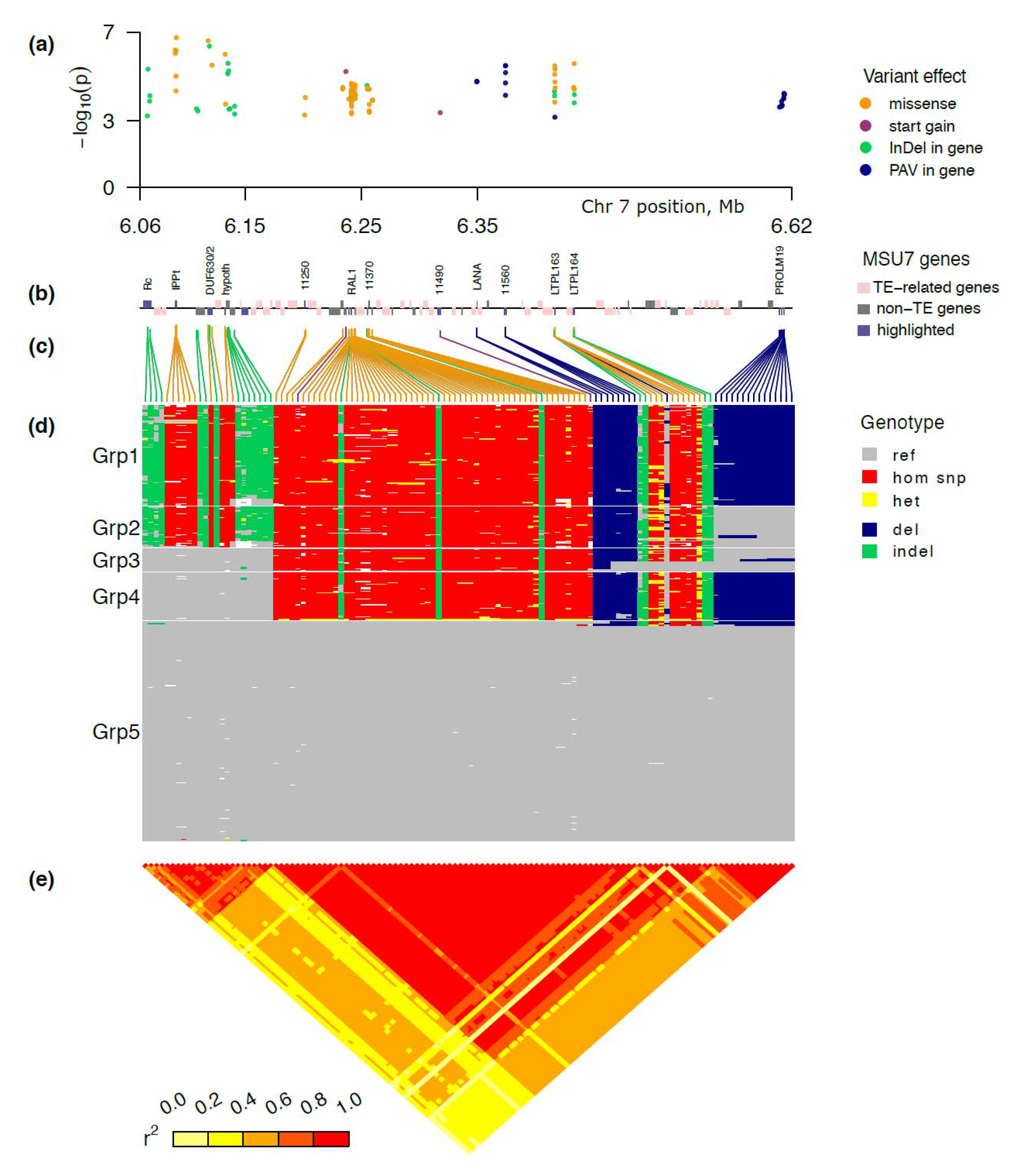
2.3. GWAS of the Selected Enriched Panel Significantly Narrowed down the AG2 Region
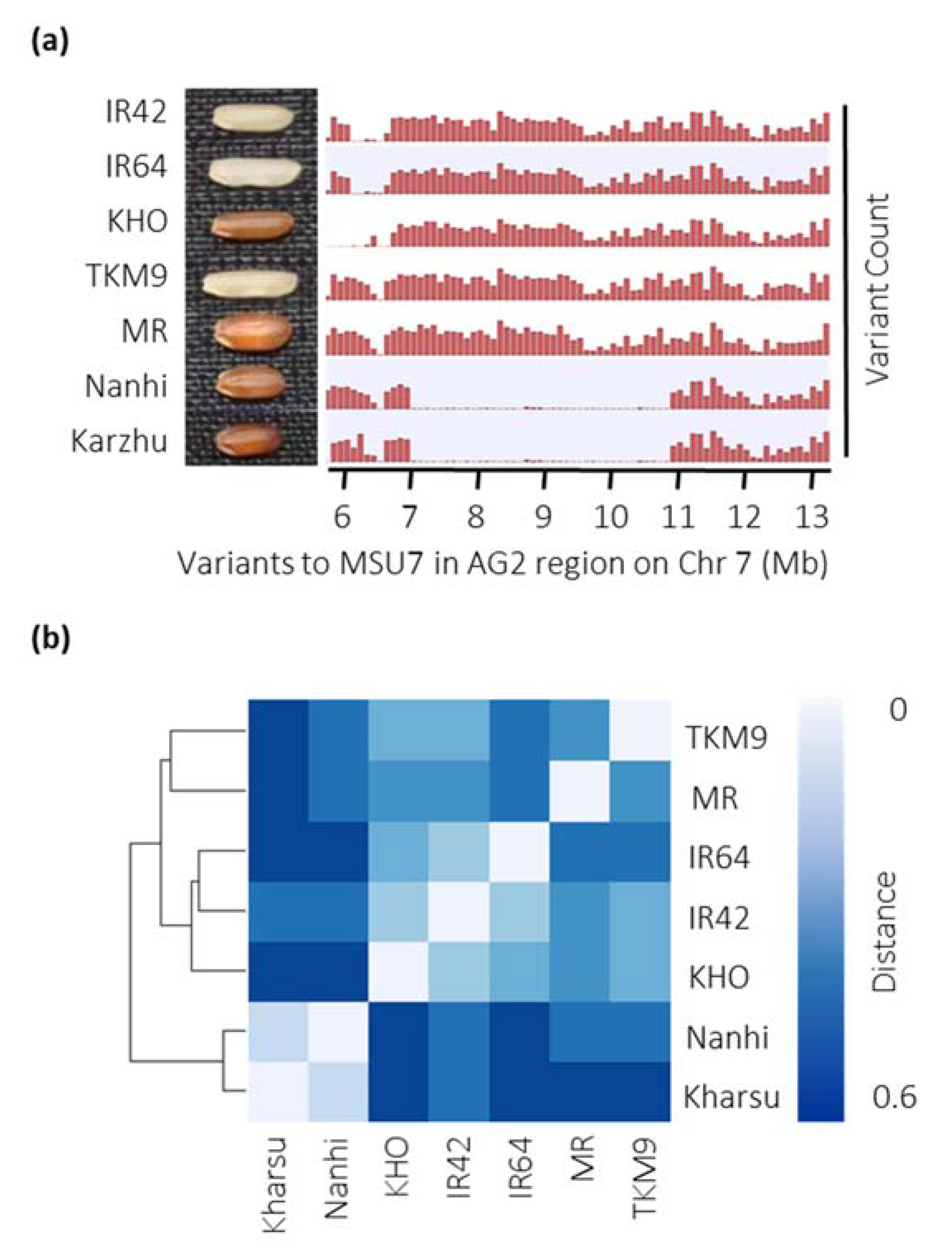
2.4. Five Major Haplotype Groups Identified within the Peak Region of AG2
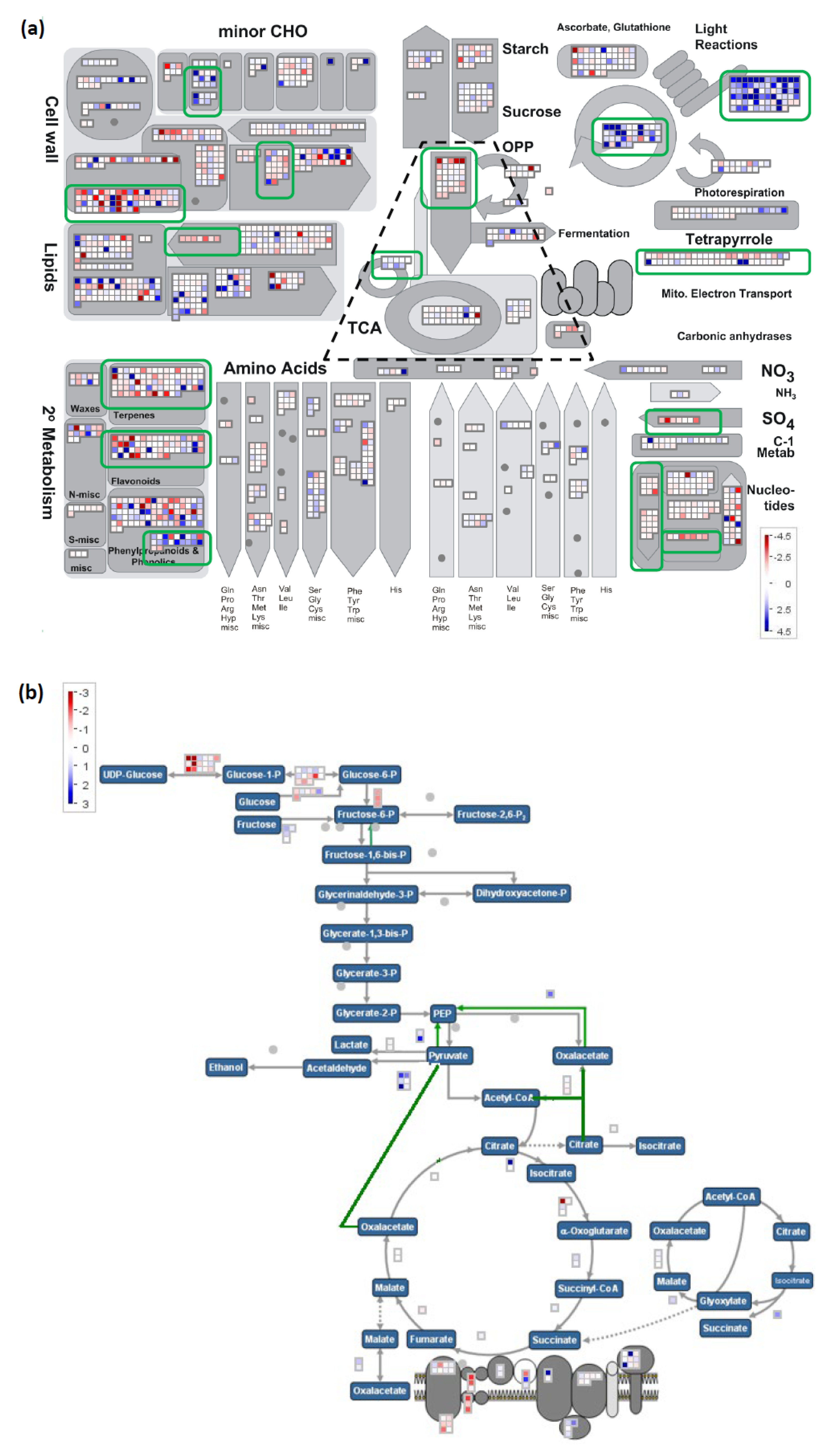
2.5. Significant Expression Patern Differences between Tolerant Donors and Susceptible Parental Genotypes Revealed by RNA-Seq Analysis
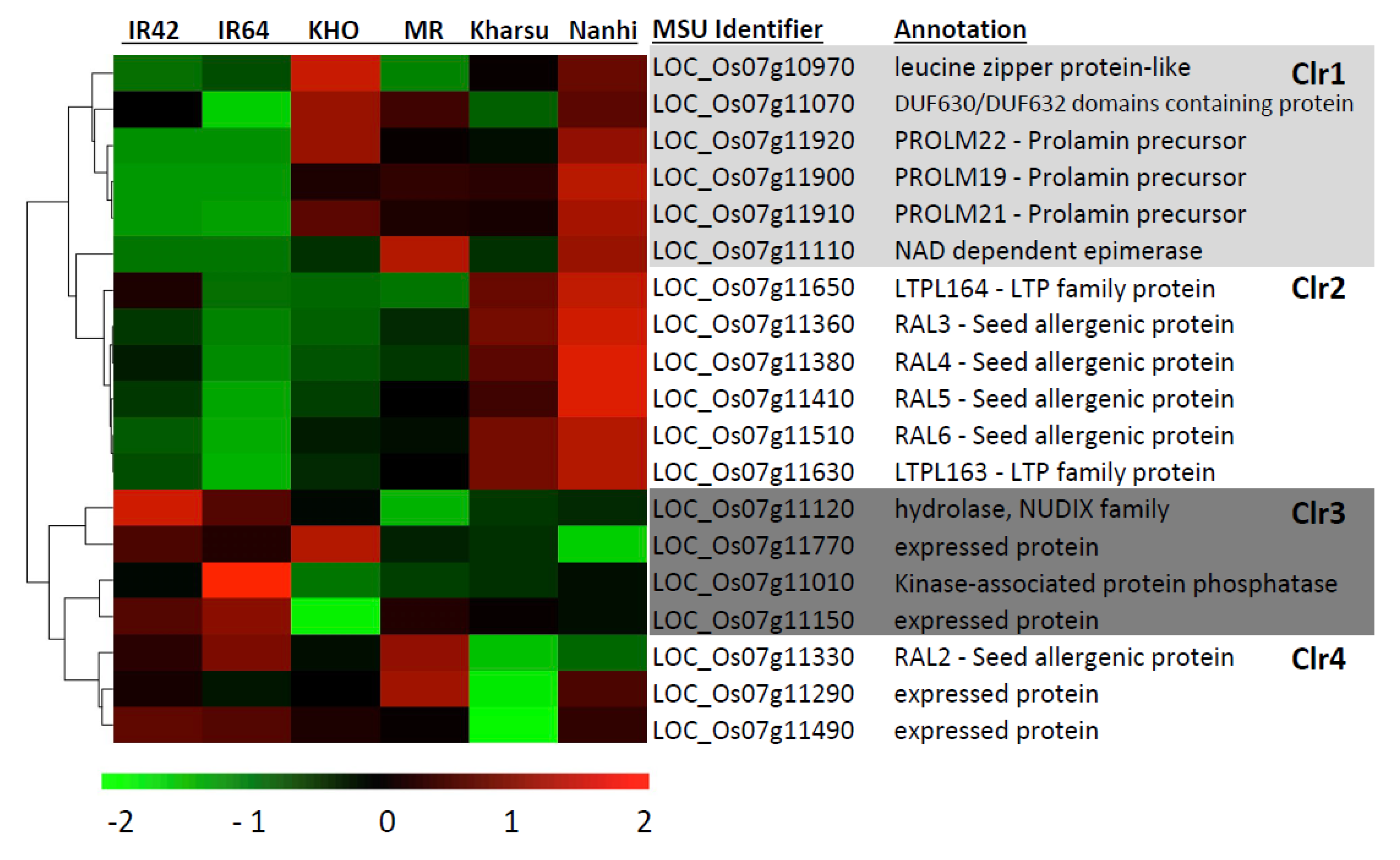
2.6. Four Distinct Clusters Identified by Regional RNAseq Analysis of the Peak Region of AG2
3. Discussion
4. Materials and Methods
4.1. Resequencing of Potential Donors and Susceptible Recipients
4.2. Development of an Enriched-GWAS Panel
4.3. Phenotyping
4.4. Association Mapping of an Enriched Panel Selection
4.5. Peak Region Analysis Using INDELs and Presence-Absence Variations (PAV)
4.6. RNAseq of AG Donors and Susceptible Parents
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AG | Anaerobic germination |
| bp | Basepairs |
| bHLH | Basic helix-loop-helix |
| dai | Days after imbibition |
| das | Days after sowing |
| DS | Direct seeding |
| DEG | Differentially expressed gene |
| DUF | Domain of unknown function |
| GEMMA | Genome-wide Efficient Mixed Model Analysis |
| GLM | Generalized linear model |
| GWAS | Genome wide association study |
| HISAT2 | Hierarchical indexing for spliced alignment of transcripts 2 |
| Indels | Insertions-deletions |
| IPP | Isopentenyl pyrophosphate |
| IRRI | International Rice Research Institute |
| IRGSP | International Rice Genome Sequencing Project |
| kb | Kilobasepairs |
| KHO | Khao Hlan On |
| LD | Linkage disequilibrium |
| LOD | Logarithm of the odds |
| LTP | Lipid transfer protein |
| Mb | Megabasepairs |
| MR | Ma-Zhan Red |
| NILs | Near isogenic lines |
| PAs | Proanthocyanidins |
| PAV | Presence-absence variant |
| PROLM | Prolamin |
| RIN | RNA integrity number |
| RNA-Seq | RNA Sequencing |
| ROS | Reactive Oxygen Species |
| SNP | Single Nucleotide Polymorphism |
| Q-Q plot | Quantile-quantile plot |
| QTL | Quantitative trait locus |
| TCA | tricarboxylic acid |
| TTP | Trehalose-6-phosphate phosphatase |
| 3K RG | 3000 Rice Genomes |
References
- Khush, G.S. What it will take to Feed 5.0 Billion Rice consumers in 2030. Plant. Mol. Biol. 2005, 59, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Muthayya, S.; Sugimoto, J.D.; Montgomery, S.; Maberly, G.F. An overview of global rice production, supply, trade, and consumption. Ann. N. Y. Acad. Sci. 2014, 1324, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.V. Global Climate Changes and Rice Food Security; FAO: Rome, Italy, 2002. [Google Scholar]
- Thornton, P.K. Recalibrating Food Production in the Developing World: Global Warming Will Change More Than Just the Climate Key Messages; CGIAR Research Program on Climate Change, Agriculture and Food Security: Wageningen, The Netherlands, 2012. [Google Scholar]
- Kumar, V.; Ladha, J.K. Direct Seeding of Rice. Recent Developments and Future Research Needs; Academic Press: Cambridge, MA, USA, 2011; Volume 111. [Google Scholar]
- Sansen, K.; Wongboon, W.; Jairin, J.; Kato, Y. Farmer-participatory evaluation of mechanized dry direct-seeding technology for rice in northeastern Thailand. Plant. Prod. Sci. 2019, 22, 46–53. [Google Scholar] [CrossRef]
- Farooq, M.; Siddique, K.H.M.; Rehman, H.; Aziz, T.; Lee, D.J.; Wahid, A. Rice direct seeding: Experiences, challenges and opportunities. Soil Tillage Res. 2011, 111, 87–98. [Google Scholar] [CrossRef]
- Tao, Y.; Chen, Q.; Peng, S.; Wang, W.; Nie, L. Lower global warming potential and higher yield of wet direct-seeded rice in Central China. Agron. Sustain. Dev. 2016, 36, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ismail, A.M.; Ella, E.S.; Vergara, G.V.; Mackill, D.J. Mechanisms associated with tolerance to flooding during germination and early seedling growth in rice (Oryza sativa). Ann. Bot. 2009, 103, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertani, A.; Brambilla, I.; Mapelli, S.; Reggiani, R. Elongation growth in the absence of oxygen: The rice coleoptile. Russ. J. Plant. Physiol. 1997, 44, 543. [Google Scholar]
- Angaji, S.A.; Septiningsih, E.M.; Mackill, D.J.; Ismail, A.M. QTLs associated with tolerance of flooding during germination in rice (Oryza sativa L.). Euphytica 2010, 172, 159–168. [Google Scholar] [CrossRef]
- Septiningsih, E.M.; Ignacio, J.C.I.; Sendon, P.M.D.; Sanchez, D.L.; Ismail, A.M.; Mackill, D.J. QTL mapping and confirmation for tolerance of anaerobic conditions during germination derived from the rice landrace Ma-Zhan Red. Theor. Appl. Genet. 2013, 126, 1357–1366. [Google Scholar] [CrossRef]
- Baltazar, M.D.; Ignacio, J.C.I.; Thomson, M.J.; Ismail, A.M.; Mendioro, M.S.; Septiningsih, E.M. QTL mapping for tolerance of anaerobic germination from IR64 and the aus landrace Nanhi using SNP genotyping. Euphytica 2014, 197, 251–260. [Google Scholar] [CrossRef]
- Baltazar, M.D.; Ignacio, J.C.I.; Thomson, M.J.; Ismail, A.M.; Mendioro, M.S.; Septiningsih, E.M. QTL mapping for tolerance to anaerobic germination in rice from IR64 and the aus landrace Kharsu 80A. Breed. Sci. 2019, 69, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myint, O.; Hernandez, J.E.; Ismail, A.M.; Septiningsih, E.M.; Cruz, P.C. Mapping QTLs for tolerance to flooding during germination and early seedling growth in rice (Oryza sativa L.) using IR64 × TKM9 population. In Proceedings of the Seventh Agricultural Research Conference, Nay Pyi Taw, Myanmar, 10–11 January 2013; pp. 81–91. [Google Scholar]
- Kretzschmar, T.; Pelayo, M.A.F.; Trijatmiko, K.R.; Gabunada, L.F.M.; Alam, R.; Jimenez, R.; Mendioro, M.S.; Slamet-Loedin, I.H.; Sreenivasulu, N.; Bailey-Serres, J.; et al. A trehalose-6-phosphate phosphatase enhances anaerobic germination tolerance in rice. Nat. Plants 2015, 1. [Google Scholar] [CrossRef]
- Toledo, A.M.U.; Ignacio, J.C.I.; Casal, C.; Gonzaga, Z.J.; Mendioro, M.S.; Septiningsih, E.M. Development of Improved Ciherang-Sub1 Having Tolerance to Anaerobic Germination Conditions. Plant. Breed. Biotechnol. 2015, 3, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Lal, B.; Gautam, P.; Nayak, A.K.; Raja, R.; Shahid, M.; Tripathi, R.; Singh, S.; Septiningsih, E.M.; Ismail, A.M. Agronomic manipulations can enhance the productivity of anaerobic tolerant rice sown in flooded soils in rainfed areas. Field Crop. Res. 2018, 220, 105–116. [Google Scholar] [CrossRef]
- Chamara, B.S.; Marambe, B.; Kumar, V.; Ismail, A.M.; Septiningsih, E.M.; Chauhan, B.S. Optimizing Sowing and Flooding Depth for Anaerobic Germination-Tolerant Genotypes to Enhance Crop Establishment, Early Growth, and Weed Management in Dry-Seeded Rice (Oryza sativa L.). Front. Plant. Sci. 2018, 9, 1654. [Google Scholar] [CrossRef]
- Mondal, S.; Khan, M.I.R.; Dixit, S.; Pompe, P.C.; Septiningsih, E.M.; Ismail, A.M. Growth, productivity and grain quality of AG1 and AG2 QTLs introgression lines under flooding in direct-seeded rice system. Field Crop. Res. 2020, 248, 107713. [Google Scholar] [CrossRef]
- Mondal, S.; Khan, M.I.R.; Entila, F.; Dixit, S.; Pompe, P.C.; Panna Ali, M.; Pittendrigh, B.; Septiningsih, E.M.; Ismail, A.M. Responses of AG1 and AG2 QTL introgression lines and seed pre-treatment on growth and physiological processes during anaerobic germination of rice under flooding. Sci. Rep. 2020, 10, 10214. [Google Scholar] [CrossRef]
- Hsu, S.K.; Tung, C.W. Genetic Mapping of Anaerobic Germination-Associated QTLs Controlling Coleoptile Elongation in Rice. Rice 2015, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, N.; Tai, S.; Wang, W.; Mansueto, L.; Palis, K.; Fuentes, R.R.; Ulat, V.J.; Chebotarov, D.; Zhang, G.; Li, Z.; et al. SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res. 2015, 43, D1023–D1027. [Google Scholar] [CrossRef]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Armitage, P.; Berry, G. Multiple Measurements. In Statistical Methods in Medical Research, 3rd ed.; Wiley: Hoboken, NJ, USA, 1994. [Google Scholar]
- Fuentes, R.R.; Chebotarov, D.; Duitama, J.; Smith, S.; De la Hoz, J.F.; Mohiyuddin, M.; Wing, R.A.; McNally, K.L.; Tatarinova, T.; Grigoriev, A.; et al. Structural variants in 3000 rice genomes. Genome Res. 2019, 29, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.T.; Thomson, M.J.; Pfeil, B.E.; McCouch, S. Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant. Cell 2006, 18, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosal, S.; Casal, C.; Quilloy, F.A.; Septiningsih, E.M.; Mendioro, M.S.; Dixit, S. Deciphering Genetics Underlying Stable Anaerobic Germination in Rice: Phenotyping, QTL Identification, and Interaction Analysis. Rice 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Jin, W.; Nagaki, K.; Tian, S.; Ouyang, S.; Buell, C.R.; Talbert, P.B.; Henikoff, S.; Jiang, J. Transcription and histone modifications in the recombination-free region spanning a rice centromere. Plant. Cell 2005, 17, 3227–3238. [Google Scholar] [CrossRef] [Green Version]
- Lasanthi-Kudahettige, R.; Magneschi, L.; Loreti, E.; Gonzali, S.; Licausi, F.; Novi, G.; Beretta, O.; Vitulli, F.; Alpi, A.; Perata, P. Transcript profiling of the anoxic rice coleoptile. Plant. Physiol. 2007, 144, 218–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narsai, R.; Howell, K.A.; Carroll, A.; Ivanova, A.; Millar, H.; Whelan, J. Defining core metabolic and transcriptomic responses to oxygen availability in rice embryos and young seedlings. Plant. Physiol. 2009, 151, 306–322. [Google Scholar] [CrossRef] [Green Version]
- Narsai, R.; Gouil, Q.; Secco, D.; Srivastava, A.; Karpievitch, Y.V.; Liew, L.C.; Lister, R.; Lewsey, M.G.; Whelan, J. Extensive transcriptomic and epigenomic remodelling occurs during Arabidopsis thaliana germination. Genome Biol. 2017, 18. [Google Scholar] [CrossRef]
- Vijayan, J.; Senapati, S.; Ray, S.; Chakraborty, K.; Molla, K.A.; Basak, N.; Pradhan, B.; Yeasmin, L.; Chattopadhyay, K.; Sarkar, R.K. Transcriptomic and physiological studies identify cues for germination stage oxygen deficiency tolerance in rice. Environ. Exp. Bot. 2018, 147, 234–248. [Google Scholar] [CrossRef]
- Min, B.; Gu, L.; McClung, A.M.; Bergman, C.J.; Chen, M.H. Free and bound total phenolic concentrations, antioxidant capacities, and profiles of proanthocyanidins and anthocyanins in whole grain rice (Oryza sativa L.) of different bran colours. Food Chem. 2012, 133, 715–722. [Google Scholar] [CrossRef]
- Gunaratne, A.; Wu, K.; Li, D.; Bentota, A.; Corke, H.; Cai, Y.Z. Antioxidant activity and nutritional quality of traditional red-grained rice varieties containing proanthocyanidins. Food Chem. 2013, 138, 1153–1161. [Google Scholar] [CrossRef]
- Goufo, P.; Trindade, H. Rice antioxidants: Phenolic acids, flavonoids, anthocyanins, proanthocyanidins, tocopherols, tocotrienols, γ -oryzanol, and phytic acid. Food Sci. Nutr. 2014, 2, 75–104. [Google Scholar] [CrossRef]
- Jia, L.G.; Sheng, Z.W.; Xu, W.F.; Li, Y.X.; Liu, Y.G.; Xia, Y.J.; Zhang, J.H. Modulation of anti-oxidation ability by proanthocyanidins during germination of arabidopsis thaliana seeds. Mol. Plant 2012, 5, 472–481. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Chen, J.; Cao, F.; Tao, Z.; Lei, T.; Tian, A.; Liu, Y.; Chen, G.; Zou, Y. Quantifying accumulation characteristics of glutelin and prolamin in rice grains. PLoS ONE 2019, 14, e0220139. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Wang, L.; Zeng, P.; He, Y.; Zhou, R.; Zhang, H.; Wang, Z. Identification of genes involved in rice seed priming in the early imbibition stage. Plant. Biol. 2017, 19, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Q.; Li, W.Q.; Miao, H.; Gan, P.F.; Qiao, L.; Chang, Y.L.; Shi, C.H.; Chen, K.M. REL2, A Gene Encoding An Unknown Function Protein which Contains DUF630 and DUF632 Domains Controls Leaf Rolling in Rice. Rice 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, M.E.; Kristensen, K.; Van Benthem, K.J.; Magnusson, A.; Berg, C.W.; Nielsen, A.; Skaug, H.J.; Machler, M.; Bolker, B.M.; Brooks, M.E.; et al. glmmTMB balances speed and flexibility among packages for zero-inflated generalized linear mixed modeling ETH Library glmmTMB Balances Speed and Flexibility Among Packages for Zero-inflated Generalized Linear Mixed Modeling. R J. 2017, 9, 378–400. [Google Scholar] [CrossRef] [Green Version]
- Mansueto, L.; Fuentes, R.R.; Borja, F.N.; Detras, J.; Abrio-Santos, J.M.; Chebotarov, D.; Sanciangco, M.; Palis, K.; Copetti, D.; Poliakov, A.; et al. Rice SNP-seek database update: New SNPs, indels, and queries. Nucleic Acids Res. 2017, 45, D1075–D1081. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4. [Google Scholar] [CrossRef]
- Zhang, L.; Pei, Y.-F.; Li, J.; Papasian, C.J.; Deng, H.-W. Improved detection of rare genetic variants for diseases. PLoS ONE 2010, 5, e13857. [Google Scholar] [CrossRef] [Green Version]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Tan, R.; Yuan, J.; Bales, C.; Du, W.; Zhang, S.; Chilvers, M.I.; Schmidt, C.; Song, Q.; Cregan, P.B.; et al. Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9. [Google Scholar] [CrossRef]
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots 2020. Available online: https://github.com/kassambara/ggpubr (accessed on 30 May 2020).
- Li, Z.; Trick, H.N. Rapid method for high-quality RNA isolation from seed endosperm containing high levels of starch. Biotechniques 2005, 38, 872–876. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 December 2016).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant. J. 2004, 37, 914–939. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tnani, H.; Chebotarov, D.; Thapa, R.; Ignacio, J.C.I.; Israel, W.K.; Quilloy, F.A.; Dixit, S.; Septiningsih, E.M.; Kretzschmar, T. Enriched-GWAS and Transcriptome Analysis to Refine and Characterize a Major QTL for Anaerobic Germination Tolerance in Rice. Int. J. Mol. Sci. 2021, 22, 4445. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094445
Tnani H, Chebotarov D, Thapa R, Ignacio JCI, Israel WK, Quilloy FA, Dixit S, Septiningsih EM, Kretzschmar T. Enriched-GWAS and Transcriptome Analysis to Refine and Characterize a Major QTL for Anaerobic Germination Tolerance in Rice. International Journal of Molecular Sciences. 2021; 22(9):4445. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094445
Chicago/Turabian StyleTnani, Hedia, Dmytro Chebotarov, Ranjita Thapa, John Carlos I. Ignacio, Walter K. Israel, Fergie A. Quilloy, Shalabh Dixit, Endang M. Septiningsih, and Tobias Kretzschmar. 2021. "Enriched-GWAS and Transcriptome Analysis to Refine and Characterize a Major QTL for Anaerobic Germination Tolerance in Rice" International Journal of Molecular Sciences 22, no. 9: 4445. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094445