
Nitrogen Signaling Genes and SOC1 Determine the Flowering Time in a Reciprocal Negative Feedback Loop in Chinese Cabbage (Brassica rapa L.) Based on CRISPR/Cas9-Mediated Mutagenesis of Multiple BrSOC1 Homologs

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
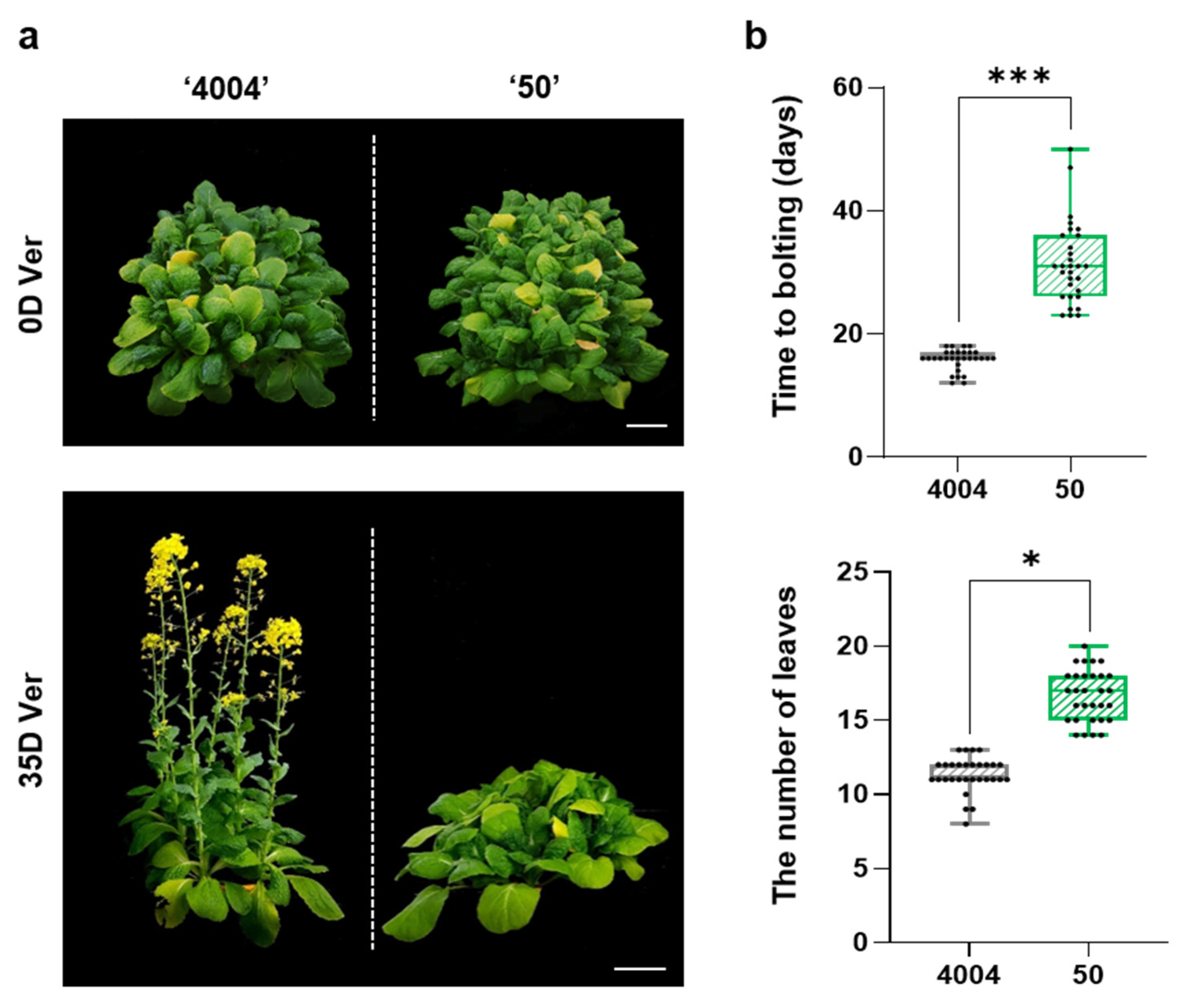
2.1. Differences in Vernalization-Induced Bolting Time between Lines “4004” and “50”
2.2. Transcriptome Profiling
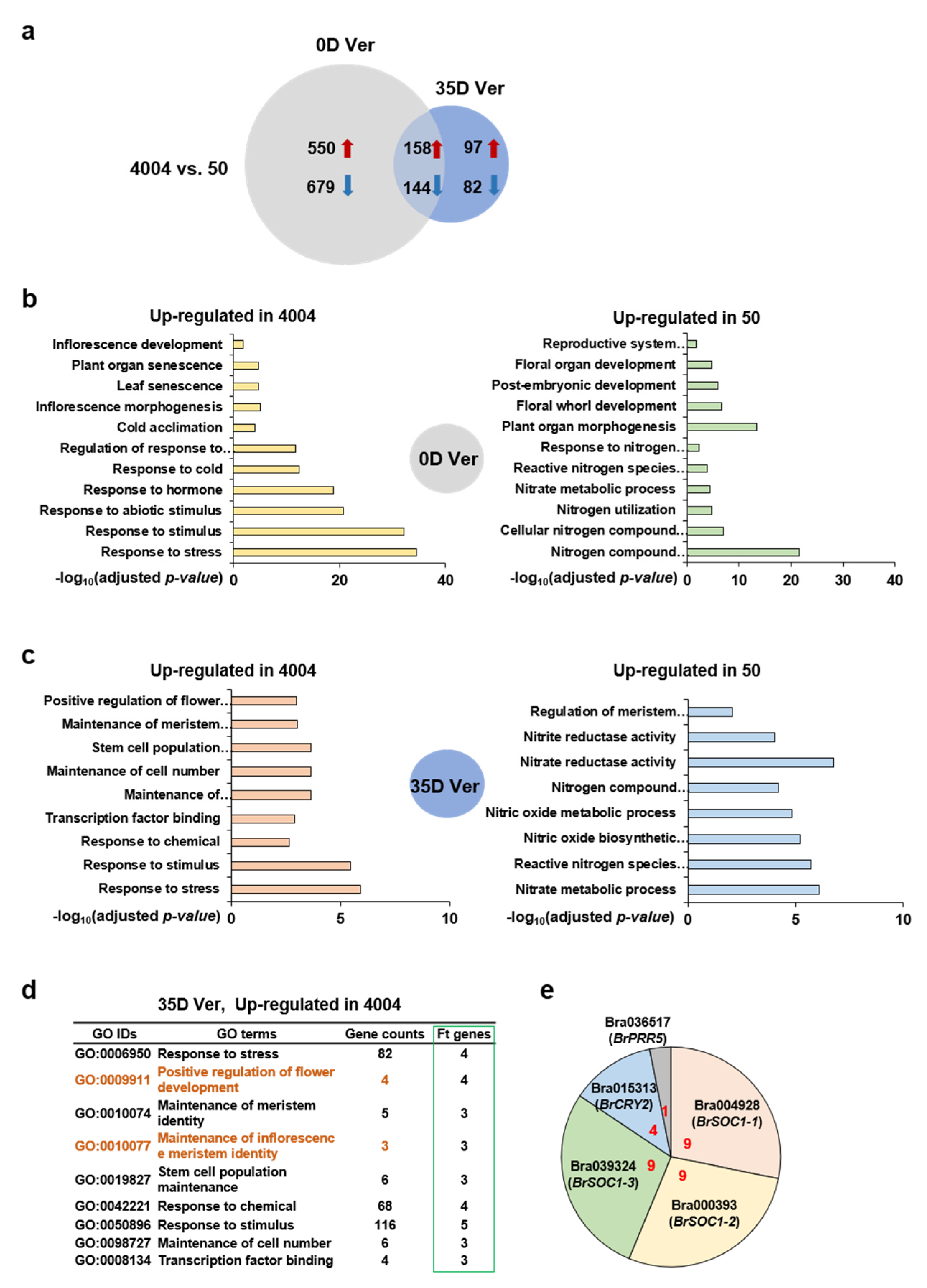
2.3. Identification of Differentially Expressed Genes (DEGs) and Functional Pathway Enrichment Analysis Comparing Lines “4004” and “50”
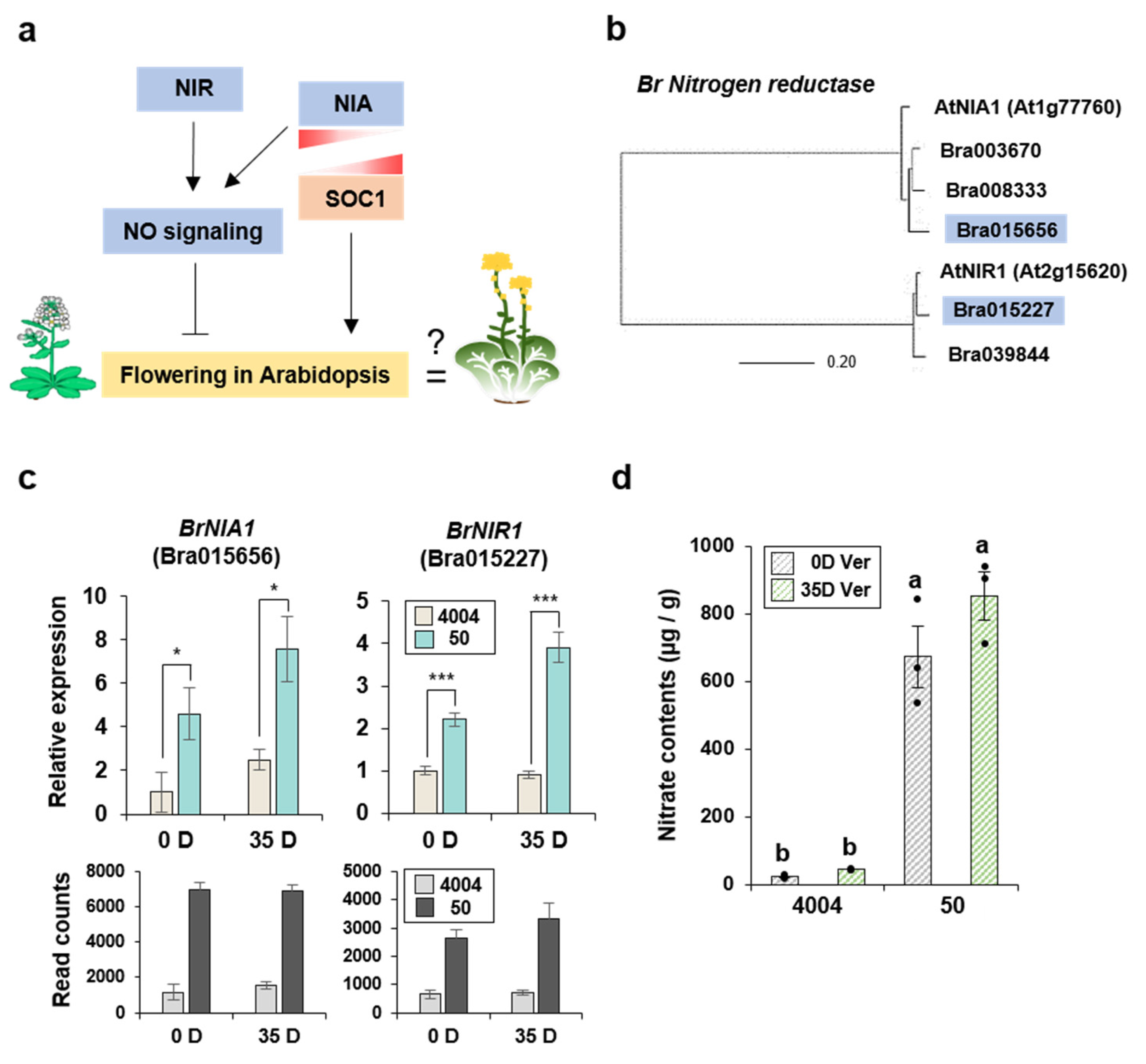
2.4. Identification of B. rapa Nitrate Reductase Genes and Relative Expression Levels Derived by Comparing Lines “4004” and “50”
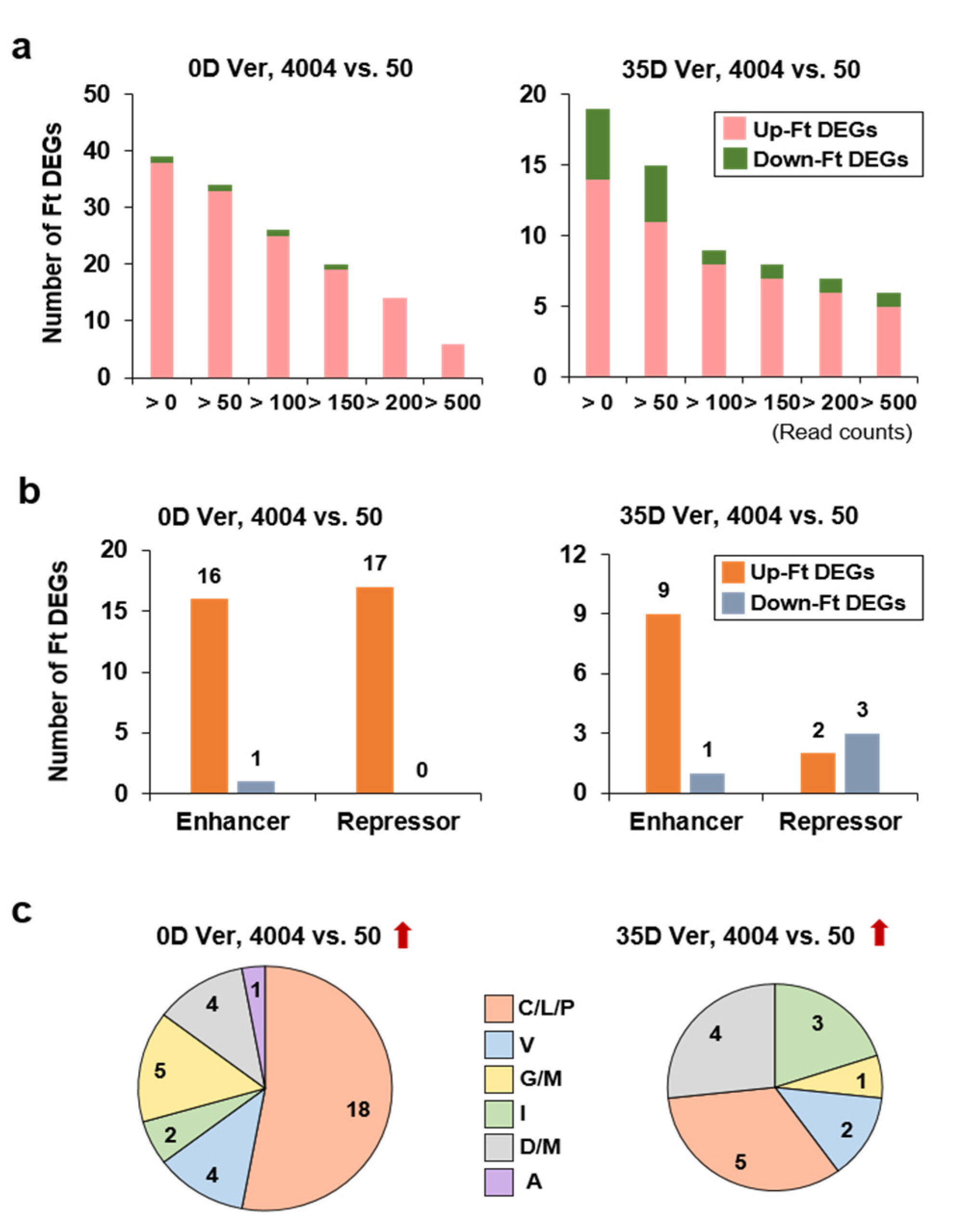
2.5. Identification and Analysis of Flowering Time Genes Differentially Expressed between the Two Inbred Lines
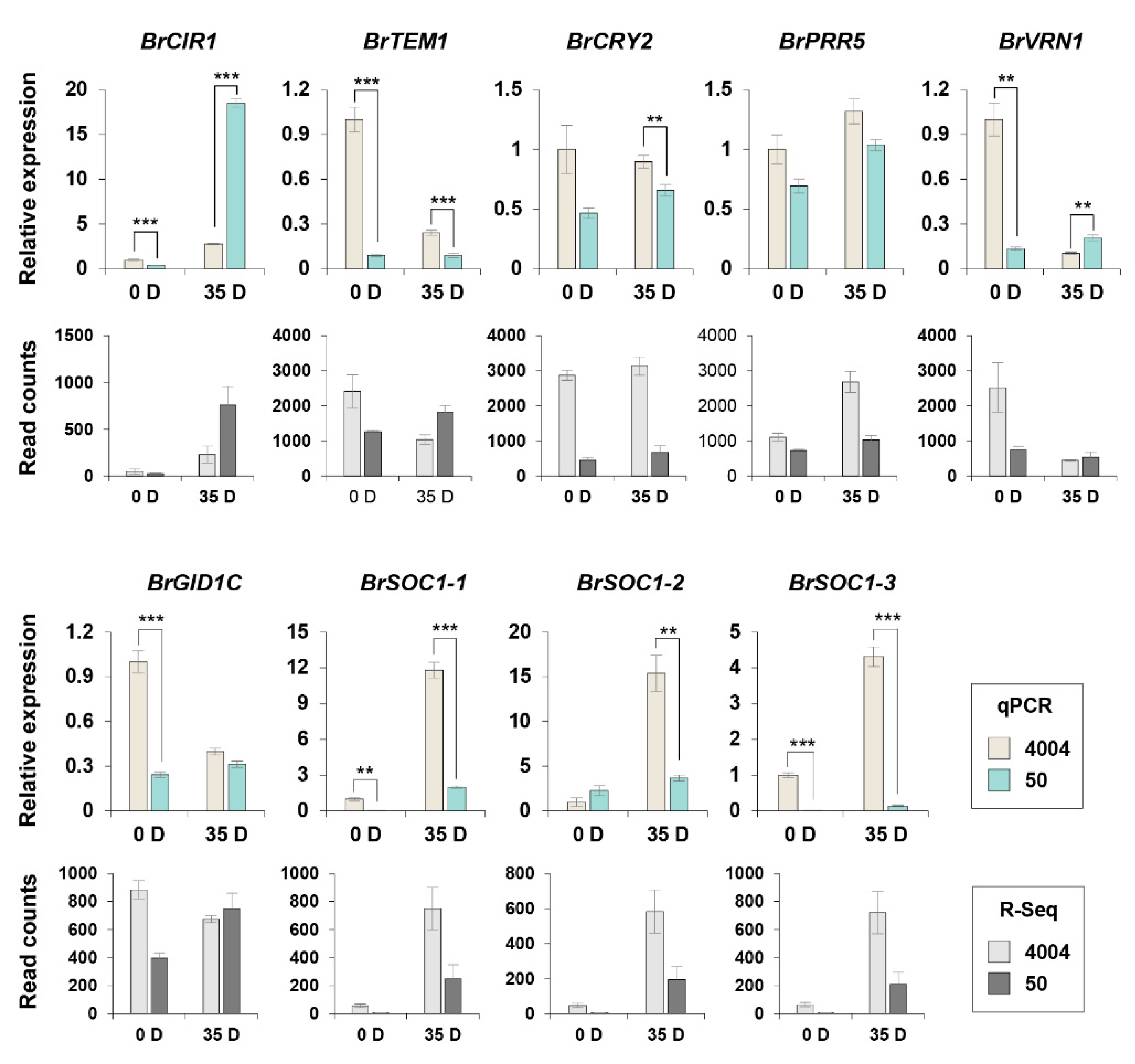
2.6. Validation of the Expression of Flowering Time Genes Involved in the Flowering Regulatory Network
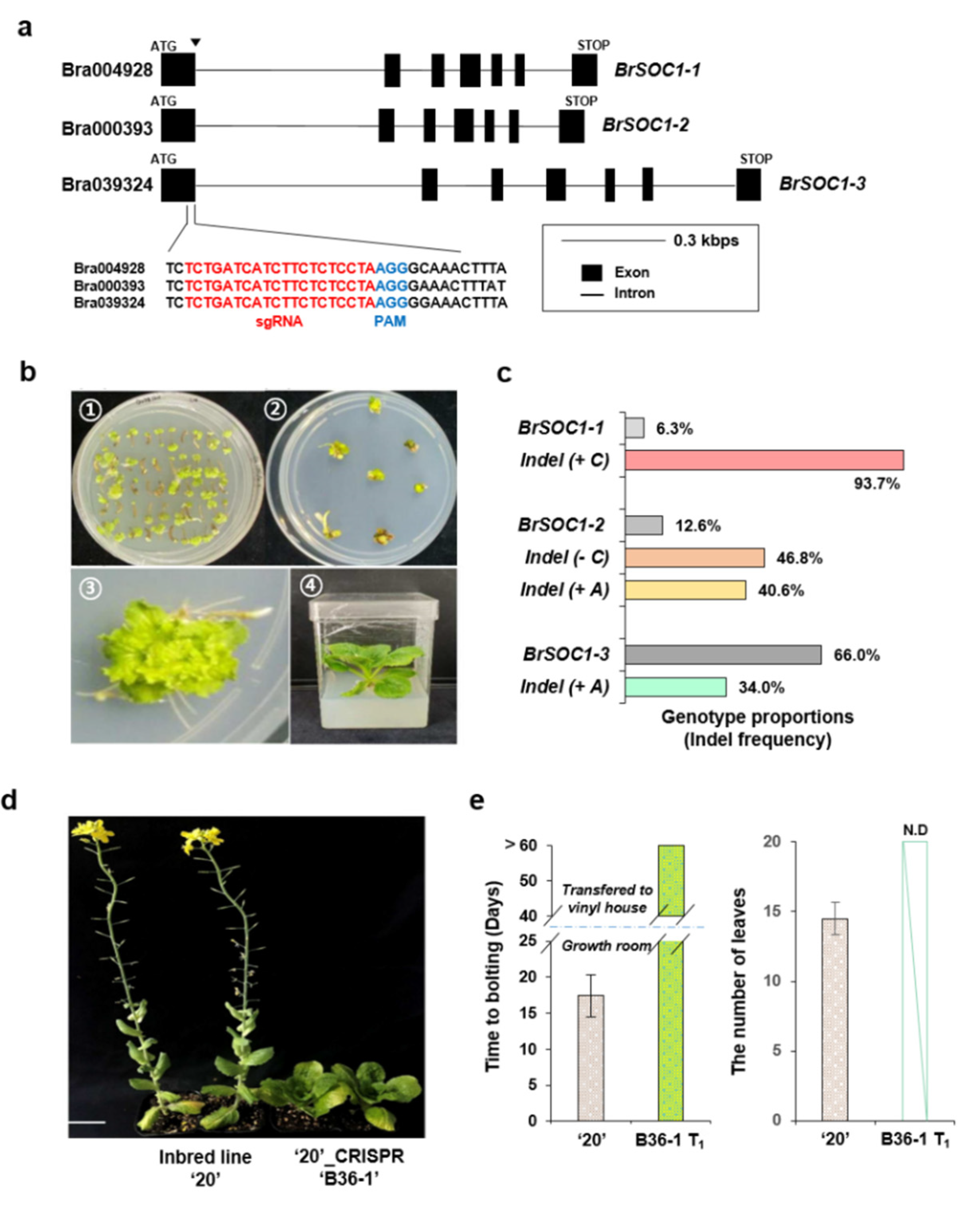
2.7. CRISPR/Cas9-Mediated Mutagenesis of Multiple BrSOC1 Homologs Introduced the Late-Bolting Trait into the Elite Line “20”
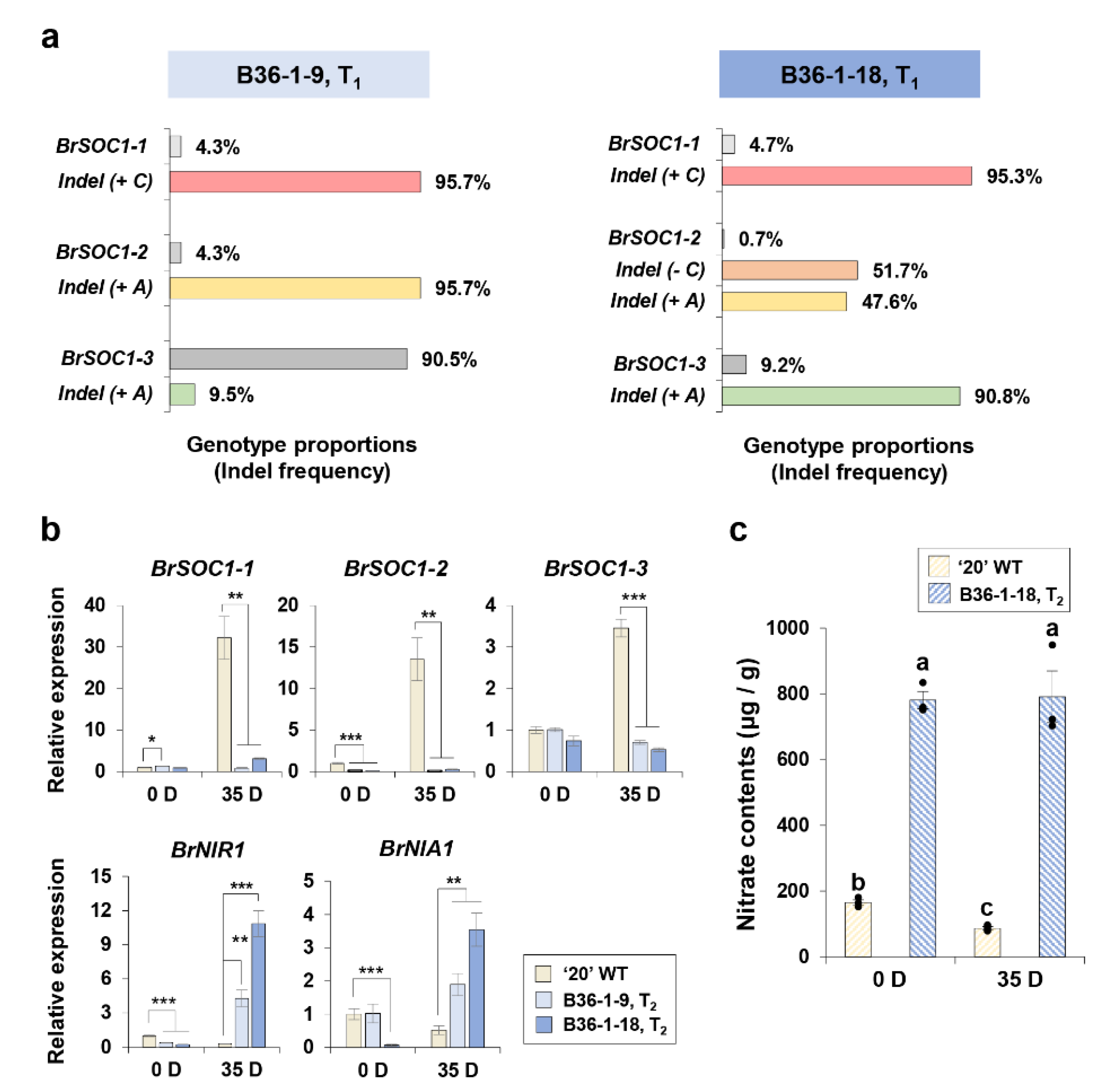
2.8. Backcross Breeding for Brsoc1/2/3 Mutagenesis Controlling the Late-Bolting Trait
2.9. Loss-of-Function BrSOC1s Showed Reciprocal Regulation with Nitrogen Signaling Genes and Nitrogen Content and Preserved Positive Feedback Regulation of the Original BrSOC1s Genes
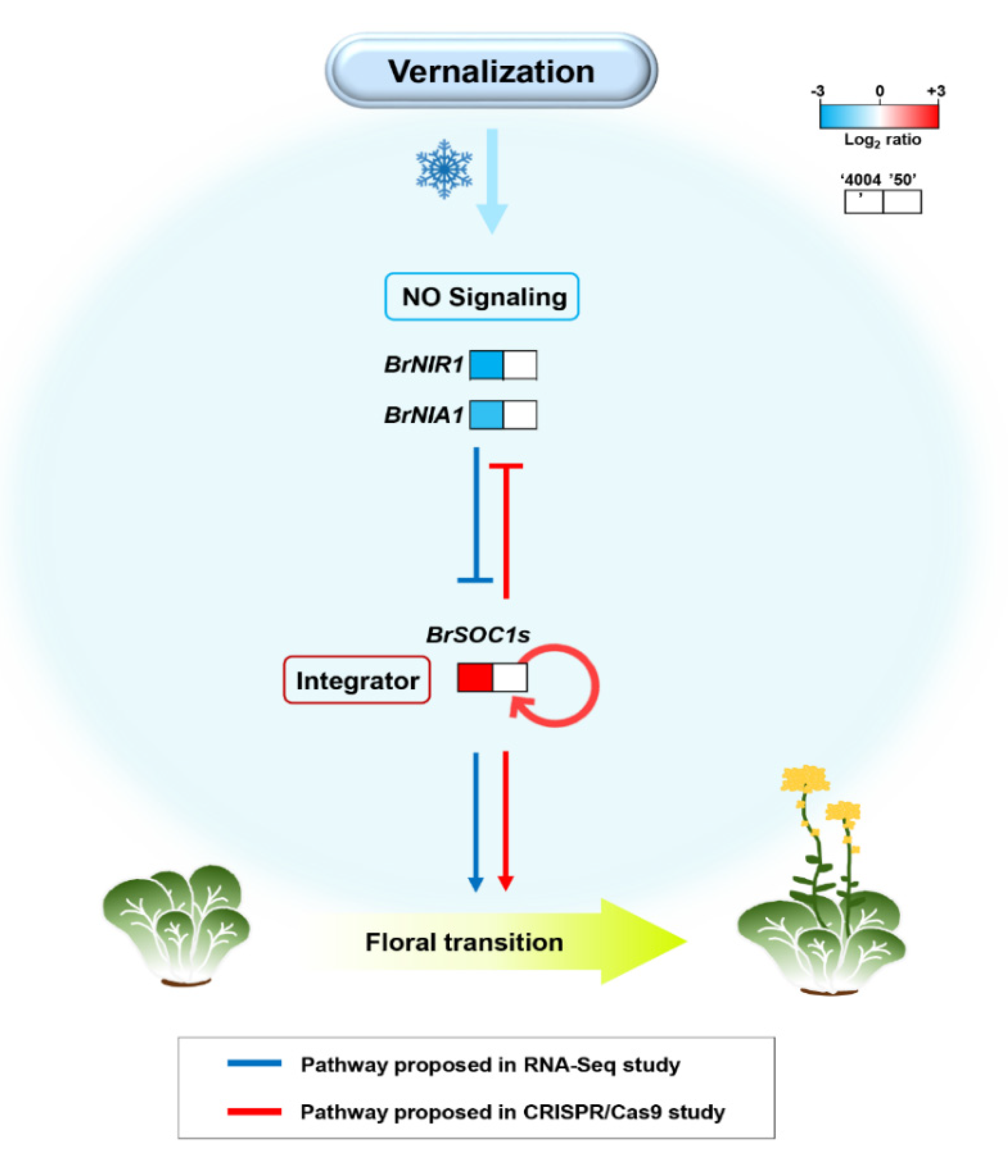
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Total RNA Isolation, Library Construction, and RNA-Seq Analysis
4.3. Transcriptome Assembly and Mapping
4.4. Identification and Analysis of DEGs
4.5. Functional Annotation of DEGs
4.6. Real-Time Reverse Transcription PCR (qPCR) Analysis
4.7. Bioinformatics Analysis
4.8. Nitrate Assay
4.9. CRISPR/Cas9-Mediated Mutagenesis of BrSOC1-1/1-2/1-3 and Genetic Transformation
4.10. Backcross Breeding for Brsoc1s
4.11. Characterization of Mutations Inherited from the T0 to T2 and the Backcrossing Generations in Transgenic Plants
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amasino, R. Seasonal and developmental timing of flowering. Plant J. 2010, 61, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Chardon, F.; Virlon, B.; Moreau, L.; Falque, M.; Joets, J.; Decousset, L.; Murigneux, A.; Charcosset, A. Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics 2004, 168, 2169–2185. [Google Scholar] [CrossRef] [PubMed]
- Ducrocq, S.; Madur, D.; Veyrieras, J.B.; Camus-Kulandaivelu, L.; Kloiber-Maitz, M.; Presterl, T.; Ouzunova, M.; Manicacci, D.; Charcosset, A. Key impact of vgt1 on flowering time adaptation in maize: Evidence from association mapping and ecogeographical information. Genetics 2008, 178, 2433–2437. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Tang, R.H.; Hao, Y.; Stevens, R.D.; Cook, C.W.; Ahn, S.M.; Jing, L.; Yang, Z.; Chen, L.; Guo, F.; et al. Nitric oxide represses the arabidopsis floral transition. Science 2004, 305, 1968–1971. [Google Scholar] [CrossRef] [PubMed]
- Seligman, K.; Saviani, E.E.; Oliveira, H.C.; Pinto-Maglio, C.A.; Salgado, I. Floral transition and nitric oxide emission during flower development in arabidopsis thaliana is affected in nitrate reductase-deficient plants. Plant Cell Physiol. 2008, 49, 1112–1121. [Google Scholar] [CrossRef]
- Weber, K.; Burow, M. Nitrogen—Essential macronutrient and signal controlling flowering time. Physiol. Plant. 2018, 162, 251–260. [Google Scholar] [CrossRef]
- Wang, W.; Hu, B.; Yuan, D.; Liu, Y.; Che, R.; Hu, Y.; Ou, S.; Liu, Y.; Zhang, Z.; Wang, H.; et al. Expression of the nitrate transporter gene osnrt1.1a/osnpf6.3 confers high yield and early maturation in rice. Plant Cell 2018, 30, 638–651. [Google Scholar] [CrossRef]
- Akhatar, J.; Goyal, A.; Kaur, N.; Atri, C.; Mittal, M.; Singh, M.P.; Kaur, R.; Rialch, I.; Banga, S.S. Genome wide association analyses to understand genetic basis of flowering and plant height under three levels of nitrogen application in brassica juncea (L.) czern & coss. Sci. Rep. 2021, 11, 4278. [Google Scholar]
- Warwick, S.I.; Francis, A.; Al-Shehbaz, I.A. Brassicaceae: Species checklist and database on cd-rom. Plant Syst. Evol. 2006, 259, 249–258. [Google Scholar] [CrossRef]
- Schiessl, S.V.; Huettel, B.; Kuehn, D.; Reinhardt, R.; Snowdon, R.J. Flowering time gene variation in brassica species shows evolutionary principles. Front. Plant Sci. 2017, 8, 1742. [Google Scholar] [CrossRef]
- Kim, S.Y.; Park, B.S.; Kwon, S.J.; Kim, J.; Lim, M.H.; Park, Y.D.; Kim, D.Y.; Suh, S.C.; Jin, Y.M.; Ahn, J.H.; et al. Delayed flowering time in arabidopsis and brassica rapa by the overexpression of flowering locus c (flc) homologs isolated from chinese cabbage (brassica rapa L. ssp. Pekinensis). Plant Cell Rep. 2007, 26, 327–336. [Google Scholar] [CrossRef]
- Jung, W.Y.; Lee, A.; Moon, H.S.; Kim, Y.; Cho, H.S. Genome-wide identification of flowering time genes associated with vernalization and the regulatory flowering networks in chinese cabbage. Plant Biotechnol. Rep. 2018, 12, 347–363. [Google Scholar] [CrossRef]
- Friend, D.J.C. Brassica. In Handbook of Flowering Volume 2; Halevy, H., Ed.; CRC Press: Boca Raton, FL, USA, 1985. [Google Scholar]
- Lin, S.I.; Wang, J.G.; Poon, S.Y.; Su, C.L.; Wang, S.S.; Chiou, T.J. Differential regulation of flowering locus c expression by vernalization in cabbage and arabidopsis. Plant Physiol. 2005, 137, 1037–1048. [Google Scholar] [CrossRef]
- Zhang, X.; Meng, L.; Liu, B.; Hu, Y.; Cheng, F.; Liang, J.; Aarts, M.G.; Wang, X.; Wu, J. A transposon insertion in flowering locus t is associated with delayed flowering in brassica rapa. Plant Sci. 2015, 241, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Shindo, C.; Aranzana, M.J.; Lister, C.; Baxter, C.; Nicholls, C.; Nordborg, M.; Dean, C. Role of frigida and flowering locus c in determining variation in flowering time of arabidopsis. Plant Physiol. 2005, 138, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Ayele, M.; Haas, B.J.; Kumar, N.; Wu, H.; Xiao, Y.; Van Aken, S.; Utterback, T.R.; Wortman, J.R.; White, O.R.; Town, C.D. Whole genome shotgun sequencing of brassica oleracea and its application to gene discovery and annotation in arabidopsis. Genome Res. 2005, 15, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Weinholdt, C.; Jedrusik, N.; Molina, C.; Zou, J.; Grosse, I.; Schiessl, S.; Jung, C.; Emrani, N. Whole-transcriptome analysis reveals genetic factors underlying flowering time regulation in rapeseed (brassica napus L.). Plant Cell Environ. 2018, 41, 1935–1947. [Google Scholar] [CrossRef]
- Jian, H.; Zhang, A.; Ma, J.; Wang, T.; Yang, B.; Shuang, L.S.; Liu, M.; Li, J.; Xu, X.; Paterson, A.H.; et al. Joint qtl mapping and transcriptome sequencing analysis reveal candidate flowering time genes in brassica napus L. BMC Genom. 2019, 20, 21. [Google Scholar] [CrossRef]
- Li, J.; Zhao, X.; Nishimura, Y.; Fukumoto, Y. Correaltion between bolting and physiological properties in chinese cabbage (brassica rapa L. Pekinensis group). J. Jpn. Soc. Hortic. Sci. 2010, 79, 294–300. [Google Scholar] [CrossRef]
- Elers, B.; Wiebe, H.-J. Flower formation of chinese cabbage. I. Response to vernalization and photoperiod. Sci. Hortic. 1984, 22, 219–231. [Google Scholar] [CrossRef]
- Mero, C.E.; Honma, S. Inheritance of bolting resistance in an intraspecific chinese cabbage x turnip cross. HortScience 1985, 20, 881–882. [Google Scholar]
- Ajisaka, H.; Kuginuki, Y.; Yui, S.; Enomoto, S.; Hirai, M. Identification and mapping of a nquantitative trait locus controlling extreme late bolting in chinese cabbage (brassica rapa L. ssp. Pekinensis syn. Campestris L.) using bulked. Euphytica 2001, 118. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.J.; Zhang, F.L.; Zou, Z.R.; Zhao, X.Y.; Zhang, D.S.; Xu, J.B. Linkage map construction and quantitative trait loci analysis for bolting based on a double haploid population of brassica rapa. J. Integr. Plant Boil. 2007, 49, 664–671. [Google Scholar] [CrossRef]
- Zhao, J.; Kulkarni, V.; Liu, N.; Del Carpio, D.P.; Bucher, J.; Bonnema, G. Brflc2 (flowering locus c) as a candidate gene for a vernalization response qtl in brassica rapa. J. Exp. Bot. 2010, 61, 1817–1825. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.; Wang, Z.; Li, K.; Lim, Y.; Piao, Z. Construction of chromosome segment substitution lines enables qtl mapping for flowering and morphological traits in brassica rapa. Front. Plant Sci. 2015, 6, 432. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Shi, X.; Feng, H.; Wang, Y. Identification of qtls with additive, epinastic, and qtl x environment intercation effects for the bolting trait in brassica rapa L. Euphytica 2016, 210, 427–439. [Google Scholar] [CrossRef]
- Liu, S.; Wang, R.; Zhang, Z.; Li, Q.; Wang, L.; Wang, Y.; Zhao, Z. High-resolution mapping of quantitative trait loci controlling main floral stalk length in chinese cabbage (brassica rapa L. ssp. Pekinensis). BMC Genom. 2019, 20, 437. [Google Scholar] [CrossRef]
- Su, T.; Wang, W.; Li, P.; Zhang, B.; Li, P.; Xin, X.; Sun, H.; Yu, Y.; Zhang, D.; Zhao, X.; et al. A genomic variation map provides insights into the genetic basis of spring chinese cabbage (brassica rapa ssp. Pekinensis) selection. Mol. Plant 2018, 11, 1360–1376. [Google Scholar] [CrossRef]
- Kong, L.; Lu, S.; Wang, Y.; Fang, C.; Wang, F.; Nan, H.; Su, T.; Li, S.; Zhang, F.; Li, X.; et al. Quantitative trait locus mapping of flowering time and maturity in soybean using next-generation sequencing-based analysis. Front. Plant Sci. 2018, 9, 995. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Duan, W.; Huang, Z.; Liu, G.; Wu, P.; Liu, T.; Li, Y.; Hou, X. Comprehensive analysis of the flowering genes in chinese cabbage and examination of evolutionary pattern of co-like genes in plant kingdom. Sci. Rep. 2015, 5, 14631. [Google Scholar] [CrossRef] [PubMed]
- Shea, D.J.; Nishida, N.; Takada, S.; Itabashi, E.; Takahashi, S.; Akter, A.; Miyaji, N.; Osabe, K.; Mehraj, H.; Shimizu, M.; et al. Long noncoding rnas in brassica rapa L. Following vernalization. Sci. Rep. 2019, 9, 9302. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Ahn, H.; Ryu, J.; Oh, Y.; Sivanandhan, G.; Won, K.-H.; Park, Y.; Kim, J.-S.; Lim, Y.; Kim, S.-G. Generation of early-flowering chinese cabbage (brassica rapa spp. Pekinensis) through crispr/cas9-mediated genome editing. Plant Biotech. Rep. 2019, 13, 491–499. [Google Scholar] [CrossRef]
- Murovec, J.; Gucek, K.; Bohanec, B.; Avbelj, M.; Jerala, R. DNA-free genome editing of brassica oleracea and b. Rapa protoplasts using crispr-cas9 ribonucleoprotein complexes. Front. Plant Sci. 2018, 9, 1594. [Google Scholar] [CrossRef] [PubMed]
- Olas, J.J.; Van Dingenen, J.; Abel, C.; Dzialo, M.A.; Feil, R.; Krapp, A.; Schlereth, A.; Wahl, V. Nitrate acts at the arabidopsis thaliana shoot apical meristem to regulate flowering time. New Phytol. 2019, 223, 814–827. [Google Scholar] [CrossRef]
- Xu, N.; Wang, R.; Zhao, L.; Zhang, C.; Li, Z.; Lei, Z.; Liu, F.; Guan, P.; Chu, Z.; Crawford, N.M.; et al. The arabidopsis nrg2 protein mediates nitrate signaling and interacts with and regulates key nitrate regulators. Plant Cell 2016, 28, 485–504. [Google Scholar] [CrossRef]
- Hsieh, P.H.; Kan, C.C.; Wu, H.Y.; Yang, H.C.; Hsieh, M.H. Early molecular events associated with nitrogen deficiency in rice seedling roots. Sci. Rep. 2018, 8, 12207. [Google Scholar] [CrossRef]
- Lee, M.-K.; Kim, H.-S.; Kim, J.-S.; Kim, S.-H.; Park, Y.-D. Agrobacterium-mediated transformation system for large-scale producion of transgenic chinese cabbage (brassica rapa L. ssp. Pekinensis) plants for insertional mutagenesis. J. Plant Biol. 2004, 47, 300–306. [Google Scholar] [CrossRef]
- Tao, Z.; Shen, L.; Liu, C.; Liu, L.; Yan, Y.; Yu, H. Genome-wide identification of soc1 and svp targets during the floral transition in arabidopsis. Plant J. 2012, 70, 549–561. [Google Scholar] [CrossRef]
- Castro Marin, I.; Loef, I.; Bartetzko, L.; Searle, I.; Coupland, G.; Stitt, M.; Osuna, D. Nitrate regulates floral induction in arabidopsis, acting independently of light, gibberellin and autonomous pathways. Planta 2011, 233, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Peng, M.; Rothstein, S.J. Genetic regulation by nla and microrna827 for maintaining nitrate-dependent phosphate homeostasis in arabidopsis. PLoS Genet. 2011, 7, e1002021. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, J.; Tankou, S. Nitrate reductase and growth of arabidopsis thaliana in solution culture. Plant Soil 2004, 266, 143–152. [Google Scholar] [CrossRef]
- Kolbert, Z.; Bartha, B.; Erdei, L. Exogenous auxin-induced no synthesis is nitrate reductase-associated in arabidopsis thaliana root primordia. J. Plant Physiol. 2008, 165, 967–975. [Google Scholar] [CrossRef]
- Liu, T.; Li, Y.; Ren, J.; Qian, Y.; Yang, X.; Duan, W.; Hou, X. Nitrate and nacl regulates floral induction in arabidopsis thaliana. Biologia 2013, 68, 215–222. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Wang, Z.Y.; Chen, Z.; Gu, H.; Qu, L.J. Constitutive expression of cir1 (rve2) affects several circadian-regulated processes and seed germination in arabidopsis. Plant J. 2007, 51, 512–525. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. Solexaqa: At-a-glance quality assessment of illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Cataldo, D.A.; Maroon, M.; Schrader, L.E.; Youngs, V.L. Rapid colorimetric determination of nitrate in plant tissue by nitration of salicylic acid. Commun. Soil Sci. Plant Anal. 1975, 6, 71–80. [Google Scholar] [CrossRef]
- Vendrell, P.F.; Zupancic, J. Determination of soil nitrate by transnitration of salicylic acid. Commun. Soil Sci. Plant Anal. 1990, 21, 1705–1713. [Google Scholar] [CrossRef]
- Kim, H.; Kim, S.T.; Ryu, J.; Choi, M.K.; Kweon, J.; Kang, B.C.; Ahn, H.M.; Bae, S.; Kim, J.; Kim, J.S.; et al. A simple, flexible and high-throughput cloning system for plant genome editing via crispr-cas system. J. Integr. Plant Biol. 2016, 58, 705–712. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.; Lee, A.; Jo, S.H.; Park, H.J.; Jung, W.Y.; Kim, H.-S.; Lee, H.-J.; Jeong, S.-G.; Kim, Y.-S.; Cho, H.S. Nitrogen Signaling Genes and SOC1 Determine the Flowering Time in a Reciprocal Negative Feedback Loop in Chinese Cabbage (Brassica rapa L.) Based on CRISPR/Cas9-Mediated Mutagenesis of Multiple BrSOC1 Homologs. Int. J. Mol. Sci. 2021, 22, 4631. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094631
Jung H, Lee A, Jo SH, Park HJ, Jung WY, Kim H-S, Lee H-J, Jeong S-G, Kim Y-S, Cho HS. Nitrogen Signaling Genes and SOC1 Determine the Flowering Time in a Reciprocal Negative Feedback Loop in Chinese Cabbage (Brassica rapa L.) Based on CRISPR/Cas9-Mediated Mutagenesis of Multiple BrSOC1 Homologs. International Journal of Molecular Sciences. 2021; 22(9):4631. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094631
Chicago/Turabian StyleJung, Haemyeong, Areum Lee, Seung Hee Jo, Hyun Ji Park, Won Yong Jung, Hyun-Soon Kim, Hyo-Jun Lee, Seon-Geum Jeong, Youn-Sung Kim, and Hye Sun Cho. 2021. "Nitrogen Signaling Genes and SOC1 Determine the Flowering Time in a Reciprocal Negative Feedback Loop in Chinese Cabbage (Brassica rapa L.) Based on CRISPR/Cas9-Mediated Mutagenesis of Multiple BrSOC1 Homologs" International Journal of Molecular Sciences 22, no. 9: 4631. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094631