
Tryptophan Metabolites at the Crossroad of Immune-Cell Interaction via the Aryl Hydrocarbon Receptor: Implications for Tumor Immunotherapy



Abstract
:1. Introduction
2. AhR Ligands: From the Discovery to the Main Emerging Role of Trp Derivatives in Immune Regulation
3. Binding Mode of Ligands to AhR: An Aspect Influencing Canonical Categorization of Agonism and Antagonism?
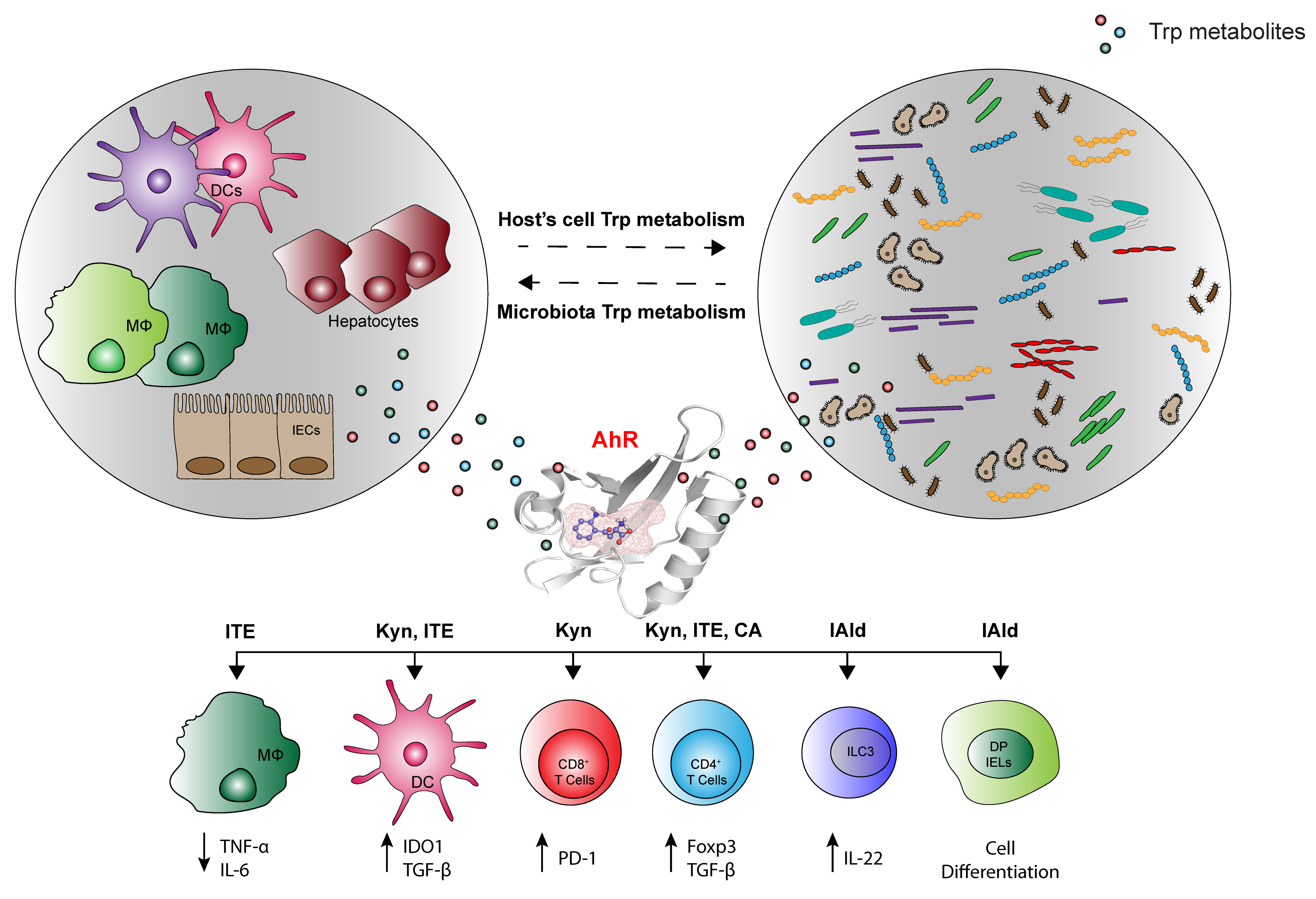
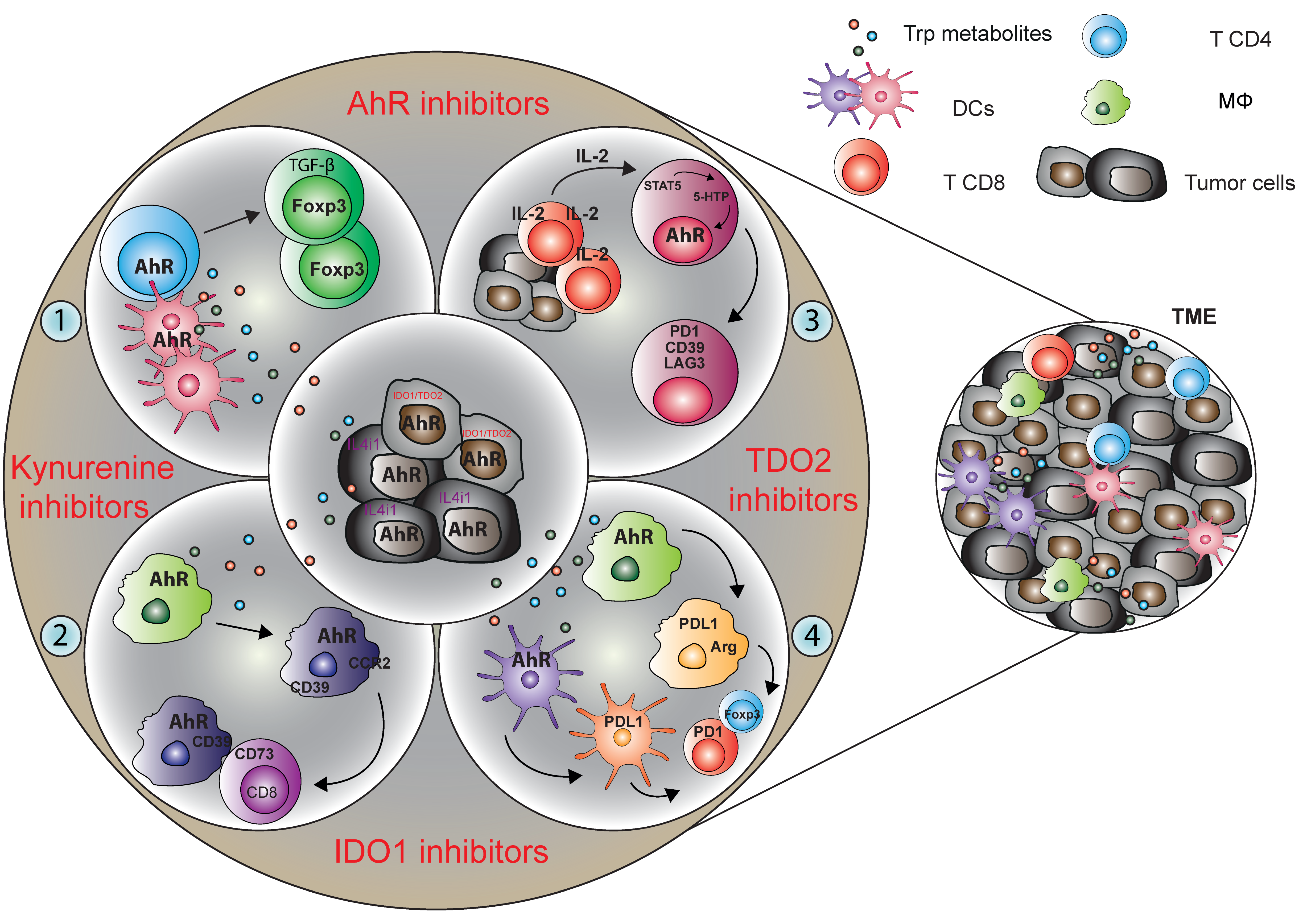
4. The Various Trp Metabolic Pathways and Metabolites in the Regulation of Immune Responses to Tumor Cells via AhR Activation
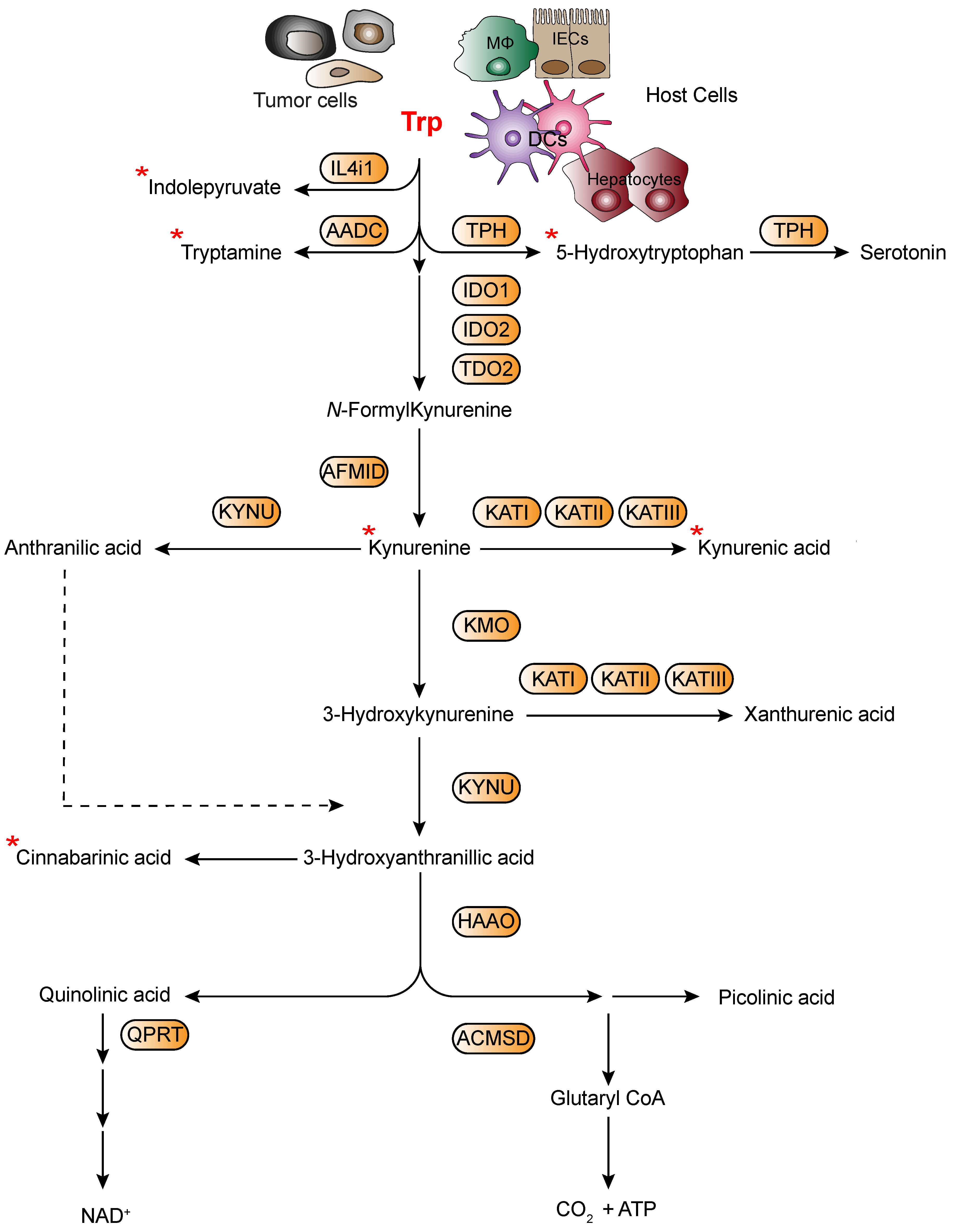
4.1. Tumor Microenvironment-Derived Trp Metabolites
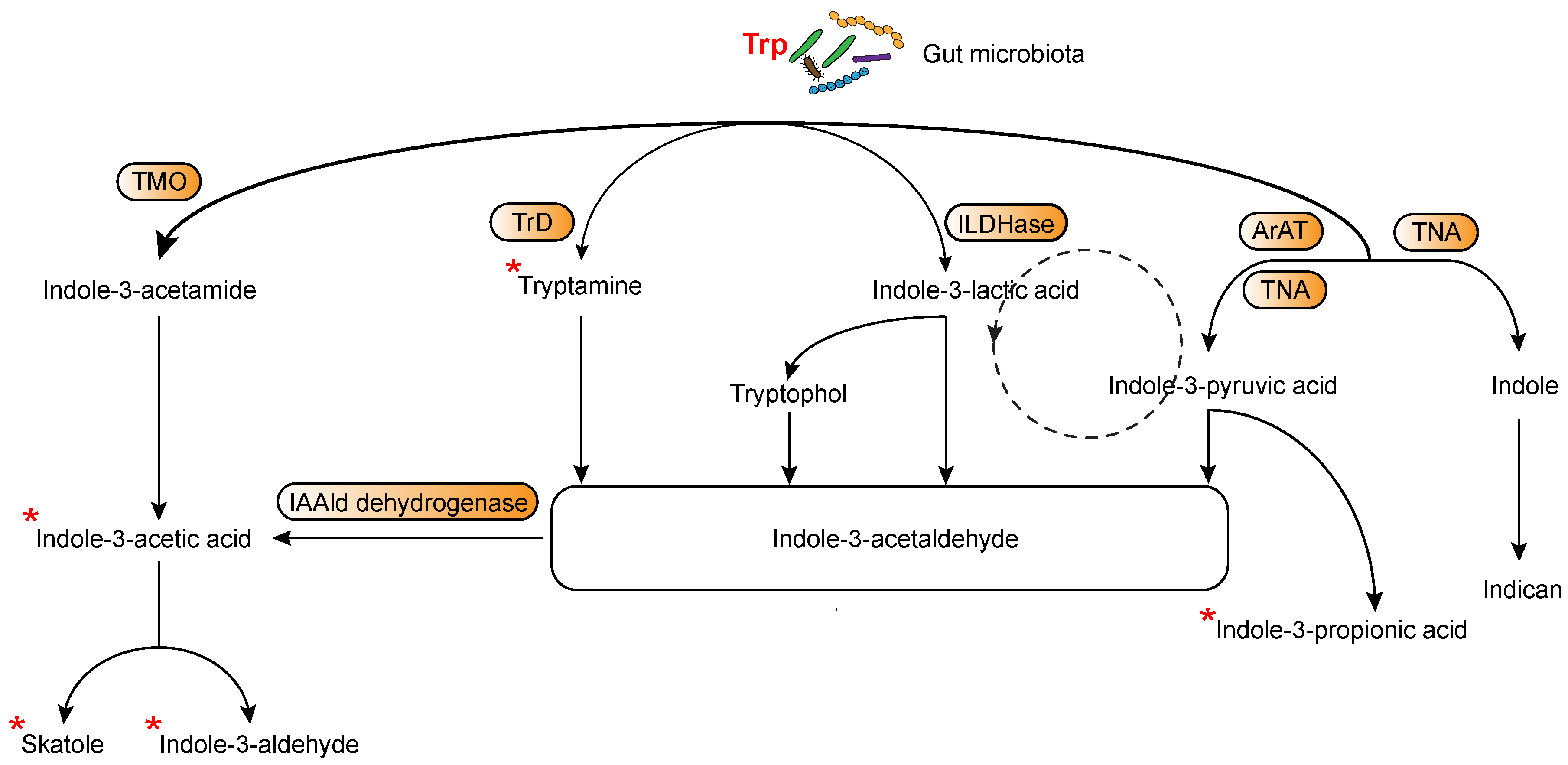
4.2. Microbially-Derived Trp Metabolites

5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Puccetti, P.; Fallarino, F.; Italiano, A.; Soubeyran, I.; MacGrogan, G.; Debled, M.; Velasco, V.; Bodet, D.; Eimer, S.; Veldhoen, M.; et al. Accumulation of an endogenous tryptophan-derived metabolite in colorectal and breast cancers. PLoS ONE 2015, 10, e0122046. [Google Scholar] [CrossRef]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Roman, A.C.; Carvajal-Gonzalez, J.M.; Merino, J.M.; Mulero-Navarro, S.; Fernandez-Salguero, P.M. The aryl hydrocarbon receptor in the crossroad of signalling networks with therapeutic value. Pharmacol. Ther. 2018, 185, 50–63. [Google Scholar] [CrossRef]
- Bock, K.W. From TCDD-mediated toxicity to searches of physiologic AHR functions. Biochem. Pharmacol. 2018, 155, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Shinde, R.; McGaha, T.L. The Aryl Hydrocarbon Receptor: Connecting Immunity to the Microenvironment. Trends Immunol. 2018, 39, 1005–1020. [Google Scholar] [CrossRef]
- Gutierrez-Vazquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Sherr, D.H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 2013, 65, 1148–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, P.; Fu, J.; Zhou, Y. The Aryl Hydrocarbon Receptor and Tumor Immunity. Front. Immunol. 2018, 9, 286. [Google Scholar] [CrossRef] [Green Version]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef]
- Wang, Z.; Snyder, M.; Kenison, J.E.; Yang, K.; Lara, B.; Lydell, E.; Bennani, K.; Novikov, O.; Federico, A.; Monti, S.; et al. How the AHR Became Important in Cancer: The Role of Chronically Active AHR in Cancer Aggression. Int. J. Mol. Sci. 2020, 22, 387. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Godschalk, R.W.L.; van Schooten, F.J. Inflammation and the chemical carcinogen benzo[a]pyrene: Partners in crime. Mutat. Res. 2017, 774, 12–24. [Google Scholar] [CrossRef]
- Donovan, M.G.; Selmin, O.I.; Romagnolo, D.F. Aryl Hydrocarbon Receptor Diet and Breast Cancer Risk. Yale J. Biol. Med. 2018, 91, 105–127. [Google Scholar] [PubMed]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolciami, D.; Gargaro, M.; Cerra, B.; Scalisi, G.; Bagnoli, L.; Servillo, G.; Fazia, M.A.D.; Puccetti, P.; Quintana, F.J.; Fallarino, F.; et al. Binding Mode and Structure-Activity Relationships of ITE as an Aryl Hydrocarbon Receptor (AhR) Agonist. ChemMedChem 2018, 13, 270–279. [Google Scholar] [CrossRef]
- Guyot, E.; Chevallier, A.; Barouki, R.; Coumoul, X. The AhR twist: Ligand-dependent AhR signaling and pharmaco-toxicological implications. Drug Discov. Today 2013, 18, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Nuti, R.; Gargaro, M.; Matino, D.; Dolciami, D.; Grohmann, U.; Puccetti, P.; Fallarino, F.; Macchiarulo, A. Ligand binding and functional selectivity of l-tryptophan metabolites at the mouse aryl hydrocarbon receptor (mAhR). J. Chem. Inf. Model. 2014, 54, 3373–3383. [Google Scholar] [CrossRef]
- Goodale, B.C.; La Du, J.; Tilton, S.C.; Sullivan, C.M.; Bisson, W.H.; Waters, K.M.; Tanguay, R.L. Ligand-Specific Transcriptional Mechanisms Underlie Aryl Hydrocarbon Receptor-Mediated Developmental Toxicity of Oxygenated PAHs. Toxicol. Sci. 2015, 147, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Poland, A.; Glover, E.; Kende, A.S. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar] [CrossRef]
- Burbach, K.M.; Poland, A.; Bradfield, C.A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. USA 1992, 89, 8185–8189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ema, M.; Sogawa, K.; Watanabe, N.; Chujoh, Y.; Matsushita, N.; Gotoh, O.; Funae, Y.; Fujii-Kuriyama, Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem. Biophys. Res. Commun. 1992, 184, 246–253. [Google Scholar] [CrossRef]
- Dolwick, K.M.; Schmidt, J.V.; Carver, L.A.; Swanson, H.I.; Bradfield, C.A. Cloning and expression of a human Ah receptor cDNA. Mol. Pharmacol. 1993, 44, 911–917. [Google Scholar] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Quintana, F.J.; Prinz, M. A gut feeling about multiple sclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, 10528–10529. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutierrez-Vazquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell. Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.A.; Macfarlane, G.T. Formation of Phenolic and Indolic Compounds by Anaerobic Bacteria in the Human Large Intestine. Microb. Ecol. 1997, 33, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Fallarino, F.; Gargaro, M.; De Luca, A.; Moretti, S.; Bartoli, A.; Romani, L. Tryptophan Feeding of the IDO1-AhR Axis in Host-Microbial Symbiosis. Front. Immunol. 2014, 5, 640. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Boadle-Biber, M.C. Regulation of serotonin synthesis. Prog. Biophys. Mol. Biol. 1993, 60, 1–15. [Google Scholar] [CrossRef]
- Yue, Y.; Huang, W.; Liang, J.; Guo, J.; Ji, J.; Yao, Y.; Zheng, M.; Cai, Z.; Lu, L.; Wang, J. IL4I1 Is a Novel Regulator of M2 Macrophage Polarization That Can Inhibit T Cell Activation via l-Tryptophan and Arginine Depletion and IL-10 Production. PLoS ONE 2015, 10, e0142979. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. Tryptophan metabolism, disposition and utilization in pregnancy. Biosci. Rep. 2015, 35, e00261. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.H.; Di Meglio, P.; Hirota, K.; Ahlfors, H.; Stockinger, B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS ONE 2013, 8, e79819. [Google Scholar] [CrossRef] [Green Version]
- Dolciami, D.; Ballarotto, M.; Gargaro, M.; López-Cara, L.C.; Fallarino, F.; Macchiarulo, A. Targeting Aryl hydrocarbon receptor for next-generation immunotherapies: Selective modulators (SAhRMs) versus rapidly metabolized ligands (RMAhRLs). Eur. J. Med. Chem. 2020, 185, 111842. [Google Scholar] [CrossRef] [PubMed]
- Romani, L.; Zelante, T.; Palmieri, M.; Napolioni, V.; Picciolini, M.; Velardi, A.; Aversa, F.; Puccetti, P. The cross-talk between opportunistic fungi and the mammalian host via microbiota’s metabolism. Semin. Immunopathol. 2015, 37, 163–171. [Google Scholar] [CrossRef]
- Vogel, C.F.; Khan, E.M.; Leung, P.S.; Gershwin, M.E.; Chang, W.L.; Wu, D.; Haarmann-Stemmann, T.; Hoffmann, A.; Denison, M.S. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: A role for nuclear factor-kappaB. J. Biol. Chem. 2014, 289, 1866–1875. [Google Scholar] [CrossRef] [Green Version]
- Di Meglio, P.; Duarte, J.H.; Ahlfors, H.; Owens, N.D.; Li, Y.; Villanova, F.; Tosi, I.; Hirota, K.; Nestle, F.O.; Mrowietz, U.; et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 2014, 40, 989–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallarino, F.; Romani, L.; Puccetti, P. AhR: Far more than an environmental sensor. Cell Cycle 2014, 13, 2645–2646. [Google Scholar] [CrossRef] [PubMed]
- Gasiewicz, T.A.; Singh, K.P.; Bennett, J.A. The Ah receptor in stem cell cycling, regulation, and quiescence. Ann. N. Y. Acad. Sci. 2014, 1310, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-Cell Transcriptome Analysis Reveals Disease-Defining T-cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2020, 181, 747. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Gellrich, F.F.; Schmitz, M.; Beissert, S.; Meier, F. Anti-PD-1 and Novel Combinations in the Treatment of Melanoma—An Update. J. Clin. Med. 2020, 9, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by l-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, J.H.; Kim, J.S.; Chon, H.J.; Kim, J.-H. A novel dual inhibitor of IDO and TDO, CMG017, potently suppresses the kynurenine pathway and overcomes resistance to immune checkpoint inhibitors. J. Clin. Oncol. 2019, 37, e14228. [Google Scholar] [CrossRef]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.C.; et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Molinier-Frenkel, V.; Prévost-Blondel, A.; Castellano, F. The IL4I1 Enzyme: A New Player in the Immunosuppressive Tumor Microenvironment. Cells 2019, 8, 757. [Google Scholar] [CrossRef] [Green Version]
- Sadik, A.; Somarribas Patterson, L.F.; Öztürk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfänder, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270.e34. [Google Scholar] [CrossRef]
- Aoki, R.; Aoki-Yoshida, A.; Suzuki, C.; Takayama, Y. Indole-3-Pyruvic Acid, an Aryl Hydrocarbon Receptor Activator, Suppresses Experimental Colitis in Mice. J. Immunol. 2018, 201, 3683–3693. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhou, N.; Zhou, L.; Wang, J.; Zhou, Y.; Zhang, T.; Fang, Y.; Deng, J.; Gao, Y.; Liang, X.; et al. IL-2 regulates tumor-reactive CD8(+) T cell exhaustion by activating the aryl hydrocarbon receptor. Nat. Immunol. 2021, 22, 358–369. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.-C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Moura-Alves, P.; Faé, K.; Houthuys, E.; Dorhoi, A.; Kreuchwig, A.; Furkert, J.; Barison, N.; Diehl, A.; Munder, A.; Constant, P.; et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature 2014, 512, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Moura-Alves, P.; Puyskens, A.; Stinn, A.; Klemm, M.; Guhlich-Bornhof, U.; Dorhoi, A.; Furkert, J.; Kreuchwig, A.; Protze, J.; Lozza, L.; et al. Host monitoring of quorum sensing during Pseudomonas aeruginosa infection. Science 2019, 366, eaaw1629. [Google Scholar] [CrossRef]
- Zhou, C.B.; Zhou, Y.L.; Fang, J.Y. Gut Microbiota in Cancer Immune Response and Immunotherapy. Trends Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with Hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [Green Version]
- Rattanaphan, P.; Mittraparp-Arthorn, P.; Srinoun, K.; Vuddhakul, V.; Tansila, N. Indole signaling decreases biofilm formation and related virulence of Listeria monocytogenes. FEMS Microbiol. Lett. 2020, 367. [Google Scholar] [CrossRef]
- Hill, D.A.; Artis, D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu. Rev. Immunol. 2010, 28, 623–667. [Google Scholar] [CrossRef] [Green Version]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Shinde, R.; Hezaveh, K.; Halaby, M.J.; Kloetgen, A.; Chakravarthy, A.; da Silva Medina, T.; Deol, R.; Manion, K.P.; Baglaenko, Y.; Eldh, M.; et al. Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat. Immunol. 2018, 19, 571–582. [Google Scholar] [CrossRef]
- Manni, G.; Mondanelli, G.; Scalisi, G.; Pallotta, M.T.; Nardi, D.; Padiglioni, E.; Romani, R.; Talesa, V.N.; Puccetti, P.; Fallarino, F.; et al. Pharmacologic Induction of Endotoxin Tolerance in Dendritic Cells by l-Kynurenine. Front. Immunol. 2020, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes-Barragan, L.; Chai, J.N.; Tianero, M.D.; Di Luccia, B.; Ahern, P.P.; Merriman, J.; Cortez, V.S.; Caparon, M.G.; Donia, M.S.; Gilfillan, S.; et al. Lactobacillus reuteri induces gut intraepithelial CD4(+)CD8αα(+) T cells. Science 2017, 357, 806–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Shen, P.; Wang, Y.; Li, Z.; Bian, J. Synthesis and in vivo antitumor evaluation of an orally active potent phosphonamidate derivative targeting IDO1/IDO2/TDO. Biochem. Pharmacol. 2019, 168, 214–223. [Google Scholar] [CrossRef]
- Dopkins, N.; Becker, W.; Miranda, K.; Walla, M.; Nagarkatti, P.; Nagarkatti, M. Tryptamine Attenuates Experimental Multiple Sclerosis Through Activation of Aryl Hydrocarbon Receptor. Front. Pharmacol. 2020, 11, 619265. [Google Scholar] [CrossRef]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Kurata, K.; Kawahara, H.; Nishimura, K.; Jisaka, M.; Yokota, K.; Shimizu, H. Skatole regulates intestinal epithelial cellular functions through activating aryl hydrocarbon receptors and p38. Biochem. Biophys. Res. Commun. 2019, 510, 649–655. [Google Scholar] [CrossRef]
- Niño-Castro, A.; Abdullah, Z.; Popov, A.; Thabet, Y.; Beyer, M.; Knolle, P.; Domann, E.; Chakraborty, T.; Schmidt, S.V.; Schultze, J.L. The IDO1-induced kynurenines play a major role in the antimicrobial effect of human myeloid cells against Listeria monocytogenes. Innate Immun. 2014, 20, 401–411. [Google Scholar] [CrossRef]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Lowe, M.M.; Mold, J.E.; Kanwar, B.; Huang, Y.; Louie, A.; Pollastri, M.P.; Wang, C.; Patel, G.; Franks, D.G.; Schlezinger, J.; et al. Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS ONE 2014, 9, e87877. [Google Scholar] [CrossRef]
- Adachi, J.; Mori, Y.; Matsui, S.; Takigami, H.; Fujino, J.; Kitagawa, H.; Miller, C.A., 3rd; Kato, T.; Saeki, K.; Matsuda, T. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J. Biol. Chem. 2001, 276, 31475–31478. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.H.; Luck, H.; Khan, S.; Schneeberger, P.H.H.; Tsai, S.; Clemente-Casares, X.; Lei, H.; Leu, Y.L.; Chan, Y.T.; Chen, H.Y.; et al. Aryl hydrocarbon receptor agonist indigo protects against obesity-related insulin resistance through modulation of intestinal and metabolic tissue immunity. Int. J. Obes. 2019, 43, 2407–2421. [Google Scholar] [CrossRef] [Green Version]
- Gillam, E.M.; Notley, L.M.; Cai, H.; De Voss, J.J.; Guengerich, F.P. Oxidation of indole by cytochrome P450 enzymes. Biochemistry 2000, 39, 13817–13824. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AhR Ligand | Source | Function | Reference |
|---|---|---|---|
| Tryptamine | Microbiota | Decreases TH17 | [79] |
| Indole-3-aldehyde | Microbiota | Increases IL-22 | [30] |
| Indole-3-acetic acid | Microbiota | Induces COX-2 | [80] |
| Indole propionic acid | Microbiota | Suppresses CNS and Gut inflammation | [28,81] |
| Indoxyl-3-sulfate | Microbiota/Host cells | Suppresses CNS Inflammation | [63] |
| Skatole | Microbiota | IEC death | [82] |
| l-kynurenine | Host cells | Antimicrobial activites, increases IDO1 and TGF-β | [76,83] |
| Kynurenic acid | Host cells | Tumor cell migration | [84] |
| Xanthurenic acid | Host cells | Tumor cell migration | [84] |
| Cinnabarinic acid | Host cells | Increases IL-22 | [85] |
| FICZ | Photo-oxidation | Enhances TH17 differentiation | [10] |
| Indirubin | Plant/Host cells | Increases CYP1A1 | [86] |
| Indigo | Plant/Host cells | Protects against HFD-induced glucose dysregulation | [86,87,88] |
| ITE | Porcine Lung | Reduces IL-6 and TNF-α | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargaro, M.; Manni, G.; Scalisi, G.; Puccetti, P.; Fallarino, F. Tryptophan Metabolites at the Crossroad of Immune-Cell Interaction via the Aryl Hydrocarbon Receptor: Implications for Tumor Immunotherapy. Int. J. Mol. Sci. 2021, 22, 4644. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094644
Gargaro M, Manni G, Scalisi G, Puccetti P, Fallarino F. Tryptophan Metabolites at the Crossroad of Immune-Cell Interaction via the Aryl Hydrocarbon Receptor: Implications for Tumor Immunotherapy. International Journal of Molecular Sciences. 2021; 22(9):4644. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094644
Chicago/Turabian StyleGargaro, Marco, Giorgia Manni, Giulia Scalisi, Paolo Puccetti, and Francesca Fallarino. 2021. "Tryptophan Metabolites at the Crossroad of Immune-Cell Interaction via the Aryl Hydrocarbon Receptor: Implications for Tumor Immunotherapy" International Journal of Molecular Sciences 22, no. 9: 4644. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094644