
Serine/Threonine Phosphatases in LTP: Two B or Not to Be the Protein Synthesis Blocker-Induced Impairment of Early Phase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
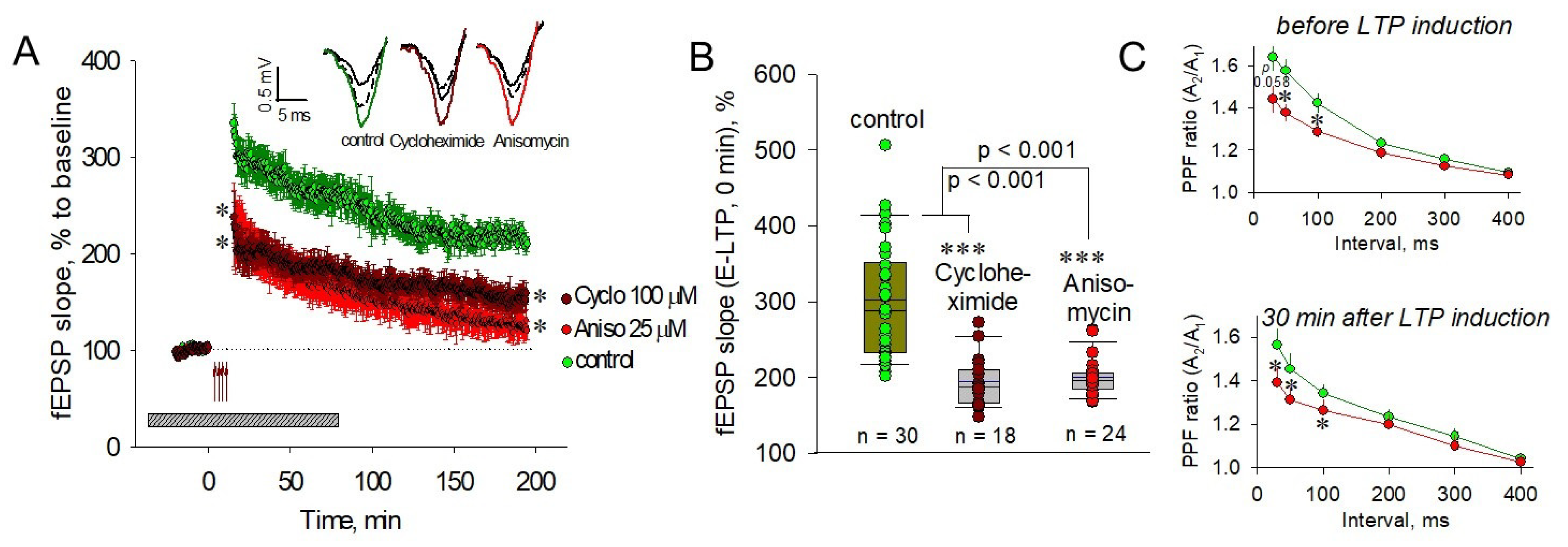
2.1. PSBs Impair Both Pre- and Postsynaptic Plasticity
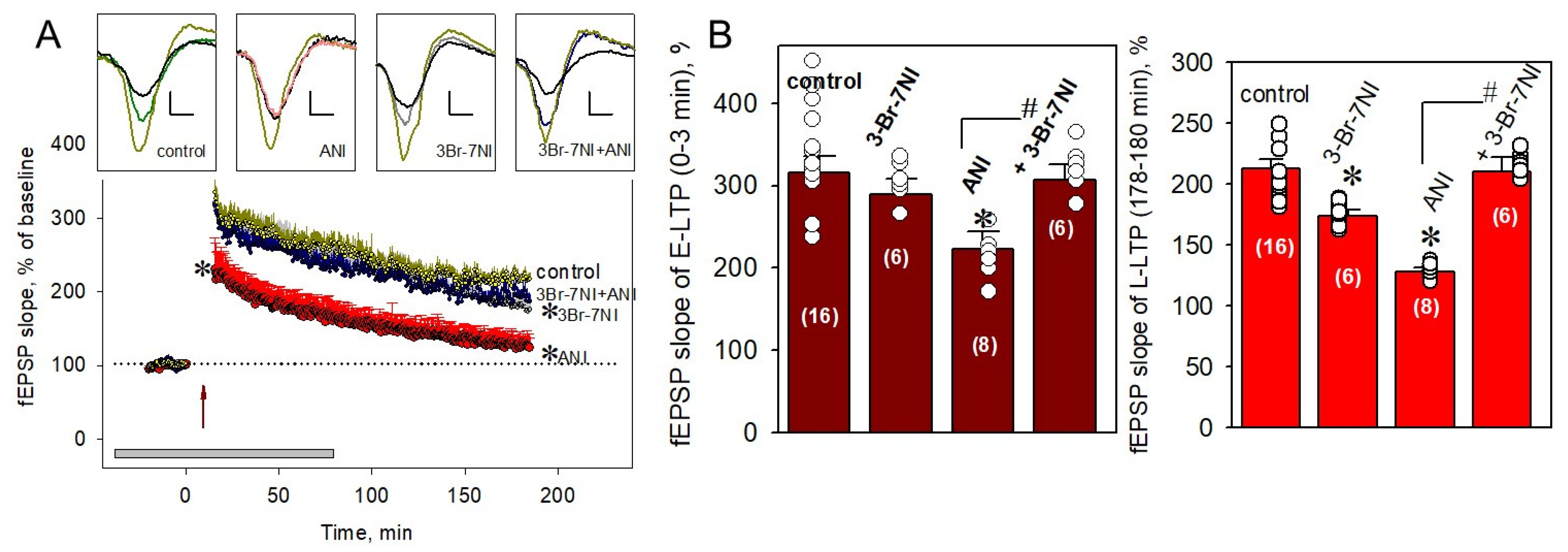
2.2. nNOS Is Involved in PSB-Induced Impairment of the E-LTP
2.3. PP2B Is Involved in the PSB-Induced Impairment of E-LTP in CA1 Neurons
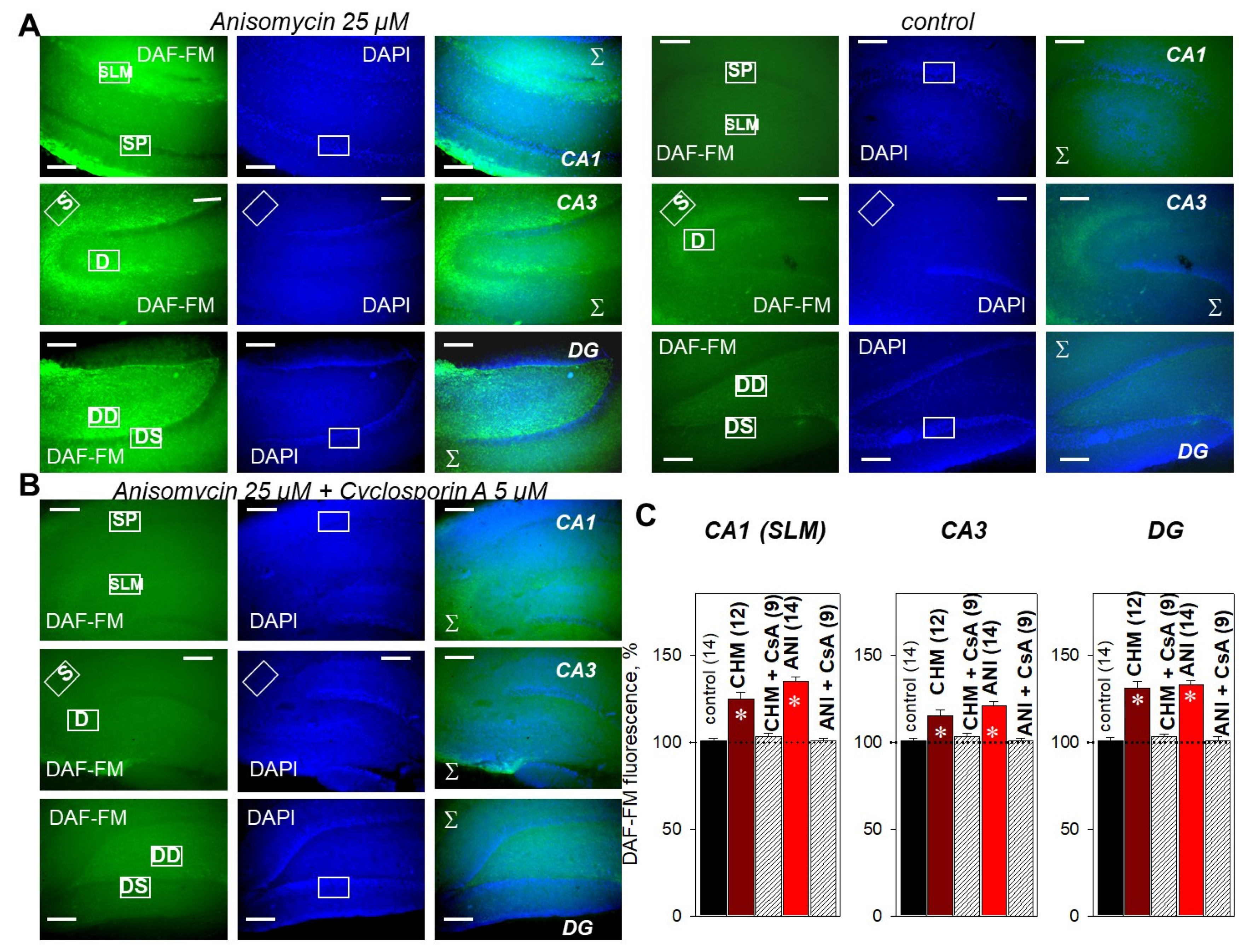
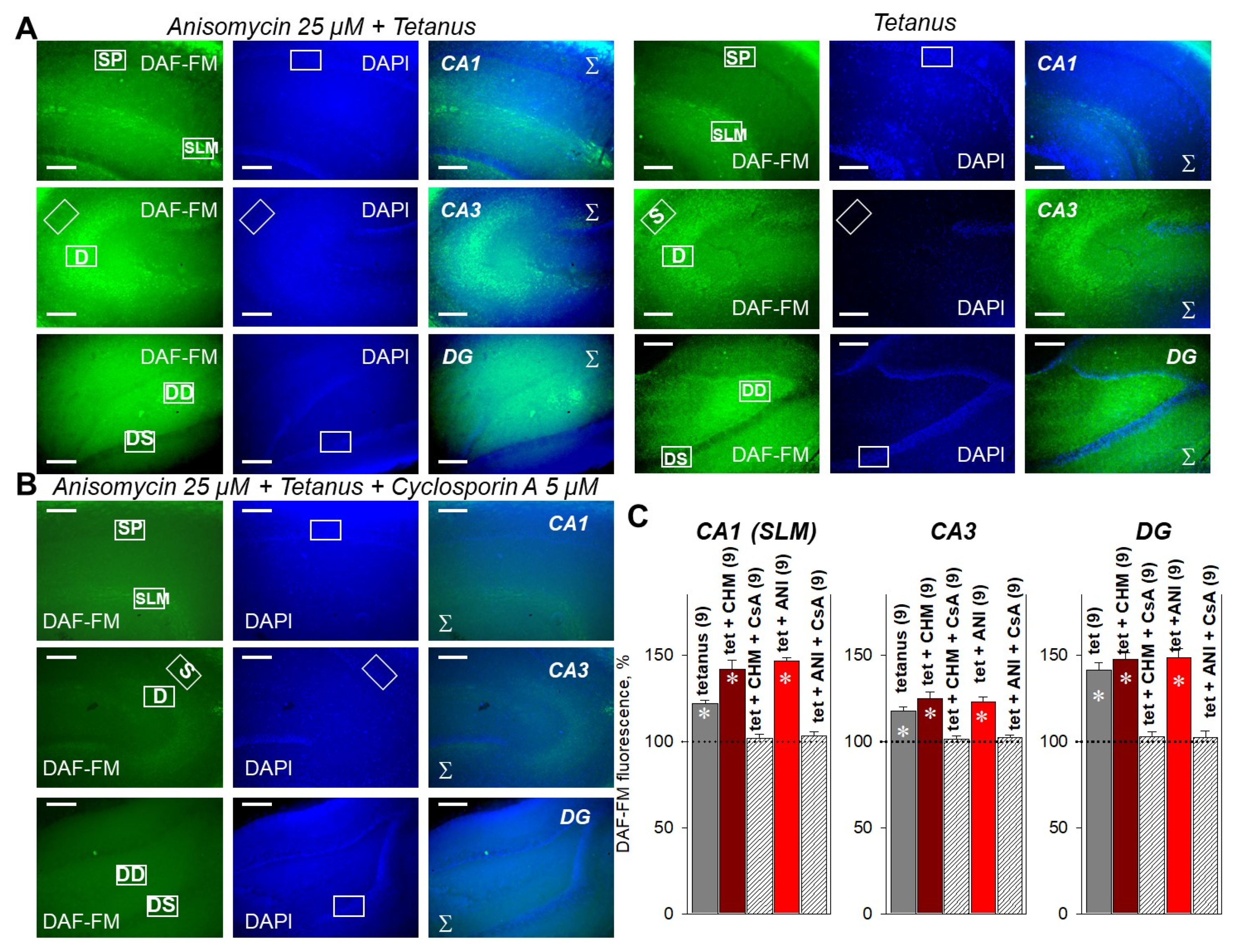
2.4. PP2B Blocker Prevents PSB-Induced NO Production in the Hippocampus
2.5. Influence of PP2B on Short-Term Plasticity
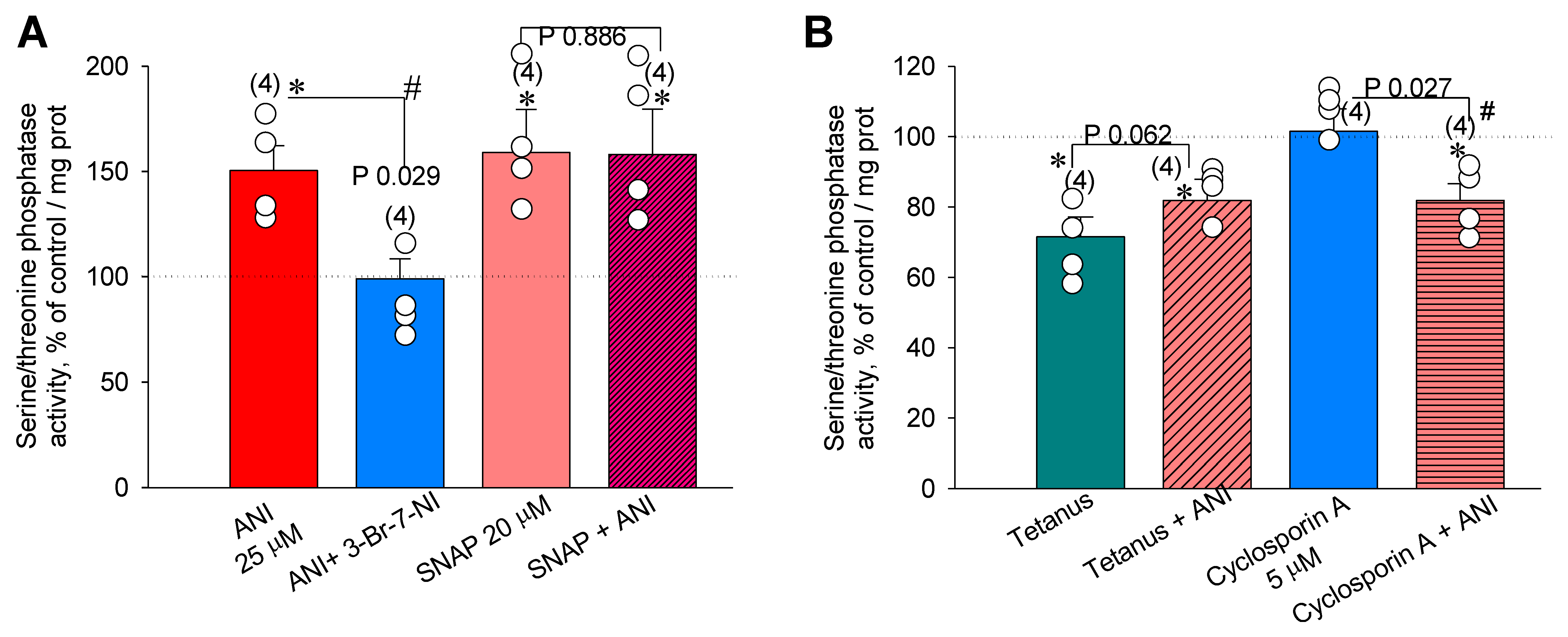
2.6. Direct Measurements of Serine/Threonine Phosphatase Activity in the Hippocampus
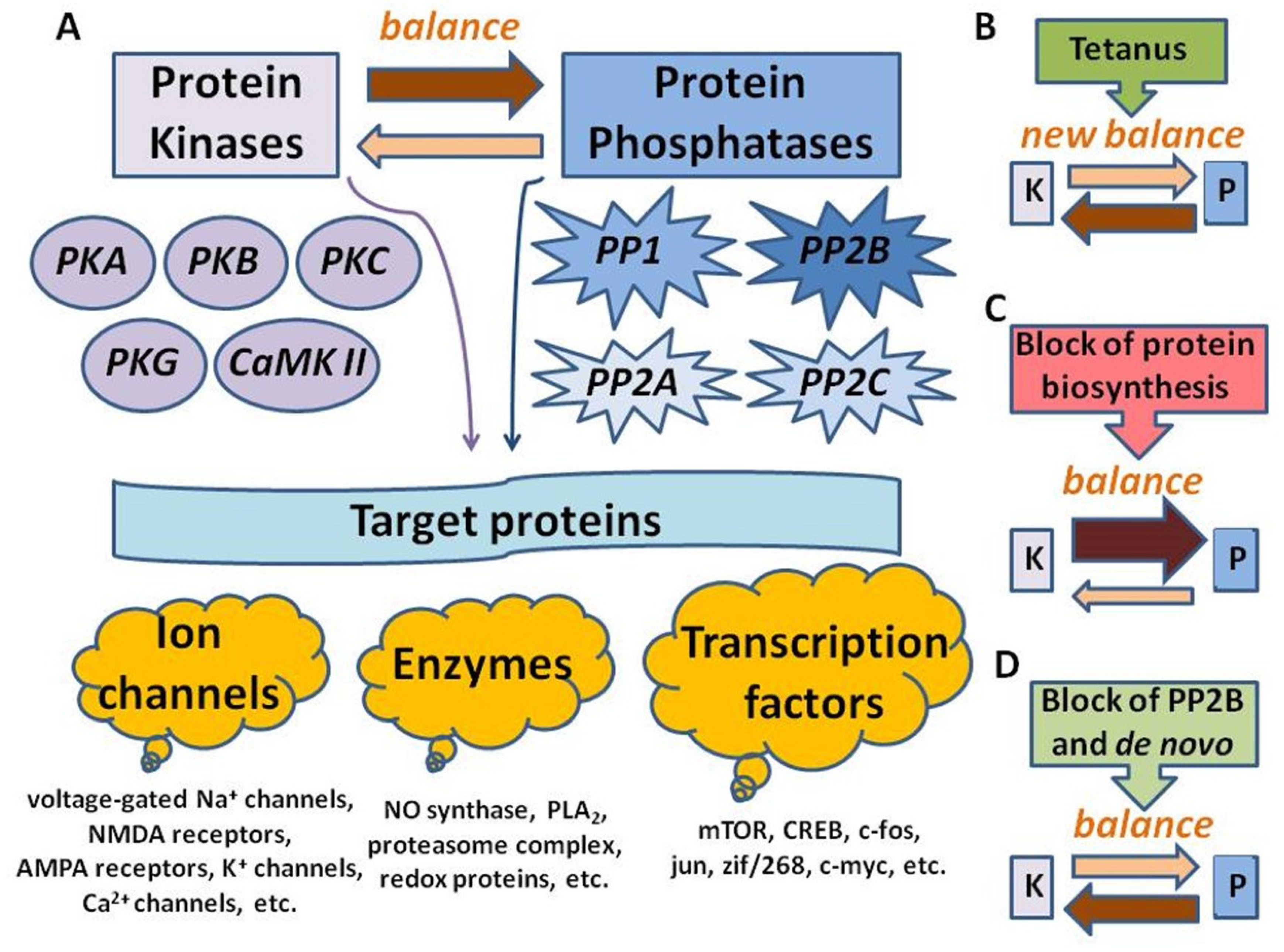
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Slice Preparation
5.2. Electrophysiology
5.3. NO Imaging in Hippocampal Slices
5.4. Phosphatase Assay
5.5. Drugs
5.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LTP | long-term potentiation |
| E-LTP | early phase of LTP |
| L-LTP | late phase of LTP |
| LTD | long-term depression |
| ACSF | artificial cerebrospinal fluid solution |
| fEPSP | field excitatory postsynaptic potential |
| PPF | paired-pulse facilitation |
| ISI | interstimulus interval |
| DAF-FM | 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate |
| 3-Br-7NI | 3-bromo-7-nitroindazole |
| NMDA | N-methyl D-aspartate |
| SNAP | (S)-nitroso-N-acetylpenicillamine |
| NO | nitric oxide |
| NOS | NO-synthase |
| nNOS | neuronal isoform of NOS |
| PP | protein phosphatase |
| PP2B | protein phosphatase 2B |
| cGMP | cyclic guanosine monophosphate |
| GC | guanylyl cyclase |
| PKG | cGMP-dependent protein kinase G |
| CaM | calmodulin |
| CaMK II | calcium/calmodulin dependent kinase II |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| SR | stratum radiatum |
| SLM | stratum lacunosum-moleculare |
| SP | stratum pyramidale |
| DG | dentate gyrus |
References
- Diamond, D.M.; Park, C.R.; Campbell, A.M.; Woodson, J.C. Competitive interactions between endogenous LTD and LTP in the hippocampus underlie the storage of emotional memories and stress-induced amnesia. Hippocampus 2015, 15, 1006–1025. [Google Scholar] [CrossRef]
- Duguid, I.; Sjöström, P.J. Novel presynaptic mechanisms for coincidence detection in synaptic plasticity. Curr. Opin. Neurobiol. 2006, 16, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.H. Cerebellar learning mechanisms. Brain Res. 2015, 1621, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Behr, J.; Wozny, C.; Fidzinski, P.; Schmitz, D. Synaptic plasticity in the subiculum. Prog. Neurobiol. 2009, 89, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Balaban, P.M.; Roshchin, M.V.; Korshunova, T.A. Two-faced nitric oxide is necessary for both erasure and consolidation of memory. Zh. Vyss. Nerv. Deiat. Im I P Pavlov 2011, 61, 274–280. [Google Scholar]
- Habib, D.; Dringenberg, H.C. Low-frequency-induced synaptic potentiation: A paradigm shift in the field of memory-related plasticity mechanisms? Hippocampus 2009, 20, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.P.; Lømo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar] [CrossRef] [PubMed]
- Lømo, T. The discovery of long-term potentiation. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 617–620. [Google Scholar] [CrossRef]
- Fonseca, R. Activity-dependent actin dynamics are required for the maintenance of long-term plasticity and for synaptic capture. Eur. J. Neurosci. 2012, 35, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Pang, P.T.; Lu, B. Regulation of late-phase LTP and long-term memory in normal and aging hippocampus: Role of secreted proteins tPA and BDNF. Ageing. Res. Rev. 2004, 3, 407–430. [Google Scholar] [CrossRef] [Green Version]
- Dolphin, A.C.; Errington, M.L.; Bliss, T.V. Long-term potentiation of the perforant path in vivo is associated with increased glutamate release. Nature 1982, 297, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, P.; Cataldi, M.; Magi, S.; Lariccia, V.; Arcangeli, S.; Amoroso, S. Role of the mitochondrial sodium/calcium exchanger in neuronal physiology and in the pathogenesis of neurological diseases. Prog. Neurobiol. 2009, 87, 58–79. [Google Scholar] [CrossRef]
- Sah, P.; Faber, E.L. Channels underlying neuronal calcium-activated potassium currents. Prog. Neurobiol. 2002, 66, 345–353. [Google Scholar] [CrossRef]
- Pittenger, C.; Kandel, E. A genetic switch for long-term memory. Comptes Rendus de l’Académie des Sciences-Series III-Sciences de la Vie 1998, 321, 91–96. [Google Scholar] [CrossRef]
- Lu, Y.F.; Hawkins, R.D. Ryanodine receptors contribute to cGMP-induced late-phase LTP and CREB phosphorylation in the hippocampus. J. Neurophysiol. 2002, 88, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Asrar, S.; Zhou, Z.; Ren, W.; Jia, Z. Ca2+ Permeable AMPA Receptor Induced Long-Term Potentiation Requires PI3/MAP Kinases but Not Ca/CaM-Dependent Kinase II. PLoS ONE 2009, 4, e4339. [Google Scholar] [CrossRef] [PubMed]
- Balaban, P.M.; Roshchin, M.; Timoshenko, A.K.; Zuzina, A.B.; Lemak, M.; Ierusalimsky, V.N.; Aseyev, N.A.; Malyshev, A.Y. Homolog of protein kinase Mζ maintains context aversive memory and underlying long-term facilitation in terrestrial snail Helix. Front. Cell. Neurosci. 2015, 9, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisman, J.E.; Zhabotinsky, A.M. A model of synaptic memory: A CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 2001, 31, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Hell, J.W. How Ca2+-permeable AMPA receptors, the kinase PKA, and the phosphatase PP2B are intertwined in synaptic LTP and LTD. Sci. Signal. 2016, 9, pe2. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, N.; Nozad, A.; Milan, H.S.; Amani, M. Okadaic acid attenuates short-term and long-term synaptic plasticity of hippocampal dentate gyrus neurons in rats. Neurobiol. Learn. Mem. 2019, 158, 24–31. [Google Scholar] [CrossRef]
- Abraham, W.C.; Dragunow, M.; Tate, W.P. The role of immediate early genes in the stabilization of long-term potentiation. Mol. Neurobiol. 1991, 5, 297–314. [Google Scholar] [CrossRef]
- Barco, A.; Alarcon, J.M.; Kandel, E.R. Expression of Constitutively Active CREB Protein Facilitates the Late Phase of Long-Term Potentiation by Enhancing Synaptic Capture. Cell 2002, 108, 689–703. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, F.; Podda, M.V.; Riccardi, E.; Frisullo, G.; Dileone, M.; Profice, P.; Pilato, F.; Di Lazzaro, V.; Grassi, C. Modulation of LTP at rat hippocampal CA3-CA1 synapses by direct current stimulation. J. Neurophysiol. 2012, 107, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Ramakers, G.M.; Biessels, G.-J.; Gispen, W.H. Effects of a phorbol ester and cyclosporin A on hippocampal synaptic plasticity in streptozotocin-induced-diabetic rats: Reduced sensitivity to phorbol esters. Neurosci. Lett. 2003, 339, 45–48. [Google Scholar] [CrossRef]
- Lin, C.H.; Lee, C.C.; Gean, P.W. Involvement of a calcineurin cascade in amygdala depotentiation and quenching of fear memory. Mol. Pharmacol. 2003, 63, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerges, N.Z.; Aleisa, A.M.; Schwarz, L.A.; Alkadhi, K.A. Chronic psychosocial stress decreases calcineurin in the dentate gyrus: A possible mechanism for preservation of early ltp. Neuroscience 2003, 117, 869–874. [Google Scholar] [CrossRef]
- Malleret, G.; Haditsch, U.; Genoux, D.; Jones, M.W.; Bliss, T.V.; Vanhoose, A.M.; Weitlauf, C.; Kandel, E.R.; Winder, D.G.; Mansuy, I.M. Inducible and Reversible Enhancement of Learning, Memory, and Long-Term Potentiation by Genetic Inhibition of Calcineurin. Cell 2001, 104, 675–686. [Google Scholar] [CrossRef]
- Terashima, A.; Taniguchi, T.; Nakai, M.; Yasuda, M.; Kawamata, T.; Tanaka, C. Rapamycin and FK506 induce long-term poten-tiation by pairing stimulation via an intracellular Ca2+ signaling mechanism in rat hippocampal CA1 neurons. Neuropharmacology 2000, 39, 1920–1928. [Google Scholar] [CrossRef]
- Zeng, H.; Chattarji, S.; Barbarosie, M.; Rondi-Reig, L.; Philpot, B.D.; Miyakawa, T.; Bear, M.F.; Tonegawa, S. Forebrain-Specific Calcineurin Knockout Selectively Impairs Bidirectional Synaptic Plasticity and Working/Episodic-like Memory. Cell 2001, 107, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Mansuy, I.M.; Mayford, M.; Jacob, B.; Kandel, E.R.; Bach, M.E. Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memory. Cell 1998, 92, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, A.V.; Bal, N.V.; Balaban, P.M. LTP suppression by protein synthesis inhibitors is NO-dependent. Neuropharmacology 2019, 146, 276–288. [Google Scholar] [CrossRef]
- Maltsev, A.V.; Balaban, P.M. PP1/PP2A inhibition-induced metaplasticity in protein synthesis blocker-treated hippocampal slices: LTP and LTD, or There and Back again. Biochem. Biophys. Res. Commun. 2021, 558, 64–70. [Google Scholar] [CrossRef]
- Okada, D. Tetrahydrobiopterin-dependent stabilization of neuronal nitric oxide synthase dimer reduces susceptibility to phosphorylation by protein kinase C. in vitro. FEBS Lett. 1998, 434, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Makino, K.; Osuka, K.; Watanabe, Y.; Usuda, N.; Hara, M.; Aoyama, M.; Takayasu, M.; Wakabayashi, T. Increased ICP promotes CaMKII-mediated phosphorylation of neuronal NOS at Ser⁸⁴⁷ in the hippocampus immediately after subarachnoid hemorrhage. Brain Res. 2015, 1616, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Pastor, A.; Llansola, M.; Felipo, V. Extracellular protein kinase A modulates intracellular calcium/calmodulin-dependent protein kinase II, nitric oxide synthase, and the glutamate-nitric oxide-cGMP pathway in cerebellum. Differential effects in hyperammonemia. ACS Chem. Neurosci. 2016, 7, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Osuka, K.; Watanabe, Y.; Usuda, N.; Atsuzawa, K.; Takayasu, M. Phosphorylation of neuronal nitric oxide synthase at Ser1412 in the dentate gyrus of rat brain after transient forebrain ischemia. Neurochem. Int. 2013, 63, 269–274. [Google Scholar] [CrossRef]
- Kotake, M.; Sato, K.; Mogi, C.; Tobo, M.; Aoki, H.; Ishizuka, T.; Sunaga, N.; Imai, H.; Kaira, K.; Hisada, T.; et al. Acidic pH increases cGMP accumulation through the OGR1/phospholipase C/Ca2+/neuronal NOS pathway in N1E-115 neuronal cells. Cell. Signal. 2014, 26, 2326–2332. [Google Scholar] [CrossRef]
- Kar, R.; Kellogg, D.L., 3rd; Roman, L.J. Oxidative stress induces phosphorylation of neuronal NOS in cardiomyocytes through AMP-activated protein kinase (AMPK). Biochem. Biophys. Res. Commun. 2015, 459, 393–397. [Google Scholar] [CrossRef] [Green Version]
- Cossenza, M.; Socodato, R.; Mejía-García, T.A.; Domith, I.; Portugal, C.C.; Gladulich, L.F.; Duarte-Silva, A.T.; Khatri, L.; Antoine, S.; Hofmann, F.; et al. Protein synthesis inhibition promotes nitric oxide generation and activation of CGKII-dependent downstream signaling pathways in the retina. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2020, 1867, 118732. [Google Scholar] [CrossRef]
- Dawson, T.M.; Steiner, J.P.; Dawson, V.L.; Dinerman, J.L.; Uhl, G.R.; Snyder, S.H. Immunosuppressant FK506 enhances phos-phorylation of nitric oxide synthase and protects against glutamate neurotoxicity. Proc. Natl. Acad. Sci. USA 1993, 90, 9808–9812. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Krukoff, T.L. Adrenomedullin stimulates nitric oxide production from primary rat hypothalamic neurons: Roles of calcium and phosphatases. Mol. Pharmacol. 2007, 72, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Li, C.; Zhang, F.; Han, N.; Zhang, G.-Y.; Yu, H.-M. Neuroprotection of γ-aminobutyric acid receptor agonists via enhancing neuronal nitric oxide synthase (Ser847) phosphorylation through increased neuronal nitric oxide synthase and PSD95 interaction and inhibited protein phosphatase activity in cerebral ischemia. J. Neurosci. Res. 2008, 86, 2973–2983. [Google Scholar] [CrossRef]
- Park, S.Y.; Kang, M.J.; Han, J.S. Neuronal NOS induces neuronal differentiation through a PKCα-dependent GSK3β inactivation pathway in hippocampal neural progenitor cells. Mol. Neurobiol. 2017, 54, 5646–5656. [Google Scholar] [CrossRef]
- Itakura, M.; Watanabe, I.; Sugaya, T.; Takahashi, M. Direct association of the unique C-terminal tail of transmembrane AMPA receptor regulatory protein γ-8 with calcineurin. FEBS J. 2014, 281, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.B. Signal-Processing Machines at the Postsynaptic Density. Sci. 2000, 290, 750–754. [Google Scholar] [CrossRef]
- Watanabe, Y.; Song, T.; Sugimoto, K.; Horii, M.; Araki, N.; Tokumitsu, H.; Tezuka, T.; Yamamoto, T.; Tokuda, M. Postsynaptic density-95 promotes calcium/calmodulin-dependent protein kinase II-mediated Ser847 phosphorylation of neuronal nitric oxide synthase. Biochem. J. 2003, 372, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Woolfrey, K.M.; Dell’Acqua, M.L. Coordination of Protein Phosphorylation and Dephosphorylation in Synaptic Plasticity. J. Biol. Chem. 2015, 290, 28604–28612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opazo, P.; Sainlos, M.; Choquet, D. Regulation of AMPA receptor surface diffusion by PSD-95 slots. Curr. Opin. Neurobiol. 2012, 22, 453–460. [Google Scholar] [CrossRef]
- Lisman, J. Glutamatergic synapses are structurally and biochemically complex because of multiple plasticity processes: Long-term potentiation, long-term depression, short-term potentiation and scaling. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160260. [Google Scholar] [CrossRef] [PubMed]
- Frey, U.; Krug, M.; Reymann, K.G.; Matthies, H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988, 452, 57–65. [Google Scholar] [CrossRef]
- Mullany, P.; Lynch, M. Changes in protein synthesis and synthesis of the synaptic vesicle protein, synaptophysin, in entorhinal cortex following induction of long-term potentiation in dentate gyrus: An age-related study in the rat. Neuropharmacology 1997, 36, 973–980. [Google Scholar] [CrossRef]
- Hu, N.-W.; Zhang, H.-M.; Hu, X.-D.; Li, M.-T.; Zhang, T.; Zhou, L.-J.; Liu, X.-G. Protein Synthesis Inhibition Blocks the Late-Phase LTP of C-Fiber Evoked Field Potentials in Rat Spinal Dorsal Horn. J. Neurophysiol. 2003, 89, 2354–2359. [Google Scholar] [CrossRef]
- Fonseca, R.; Nägerl, U.V.; Bonhoeffer, T. Neuronal activity determines the protein synthesis dependence of long-term potentiation. Nat. Neurosci. 2006, 9, 478–480. [Google Scholar] [CrossRef]
- Mansuy, I.M.; Shenolikar, S. Protein serine/threonine phosphatases in neuronal plasticity and disorders of learning and memory. Trends Neurosci. 2006, 29, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L.L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; E Costello, C.; Cravatt, B.F.; Fenselau, C.; A Garcia, B.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Coba, M.P. Regulatory mechanisms in postsynaptic phosphorylation networks. Curr. Opin. Struct. Biol. 2019, 54, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.O.; Yu, L.; Coba, M.P.; Husi, H.; Campuzano, I.; Blackstock, W.P.; Choudhary, J.S.; Grant, S.G.N. Proteomic Analysis of in Vivo Phosphorylated Synaptic Proteins. J. Biol. Chem. 2005, 280, 5972–5982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.J.; Sharma, K.; Zangrandi, L.; Chen, C.; Humphrey, S.J.; Chiu, Y.-T.; Spetea, M.; Liu-Chen, L.-Y.; Schwarzer, C.; Mann, M. In vivo brain GPCR signaling elucidated by phosphoproteomics. Science 2018, 360, eaao4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belmeguenai, A.; Hansel, C. A Role for Protein Phosphatases 1, 2A, and 2B in Cerebellar Long-Term Potentiation. J. Neurosci. 2005, 25, 10768–10772. [Google Scholar] [CrossRef] [Green Version]
- Onuma, H.; Lu, Y.-F.; Tomizawa, K.; Moriwaki, A.; Tokuda, M.; Hatase, O.; Matsui, H. A calcineurin inhibitor, FK506, blocks voltage-gated calcium channel-dependent LTP in the hippocampus. Neurosci. Res. 1998, 30, 313–319. [Google Scholar] [CrossRef]
- Schonewille, M.; Belmeguenai, A.; Koekkoek, S.; Houtman, S.; Boele, H.; van Beugen, B.; Gao, Z.; Badura, A.; Ohtsuki, G.; Amerika, W.; et al. Purkinje Cell-Specific Knockout of the Protein Phosphatase PP2B Impairs Potentiation and Cerebellar Motor Learning. Neuron 2010, 67, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Zorumski, C.F.; Izumi, Y. Nitric oxide and hippocampal synaptic plasticity. Biochem. Pharmacol. 1993, 46, 777–785. [Google Scholar] [CrossRef]
- Ko, G.Y.; Kelly, P.T. Nitric oxide acts as a postsynaptic signaling molecule in calcium/calmodulin-induced synaptic potentiation in hippocampal CA1 pyramidal neurons. J. Neurosci. 1999, 19, 6784–6794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossenza, M.; Cadilhe, D.V.; Coutinho, R.N.; Paes-De-Carvalho, R. Inhibition of protein synthesis by activation of NMDA receptors in cultured retinal cells: A new mechanism for the regulation of nitric oxide production. J. Neurochem. 2006, 97, 1481–1493. [Google Scholar] [CrossRef]
- Hardingham, N.; Dachtler, J.; Fox, K. The role of nitric oxide in presynaptic plasticity and homeostasis. Front. Cell. Neurosci. 2013, 7, 190. [Google Scholar] [CrossRef] [Green Version]
- Bal, N.V.; Rysakova, M.P.; Vinarskaya, A.K.; Ivanova, V.; Zuzina, A.B.; Balaban, P.M. Cued memory reconsolidation in rats requires nitric oxide. Eur. J. Neurosci. 2017, 45, 643–647. [Google Scholar] [CrossRef]
- Solà, C.; Tusell, J.M.; Serratosa, J. Comparative study of the distribution of calmodulin kinase II and calcineurin in the mouse brain. J. Neurosci. Res. 1999, 57, 651–662. [Google Scholar] [CrossRef]
- Foster, T.C.; Sharrow, K.M.; Masse, J.R.; Norris, C.M.; Kumar, A. Calcineurin links Ca2+ dysregulation with brain aging. J. Neurosci. 2001, 21, 4066–4073. [Google Scholar] [CrossRef] [Green Version]
- Giri, P.R.; Marietta, C.A.; Higuchi, S.; Kincaid, R.L. Molecular and Phylogenetic Analysis of Calmodulin-Dependent Protein Phosphatase (Calcineurin) Catalytic Subunit Genes. DNA Cell Biol. 1992, 11, 415–424. [Google Scholar] [CrossRef]
- Crabtree, G.R. Generic Signals and Specific Outcomes: Signaling through Ca2+, Calcineurin, and NF-AT. Cell 1999, 96, 611–614. [Google Scholar] [CrossRef] [Green Version]
- Baumgärtel, K.; Mansuy, I.M. Neural functions of calcineurin in synaptic plasticity and memory. Learn. Mem. 2012, 19, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Kipanyula, M.J.; Kimaro, W.H.; Etet, P.F.S. The Emerging Roles of the Calcineurin-Nuclear Factor of Activated T-Lymphocytes Pathway in Nervous System Functions and Diseases. J. Aging Res. 2016, 2016, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Loh, C.; Shaw, K.T.; Carew, J.; Viola, J.P.; Luo, C.; Perrino, B.A.; Rao, A. Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J. Biol. Chem. 1996, 271, 10884–10891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graef, I.A.; Wang, F.; Charron, F.; Chen, L.; Neilson, J.; Tessier-Lavigne, M.; Crabtree, G.R. Neurotrophins and Netrins Require Calcineurin/NFAT Signaling to Stimulate Outgrowth of Embryonic Axons. Cell 2003, 113, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, J.-L.; Xu, X.; Zhou, X.-P.; Du, J.; Wang, X.; Zhou, Y.; Zhu, Q.; Yao, L.-L.; Wang, Y.-G.; et al. NMDA receptors inhibit axonal outgrowth by inactivating Akt and activating GSK-3β via calcineurin in cultured immature hippocampal neurons. Exp. Cell Res. 2018, 371, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Choi, H.-S.; Choi, H.J. Estrogen-related receptor gamma regulates dopaminergic neuronal phenotype by activating GSK3β/NFAT signaling in SH-SY5Y cells. J. Neurochem. 2015, 133, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Weider, M.; Starost, L.J.; Groll, K.; Küspert, M.; Sock, E.; Wedel, M.; Fröb, F.; Schmitt, C.; Baroti, T.; Hartwig, A.C.; et al. Nfat/calcineurin signaling promotes oligodendrocyte differentiation and myelination by transcription factor network tuning. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Hurwitz, M.Y.; Putkey, J.A.; Klee, C.B.; Means, A.R. Domain II of calmodulin is involved in activation of calcineurin. FEBS Lett. 1988, 238, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Kakalis, L.T.; Kennedy, M.; Sikkink, R.; Rusnak, F.; Armitage, I.M. Characterization of the calcium-binding sites of calcineurin B. FEBS Lett. 1995, 362, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Sikkink, R.; Haddy, A.; MacKelvie, S.; Mertz, P.; Litwiller, R.; Rusnak, F. Calcineurin subunit interactions: Mapping the calcineurin B binding domain on calcineurin A. Biochemistry 1995, 34, 8348–8356. [Google Scholar] [CrossRef]
- Kissinger, C.R.; Parge, H.E.; Knighton, D.R.; Lewis, C.T.; Pelletier, L.A.; Tempczyk, A.; Kalish, V.J.; Tucker, K.D.; Showalter, R.E.; Moomaw, E.W.; et al. Crystal structures of human calcineurin and the human FKBP12–FK506–calcineurin complex. Nat. Cell Biol. 1995, 378, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Norris, C.M.; Kadish, I.; Blalock, E.M.; Chen, K.C.; Thibault, V.; Porter, N.M.; Landfield, P.W.; Kraner, S.D. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J. Neurosci. 2005, 25, 4649–4658. [Google Scholar] [CrossRef] [Green Version]
- Reese, L.C.; Taglialatela, G. A Role for Calcineurin in Alzheimers Disease. Curr. Neuropharmacol. 2011, 9, 685–692. [Google Scholar] [CrossRef]
- Klee, C.B.; Crouch, T.H.; Krinks, M.H. Calcineurin: A calcium- and calmodulin-binding protein of the nervous system. Proc. Natl. Acad. Sci. USA 1979, 76, 6270–6273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bales, J.W.; Ma, X.; Yan, H.Q.; Jenkins, L.W.; Dixon, C.E. Regional calcineurin subunit B isoform expression in rat hippocampus following a traumatic brain injury. Brain Res. 2010, 1358, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth, R.D.; Dunbar, R.L.; Mermelstein, P.G. Calcineurin regulation of neuronal plasticity. Biochem. Biophys. Res. Commun. 2003, 311, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Jurado, S.; Biou, V.; Malenka, R.C. A calcineurin/AKAP complex is required for NMDA receptor–dependent long-term depression. Nat. Neurosci. 2010, 13, 1053–1055. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-R.; Hu, Y.-M.; Chen, H.; Pan, H.-L. Calcineurin inhibitor induces pain hypersensitivity by potentiating pre- and postsynaptic NMDA receptor activity in spinal cords. J. Physiol. 2013, 592, 215–227. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Ren, Y.; Yan, Z. Postsynaptic Density-95 (PSD-95) and Calcineurin Control the Sensitivity of N-Methyl-d-aspartate Receptors to Calpain Cleavage in Cortical Neurons. Mol. Pharmacol. 2008, 74, 360–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, J.L.; Scott, J.D.; Dell’Acqua, M.L. Control of Homeostatic Synaptic Plasticity by AKAP-Anchored Kinase and Phosphatase Regulation of Ca2+-Permeable AMPA Receptors. J. Neurosci. 2018, 38, 2863–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachser, R.M.; Santana, F.; Crestani, A.P.; Lunardi, P.; Pedraza, L.K.; Quillfeldt, J.A.; Hardt, O.; Alvares, L.D.O. Forgetting of long-term memory requires activation of NMDA receptors, L-type voltage-dependent Ca2+ channels, and calcineurin. Sci. Rep. 2016, 6, 22771. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-S.; Zhang, Z.; Zhao, W.-D.; Wang, D.; Luo, F.; Wu, L.-G. Calcineurin Is Universally Involved in Vesicle Endocytosis at Neuronal and Nonneuronal Secretory Cells. Cell Rep. 2014, 7, 982–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumashiro, S.; Lu, Y.-F.; Tomizawa, K.; Matsushita, M.; Wei, F.-Y.; Matsui, H. Regulation of synaptic vesicle recycling by calcineurin in different vesicle pools. Neurosci. Res. 2005, 51, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Winder, D.G.; Sweatt, J.D. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat. Rev. Neurosci. 2001, 2, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Ishii, N.; Shibasaki, F. Calcineurin and cyclophilin D are differential targets of neuroprotection by immunosuppressants CsA and FK506 in ischemic brain damage. Acta Neurochir. Suppl. 2003, 86, 105–111. [Google Scholar] [PubMed]
- Cheng, P.; Chen, K.; Yu, W.; Gao, S.; Hu, S.; Sun, X.; Huang, H. Protein phosphatase 2A (PP2A) activation promotes axonal growth and recovery in the CNS. J. Neurol. Sci. 2015, 359, 48–56. [Google Scholar] [CrossRef]
- Rosenstock, T.R.; de Brito, O.M.; Lombardi, V.; Louros, S.; Ribeiro, M.; Almeida, S.; Ferreira, I.L.; Oliveira, C.R.; Rego, A.C. FK506 ameliorates cell death features in Huntington’s disease striatal cell models. Neurochem. Int. 2011, 59, 600–609. [Google Scholar] [CrossRef]
- Maltsev, A.; Funikov, S.; Burov, A.; Spasskaya, D.; Ignatyuk, V.; Astakhova, T.; Lyupina, Y.; Deikin, A.; Tutyaeva, V.; Bal, N.; et al. Immunoproteasome Inhibitor ONX-0914 Affects Long-Term Potentiation in Murine Hippocampus. J. Neuroimmune Pharmacol. 2021, 16, 7–11. [Google Scholar] [CrossRef]
- Maltsev, A.; Evdokimovskii, E.; Kokoz, Y. Synergism of myocardial β-adrenoceptor blockade and I1-imidazoline receptor-driven signaling: Kinase-phosphatase switching. Biochem. Biophys. Res. Commun. 2019, 511, 363–368. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maltsev, A.V.; Bal, N.V.; Balaban, P.M. Serine/Threonine Phosphatases in LTP: Two B or Not to Be the Protein Synthesis Blocker-Induced Impairment of Early Phase. Int. J. Mol. Sci. 2021, 22, 4857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094857
Maltsev AV, Bal NV, Balaban PM. Serine/Threonine Phosphatases in LTP: Two B or Not to Be the Protein Synthesis Blocker-Induced Impairment of Early Phase. International Journal of Molecular Sciences. 2021; 22(9):4857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094857
Chicago/Turabian StyleMaltsev, Alexander V., Natalia V. Bal, and Pavel M. Balaban. 2021. "Serine/Threonine Phosphatases in LTP: Two B or Not to Be the Protein Synthesis Blocker-Induced Impairment of Early Phase" International Journal of Molecular Sciences 22, no. 9: 4857. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094857